

Abstract
The central problem of complex inheritance is to map oligogenes for disease susceptibility, integrating linkage and association over samples that differ in several ways. Combination of evidence over multiple samples with 1,037 families supports loci contributing to asthma susceptibility in the cytokine region on 5q [maximum logarithm of odds (lod) = 2.61 near IL-4], but no evidence for atopy. The principal problems with retrospective collaboration on linkage appear to have been solved, providing far more information than a single study. A multipoint lod table evaluated at commonly agreed reference loci is required for both collaboration and metaanalysis, but variations in ascertainment, pedigree structure, phenotype definition, and marker selection are tolerated. These methods are invariant with statistical methods that increase the power of lods and are applicable to all diseases, motivating collaboration rather than competition. In contrast to linkage, positional cloning by allelic association has yet to be extended to multiple samples, a prerequisite for efficient combination with linkage and the greatest current challenge to genetic epidemiology.
The central problem of complex inheritance is to map oligogenes for disease susceptibility. Even the largest study has low power to map genes of small effect by the complementary methods of linkage and allelic association, and a confirmatory sample is necessary even for genes of large effect. Often the significance of multiple samples is controversial. These considerations favor combination of evidence, for which there are three approaches called meta-analysis, prospective collaboration, and retrospective collaboration.
Meta-analysis typically uses published summaries and so is comprehensive. It would be the method of choice if publication did not favor positive results and the data were summarized in a way that distinguishes information, effect, and location, like lod scores for major loci. Unfortunately, studies of complex inheritance differ widely in phenotype definition, selection of families or cases and controls, choice of marker loci, and statistical analysis. The only common denominator is a nominal significance level, which is usually not specified unless it lies beyond an arbitrary threshold. Such material, although peer-reviewed, is subject to many errors and biases but can be used to define candidate regions (1).
Prospective collaboration is typified by national consortia like the Collaborative Study on the Genetics of Asthma (2). Each member of the consortium tries to follow the same protocol, including phenotype definitions, sampling scheme, and markers. This effort assures uniformity but makes no use of the many studies with different but equally defensible protocols.
Retrospective collaboration is typified by the international Consortium on Asthma Genetics (COAG). Each member of the consortium provides data published independently with no agreement about protocol. The analysis must be coherent, or else would be no more than a meta-analysis of a subset of data. Weakly parametric analysis achieves coherence by simultaneous estimation of parameters for information, effect, and location in each study. Synthesis is realized by a lod table at fixed points or by averaging estimates of location over studies, weighted by information. Both approaches give a pooled estimate, its significance, and a test of heterogeneity among studies. Because location is common to linkage and allelic association, the two are efficiently combined without prejudging which is more informative in a particular case.
Election of weakly parametric analysis has both advantages and disadvantages. Dominance, penetrance, and gene frequency are jointly summarized by a single parameter for effect. Model misspecification is much less of a problem than for strongly parametric segregation and linkage analysis where the model has low credibility if estimates of its parameters are unreliable, and multiple markers are usually not accommodated. On the other hand, weakly parametric analysis cannot estimate all genetic parameters and therefore can allow only roughly for different types of ascertainment. The optimal method must be determined by data analysis. We expect weakly parametric analysis to be preferable for coarse localization with multiple markers, but fully parametric analysis under a two-locus or mixed model is required to characterize a candidate locus.
Study Design
The cytokine region on chromosome 5q31–33 is one of the most promising candidates for asthma and atopy. IL-4 gives evidence of linkage and association to noncognate IgE (3, 4) and weak association to specific IgE (5). A locus for familial eosinophilia maps to D5S816 (6), and this assignment is supported by a sample of discordant sib pairs (7). ADRB2 may be associated with asthma and bronchial reactivity (8). IL-12B shows linkage with atopy in the mouse (9), but not in the human (10). It would not be surprising if more than one locus in the cytokine region influences susceptibility to asthma or atopy. However, the published samples are small, and phenotypes are variously defined. Significance is rarely strong and often absent.
These inconsistencies motivated the Consortium on Asthma Genetics (COAG), in which participation was solely on the basis of good will and contractual freedom, with no intentional bias for or against negative reports. The studies differ in all possible respects, giving a fair test of retrospective collaboration among a modest number of centers (Table 1). Asthma prevalence was estimated for children in the general population, by using the random sample, if one was available, or otherwise a closely related population chosen by the contributing investigator. These prevalences were used to estimate phenotype means in the population as described in Statistical Methods. The minimal criterion for affection was a positive answer to the question “ever had asthma,” but some studies used more stringent criteria for probands and sometimes secondary cases based on medical records, bronchial hyperreactivity, medication, or symptoms ascertained by questionnaire (19). Each study reported asthma and one or more other variables related to allergy (Table 2). Details are given in the cited references.
Table 1.
Populations and samples
| Center | Code | Reference | Number
of families
|
Asthmatic sib pairs | Asthma prevalence* | Number of markers | ||
|---|---|---|---|---|---|---|---|---|
| r | m | p | ||||||
| Southampton | UKS | 11 | 131 | 60 | 49 | 112 | .160 | 14 |
| Oxfordshire | UKO | 12 | — | — | 80 | 58 | .160 | 8 |
| Busselton | AuB | 12 | 80 | — | — | 4 | .070 | 10 |
| Perth | AuP | 13 | 98 | — | 25 | 77 | .240 | 2 |
| Finland | Fin | 14 | — | 39 | 15 | 46 | .070 | 17 |
| Munchen | GerM | 15 | — | 97 | — | 109 | .075 | 22 |
| Freiburg | GerF | 16 | 72# | — | — | 18 | .075 | 5 |
| Barbados | Bar | 17 | — | 33 | — | 63 | .165 | 5 |
| U.S. African-American | USAA | 2 | — | 114 | — | 195 | .300 | 18 |
| U.S. Caucasian | USC | 2 | — | 112 | — | 183 | .200 | 19 |
| Fujian | China | 18 | — | 9 | 23 | 32 | .050 | 3 |
r, random nuclear; m, multiplex asthma pedigree; p, nuclear with asthma proband; #, selection on atopic proband.
In general population, estimated by each center from its random sample or secondary sources.
Table 2.
Variables used in this study by population
| Population | Asthma | Bronchial response | Wheeze history | FEV1 | Eosinophils | Atopy history | Total IgE | Specific IgE | Skin prick |
|---|---|---|---|---|---|---|---|---|---|
| UKS | + | + | + | + | − | + | + | + | + |
| UKO | + | − | − | − | − | − | + | + | + |
| AuB | + | + | − | + | + | − | + | + | + |
| AuP | + | + | + | + | + | − | + | + | − |
| Fin | + | − | − | − | − | − | + | − | − |
| GerM | + | + | + | − | − | − | + | + | + |
| GerF | + | − | − | − | − | − | + | + | + |
| Bar | + | − | − | − | − | + | + | + | − |
| USAA | + | − | − | − | − | − | + | − | − |
| USC | + | − | − | − | − | − | + | − | − |
| China | + | + | − | − | − | − | + | − | + |
Not all typing errors are detected by apparent parentage exclusion. To accommodate residual errors, we constructed a map for each sample, constraining order but not distance. All lods are multipoint (20), and so they could be evaluated at an interpolated location with no marker in that sample. Eight locations in the cytokine cluster were recognized, subequally spaced, corresponding to markers in the location database LDB (21) and termed reference loci (Table 3). A multipoint lod table evaluated at commonly agreed reference loci is required by the methods of this paper, whereas variations in ascertainment, pedigree structure, phenotype definition, and marker selection are tolerated.
Table 3.
Marker loci
| Locus | Mb | cM (M) | cM (F) | cM (ave) | band | UKS | UKO | AuB | AuP | Fin | GerM | GerF | Bar | USAA | USC | China |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ptr | 0.000 | 0.00 | 0.00 | 0.00 | p15.33 | − | − | − | − | − | − | − | − | − | − | − |
| D5S2501 | 122.058 | 109.49 | 163.26 | 136.38 | q22.3 | − | − | − | − | − | − | − | − | + | + | − |
| *D5S421 | 124.957 | 110.94 | 169.91 | 140.43 | q23.1 | + | − | − | − | − | + | − | − | − | − | − |
| D5S404 | 133.190 | 114.60 | 180.24 | 147.42 | q23.3 | − | − | − | − | + | + | − | − | − | − | − |
| D5S622 | 135.548 | 115.95 | 182.97 | 149.46 | q23.3 | − | − | − | − | + | + | − | − | − | − | − |
| *D5S1505 | 135.564 | 115.96 | 182.97 | 149.47 | q23.3 | − | − | − | − | − | − | − | − | + | + | − |
| D5S490 | 136.850 | 117.20 | 184.72 | 150.96 | q23.3 | − | + | + | − | + | − | − | − | − | − | − |
| D5S642 | 138.240 | 119.23 | 187.56 | 153.40 | q23.3 | + | − | − | − | + | − | − | + | − | − | − |
| D5S808 | 138.347 | 119.41 | 187.57 | 153.49 | q23.3 | − | − | − | − | − | − | − | + | − | − | − |
| D5S666 | 139.180 | 120.56 | 187.74 | 154.15 | q31.1 | + | − | − | − | − | − | − | + | − | − | − |
| *IL4 | 139.350 | 120.56 | 187.90 | 154.23 | q31.1 | + | + | + | − | + | + | + | + | − | − | + |
| D5S1984 | 140.372 | 120.58 | 188.84 | 154.71 | q31.1 | − | + | + | − | − | − | − | − | − | − | − |
| IRF1 | 140.430 | 120.58 | 188.89 | 154.73 | q31.1 | − | − | − | − | + | − | + | − | − | − | − |
| D5S1995 | 140.711 | 120.59 | 189.15 | 154.87 | q31.1 | − | − | − | − | + | − | − | − | − | − | − |
| D5S2117 | 140.880 | 120.59 | 189.31 | 154.95 | q31.1 | − | + | + | − | − | − | − | − | − | − | − |
| D5S2115 | 142.430 | 121.84 | 192.68 | 157.26 | q31.1 | − | − | − | − | + | − | − | − | − | − | − |
| *IL9 | 142.648 | 121.88 | 193.41 | 157.64 | q31.1 | + | + | + | − | + | + | + | − | − | − | − |
| D5S816 | 142.743 | 121.90 | 193.72 | 157.81 | q31.1 | − | − | − | − | + | − | − | − | + | + | − |
| D5S393 | 142.860 | 121.93 | 194.11 | 158.02 | q31.1 | + | − | + | + | + | − | + | − | − | − | + |
| D5S500 | 144.453 | 122.62 | 196.46 | 159.54 | q31.2 | − | + | − | − | + | − | − | − | − | − | − |
| D5S414 | 144.640 | 122.63 | 196.60 | 159.62 | q31.2 | − | − | − | − | − | + | − | − | − | − | − |
| D5S399 | 144.876 | 122.70 | 196.90 | 159.80 | q31.2 | + | − | + | + | + | − | + | − | − | − | − |
| D5S658 | 147.010 | 123.38 | 199.57 | 161.47 | q31.2 | + | − | − | − | − | − | − | − | − | − | − |
| D5S1480 | 151.313 | 125.31 | 208.47 | 166.89 | q31.3 | − | − | − | − | − | − | − | − | + | + | − |
| *D5S436 | 152.500 | 125.36 | 208.84 | 167.10 | q31.3 | + | + | + | − | + | − | − | + | − | − | + |
| D5S210 | 152.609 | 125.44 | 209.15 | 167.30 | q31.3 | + | − | + | − | − | − | − | − | − | − | − |
| D5S434 | 155.051 | 126.52 | 213.91 | 170.22 | q32 | − | − | − | − | + | − | − | − | − | − | − |
| D5S413 | 156.380 | 126.65 | 215.24 | 170.94 | q32 | − | − | − | − | + | − | − | − | − | − | − |
| *ADRB2 | 156.380 | 126.65 | 215.24 | 170.94 | q32 | + | − | − | − | − | − | − | − | − | − | − |
| D5S640 | 158.081 | 126.83 | 216.95 | 171.89 | q32 | − | − | − | − | − | + | − | − | − | − | − |
| CSF1R | 158.151 | 126.83 | 217.02 | 171.93 | q32 | − | + | − | − | − | − | − | − | − | − | − |
| D5S470 | 159.849 | 127.02 | 218.85 | 172.94 | q33.1 | + | − | − | − | − | − | − | − | − | − | − |
| *D5S410 | 163.279 | 127.50 | 223.98 | 175.74 | q33.1 | + | − | + | − | − | + | − | − | − | − | − |
| D5S820 | 166.497 | 130.18 | 225.18 | 177.68 | q33.3 | − | − | − | − | − | − | − | − | + | + | − |
| *IL12B | 171.590 | 134.30 | 227.98 | 181.14 | q34 | − | − | − | − | − | − | − | − | − | − | − |
| D5S422 | 174.370 | 135.74 | 229.83 | 182.79 | q34 | − | − | − | − | − | + | − | − | − | − | − |
| Total markers | – | – | – | – | 13 | 8 | 10 | 2 | 16 | 22 | 5 | 5 | 18 | 18 | 3 | |
| Markers in region | – | – | – | – | 13 | 8 | 10 | 2 | 16 | 9 | 5 | 5 | 5 | 5 | 3 | |
Reference loci.
Statistical Methods.
Our sib-pair linkage analysis uses the β model, in which risk factors
are multiplicative and the probability of identity by descent is
conditional on phenotypes, making the model robust to incomplete
ascertainment (22). Under the null hypothesis that β = 0, the
probability of k alleles identical by descent is
ck and the corresponding probability under the
alternative hypothesis is
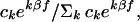 where f = 1 for pairs of affected sibs when normals are
excluded. When normals are included we take f =
(x − μ) (x′ − μ)/U, where
x,x′ are the scores for a sib pair and μ is the
mean for a random sample if known. Otherwise, we approximated a random
sample as μ = pμA + (1
− p)μN, where p is the
population prevalence of asthma (Table 1) and
μA, μN are the means of
asthmatics and nonasthmatics in the sample. U is the mean of
(x − μ)(x′ − μ) for affected pairs,
which therefore have E(f) = 1. This
scaling by U is irrelevant for methods that depend only on
χ2, but it gives a meaningful estimate of β
and may reduce heterogeneity among studies by making values of β more
comparable. Simulation studies have shown good power of the β model
under incomplete ascertainment (which is typical of asthma studies),
although other methods are more powerful in random samples. Variables
like total IgE that are positively skewed were transformed to natural
logarithms and standardized as above.
where f = 1 for pairs of affected sibs when normals are
excluded. When normals are included we take f =
(x − μ) (x′ − μ)/U, where
x,x′ are the scores for a sib pair and μ is the
mean for a random sample if known. Otherwise, we approximated a random
sample as μ = pμA + (1
− p)μN, where p is the
population prevalence of asthma (Table 1) and
μA, μN are the means of
asthmatics and nonasthmatics in the sample. U is the mean of
(x − μ)(x′ − μ) for affected pairs,
which therefore have E(f) = 1. This
scaling by U is irrelevant for methods that depend only on
χ2, but it gives a meaningful estimate of β
and may reduce heterogeneity among studies by making values of β more
comparable. Simulation studies have shown good power of the β model
under incomplete ascertainment (which is typical of asthma studies),
although other methods are more powerful in random samples. Variables
like total IgE that are positively skewed were transformed to natural
logarithms and standardized as above.
Because protocols were not uniform, we used principal component analysis to reduce the reported variables to a smaller number. Taking parents and children together in family studies, we extracted the first two principal components of standardized variables in each study (Table 2). Where feasible, we adjusted the principal components by stepwise cubic regression on age and sex, retaining terms significant at the 0.05 level. Age was not reported for two samples, but its effect is so small that these samples were retained. The adjusted principal components are termed scores. The atopy score (first component), a general factor that spans allergy and inflammation, assigns nearly equal weight to each standardized variable. The asthma score (second component) assigns positive weights to asthma variables and negative weights to allergy variables, thereby giving the highest score to severe intrinsic asthma and the lowest score to severe allergy without asthma, unaffected individuals being intermediate. The advantage of these scores is that studies with different phenotype variables may be scored nearly orthogonally, so that loci acting on allergy and pulmonary inflammation are discriminated. However, no increase in power was observed.
Multivariate analysis provides a large number of scores, orthogonal or correlated, each optimal under assumptions that may be violated and in any case are not directly related to identification of oligogenes. Therefore, the biological validity of scores will be controversial until susceptibility genes are identified. Although selection of multiplex families through asthmatic probands must increase the power of linkage and association tests, the function of a particular gene may be more closely related to a quantitative trait like atopy, inflammation, or eosinophilia than to an arbitrary dichotomy between asthma and normal. Because of this uncertainty, we considered other variables of possibly greater specificity than the scores: asthma status as a diagnosis of asthma, omitting normals; asthma dichotomy as a 0,1 score for normals and affected; IgE as the logarithm of total IgE adjusted for covariance with cubic terms in age and sex; asthma index as the mean of atopy and asthma scores; and atopy index as the mean difference of those scores. These orthogonal indices are specific for asthma and atopy, whereas the scores provide a general factor and a contrast between asthma and atopy.
Interpolation in a dense multilocus map is reliable, but extrapolation beyond flanking markers cannot be trusted. We therefore censored lods in each sample beyond the flanking reference loci, reducing proximal and distal information. The utility of markers outside the candidate region is now evident. An additional constraint is that power to detect allelic association is low because allele codes and marker loci are study specific, markers are spaced at mean distances exceeding 1 cM, and multiple alleles may be grouped in different ways. Pending a dense map of diallelic polymorphisms, little use can be made of allelic association.
Methods for combination of samples are based on simulation studies that favor three approaches. Self and Liang (23) considered a commingled χ2 distribution that under H0 is expected to give evidence against linkage in half the samples, which are assigned χ2 = 0. This method appears to be more powerful than maximum likelihood (ML) scores with heterogeneous data (24). If ui is the ML score for the ith sample with information ki, with U = Σui and K = Σki, then the corresponding lod is Z0 = U2/K (2 ln 10), and the first step in Newton-Raphson iteration gives a rough estimate of the mean effect β as U/K. On the assumption that β is constant among samples, iteration over all samples gives β̂ and likelihood-based lod Ẑ. Under the β model for pairs of affected sibs, the ith region contributes βi to the total genetic effect Σβi, where the risks to sibs relative to the general population are λi = exp(βi) and λ = exp (Σβi), respectively (25). We do not use the sum of lods over n samples, because estimation of β in each of n samples accounts for n degrees of freedom and makes this test least powerful even in replicate samples (24).
Asthma.
In a population with prevalence 0.038 for asthma, the value of β estimated from recurrence in relatives was 1.06 ± .14 (25). Under a multiplicative model, specific loci contribute additively to β. Even large samples do not have high power to detect through linkage a locus-specific effect as small as 0.1 (22). It would therefore be surprising if more than a handful of candidate loci could be convincingly demonstrated by linkage.
As expected, evidence from asthmatic sib pairs for an effect of the cytokine cluster is modest (Table 4). Two small samples were omitted: GerF with 18 pairs and AuB with four pairs. Six samples give positive lods over all or most of the region, whereas three samples give no positive lods within flanking markers. This evidence can be assessed in several ways (Table 5). Linkage of asthma status to the proximal region is significant by the Self and Liang approach that is favored in simulation studies (24). Fitting a parabola to the values for the first three reference loci, the maximal lod is 2.61, with conservative significance level less than 1/antilog = .0025. Asymptotic theory gives χ21 = (2 ln 10)2.61 = 12.00, with corresponding 1-tailed significance level 0.00027. As usual, this asymptotic level is an order of magnitude less than the conservative test (26). Although the data do not reach the conventional lod of 3, they confirm other claims of linkage to this region. A conservative 90% confidence interval is given by all lods above 1.61, and therefore includes IL-4 and IL-13 but not the more distal IL-9. Affected sib pairs give stronger evidence than any of the dichotomies or quantitative traits that we analyzed.
Table 4.
lods for asthmatic sib pairs by sample
| Reference locus | UKS | UKO | AuP | Fin | GerM | Bar | USAA | USC | China |
|---|---|---|---|---|---|---|---|---|---|
| D5S421 | 1.928 | 2.452* | −0.092* | −0.057* | 0.005 | 0.092* | 0.004 | 0.306 | 0.021* |
| D5S1505 | 1.151 | 2.452* | −0.092 | −0.066 | −0.002 | 0.092* | 0.364 | 0.095 | 0.021* |
| IL-4 | 0.508 | 1.948 | −0.092 | −0.027 | −0.123 | 0.118 | 0.793 | 0.183 | 0.021 |
| IL-9 | 0.070 | 1.372 | −0.049 | −0.014 | −0.141 | 0.201 | 0.677 | 0.154 | −0.027 |
| DSS436 | 0.111 | 2.088 | −0.007* | 0.000 | −0.595 | 0.071 | 0.318 | 0.321 | 0.042 |
| ADRB2 | 0.309 | 2.112 | −0.007* | −0.007 | −0.732 | 0.063* | 0.080 | 0.207 | 0.042* |
| D5S410 | 0.616 | 2.113* | −0.007* | −0.009* | −0.469 | 0.063* | −0.009 | 0.013 | 0.042* |
| IL-12B | 0.616 | 2.113* | −0.007* | −0.009* | −0.008 | 0.063* | −0.037 | −0.000 | 0.042* |
Not used for combination of evidence because beyond flanking markers.
Table 5.
Summary of lods for asthma variables (S and L, Self and Liang)
| Reference locus | Asthma
status
|
Asthma dichotomy
|
Score S and L | Index S and L | |||||
|---|---|---|---|---|---|---|---|---|---|
| S and L | Zo | Ẑ | β0 | β̂ | S and L | Z0 | |||
| D5S421 | 1.497 | 0.796 | 0.804 | 0.123 | 0.124 | 1.518 | 1.968 | 0.822 | 0.635 |
| D5S1505 | 2.552 | 1.376 | 1.353 | 0.152 | 0.150 | 2.090 | 1.831 | 0.400 | 1.719 |
| IL-4 | 1.778 | 1.559 | 1.485 | 0.164 | 0.156 | 1.457 | 1.086 | 0.178 | 1.493 |
| IL-9 | 0.889 | 0.666 | 0.663 | 0.091 | 0.091 | 0.747 | 0.266 | 0.039 | 0.887 |
| D5S436 | 1.331 | 0.926 | 0.940 | 0.117 | 0.119 | 0.846 | 0.344 | 0.295 | 1.643 |
| ADRB2 | 1.473 | 0.414 | 0.421 | 0.090 | 0.092 | 0.933 | 0.402 | 0.964 | 2.090 |
| D5S410 | 0.289 | −0.001 | −0.002 | 0.003 | 0.010 | 0.085 | 0.133 | 1.150 | 0.985 |
| IL-12B | 0.005 | −0.024 | −0.024 | −0.024 | 0.010 | 0.005 | −0.010 | 0.187 | 0.632 |
The distal part of the cytokine region gives a smaller maximal lod, estimated by quadratic interpolation to be 1.53, with conservative significance level <0.029 and asymptotic level 0.0039. The asthma index gives by interpolation a higher lod of 2.10, corresponding to a conservative significance level <0.0080 and asymptotic level 0.00094. This index is selected as an extremum, and so there is insufficient reason to prefer it to asthma status. Together, they provide confirmation of one or more oligogenes near ADRB2 with modest effects on asthma susceptibility.
Atopy.
The logarithm of total IgE and other measures of atopy do not approach significance by any method in any study (data not shown) nor overall (Table 6). Several metrics have a maximum proximal to the recognized cytokine interval, although we did not extrapolate beyond tested markers. Study-specific genetic maps were used to minimize effects of typing errors, which tend to displace maxima toward flanking markers. Because maxima distal to ADRB2 were not observed, the proximal maxima may be a chance variation among very small lods. The IL-4 region is farthest from significance.
Table 6.
Summary of lods for atopy variables (S and L, Self and Liang)
| Reference locus | IgE
|
Atopy
score
|
Atopy index
|
||||
|---|---|---|---|---|---|---|---|
| S and L | Z0 | Ẑ | S and L | Z0 | S and L | Z0 | |
| D5S421 | .200 | .340 | .315 | 1.045 | .859 | .379 | .401 |
| D5S1505 | .118 | .455 | .432 | .089 | .259 | .017 | .036 |
| IL-4 | .019 | .028 | .028 | .028 | .025 | .014 | −.020 |
| IL-9 | .035 | −.022 | −.098 | .067 | .008 | .027 | .001 |
| D5S436 | .113 | .000 | −.030 | .186 | .000 | .083 | .000 |
| ADRB2 | .314 | .160 | .164 | .137 | .071 | .131 | .124 |
| D5S410 | .183 | .082 | .085 | .044 | .090 | .128 | .173 |
| IL-12B | .124 | .011 | .013 | .119 | .008 | .013 | −.056 |
Discussion
Disagreement about study design is characteristic of complex inheritance. Probands may be children (12, 15) or adults (14, 16), selected through affection or a correlated trait (12, 16). Some studies concentrate on affected sib pairs or extended pedigrees. Ostensibly random families may be preferred in populations with high prevalence (13). Association tests introduce case-control and other study designs. Measurement and interpretation of quantitative traits such as serum total IgE, specific IgE, skin prick tests, and bronchial hyperreactivity often differ among studies. These factors could create heterogeneity, favoring statistical methods that stratify by sample. The only suggestion of heterogeneity is that most of the evidence for asthma susceptibility comes from the two British samples (Table 4). We have no convincing explanation for this possible difference. Because this is only one partition of the samples among many, we do not attempt to test its significance. However, future studies of linkage and especially association should distinguish the British samples.
Notwithstanding possible heterogeneity, we have succeeded in combining evidence over samples, confirming that the 5q candidate region has oligogenes affecting asthma susceptibility. We failed to identify a candidate locus for atopy, which has three possible explanations. First, there may be no gene in the 5q cytokine cluster with an effect on atopy large enough to be detected by linkage, even when 10 samples are pooled. Second, the genes implicated in atopy may differ so much among populations that combined analysis must fail. This extreme diversity is not the case for ABO and HLA associations with disease nor for malaria-dependent polymorphisms; it does not seem to be true for asthma, and no study approaches significance for atopy. Third, although participation in COAG was intended to be unbiased, there may have been selective nonparticipation of studies with significant results (3, 7, 27, 28). However, many negative studies also did not join this consortium, which at the outset did not have convincing evidence that retrospective collaboration would add value to reports of single studies. Estimated effects β are subject to large error, but β̂ in Table 5 is 0.156 at IL-4 and 0.092 at ADRB2, corresponding to substantial risk for asthma attributable to the 5q cytokine region. The contribution to genetic effects on asthma is estimated to lie between 15% and 23% (25), which is as great as can reasonably be expected in a sample without participation bias. It would be surprising if there were such bias for atopy in studies ascertained mostly through asthma.
Finally, another analysis might be more powerful through an alternative metric such as eosinophilia, forced expiratory volume (FEV1), or noncognate IgE (3), or through a different statistical method that might provide better control over typing error or use the data more efficiently. Study-specific map distances may not be the best way to control spurious crossing over because of typing error, but limited experience suggests that exclusion of loci with notable typing error does not increase power (29), and locus-specific error filtration is in its infancy. Zhang et al. (24) did not find large differences in power by using alternative linkage methods. Refinement of lods to increase power would not alter methods to combine linkage evidence over studies.
With dense markers, allelic association (linkage disequilibrium) might be more powerful than linkage, as Risch and Merikangas (30) have suggested. Their calculations assume that a causal polymorphism is included among several hundred thousand polymorphisms in a genome scan. Methods for positional cloning by allelic association are a topic of active and perhaps frenetic research, because linkage has been disappointing. Allelic association is robust to ascertainment bias and marker mistyping that distorts the linkage map, but methods to exploit allelic association are primitive and largely untested. Should the unit of analysis be an allele, a set of alleles pooled by a test of significance, or all alleles at a locus without regard to individual significance? Which of many metrics is optimal? How can a large number of loci be represented by a composite likelihood that will evade a heavy Bonferroni correction for the multitude of polymorphisms required for an efficient genome scan by allelic association, estimated to be from 30,000 (31) to 500,000 (32)? Because an allele may be either positively or negatively associated with a disease, the Self and Liang approach that is perhaps optimal for linkage is not applicable to combination of evidence for linkage disequilibrium. What approach is most suitable? Although progress is being made in positional cloning of oligogenes by allelic association, these questions have not been answered. Both simulated and real data like the COAG collaboration will help resolve these issues. As candidate loci are confirmed and elucidated there will be progress in phenotype definition, error filtration, and efficient synthesis of linkage and association.
Acknowledgments
We are grateful to the members of the Collaborative Study on the Genetics of Asthma (CSGA), who chose not to join the international consortium but generously contributed their data. Many people in the research groups of the authors participated in this collaboration. They are cited in the references, which report the grants that supported their work. COAG was founded under a grant from the National Asthma Campaign, and the analysis presented here was supported by the Medical Research Council.
Abbreviation
- lod
logarithm of odds
References
- 1.Collins A, Ennis S, Tapper W, Morton N E. Genet Mol Biol. 2000;23:1–10. [Google Scholar]
- 2.CSGA. Nat Genet. 1997;15:389–392. doi: 10.1038/ng0497-389. [DOI] [PubMed] [Google Scholar]
- 3.Marsh D G, Neely J D, Breazeale D R, Ghosh B, Freidhoff L R, Ehrlich-Kautzky E, Schou C, Krishnaswamy G, Beaty T H. Science. 1994;264:1152–1156. doi: 10.1126/science.8178175. [DOI] [PubMed] [Google Scholar]
- 4.Dizier M-H, Sandford A, Walley A, Philippi A, Cookson W, Demenais F. Genet Epidemiol. 1999;16:84–94. doi: 10.1002/(SICI)1098-2272(1999)16:1<84::AID-GEPI7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 5.Walley A J, Cookson W O C M. J Med Genet. 1996;33:689–692. doi: 10.1136/jmg.33.8.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rioux J D, Stone V, Daly M, Cargill M, Green T, Nguyen H, Nutman T, Zimmerman P A, Tucker M A, Hudson T, et al. Am J Hum Genet. 1998;63:1086–1094. doi: 10.1086/302053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez F D, Solomon S, Holberg C J, Graves P E, Baldwin M, Erickson R P. Am J Respir Crit Care Med. 1998;158:1739–1744. doi: 10.1164/ajrccm.158.6.9712040. [DOI] [PubMed] [Google Scholar]
- 8.D'Amato M, Vitiani L R, Petrelli O, Ferrigno L, di Pietro A, Trezza R, Matricardi P M. Am J Respir Crit Care Med. 1998;158:1968–1973. doi: 10.1164/ajrccm.158.6.9804126. [DOI] [PubMed] [Google Scholar]
- 9.Gorham J D, Guler M L, Steen R G, Mackay A J, Daly M J, Frederick K, Dietrich M F, Murphy K M. Proc Natl Acad Sci USA. 1996;93:12667–12472. doi: 10.1073/pnas.93.22.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyers D A, Postma D S, Panhuyen C I M, Amelung P J, Levitt R C, Bleecker E R. Genomics. 1994;23:464–470. doi: 10.1006/geno.1994.1524. [DOI] [PubMed] [Google Scholar]
- 11.Holloway, J., Beghe, B., Lonjou, C., Peng, Q., Gaunt, T. R., Gomez, I., Thomas, N. S., Holgate, S. T. & Morton, N. E. (2000) Genes Immun., in press. [DOI] [PubMed]
- 12.Daniels S E, Bhattacharrya S, James A, Leaves N I, Young A, Hill M R, Faux J A, Ryan G F, le Söuef P N, Lathrop G M, et al. Nature (London) 1996;383:247–250. doi: 10.1038/383247a0. [DOI] [PubMed] [Google Scholar]
- 13.Palmer L J, Daniels S E, Rye P J, Gibson N A, Tay G K, Cookson W O C M, Goldblatt J, Burton P R, Le Söuef P. Am J Respir Crit Care Med. 1998;158:1825–1830. doi: 10.1164/ajrccm.158.6.9804037. [DOI] [PubMed] [Google Scholar]
- 14.Laitinen T, Kauppi P, Ignatius J, Ruotsalainen T, Daly M J, Kääriäinen H, Kruglyak L, Laitinen H, de la Chapelle A, Lander E S, et al. Hum Mol Genet. 1997;6:2069–2076. doi: 10.1093/hmg/6.12.2069. [DOI] [PubMed] [Google Scholar]
- 15.Wjst M, Fischer G, Immervoll T, Jung M, Saar K, Rueschendorf F, Reis A, Ulbrecht M, Gomolka M, Weiss E H, et al. Genomics. 1999;58:1–8. doi: 10.1006/geno.1999.5806. [DOI] [PubMed] [Google Scholar]
- 16.Deichmann K A, Heinzmann A, Forster J, Dischinger S, Mehl C, Brueggenolte E, Hildebrandt F, Moseler M, Kuehr J. Clin Exp Allergy. 1998;28:151–155. doi: 10.1046/j.1365-2222.1998.00159.x. [DOI] [PubMed] [Google Scholar]
- 17.Barnes K C, Neely J D, Duffy D, Freidhoff L R, Breazeale D R, Shou C, Naidu R P, Levett P N, Renault B, Kucherlapati R, et al. Genomics. 1996;37:41–50. doi: 10.1006/geno.1996.0518. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Du X, Lin L, Jiang J, Hu L, Fu J, Zhang H, Chen Y. Ching Hua I Hsueh I Chuan Hsueh Tsa Chih. 1999;16:307–310. [PubMed] [Google Scholar]
- 19.ISAAC. Eur Respir J. 1998;12:315–335. doi: 10.1183/09031936.98.12020315. [DOI] [PubMed] [Google Scholar]
- 20.Lio P, Morton N E. Proc Natl Acad Sci USA. 1997;94:5344–5348. doi: 10.1073/pnas.94.10.5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins A, Frezal J, Teague J, Morton N E. Proc Natl Acad Sci USA. 1996;93:14771–14775. doi: 10.1073/pnas.93.25.14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morton N E. Proc Natl Acad Sci USA. 1996;93:3471–3476. doi: 10.1073/pnas.93.8.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Self S G, Liang K G. J Am Stat Assoc. 1987;82:605–610. [Google Scholar]
- 24.Zhang, W., Collins, A. & Morton, N. E. (2000) Hum. Hered., in press.
- 25.Collins A, MacLean C J, Morton N E. Proc Natl Acad Sci USA. 1996;93:9177–9181. doi: 10.1073/pnas.93.17.9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morton N E. Proc Natl Acad Sci USA. 1998;62:690–697. [Google Scholar]
- 27.Postma D S, Bleecker E R, Amelung P J, Holroyd K J, Xu J, Panhuysen C I M, Meyers D A, Levitt R C. New Engl J Med. 1995;333:894–900. doi: 10.1056/NEJM199510053331402. [DOI] [PubMed] [Google Scholar]
- 28.Ober C, Cox N J, Abney M A, Di Rienzo A, Lander E S, Changyaleket B, Gidley H, Kurtz B, Lee J, Nance M, et al. Hum Mol Genet. 1998;7:1393–1398. doi: 10.1093/hmg/7.9.1393. [DOI] [PubMed] [Google Scholar]
- 29.Gomes I, Collins A, Lonjou C, Thomas N S, Wilkinson J, Watson M, Morton N E. Ann Hum Genet. 1999;63:535–538. doi: 10.1017/S0003480099007824. [DOI] [PubMed] [Google Scholar]
- 30.Risch N, Merikangas K. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 31.Collins A, Lonjou C, Morton N E. Proc Natl Acad Sci USA. 1999;96:15173–15177. doi: 10.1073/pnas.96.26.15173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kruglyak L. Nat Genet. 1999;22:139–144. doi: 10.1038/9642. [DOI] [PubMed] [Google Scholar]