

Abstract
Despite the high clinical impact of established and emerging respiratory viruses, some critical aspects of the host response to these pathogens still need to be defined. In that context, we aimed at two major issues: first, what are the innate immune mechanisms that control common respiratory viral infections; and second, whether these mechanisms also cause long-term airway disease. Using a mouse model of viral bronchiolitis, we found that antiviral defense depends at least in part on a network of mucosal epithelial cells and macrophages specially programmed for immune-response gene expression. When this network is compromised, the host is highly susceptible to infection, but network components can be engineered to provide increased resistance to infection. Similar alterations appear in asthma and chronic bronchitis/chronic obstructive pulmonary disease, suggesting that evolving attempts to improve antiviral defense may also lead to inflammatory airway disease. Indeed, in genetically susceptible mice, respiratory paramyxoviruses cause a “hit and run” phenomenon that is manifested by the development of a permanent airway disease phenotype long after the infection has cleared. The phenotype can be segregated into individual traits to achieve more precise definition of just how viruses reprogram host behavior. Identifying specific components of the mucosal immune system that manifest an aberrant antiviral response may thereby allow for adjusting this response to improve acute and chronic outcomes after viral infection.
Keywords: airway hyperreactivity, apoptosis, chemokine, interferon signal transduction, mucosal immunity, mucous cell metaplasia
Similar to other chronic inflammatory diseases, the concept has gradually evolved that chronic airway diseases, typified by asthma and chronic obstructive pulmonary disease (COPD), are driven by a detrimental inflammatory response, and furthermore, that this mechanism represents an aberration or extension of the normal immune response. Despite the complexity of the immune response (or perhaps because of it), a relatively simple scheme was developed for asthma pathogenesis that rests on the classification of the adaptive immune system, and especially the T-cell responses to allergic and nonallergic stimuli that enter the airway. This scheme was devised largely in murine models of the immune response in which CD4+ T-cell–dependent responses may be classified into T-helper type 1 (Th1) or Th2, where Th1 cells characteristically mediate delayed-type hypersensitivity reactions and Th2 cells promote B-cell–dependent humoral immunity (1). In this system, interleukin (IL)-4, IL-5, IL-9, and IL-13 promote Th2 cell differentiation and B-cell–dependent IgE production, tissue eosinophilia, airway hyperreactivity, and goblet cell metaplasia (2). Furthermore, these responses are downregulated by Th1 cytokines, such as interferon (IFN) γ and IL-12, and are inversely correlated with the general level of Th1 cell responses (3–6). By extension, the traditional scheme for asthma pathogenesis is based on a relative increase in Th2 responses in combination with a decrease in Th1 cellular responses. More recently, murine models indicate that Th1 cells may still be necessary for the development of a Th2 response (7, 8) and that CD8+ T cells, natural killer T cells, and regulatory T cells may also contribute to the allergic response (9–11). In all cases, however, the scheme rests chiefly on the hypothesis that asthma pathogenesis depends critically if not solely on the overproduction of Th2 cytokines (12–17).
However, several lines of evidence in model systems and in humans raise questions for the Th2 hypothesis as a full explanation for asthma. Thus, as noted above, Th1 as well as antigen-specific Th2 cells may be necessary for initiating the allergic response, even in mouse models of asthma (7, 8). Furthermore, endpoints of the allergic response, such as airway hyperrreactivity and mucus production, may develop without IgE production or eosinophil influx (18–20). In some cases, airway reactivity may be dissociated from eosinophilic inflammation on the basis of genetic background (21, 22). In fact, in human subjects, the development of allergy and asthma is often dissociated as well (23), and linkage between asthma and candidate genes for atopy has not been found in large population studies (24). Moreover, treatment aimed at selective blockade of Th2 pathways has not yet proven to be efficacious in asthma (17). Indeed, the type of inflammation found in early childhood asthma and severe asthma may show little evidence of a traditional allergic response. Nonetheless, these discrepancies are generally ascribed to the complexity of the allergic response, so that other features of the response may still lead to the asthma phenotype (25).
These discrepancies are also relevant to the pathogenesis of COPD, particularly because the chronic bronchitis phenotype overlaps significantly with the one for asthma. In fact, some have argued that asthma pathogenesis may overlap with the development of the COPD phenotype (26). Shared mechanisms are highlighted by the association of abnormal airway inflammation with progression of COPD (27). Immune cell infiltration in asthma (typically by eosinophils, mast cells, and CD4+ Th2 cells) is often contrasted with inflammation in COPD (often characterized by neutrophils, macrophages, and CD8+ T cells), but CD4+ T cells appear prominently in COPD and CD8+ T cells may contribute to asthma (27, 28), and, as discussed below, activated macrophages may be found in both conditions. In either case, the same mediator profile may be produced by more than one cellular source, so even with a distinct inflammatory cell profile, it is still possible that asthma and COPD share similar molecular mechanisms and consequent overlap in end-organ dysfunction. In that regard, genetic analysis indicates that candidate genes, such as IL-13, yield similar susceptibility profiles in asthma and COPD cohorts (29), and overexpression of IL-13 in mice may lead to a phenotypes common to asthma and COPD (i.e., mucous cell metaplasia and airway hyperreactivity) (30). In the absence of allergy, is it possible, for example, that another component of the immune response could lead to Th2 cytokine production and consequent airway disease?
AN ALTERNATIVE IMMUNE MODEL: AIRWAY RESPONSES TO VIRUS
To resolve these outstanding issues for asthma and COPD pathogenesis, we focused on related but distinct aspects of airway immunity that are particularly relevant to the host response against microbial pathogens (especially respiratory viruses). Our initial reasoning was related to the concept that the adaptive immune system (manifested by the diverse repertoire of T and B cells) requires signals about the origin of the antigen and the type of response to be induced. Furthermore, these signals must be provided by the innate immune system. In this general context, and in the particular context of the response to inhaled agents, we proposed that the epithelial cells lining the airway surface represent an ideal candidate to act as a primary sentinel in innate immunity (31). In particular, our observations in models of airway inflammation and in human subjects with inflammatory airway disease indicated that the immune cell response to inhaled agents was topographically directed toward and then through the airway epithelium. In turn, this finding suggested that the epithelium was charged with directing immune cell traffic to the airway lumen, so that initial studies were directed toward defining the underlying mechanism for transepithelial traffic of immune cells. In the course of our studies, we learned that the role of airway epithelial cells is complemented by the actions of airway macrophages, perhaps in line with observations that macrophages also patrol the airway and, together with airway epithelial cells, are the primary sites for replication of common respiratory viruses (32). Here, we begin to develop the concept that airway epithelial cells and macrophages appear to constitute critical (but not the only) components of an innate antiviral immune system, and their roles can be defined by studies of viral infection in vitro (using physiologic cell culture systems) and in vivo (using high-fidelity mouse models).
Studies In Vitro Using Airway Epithelial Cells and Macrophages
For studies of isolated cells, special care was taken to preserve the native properties of airway epithelial and immune cells. Airway epithelial cell cultures were modified from ones that are appropriate for studying immune cell traffic out of the circulation (where cells must be slowed and activated and often move from a lumenal to ablumenal compartment). In the case of the airway epithelium, immune cells are directed from an ablumenal to a lumenal location and initial immune cell slowing (i.e., rolling) is not necessarily a feature of this system. Indeed, it became apparent that airway epithelial cells use a special program for directing immune cell traffic that depends on a two-step area code for first, cell adhesion, and second, chemotaxis (33–35). In the case of T-cell movement, these two steps are typified by distinct expression of intercellular adhesion molecule 1 (ICAM-1) and secretion of CC-chemokine ligand 5 (CCL5; formerly designated as RANTES [regulated upon activation, normal T-cell expressed and secreted]). For ICAM-1, distribution on both apical and basolateral cell surfaces mediates efficient cell adhesion at the basal cell surface (to aid in transmigration) and at the apical cell surface (for retention and movement along the airway). For CCL5, a pattern of polarized apical secretion provides for a soluble chemical gradient for T-cell movement from the subepithelium (where levels are low) to the mucosal surface (where levels are higher) and retention at that lumenal site. We have presented a scheme for how these two pathways for controlling cell adhesion and chemotaxis may be coordinated to explain epithelial cell–dependent transmigration of T cells in a basal-to-apical direction (36). Here we suggest that these patterns also fit well with available data for ICAM-1 and CCL5 expression in airway epithelium in vivo on the basis of functional data from ICAM-1– and CCL5-null mice. In the case of CCL5, however, a nonredundant capacity for mediating macrophage survival was also uncovered that was distinct from directing immune cell traffic (described below).
Analogous to how ICAM-1 and CCL5 complement each other to direct immune cell traffic, these epithelial immune-response products also typify how transcriptional and post-transcriptional mechanisms synergize to control gene expression (Figure 1). Thus, epithelial ICAM-1 gene expression is regulated by IFN-γ signal transduction featuring the Stat1 transcription factor as a critical regulator of transcription (37–41). CCL5 gene expression is also inducible by IFN-γ but is exquisitely sensitive to direct induction by viral replication (thereby bypassing the need for immune cell IFN production). In this case, the virus (and IFN-γ) relies on regulation at the post-transcriptional level by stabilization of CCL5 mRNA (42). These virus-driven events may also be complemented by nuclear factor-κB–dependent transcriptional activity due either to cytokine (tumor necrosis factor) or Toll-like receptor actions. This transcriptional/post-transcriptional synergy may explain why CCL5 exhibits greater induction than any other chemokine (and perhaps any other known gene) after respiratory viral infection. As discussed below, another important response of the airway epithelial cell to cytosolic detection of viral replication is the generation of type I IFN (i.e., IFN-β). On the basis of in vitro (and later in vivo) studies of IFN- and Stat1-null mice, IFN-β–dependent signaling appears to be a critical line of antiviral defense (43, 44). In this case, other transcription factors (e.g., interferon regulatory factor-7) may also be required (45), although this pathway has not been defined as yet for airway cells or common respiratory viruses. Of note, respiratory syncytial virus (RSV) has devised a special mechanism to disrupt type I IFN signaling based on the expression of distinct nonstructural proteins (NS1 and NS2) (46). These gene products appear to successfully compromise IFN signaling in human but not other species of host cells, thereby providing a mechanism for host range as well.
Figure 1.
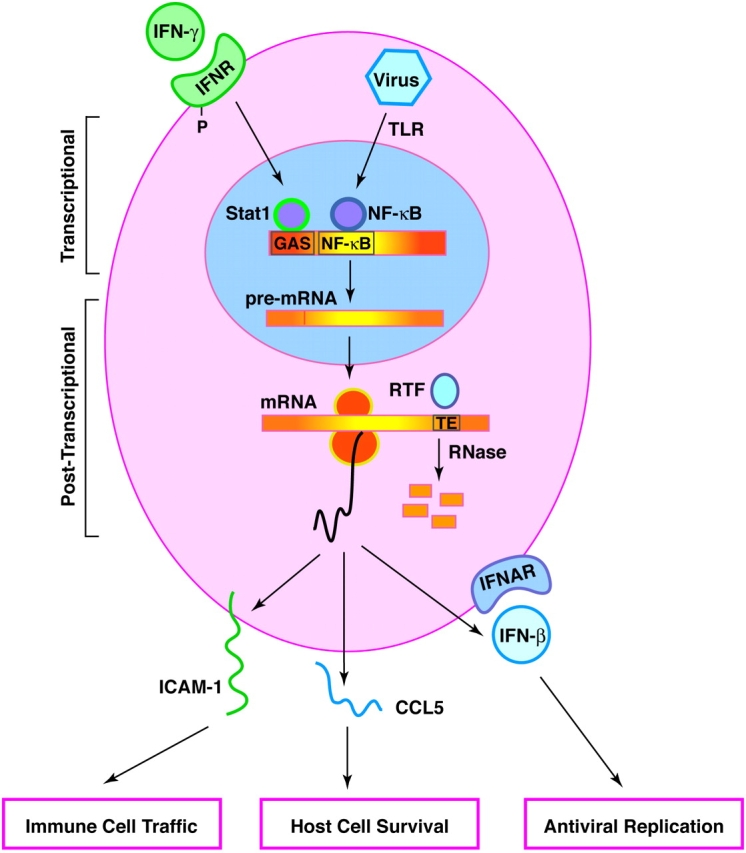
Critical aspects of the antiviral response in the host cell. For intercellular adhesion molecule 1 (ICAM-1) and related genes, expression is controlled at the transcriptional level via IFN-γ signaling. Sequential steps include IFN-γR activation/phosphorylation, Stat1 translocation and interaction at γ-activation site (GAS) in the gene promoter region, generation of pre-mRNA transcripts, and mRNA maturation and release to ribosomal machinery for translation and then processing of protein for expression at the appropriate cellular site. For CC-chemokine ligand 5 (CCL5) and related genes, expression is controlled at the transcriptional level by a direct effect of the virus. For example, virus (e.g., respiratory syncytial virus [RSV] or Sendai virus) may interact with Toll-like receptors (TLR) and so activate nuclear factor (NF)-κB and consequent NF-κB–dependent gene transcription. In addition, cytosolic viral replication may act downstream at the post-transcriptional level and alter gene expression by stabilizing mRNA, likely by influencing mRNA turnover factors (RTF) that bind to RNA turnover elements and mediate RNase-dependent degradation. Expression of ICAM-1, CCL5, and IFN-β allows for control of immune cell traffic, host cell survival, and viral replication, respectively. This scheme was defined in airway epithelial cells but is also active in macrophages that are also similarly but less effectively infected by common respiratory viruses.
By comparison to airway epithelial cell studies, the effect of respiratory viruses on lung macrophage behavior is poorly characterized. Previous reports document the capacity of alveolar macrophages to be infected by respiratory viruses (e.g., RSV), although infection may require a higher-level inoculum (47). Similarly, virus can be detected in alveolar macrophages in vivo, but less commonly than in airway epithelial cells (32). In this setting, it is not clear whether viruses primarily infect the macrophages, or perhaps more commonly, if they become infected by phagocytosis of apoptotic epithelial cells left in the wake of viral cytopathic effects. As developed below, this phenomenon raises the possibility that macrophage scavenging is another critical aspect of the antiviral response in the lung.
Studies In Vivo Using Mice and Viruses
Our interest in antiviral responsiveness of airway epithelial cells and macrophages was next extended to studies of mucosal immune function in vivo. For these experiments, we developed a mouse model to take further advantage of genetic and genomic strategies. To achieve epithelial and macrophage activation (which we considered essential features of chronic airway disease), respiratory viral infection was chosen as a natural stimulus to model. Because we were interested in asthma and COPD pathogenesis, we focused on common respiratory viruses that target the airways and have been linked to development or at least exacerbation of asthma and COPD. This raised the possibility of using a paramyxovirus, and within this family of negative-strand RNA viruses, there are several common human and mouse pathogens available for laboratory use (Figure 2). Among these viruses, RSV would have been an ideal choice because it is the most common cause of serious respiratory illness in early childhood and is most often associated with the development of childhood asthma. Unfortunately, mice are relatively resistant to infection with RSV, so a high-threshold inoculum of virus must be given and the resulting all-or-none pattern of illness manifests primarily as alveolitis with viral localization to type I alveolar epithelial cells (48). Thus, although RSV was particularly useful in studies of isolated human cells, its utility was limited for experimental studies done in vivo. We also considered using a newly identified human pathogen, human metapneumovirus (hMPV), which appears to be associated with asthma, but the native virus is also weakly pathogenic in immunocompetent mice (unpublished observations, E.A. and M.J.H.). In immune-compromised animals and humans, hMPV causes more severe infection (49), but implications for airway disease are still being defined. Recently, another hMPV isolate has been reported to cause persistent infection and Th2 cytokine response in mice (50). This type of variability has also been found for RSV and pneumonia virus of mice, so in each case it will be of interest to define the genetic differences between viral isolates (or with viral passage) that account for a change in host range and response.
Figure 2.

Genomic organization for paramyxoviruses used for studies of acute viral bronchiolitis in mice. Some viral genes (e.g., NS1 and NS2) are distinct for RSV and help viral infectivity by blocking IFN signaling. Other viral genes (e.g., F gene) require specific host factors to enable viral entry into the host cell. Each of these strategies thereby confers a degree of species specificity and so limits host range. The functional impact of individual viral genes for causing acute infection and for conferring the chronic airway disease phenotype still needs to be more completely defined.
Although we continue to study these issues, our in vivo studies were initiated by substituting a closely related paramyxovirus—that is, mouse parainfluenza type I or Sendai virus (SeV)—for RSV in the mouse model. SeV was isolated initially from humans and exhibits natural pathogenicity in immunocompetent mice. Delivery of intranasal SeV in the proper inoculum (e.g., 2 × 105 plaque-forming units) and mouse strain (e.g., C57BL/6J) allows for the development of viral bronchiolitis that maintains high fidelity with what we observe in humans (51). In particular, there is reversible illness with infection limited to the airway mucosa and inflammation largely restricted to peribronchial and bronchiolar tissues. At higher inoculum, there is propagation of infection to distal airspaces with bronchopneumonia that, if severe enough, can lead to respiratory death. At either inoculum, viral replication is localized primarily to airway epithelial cells (although detectable in airspace macrophages as well), thereby resembling the pattern that is found in children with severe paramyxoviral infection due to RSV (52). Moreover, the infected epithelial cell population expresses a profile of immune-response gene expression similar to the one found in cultured airway epithelial cells infected with RSV or SeV, notably including inducible expression of ICAM-1, CCL5, IL-12p40, as well as Stat1, which appears to be autoamplified under conditions of IFN production.
The choice of C57BL/6J mice was based on experiments indicating that the humanlike pattern of illness was not found in several other genetic backgrounds, but this choice also afforded an opportunity to study genetically modified mice that are commonly generated on this same background. Comparison of mice with targeted null mutations to wild-type control mice thereby allowed us to define the function of immune-response genes important for antiviral host defense. Table 1 summarizes findings with some of the genotypes that we have recently studied in the SeV bronchiolitis model. The results indicate that null mutations of the Th1 immune cell network for antiviral defense (e.g., IFN-γ or IL-12p40) did not by themselves adversely affect the host by increasing inflammation or decreasing viral clearance. By contrast, mutations that alter the expression of epithelial immune-response genes invariably alter inflammation and eventual outcome. In particular, loss of ICAM-1 decreases airway inflammation (as predicted from in vitro studies) and blocks airway hyperreactivity. Loss of IL-12p35 causes overproduction of epithelial IL-12p40, which in turn increases macrophage-dependent inflammation and consequent morbidity (51). This finding highlights the role of IL-12p80 as a macrophage chemoattractant (53). Loss of CCL5 causes an increase in epithelial and macrophage apoptosis in the face of viral infection and thereby leads to impaired viral clearance due to a compromise in the macrophage scavenging system (54). The defect depends on CCL5/CCR5 signaling and can therefore be found in CCR5-null mice as well. Thus, macrophages isolated from CCL5- or CCR5-null mice as well as human macrophages treated with anti-CCR5 blocking antibody exhibit increased apoptosis after viral infection. These findings served to highlight the role of the macrophage in antiviral defense, and the special need for airway macrophage survival in the face of viral infection. Moreover, CCR5 allelic variation is known to alter susceptibility to devastating pathogens (e.g., HIV) but may also influence the acute and chronic responses to common respiratory viruses (55).
TABLE 1.
Role of immune-response genes in viral bronchiolitis based on behavior of inbred (C57bl/6J) mice with targeted null mutations and paramyxoviral (sendai virus) infection
| Gene Target | Viral Clearance | Inflammation | Mortality | Airway Reactivity |
|---|---|---|---|---|
| IFN-γ | No change | No change | No change | No change |
| IL-12p40* | No change | No change | No change | No change |
| ICAM-1* | No change | Decreased | Decreased | Decreased |
| IL-12p35* | No change | Increased | Increased | No change |
| Stat1* | Decreased | Increased | Increased | Not determined† |
| CCL5* | Prolonged | Increased | Increased | Not determined† |
Definition of abbreviations: CCL = CC-chemokine ligand; ICAM = intercellular adhesion molecule; IL = interleukin.
Targets that alter gene expression in airway epithelial cells as well as immune cells.
Increased susceptibility to viral infection interferes with comparison to wild-type control mice.
Among the immune-targeted mice that we studied, the most devastating compromise in antiviral defense is found after the loss of Stat1 function. In Stat1-null mice, there is a marked increase in susceptibility to SeV (as well as RSV or influenza virus) infection, with lethal consequences (43, 56, 57). The loss of type I IFN signaling is likely the main determinant of the Stat1-null defect, because the same phenotype develops in mice with a null mutation of the receptor for type I IFN (i.e., IFNAR), whereas, as noted above, the loss of IFN-γ signaling alone has little impact on the antiviral response, at least in response to influenza virus. Mice bred to be both IFNAR and IFN-γ null exhibit a slightly more severe defect than IFNAR null alone, so IFN-γ has some contribution when the type I IFN system is lost. The same profile of immune compromise appears to be manifest in humans with defective IFN signaling. Thus, humans with Stat1 alleles that disrupt type I and II IFN signaling suffer lethal viral infections in infancy, whereas alleles that disrupt only IFN-γ signaling appear to cope with viral infections but may later manifest a defect in host defense against mycobacterial infection (58). As noted above, RSV contains distinct genetic components to disrupt type I IFN signaling in host cells (46), providing at least a partial explanation for continued success of this virus to achieve infection in humans.
In the context of this work, we have aimed to define the role of epithelial Stat1 in defense against respiratory viruses. One approach depends on targeted loss of Stat1 function in the mucosal epithelial cell versus immune cells. Another approach depends on targeted enhancement of Stat1 signaling function. Our work on these issues is still in progress, but initial findings indicate that a Stat1 modified for hyperresponsiveness to IFN can mediate more effective viral clearance (59). As developed below in the section on human studies, the apparent hyperresponsiveness of Stat1 found in inflammatory disease may therefore represent an attempt to evolve Stat1 signaling capacity to afford a host advantage in the native setting as well. Definitive proof of concept awaits the completion of ongoing studies of bone marrow–chimeric mice as well as mice with epithelial-specific targeting of Stat1 expression. A particular challenge is the need to define new transgenic systems specific for ciliated airway epithelial cells, because this subpopulation appears to be the primary target of paramyxoviral infection. In that regard, chimeric and transgenic systems to specifically target the macrophage population need to be studied in this context as well. Nonetheless, the results to date already suggest a critical role for airway epithelial cells and macrophages for optimal defense against the types of respiratory viruses that are linked to the development of asthma and possibly other forms of chronic airway disease.
CHRONIC AIRWAY RESPONSES TO VIRUS
Our initial studies of paramyxoviral infection provided a useful framework for new observations on innate antiviral immunity, and the particular contributions of airway epithelial cells and macrophages, but the work focused largely on the acute response to viral infection. Because asthma and chronic bronchitis are often lifelong diseases and are strongly influenced by genetic background, we next questioned whether our experimental approach could be further developed to address the critical issues of chronicity and susceptibility. We therefore next focused on whether we could detect a long-term effect of respiratory viral infection on airway behavior, and if so, whether we could segregate this chronic change from the acute events that surround viral infection. We reasoned that inhibition of the acute inflammatory response could be achieved by targeted disruption of airway epithelial immune-response genes and so influence acute and but not chronic inflammatory disease phenotypes. As noted above, among candidate genes that might mediate immune cell traffic, ICAM-1 is the predominant determinant for adhesion of immune cells to epithelial cells in vitro (33–35). Thus, loss of ICAM-1 function would likely lead to decreased airway inflammation. Indeed, we found that ICAM-1 expression is induced primarily on host airway epithelial cells by viral infection and is necessary for the full development of acute inflammation and concomitant postviral airway hyperreactivity (60). These findings finally established a cause-and-effect relationship between acute airway inflammation and hyperreactivity, which had been proposed in our initial report in 1983 (61).
Although these findings linking inflammation and hyperreactivity were perhaps expected, the next results were surprising. Thus, as we monitored host response over time, we discovered that a single, primary viral infection also caused airway hyperreactivity and goblet cell metaplasia that lasted for at least a year after complete clearance of virus (60). This long-term (essentially permanent) phenotype developed regardless of ICAM-1 deficiency and ICAM-1–dependent alteration of the acute inflammatory response, thereby indicating distinct determinants for acute and chronic airway disease phenotypes. None of the long-term virus-induced abnormalities were influenced by IFN-γ deficiency, because each developed in IFN-γ–null mice as well. Each of these features is distinct from allergen-induced airway hyperreactivity and goblet cell metaplasia in the same mouse system. Thus, airway hyperreactivity and goblet cell metaplasia are also inducible by allergen challenge in this genetic background, but in the case of allergy, the phenotypes resolve spontaneously with time (in the absence of treatment or additional challenges) and are sensitive to the levels of IFN-γ. Indeed, the IFN-independent nature of virus-induced phenotypes in this experimental setting is reminiscent of the low levels of IFN-γ or IFN-γ–producing cells found in subjects with asthma (62). Whether IFN production is similarly low in patients with COPD and whether other features of this inflammatory response are similar to the pattern in subjects with asthma and COPD are still under study. Although we define the mechanism for development of disease traits in this mouse model, we have designated the phenotype as “asthmitis,” recognizing that the type of inflammation is distinct from the allergic response and (as developed below) likely shares cellular and molecular features with ones found in asthma and chronic bronchitis.
The main impact of this initial work, however, is that a single paramyxoviral infection can cause both acute and chronic manifestations of asthma and chronic bronchitis. These findings therefore raise the possibility that asthma not only resembles a persistent antiviral response (51, 62) but may also be caused by such a response, and so provide the experimental link between paramyxoviral infection in early life with subsequent asthma in childhood and perhaps adulthood. The findings also provide a substrate for segregating a complex disease into individual traits that can then be linked to the expression of specific host genes. Thus, ICAM-1 expression appears capable of regulating the acute but not necessarily the chronic phenotype, whereas additional determinants may confer individual traits (i.e., airway hyperreactivity and goblet cell metaplasia) that constitute the chronic phenotype. The mouse model is particularly suited to defining genetic and immunologic determinants that can then be tested for mechanistic relevance in subjects with asthma or COPD. Critical questions include the following: how do specific viral genes confer the chronic disease phenotype, how do host genetics determine susceptibility to developing the phenotype, how is memory for the phenotype preserved, and how do we segregate the individual traits that comprise the chronic phenotype? These issues can be addressed both from the perspective of the virus as well as the host.
Viral Determinants
From the microbial perspective, the viral pathogen must be equipped with specific components to confer a long-term airway disease phenotype. At the least, this will require viral genes encoding for products that target successful infection to the airways and to the proper host species. For example, the Sendai viral F (fusion) gene product contributes to pathogenicity in mice because this species expresses a specific airway protease required for proteolytic cleavage and activation of the protein needed to achieve viral entry into the host cell (Figure 2). As another example, RSV nonstructural proteins NS1 and NS2 are able to inactivate human but not mouse Stat2 and thereby inhibit IFN signaling in the host cell (46). Indeed, siRNA-dependent inhibition of NS1/NS2 expression allowed for an antiviral strategy in a mouse model (63). These studies, as well as those noted above for enhanced Stat1 function, raise the possibility of decreasing the severity of acute viral infection to also decrease the subsequent long-term sequelae.
On a more complex level, it is also useful to consider the viral determinants for how respiratory viral infections can cause permanent abnormalities in airway behavior in the experimental setting and perhaps in humans. We recognized at least three possibilities to explain long-term actions of paramyxoviruses: (1) persistent infection (similar to hepatitis C virus), (2) mutant quasi-species (similar to coronaviruses), or (3) a hit-and-run phenomenon (similar to adenoviruses). Experiments aimed at monitoring tissue (especially lung) levels of virus with immunoblotting, immunostaining, plaque-forming assay, and real-time polymerase chain reaction all indicated that SeV was completely cleared from the organism by 2 weeks after inoculation. This finding indicated that the actions of SeV to cause a chronic asthma phenotype are based on a hit-and-run strategy because viral effects persist after clearance. Indeed, the results provide evidence of the capacity for nononcogenic RNA viruses to irreversibly reprogram host cell behavior in a manner previously restricted to oncogenic DNA viruses (64, 65). Further proof and understanding of this part of the process will depend on identifying specific viral gene products responsible for altering host gene expression and consequent phenotype. Of note, we found that viral clearance was just as efficient in ICAM-1–null animals, so the antiinflammatory effect of losing ICAM-1 is protective against acute hyperreactivity but does not interfere with efficient viral clearance. This outcome raises a novel strategy for therapeutic development based on decreasing inflammation while preserving or even enhancing immunity.
Host Determinants
Mechanisms of virus-induced airway disease are also linked to the host response, and the mouse model specially lends itself to genetic and immunologic strategies for defining the basis for the response that leads to a disease phenotype. Because it was not possible to separate goblet cell metaplasia from airway hyperreactivity within a single host genetic background, we identified parental strains of mice that were matched for a similar acute response, with similar histopathology and viral clearance rates, but for a divergent chronic response (66). This divergence in genetic susceptibility, and the fact that the F1 children showed an intermediate phenotype, has allowed us to begin mapping the genetic basis for these traits in an F2 intercross generation, as recently reviewed (67). Initial results indicate that asthma traits are linked to distinct genetic loci. Additional genetic analysis will be needed to determine how these chronic phenotypes (i.e., airway hyperreactivity and goblet cell metaplasia) segregate in mice and in humans and to define the relevant genes for susceptibility at each time point. These genetic studies still need to be completed, but taken together with the earlier work on ICAM-1–null mice, we can start to build a model for how transient viral replication can cause acute inflammation/hyperreactivity as well as chronic goblet cell metaplasia/airway hyperreactivity, likely driven by ongoing airway stimulation and concomitant pressure from the innate and adaptive immune systems (summarized in Figure 3).
Figure 3.

Model for acute and chronic airway responses to respiratory viruses. The model is based on the time course of quantitative traits that develop after primary paramyxoviral infection in mice. Events begin with viral replication (which peaks at 3–5 days after inoculation), which is later cleared from the lung (by 2 weeks after inoculation). This initial infection is followed by induction of epithelial immune-response gene expression (which peaks at 5 days after inoculation) and is followed by immune cell infiltration (which peaks at various times depending on cell type, e.g., 3 days for neutrophils, 8 days for macrophages, and 12 days for lymphocytes). Each of these events is linked to the subsequent development of acute airway hyperreactivity that depends on ICAM-1 gene expression and peaks at approximately 21 days after inoculation. After this time, there is progressive and chronic goblet cell metaplasia and hyperreactivity that persist indefinitely after infection and are likely driven by ongoing pressure from inhaled inflammatory stimuli and a reprogrammed innate and adaptive immune system. These chronic disease traits can be segregated by choice of mouse strain or breeding for susceptible and resistant offspring. A similar set of events may follow respiratory viral infection (e.g., due to RSV) in children that develop a persistent or recurrent asthma phenotype. Modified by permission from Reference 31.
STUDIES IN HUMANS WITH ASTHMA AND COPD
A critical aspect of our work has been to validate findings from in vitro and in vivo models of airway disease by performing studies of the innate antiviral network in human subjects with asthma or chronic bronchitis/COPD. Remarkably, select components of the antiviral network are commonly activated in subjects with asthma as well as COPD even in the apparent absence of viral infection. In the case of asthma, we found selective activation of epithelial Stat1 in the absence of any change in the airway tissue levels of IFN-γ, the cytokine that is normally capable of driving epithelial Stat1 activation and consequent ICAM-1 gene expression (62). Levels of Stat1 activation correlated with increases in expression of ICAM-1, interferon regulatory factor-1, and Stat1 itself, suggesting that activation drives transcription of downstream target genes. Together, the findings implied that asthma is characterized by an abnormality in cytokine signaling that is based in airway epithelial cells versus the traditional abnormality in cytokine production from immune cells. In a subsequent study of human subjects, we found that expression of another virus-inducible product (i.e., IL-12p80) was also increased in airway epithelial cells of asthmatic airways (51). Similar to the case for the Stat1-dependent gene network, induction of IL-12p40 gene is unaffected by glucocorticoid treatment, unrelated to allergy, and is present in a pattern similar to the one found in paramyxoviral infection.
In addition to our studies of subjects with stable asthma (often being treated with glucocorticoids), we have also monitored subjects with asthma after withdrawal from a controlled period of treatment with inhaled glucocorticoids. Within the glucocorticoid-withdrawal group, we compared subjects who experienced a flare of their disease to those that did not change (on the basis of symptoms, bronchodilator use, and measurements of maximal airflow). Although these flares could have been driven by increased allergen exposure or heightened signaling responses to viral infection, we reasoned that the distinct cell and molecular biology of the CCL5 system might also make it a candidate for mediating flares of asthma. If this were the case, we might expect CCL5 expression to be increased selectively in those with flares of their disease. Indeed, we found that expression of the epithelial CCL5 gene is inducible by glucocorticoid withdrawal, and induction occurs in conjunction with airway T-cell infiltration, obstruction, and hyperreactivity (35, 68). The precise mechanism for increased CCL5 gene expression in vivo still needs to be determined, but the absence of notable and relevant transcription factor (especially nuclear factor-κB) activation suggests that expression may depend on the same mRNA stabilization mechanism that is characteristic of the response to paramyxoviral replication. Thus, for each of three separate epithelial immune-response genes (i.e., Stat1, IL-12p40, and CCL5), activation is characteristic of the normal response to viral infection and is a signature of the presumably abnormal response in asthma. Taken together, these findings bolster the role of the epithelial antiviral network in asthma and led us to propose an alternative paradigm for the disease that includes epithelial, viral, and allergic components (Figure 4) (31).
Figure 4.

Scheme for integrating the innate antiviral response into the development of asthma and chronic bronchitis. In the case of asthma (designated by “A”), increases in Th1-like antiviral signals (e.g., Stat1 activation as well as interleukin-12p40 [IL-12p40] and CCL5 expression) in airway epithelial cells occur in concert with allergen-driven production of Th2 cytokines (e.g., IL-4, IL-5, IL-9, and IL-13) in immune cells (e.g., T cells, eosinophils, and mast cells). Macrophage activation has been detected but molecular characteristics are less well defined. The combination of epithelial, viral, and allergic components led to designation of this pathogenesis scheme as an epi-vir-all paradigm. In the case of chronic bronchitis (designated by “CB”), there is also evidence of abnormalities in airway epithelial cell, T-cell, and macrophage behavior as well as interaction between disease triggers (i.e., viral infection and cigarette smoking), but the antiviral characteristics of these cellular and molecular events still need to be defined. For each disease phenotype, these events depend on genetic susceptibility to develop a chronic airway response. Environmental and genetic variation therefore allow for overlap between disease phenotypes (i.e., asthmatic bronchitis).
In comparative studies of subjects with COPD, we did not detect constitutive activation of epithelial immune-response genes, at least for Stat1 and IL-12p40 (51, 62). This difference in epithelial cell behavior suggests that any epithelial alterations might occur in smaller airways distal to the sampling site or that the cellular site of activation was contained in another component of the innate immune system (e.g., the airway macrophage). Studies are still underway, but initial work (and previous reports by others) suggests that macrophage activation is a consistent feature of subjects with chronic bronchitis (27). This activation likely overlaps with activation found by our group and others in asthma (51, 53). Moreover, it appears that a critical site for mucous cell metaplasia is in the very distal airways (27), precisely the site of greatest epithelial metaplasia during paramyxoviral bronchiolitis. A particular challenge is to define the full behavior of epithelial and macrophage behavior under normal conditions versus viral infection or chronic airway disease, under the premise that an exaggerated response may drive clinically significant disease traits—namely, airway hyperreactivity and mucous cell metaplasia—that are common to both. Indeed, we might expect considerable synergy between the susceptibility to cigarette smoke– and virus-induced bronchiolitis in genetically susceptible individuals. Variation in environmental stimuli (i.e., allergen, virus, and cigarette smoke) and genetic susceptibility may thereby account for overlap between disease phenotypes (i.e., asthmatic bronchitis) as well.
CONCLUSIONS AND FUTURE DIRECTIONS
Abnormal immune cell accumulation in the airways is characteristic of patients with asthma and COPD. Traditionally, at least in the case of asthma, the abnormal inflammation has been linked to excessive Th2 cytokine production in the context of a skewed response of the adaptive immune system. An overlapping phenotype in chronic bronchitis suggests that this immune mechanism may contribute to airway hyperreactivity and mucous cell metaplasia in that setting as well. We submit that this immune cell behavior (a manifestation of adaptive immunity), although often a response to allergen, can also be regulated by the primary response of airway epithelial cells and macrophages (critical components of innate immunity). We have presented evidence that a subset of epithelial immune-response genes may be critical for antiviral defense and may exhibit aberrant activation in asthma. Thus, paramyxoviral infection and asthma may share a propensity to activate a network of epithelial immune-response genes that are part of the innate immune response (35, 51, 62). Macrophage activation is also found in subjects with asthma but may be even more prominent in subjects with chronic bronchitis (27, 51), so current studies aim to further characterize the behavior of the antiviral response in airway epithelial cells and macrophages in both types of subjects.
These studies will further serve to associate the innate antiviral immune response with asthma and chronic bronchitis, but will not directly address the issues of the underlying mechanisms for chronicity and susceptibility to the asthma or chronic bronchitis phenotype. Our more recent approach has therefore analyzed whether a persistent antiviral response can drive the airway disease phenotype, at least experimentally. In that regard, we have used a mouse model of viral bronchiolitis to establish the capacity of a single, transient paramyxoviral infection to permanently change airway epithelial and smooth muscle behavior toward disease traits (i.e., airway hyperreactivity and goblet cell metaplasia) found in asthma and chronic bronchitis. By taking advantage of genetic approaches, this chronic phenotype can be segregated from the acute antiviral response in a single strain of mice and can be mapped by comparing susceptible to resistant strains. The teleology of this chronic response is uncertain, but may represent an evolving but maladaptive attempt to improve antiviral host defense. Similarly, we speculate that airway hyperreactivity and goblet cell metaplasia may ordinarily offer some advantage to host defense and become disease producing only if manifest in excess for prolonged time periods. Further studies will be required to precisely identify the genes responsible for epithelial remodeling and chronic hyperreactivity in response to paramyxoviral infection, but the lack of IFN-γ–dependent regulation in this setting implies already that the viral pathway is distinct from ones driven by allergen. The interaction of viral infection with other stimuli of chronic airway disease (especially cigarette smoke) also needs to be defined. On the basis of what we have learned about the innate antiviral network, it may be feasible to adjust antiviral responses in a way that improves immunity and decreases inflammation. Examples of this approach include modifying viral genes for pathogenicity (e.g., NS1/NS2) or host genes for antiviral defense (e.g., Stat1 and CCL5/CCR5). We submit that further effort in this regard provides an opportunity to improve therapeutics for viral infections and virus-induced diseases of the airway.
Acknowledgments
The authors thank their laboratory colleagues for valuable assistance and advice in the course of this work.
Supported by grants from the National Institutes of Health (Heart, Lung, and Blood Institute), the Martin Schaeffer Fund, and the Alan A. and Edith L. Wolff Charitable Trust.
Conflict of Interest Statement: M.J.H. and Washington University received $105,000 from 2002–2005 from Roche Biosciences as a research grant to study models of airway disease. J.W.T. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. E.Y.K. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. M.S.L. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. A.C.P. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. L.P.S. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. E.A. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. Y.Z. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Murphy KM, Ouyang W, Farrar JD, Yang J, Ranganath S, Asnagli H, Afkarian M, Murphy TL. Signaling and transcription in T helper development. Annu Rev Immunol 2000;18:451–494. [DOI] [PubMed] [Google Scholar]
- 2.Holtzman MJ, Look DC, Iademarco MF, Dean DC, Sampath D, Castro M. Asthma. In: Jameson JL, editor. Principles of molecular medicine. Totawa, NJ: Humana Press; 1998. pp. 319–327.
- 3.Gavett SH, O'Hearn DJ, Li X, Huang S-K, Finkelman FD, Wills-Karp M. Interleukin 12 inhibits antigen-induced airway hyperresponsiveness, inflammation, and Th2 cytokine expression in mice. J Exp Med 1995;182:1527–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lack G, Bradley KL, Hamelmann E, Renz H, Loader J, Leung DYM, Larsen GL, Gelfand EW. Nebulized IFN-γ inhibits the development of secondary allergic responses in mice. J Immunol 1996;157:1432–1439. [PubMed] [Google Scholar]
- 5.Sur S, Wild JS, Choudhury BK, Sur N, Alam R, Klinman DM. Long term prevention of allergic lung inflammation in a mouse model of asthma by CpG oligodeoxynucleotides. J Immunol 1999;162:6284–6293. [PubMed] [Google Scholar]
- 6.Finotto S, Neurath NF, Glickman JN, Qin S, Lehr HA, Green FHY, Ackerman K, Haley K, Galle PR, Szabo SJ, et al. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science 2002;295:336–338. [DOI] [PubMed] [Google Scholar]
- 7.Randolph DA, Stephens R, Carruthers CJL, Chaplin DD. Cooperation between Th1 and Th2 cells in a murine model of eosinophilic airway inflammation. J Clin Invest 1999;104:1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen G, Berry G, DeKruyff R, Umetsu D. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J Clin Invest 1999;103:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyahara N, Swanson BJ, Takeda K, Taube C, Miyahara S, Kodama T, Dakhama A, Ott VL, Gelfand EW. Effector CD8+ T cells mediate inflammation and airway hyper-responsiveness. Nat Med 2004;10:865–869. [DOI] [PubMed] [Google Scholar]
- 10.Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, Taniguchi M, Grusby MJ, Dekruyff RH, Umetsu DH. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyper-reactivity. Nat Med 2003;9:582–588. [DOI] [PubMed] [Google Scholar]
- 11.Stock P, Akbari O, Berry G, Freeman GJ, Dekruyff RH, Umetsu DH. Induction of T helper type 1-like regulatory cells that express Foxp3 and protect against airway hyper-reactivity. Nat Immunol 2004;5:1149–1156. [DOI] [PubMed] [Google Scholar]
- 12.Kay AB. Allergy and allergic diseases. N Engl J Med 2001;344:30–37. [DOI] [PubMed] [Google Scholar]
- 13.Berry MA, Parker D, Neale N, Woodman L, Morgan A, Monk P, Bradding P, Wardlaw AJ, Pavord ID, Brightling CE. Sputum and bronchial submucosal IL-13 expression in asthma and eosiniophililc bronchitis. J Allergy Clin Immunol 2004;114:1106–1109. [DOI] [PubMed] [Google Scholar]
- 14.Ober C, Moffatt MF. Contributing factors to the pathobiology: the genetics of asthma. Clin Chest Med 2000;21:245–261. [DOI] [PubMed] [Google Scholar]
- 15.Willliams CMM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J Exp Med 2000;192:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bandeira-Melo C, Herbst A, Weller PF. Eotaxins: contributing to the diversity of eosinophil recruitment and activation. Am J Respir Cell Mol Biol 2001;24:653–657. [DOI] [PubMed] [Google Scholar]
- 17.Grayson MH, Holtzman MJ. Lessons from allergic rhinitis versus asthma pathogenesis and treatment. Immunol Allergy Clin N Am 2002;22:845–869. [Google Scholar]
- 18.Melhop PD, van de Rijn M, Goldberg AB, Brewer JP, Kurup VP, Martin TR, Oettgen HC. Allergen-induced bronchial hyperreactivity and eosinophilic inflammation occur in the absence of IgE in a mouse model of asthma. Proc Natl Acad Sci USA 1997;94:1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hogan SP, Mould A, Kikutani H, Ramsay AJ, Foster PS. Aeroallergen-induced eosinophilic inflammation, lung damage, and airways hyperreactivity in mice can occur independently of IL-4 and allergen-specific immunoglobulins. J Clin Invest 1997;99:1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science 1998;282:2258–2261. [DOI] [PubMed] [Google Scholar]
- 21.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. IL-5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med 1995;183:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corry DB, Folkesson HG, Warnock ML, Erle DJ, Matthay MA, Wiener-Kronish JP, Locksley RC. Interleukin 4, but not interleukin 5 or eosinophils, is required in a murine model of acute airway hyperreactivity. J Exp Med 1995;183:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holtzman MJ, Sampath D, Castro M, Look DC, Jayaraman S. The one-two of T helper cells: does interferon-γ knockout the Th2 hypothesis for asthma? Am J Respir Cell Mol Biol 1996;14:316–318. [DOI] [PubMed] [Google Scholar]
- 24.Hakonarson H, Bjornsdottir US, Ostermann E, Arnason T, Adalsteinsdottir AE, Halapi E, Shkolny D, Kristjansson K, Gudnadottir SA, Frigge ML, et al. Allelic frequencies and patterns of single-nucleotide polymorphisms in candidate genes for asthma and atopy in Iceland. Am J Respir Crit Care Med 2001;164:2036–2044. [DOI] [PubMed] [Google Scholar]
- 25.Drazen JM, Arm JP, Austen KF. Sorting out the cytokines of asthma. J Exp Med 1996;183:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Postma DS, Boezen HM. Rationale for the Dutch hypothesis. Chest 2004;126:96S–104S. [DOI] [PubMed] [Google Scholar]
- 27.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004;350:2645–2653. [DOI] [PubMed] [Google Scholar]
- 28.O'Sullivan S, Cormican L, Faul JL, Ichinohe S, Johnston SL, Burke CM, Poulter LW. Activated, cytotoxic CD8+ T lymphocytes contribute to the pathology of asthma death. Am J Respir Crit Care Med 2001;164:560–564. [DOI] [PubMed] [Google Scholar]
- 29.Meyers DA, Larj MJ, Lange L. Genetics of asthma and COPD. Chest 2004;126:105S–110S. [DOI] [PubMed] [Google Scholar]
- 30.Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, Chen Q, Homer RJ, Wang J, Rabach LA, et al. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodeling. J Clin Invest 2002;110:463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holtzman MJ, Morton JD, Shornick LP, Tyner JW, O'Sullivan MP, Antao A, Lo M, Castro M, Walter MJ. Immunity, inflammation, and remodeling in the airway epithelial barrier: epithelial-viral-allergic paradigm. Physiol Rev 2002;82:19–46. [DOI] [PubMed] [Google Scholar]
- 32.Midulla F, Villani A, Panuska JR, Dab I, Kolls JK, Merolla R, Ronchetti R. Respiratory syncytial virus lung infection in infants: immunoregulatory role of infected alveolar macrophages. J Infect Dis 1993;168:1515–1519. [DOI] [PubMed] [Google Scholar]
- 33.Nakajima S, Look DC, Roswit WT, Bragdon MJ, Holtzman MJ. Selective differences in vascular endothelial- vs. airway epithelial-T cell adhesion mechanisms. Am J Physiol 1994;267:L422–L432. [DOI] [PubMed] [Google Scholar]
- 34.Nakajima S, Roswit WT, Look DC, Holtzman MJ. A hierarchy for integrin expression and adhesiveness among T cell subsets that is linked to TCR gene usage and emphasizes Vδ1+ γδ T cell adherence and tissue retention. J Immunol 1995;155:1117–1131. [PubMed] [Google Scholar]
- 35.Taguchi M, Sampath D, Koga T, Castro M, Look DC, Nakajima S, Holtzman MJ. Patterns for RANTES secretion and intercellular adhesion molecule-1 expression mediate transepithelial T cell traffic based on analyses in vitro and in vivo. J Exp Med 1998;187:1927–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holtzman MJ, Castro M, Look DC, O'Sullivan M, Walter MJ. Regulation of epithelial-leukocyte interaction and epithelial immune-response genes. In: Busse WW, Holgate ST, editors. Asthma and rhinitis. Cambridge, MA: Blackwell Scientific; 2000. pp. 784–800.
- 37.Look DC, Keller BT, Rapp SR, Holtzman MJ. Selective induction of intercellular adhesion molecule-1 by interferon-γ in human airway epithelial cells. Am J Physiol 1992;263:L79–L87. [DOI] [PubMed] [Google Scholar]
- 38.Look DC, Pelletier MR, Holtzman MJ. Selective interaction of a subset of interferon-γ response element binding proteins with the intercellular adhesion molecule-1 (ICAM-1) gene promoter controls the pattern of expression on epithelial cells. J Biol Chem 1994;269:8952–8958. [PubMed] [Google Scholar]
- 39.Look DC, Pelletier MR, Tidwell RM, Roswit WT, Holtzman MJ. Stat1 depends on transcriptional synergy with Sp1. J Biol Chem 1995;270:30264–30267. [DOI] [PubMed] [Google Scholar]
- 40.Walter MJ, Look DC, Tidwell RM, Roswit WT, Holtzman MJ. Targeted inhibition of interferon-γ-dependent ICAM-1 expression using dominant-negative Stat1. J Biol Chem 1997;272:28582–28589. [DOI] [PubMed] [Google Scholar]
- 41.Look DC, Roswit WT, Frick AG, Gris-Alevy Y, Dickhaus DM, Walter MJ, Holtzman MJ. Direct suppression of Stat1 function during adenoviral infection. Immunity 1998;9:871–880. [DOI] [PubMed] [Google Scholar]
- 42.Koga T, Sardina E, Tidwell RM, Pelletier MR, Look DC, Holtzman MJ. Virus-inducible expression of a host chemokine gene relies on replication-linked mRNA stabilization. Proc Natl Acad Sci USA 1999;96:5680–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, Levy DE, Durbin JE. The role of interferon in influenza virus tissue tropism. J Virol 1998;72:8550–8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durbin JE, Johnson TR, Durbin RK, Mertz SE, Morotti RA, Peebles RS, Graham BS. The role of IFN in respiratory syncytial virus pathogenesis. J Immunol 2002;168:2944–2952. [DOI] [PubMed] [Google Scholar]
- 45.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, et al. IRF-7 is the master regulator of type-1 interferon-dependent immune responses. Nature 2005;434:772–777. [DOI] [PubMed] [Google Scholar]
- 46.Lo MS, Brazas RM, Holtzman MJ. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and type I interferon responsiveness. J Virol 2005;79:9315–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pirhonen J, Sareneva T, Kurimoto M, Julkunen I, Matikainen S. Virus infection activates IL-1b and IL-18 production in human macrophages by a caspase-1-dependent pathway. J Immunol 1999;162:7322–7329. [PubMed] [Google Scholar]
- 48.Graham BS, Perkins MD, Wright PF, Karzon DT. Primary respiratory syncytial virus infection in mice. J Med Virol 1988;26:153–162. [DOI] [PubMed] [Google Scholar]
- 49.Sumino KC, Agapov E, Pierce RA, Trulock EP, Pfeifer JD, Ritter JH, Gaudreault-Keener M, Storch GA, Holtzman MJ. Detection of severe human metapneumovirus infection by real-time polymerase chain reaction and histopathological assessment. J Infect Dis (In press) [DOI] [PMC free article] [PubMed]
- 50.Alavarez R, Tripp RA. The immune response to human metapneumovirus is associated with aberrant immunity and impaired virus clearance in BALB/c mice. J Virol 2005;79:5971–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walter MJ, Kajiwara N, Karanja P, Castro M, Holtzman MJ. IL-12 p40 production by barrier epithelial cells during airway inflammation. J Exp Med 2001;193:339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Domachowske JB, Rosenberg HF. Respiratory syncytial virus infection: immune response, immunopathogenesis, and treatment. Clin Microbiol Rev 1999;12:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walter MJ, Holtzman MJ. Epithelial production of IL-12 p80 during viral infection and asthma supports an altered paradigm for airway inflammation. In: Eissa NT, Huston DP, editors. Biology of airway inflammation: therapeutic targets, lung biology in health and disease. New York: Marcel Dekker; 2002.
- 54.Tyner JW, Uchida O, Kajiwara N, Kim EY, O'Sullivan MP, Walter MJ, Cook DN, Danoff TM, Holtzman MJ. CCL5/CCR5 interaction provides anti-apoptotic signals for macrophage survival during viral infection. Nat Med (In press). [DOI] [PMC free article] [PubMed]
- 55.Hull J, Rowlandds K, Lockhart E, Moore C, Sharland M, Kwiatkowski D. Variants of the chemokine receptor CCR5 are associatd with severe bronchiolitis caused by respiratory syncytial virus. J Infect Dis 2003;188:904–907. [DOI] [PubMed] [Google Scholar]
- 56.Durbin JE, Fernandez-Sesma A, Lee C-K, Dharma Rao T, Frey AB, Moran TM, Vukmanovic S, Garcia-Sastre A, Levy DE. Type I IFN modulates innate and specific antiviral immunity. J Immunol 2000;164:4220–4228. [DOI] [PubMed] [Google Scholar]
- 57.Johnson TR, Mertz SE, Gitiban N, Hammond S, LeGallo R, Durbin RK, Durbin JE. Role for innate IFNs in determining respiratory syncytial virus immunopathology. J Immunol 2005;174:7234–7241. [DOI] [PubMed] [Google Scholar]
- 58.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, et al. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat Genet 2003;33:388–391. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Takami K, Lo MS, Patel AC, Huang G, Yu Q, Roswit WT, Holtzman MJ. Improved interferon efficacy and viral clearance by engineering a super-Stat1 transgene that drives transcriptosome assembly [abstract]. Proc Am Thorac Soc 2005;2:A308. [Google Scholar]
- 60.Walter MJ, Morton JD, Kajiwara N, Agapov E, Holtzman MJ. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J Clin Invest 2002;110:165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holtzman MJ, Fabbri LM, O'Byrne PM, Gold BD, Aizawa H, Walters EH, Alpert SE, Nadel JA. Importance of airway inflammation for hyperresponsiveness induced by ozone in dogs. Am Rev Respir Dis 1983;127:686–690. [DOI] [PubMed] [Google Scholar]
- 62.Sampath D, Castro M, Look DC, Holtzman MJ. Constitutive activation of an epithelial signal transducer and activator of transcription (Stat1) pathway in asthma. J Clin Invest 1999;103:1353–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang W, Yang H, Kong X, Mohapatra S, Juan-Vergara HS, Hellermann G, Behera S, Singam R, Lockey RF, Mohapatra SS. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat Med 2005;11:56–62. [DOI] [PubMed] [Google Scholar]
- 64.Shen Y, Zhu H, Shenk T. Human cytomegalovirus IE1 and IE2 proteins are mutagenic and mediate “hit and run” oncogenic transformation in cooperation with the adenovirus E1A proteins. Proc Natl Acad Sci USA 1997;94:3341–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nevels M, Tauber B, Spruss T, Wolf H, Dobner T. “Hit and run” transformation by adenovirus oncogenes. J Virol 2001;75:3089–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morton JD, Alevy Y, Peltz G, Holtzman MJ. Functional genomics of asthma: role of CLCA3 in goblet cell metaplasia but not airway hyperreactivity. J Allergy Clin Immunol 2004;113:S205. [Google Scholar]
- 67.Holtzman MJ, Kim EY, Morton JD. Genetic and genomic approaches to complex lung diseases using mouse models. In: Peltz G, editor. Computational genetics and genomics: tools for understanding disease. Totawa, NJ: Humana Press; 2005. pp. 99–141.
- 68.Castro M, Block SR, Jenkersen MV, De S, Martino DL, Hamilos RB, Cochran XE, Zhang L, Wang H, Bradley JP, et al. Asthma exacerbation after glucocorticoid withdrawal reflects T cell recruitment to the airway. Am J Respir Crit Care Med 2004;169:842–849. [DOI] [PubMed] [Google Scholar]