

Abstract
Baeyer-Villiger monooxygenases (BVMOs), mostly flavoproteins, were shown to be powerful biocatalysts for synthetic organic chemistry applications and were also suggested to play key roles for the biosyntheses of various natural products. Here we present the three-dimensional structure of MtmOIV, a 56 kD homo-dimeric FAD- and NADPH-dependent monooxygenase, which catalyzes the key frame-modifying step of the mithramycin biosynthetic pathway and currently the only BVMO proven to react with its natural substrate via a Baeyer-Villiger reaction. MtmOIV’s structure was determined by X-ray crystallography using molecular replacement to a resolution of 2.9Å. MtmOIV cleaves a C-C bond, essential for the conversion of the biologically inactive precursor, premithramycin B, into the active drug mithramycin. The MtmOIV structure combined with substrate docking calculations and site-directed mutagenesis experiments implicate several residues to participate in co-factor and substrate binding. Future experimentation aimed at broadening the substrate specificity of the enzyme could facilitate the generation of chemically diverse mithramycin analogues through combinatorial biosynthesis.
Baeyer-Villiger monooxygenases (BVMOs)& were described and classified as a relatively new sub-class of flavoprotein monooxygenases (1-4). BVMOs are powerful biocatalysts for synthetic organic chemistry applications(5-7), were also suggested to play key roles for the biosyntheses of various natural products(8-13), and are involved in pro-drug activation (14, 15) as well as in biodegradation reactions (16-18).
Mithramycin (MTM, also known as aureolic acid, mithracin, LA-7017, PA-144, and plicamycin) is an aureolic acid-type polyketide anticancer antibiotic produced by the soil bacterium Streptomyces argillaceus (ATCC 12956) and various other streptomycetes. The small distinct group of aureolic acid-type anticancer antibiotics includes MTM, chromomycin, olivomycin, UCH9 and durhamycin. All of these drugs contain the same polyketide derived tricyclic aromatic core with a highly functionalized pentyl side chain attached at C-3, but vary with respect to their 7-side chains and their saccharide patterns. MTM exhibits anticancer activity by inhibiting replication and transcription via cross-linking of DNA strands, affecting predominantly GC-rich regions normally used by Sp-promoters (19-23). MTM has been used clinically to treat certain cancers, such as testicular carcinoma (24), and bone diseases, such as Paget’s disease (25), and particularly to address cancer-related hypercalcemia (26). MTM was also recently identified as a lead drug for a potential therapy for Huntington’s disease (27). However, the clinical use of MTM is currently limited due to its high toxicity, which potentially could be reduced with the generation of new analogues. MTM consists of a polyketide-derived tricyclic core with a highly functionalized pentyl side chain attached in 3-position and five deoxysugars linked as trisaccharide and disaccharide chains in 2- and 6- positions, respectively (28) (Figure 1). The MTM biosynthesis (Figure 1) proceeds through the polyketide derived tetracyclic premithramycinone, to which five sugar moieties and one C-methyl group in 9-position are added leading to premithramycin B (29). Then, MtmOIV oxidatively cleaves the fourth ring via a Baeyer-Villiger reaction generating MTM’s characteristic tricyclic aglycone core and highly functionalized pentyl side chain in 3-position. The Baeyer-Villiger reaction precedes lactone opening, decarboxylation, and the final step of MTM biosynthesis, reduction of the 4′-keto group catalyzed by ketoreductase MtmW (30).
Figure 1.

Biosynthetic pathway to mithramycin. The reaction catalyzed by BVMO MtmOIV yields premithramycin B-lactone, validating the BV mechanism, and is further converted to mithramycin DK (presumably spontaneously). The ketoreductase MtmW catalyzes the final step of mithramycin biosynthesis
The function of MtmOIV as a BVMO was proven through studies with the isolated and overexpressed enzyme, allowing the isolation and characterization of key reaction intermediates, such as premithramycin B-lactone, the product mithramycin DK and various shunt products (31).
Only a few crystal structures of Baeyer-Villiger reaction performing enzymes are available to date, including phenylacetone monooxygenase from the thermophilic bacterium Thermobifida fusca and the oxygenating component of 3,6-diketocamphane monooxygenase (32, 33). Various BVMOs were described and studied, and most of these seem to be involved in oxidative degradation pathways, with substrates ranging from simple ketones, such as acetone or cyclohexanone, to steroids, such as progesterone (34, 35). However, in many cases it remains unclear whether the used substrates are the true natural substrates of such BV-reaction performing enzymes, and one could argue in several cases that the enzymes have no other choice but to perform a BV-reaction when confronted with a relatively simple ketone-substrate. In contrast, the MtmOIV substrate premithramycin B is a highly functional and decorated molecule, and so far the most complex compound to date converted by a BV-reaction. Thus, MtmOIV might be arguably the only BVMO, which was investigated and kinetically characterized with its proven natural substrate (31, 36-38).
Combinatorial biosynthesis produces new natural product drug analogues by recombining biosynthetic gene clusters with genes from other pathways and has been used recently to generate new MTM analogues, some with improved biological activities (20, 39-42). Recent efforts were directed toward the design of new MTM analogues with altered saccharide patterns. However, despite some success, the number of such analogues is limited, partly due to restrictions imposed by the bottleneck-enzyme MtmOIV, as the accumulation of various premithramycins suggested (41, 42).
It was expected that the crystal structure of MtmOIV would give insight into the intriguing enzymatic BV mechanism and would pave the way to alter the substrate specificity of MtmOIV necessary for the generation of further mithramycin analogues. In its current state MtmOIV appears to tolerate only some changes to the substrate, and compounds with a drastically altered, complete saccharide pattern cannot be converted, and remain biologically inactive premithramycins. We hypothesized that if the binding properties of substrate premithramycin B in MtmOIV’s active site were better understood, it would be feasible to engineer selected changes to residues involved in substrate binding, potentially leading to an enzyme with altered substrate specificity useful for the generation of further new bioactive analogues of MTM. To this end, we report here the crystal structure of MtmOIV, its putative substrate binding cavity and residues likely involved in substrate binding.
Based on function and sequence similarity, MtmOIV was previously characterized as a member of the GR2 subfamily of FAD-dependent enzymes (37, 43). Recent additions to this family include PgaE and CabE, hydroxylases involved in angucycline biosynthesis pathways that have high sequence similarity (>45%) to MtmOIV. The crystal structures of PgaE and CabE were recently reported (44), and given the high sequence conservation, we were able to use these structures as models for molecular replacement. Previously we reported the overexpression, crystallization, and preliminary structure determination of MtmOIV (37). Here, we present the refined 2.9 Å crystal structure of MtmOIV enabling characterization of the FAD binding site and identification of the putative substrate binding pocket. Mutations to residues thought to be involved in FAD binding reduced the activity of the enzyme, as expected. Furthermore, co-crystallization and docking experiments with substrate premithramycin B provides some preliminary insight into the substrate binding, for which further evidence was gathered from selected mutations.
EXPERIMENTAL PROCEDURES
Protein expression, purification, and crystallization
The cloning, overexpression, purification, crystallization, and initial data collection have previously been reported (37). Briefly, the mtmOIV coding sequence (1.5 kb) was amplified and subcloned into pRSETb and expressed with an N-terminal His6 fusion tag for purification. This construct was then transformed into BL21(DE3)pLysS cells. A 1-liter culture of NZCYM medium (Fisher) supplemented with 1 mM ampicillin and 1 mM chloramphenicol was grown at 37 °C until an OD600 of 0.5-0.7 and then IPTG was added to a final concentration of 1 mM and grown at 20 °C for an additional 12 hours, which led to a higher protein yield of 14 mg/L culture. Protein concentration was determined by Bradford Assay (Sigma) for crystallization studies and BCA Assay (Pierce) for kinetic studies. Using 1 mL well solution volume and drops consisting of 1 μL protein and 1 μL well solution, crystallization of MtmOIV was accomplished at room temperature using the hanging drop vaporization method with protein at 8 mg/mL and well solution consisting of 0.1 M Na-HEPES pH 7.5, 10%(v/w) PEG 8000, and 8%(v/v) ethylene glycol. The best crystals typically grew within 3-5 days, were harvested directly from the crystallization drop, and then plunged into liquid nitrogen for storage until data collection. Co-crystallization was performed by pre-incubation of protein with premithramycin B (2:1 molar ratio with protein) for at least 1 hour before setting crystallization trays.
Data collection and structure determination
Crystals were initially screened using an in-house X-ray source (Rigaku Cu-rotating anode X-ray generator, R-AXIS 4++ detector, CrystalClear software) with final data collected on the SER-CAT (ID-22) beamline at the Advanced Photon Source of the Argonne National Laboratory. The data were processed using HKL2000 (45) to space group C2 with the cell dimensions a = 145.27, b = 114.44, c = 138.56, α=90.00°, β=103.03°, γ=90.00°. Initial cell content analysis using CCP4 (46) suggested 4 molecules per ASU with a solvent content of 51% (37). However, we only observed 3 molecules in the ASU, yielding a relatively high solvent content of 64%. Initial phases were determined by molecular replacement with PHASER within the CCP4 Software package (46) using PgaE (PDB code 2QA1) as a search model where ClustalW (47) was used to create a sequence alignment between MtmOIV and PgaE which was then input into the program Chainsaw/CCP4 (48) to create an initial model. Model building was performed using PHENIX (49) and subsequent rebuilding and refinement, as well as, water assignments were performed using COOT(50), CNS (51), and PHENIX (49) with restrained NCS averaging and TLS refinement. The final structure of MtmOIV was refined to 2.9 Å resolution with a final R/Rfree of 0.24/0.29 and contained 494 of 533 residues. The N-terminal His6-tag was not observed within the crystal structure and assumed to be disordered since it was not removed during purification. The final structure was analyzed using Molprobity (52) and figures created using PyMOL (53). RMSD analysis was performed using STRAP (54).
MtmOIV mutagenesis
The putative FAD binding site mutants F180A and F272A were prepared and analyzed (Table 2). Due to high GC content (>80%), the Stratagene QuikChange kit was utilized with primers designed for site saturation (55) with a slow down PCR technique incorporated in the design of the temperature cycles (56). The mutagenesis primers used for F180A: (forward) 5′ - GAT CGG GCT CCC GGC ACC GAG GCC ACC GTC CGC GCC - 3′ and (reverse) 5′ - GGT GCC GGG AGC CCG ATC CGA GGC CAG CCG CCA CAC - 3′; F272A: (forward) 5′ - TCC CGG GCA GGA GAC GCG AGC CGC CAG GCG AAG CGC TAC - 3′ and (reverse) 5′ - CGC GTC TCC TGC CGC GGA GAG CCA CGA CAC CGA TTC - 3′. Substrate binding site mutants and R52A were prepared with Stratagene QuikChangeXL kit following standard protocol. Mutagenesis primers used for F89A: (forward) 5′ - C TTC GCC GGG ATC GCC ACC CAG GGC CTG - 3′ and (reverse) 5′ - CAG GCC CTG GGT GGC GAT CCC GGC GAA G - 3′; L107A: (forward) 5′ - C CCG TAC ACG GGC GCG GTG CCG CAG TCG - 3′ and (reverse) 5′ - CGA CTG CGG CAC CGC GCC CGT GTA CGG G - 3′; R204A: (forward) 5′ - GAG GTG CCG CGC GCC TGG GAG CGC AC - 3′ and (reverse) 5′ - GT GCG CTC CCA GGC GCG CGG CAC CTC - 3′. R52A: (forward) 5′ - C GGC CAC GAC GCG GCG GGG GCC - 3′ and (reverse) 5′ - GGC CCC CGC CGC GTC GTG GCC G - 3′.
Table 2.
Activity comparison of MtmOIV, the substrate binding site mutants F89A, L107A and R204A (blue), and the FAD binding site mutants F180A, F272A and R52A (red), compared to the wild-type enzyme (WT) using NADPH (preferred) or NADH (less preferred) as co-factor
| Km (μM) PreB | kcat (s-1) | kcat/Km (s-1 mM-1) | FAD mole ratio (molMtmOIV/molFAD) | Uncoupling ratio (102 μM PreB, 10 min); (molproduct/molH2O2) | |
|---|---|---|---|---|---|
| NADPH | |||||
| WT | 31 ± 10 | 1.10 ± 0.11 | 35 | 0.56 | 15 |
| F89A | 27 ± 9 | 1.27 ± 0.11 | 47 | 0.42 | >80 |
| L107A | --- | --- | --- | <0.01 | --- |
| R204A | 18 ± 3 | 0.73 ±0.02 | 41 | 0.24 | >300 |
| R52A | 79 ± 10 | 0.21 ± 0.01 | 2.7 | 0.07 | >50 |
| F180A | --- | --- | --- | < 0.01 | --- |
| F272A | --- | --- | --- | < 0.01 | --- |
| NADH | |||||
| WT | 38 ± 18 | 0.97 ± 0.14 | 26 | 0.56 |
FAD Content
Each protein preparation of wild type and mutant MtmOIV enzymes contained non-covalently bound FAD molecules demonstrated by the bright to faint yellow color of the preparations following purification. The concentration of FAD was determined using a Shimadzu UV-1800 spectrophotometer. Initially, approximately 100 μg of protein (100 μL) was diluted 1:3 in 20 mM potassium phosphate (pH 8.25) and 25 μL of 50% trichloroacetic acid (TCA, Fisher) was added to bring the final concentration of the solution to 3% TCA. The protein was precipitated on ice for 30 min and spun at 10,000 g for 5 min. The supernatant was removed and its absorbance was measured. FAD content was determined by absorbance at 450 nm (ε450 = 11,300 M-1 cm-1).
Kinetic analysis of MtmOIV
A kinetic assay has been previously reported (31). Briefly, we monitored the conversion (pH 8.25, 30 °C, 10 min) of premithramycin B by MtmOIV in a continuous assay measuring the oxidation of NADPH at 340 nm (ε340 = 6220 m-1 cm-1) in the presence of FAD and O2 (0.25 mm NADPH/NADH; 0.1 mm FAD added; open cuvette). The reactions were initiated by addition of 1 μm wild type or mutant MtmOIV. Kinetic parameters were determined by fitting with Kaleidagraph 4.0 (Synergy).
Uncoupling Assays
To estimate reaction uncoupling, production of hydrogen peroxide was measured. At the termination of a 10 minute kinetic reaction (1 μM enzyme, 102 μM substrate, 250 μM NADPH, 100 μM FAD, open cuvette), a 100 μL sample was withdrawn and used in the following assay. Briefly, 700 μL of 5mm 4-aminoantipyrene, 10mm vanillic acid, and 40 U/mL horseradish peroxidase in 0.2 m sodium chloride, 0.2 m MOPS, pH 7.5 was combined with 100 μL of enzyme reaction. In this assay, horseradish peroxidase uses hydrogen peroxide to oxidize 4-aminoantipyrene creating a product that condenses with vanillic acid to produce a red quinone imine dye. The dye has a broad absorbance spectrum peaking at ∼490 nm. A 30% (w/w) H2O2 stock (Mallinckrodt) was used to create a standard curve from 0 - 1 mm and the absorbance at 490 nm was measured to determine hydrogen peroxide content in reaction samples (57, 58).
Premithramycin B docking experiments
The structure of premithramycin B was created using the PRODRG2 Server (59, 60), and docking experiments were performed using AutoDock 4 (61) and an Autogrid starting location was based on difference density observed in a low resolution co-crystal structure from a MtmOIV-premithramycin B co-crystallization attempt (data not shown).
RESULTS
Overall Structure
MtmOIV crystallized in space group C2. The structure was refined to a resolution of 2.9 Å with 3 molecules in the asymmetric unit. The final structure contains residues 14-508 of 533 total residues for MtmOIV including the FAD cofactor (Figure 2A). The FAD cofactor was found non-covalently bound in the crystal structure (Figure 2B). Additionally, the N-terminal His6-tag used for purification and the last 25 residues of the C-terminus were found to be disordered, and not present. About 15% of the side chains were not visible in the electron density maps and were therefore left out of the final structure. This observation is likely attributed to both the high solvent content of the crystals (64%) and low resolution of the crystal structure. There is an absent loop along residues 218-226 in the MtmOIV structure which corresponds to a previously reported disordered surface loop in PgaE, likely attributed to mobility and shown to be involved in the association with NADPH (44), which also is a co-factor of MtmOIV.
Figure 2.

A. Overall structure of MtmOIV. The FAD binding domain is shown in gold, the middle domain in blue, and the C-terminal domain in green. The FAD cofactor is shown as spheres. The Rossman-type fold and the substrate/co-factor binding cavity are indicated accordingly. B. View of FAD binding with FAD shown in stick figure and simulated annealing omit |Fo-Fc| electron density (3.5σ) for FAD in green mesh. The FAD binding domain is in gold and the middle domain is in blue. C. Superposition of the crystal structure of MtmOIV (gold) and PgaE (green). The FAD cofactor is shown as spheres. D. FAD binding with involved residues and distances. Figure was created using LIGPLOT (62).
As expected, based on high sequence identity (∼43%), the structure of MtmOIV is very similar to the structure of PgaE with an RMSD of 2.38 Å for 463 aligned residues (Figure 2C). In total, 98.3% of the MtmOIV residues were in the Ramachandran allowed regions with 78% in most favored regions. Final data collection and refinement statistics are summarized in Table 1. Figure 2A shows that MtmOIV contains three domains: the FAD binding domain (residues 14-183 and 276-394), the middle domain (residues 184-275), and the C-terminal domain (residues 395-508). The FAD binding domain contains two Rossman-like βαβ-folds (G19-G24 and G162-G167) and forms the central hub of the enzyme’s structure.
Table 1.
MtmOIV data collection and refinement summary
| Data Collection and Refinement Statistics for MtmOIV | |
|---|---|
| Native | |
| Space group | C2 |
| Mol/ASU | 3 |
| λ (Å) | 1.0 |
| Matthew’s coefficient | 3.91 |
| Solvent content (%) | 68.34 |
| a (Å) | 145.24 |
| b (Å) | 114.48 |
| c (Å) | 138.61 |
| α (°) | 90.00 |
| β (°) | 103.03 |
| γ (°) | 90.00 |
| Resolution (Å) * | 2.9 - 30 (2.9 - 3.0) |
| Completeness (%)* | 96.3 (91.7) |
| Redundancy* | 3.5 (2.5) |
| Rsym *† | 6.8 (46.1) |
| I / σ * | 22.4 (2.1) |
| R§/Rfree¶ | 0.24/0.29 |
| # of unique reflections | 47,403 |
| Wilson B (Å2) | 79.08 |
| Average B-factors (Å2) | 92.3 |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.908 |
| # water molecules | 6 |
| Ramachandran most favored (%) | 78.0 |
| Ramachandran additionally allowed (%) | 17.3 |
| Ramachandran generously allowed (%) | 3.0 |
| Ramachandran disallowed (%) | 1.7 |
| PDB ID code | 3FMW |
Rsym = Σhkl,j (|Ihkl-<Ihkl>|) / Σhkl,j Ihkl, where <Ihkl> is the average intensity for a set of j symmetry related reflections and <Ihkl> is the value of the intensity for a single reflection within a set of symmetry-related reflections.
R factor = Σhkl(||Fo| - |Fc||)/Σhkl|Fo| where Fo is the observed structure factor amplitude and Fc is the calculated structure factor amplitude.
Rfree = Σhkl,T(||Fo| - |Fc||)/Σhkl,T|Fo|, where a test set, T (5% of the data), is omitted from the refinement.
indicates statistics in last resolution shell shown in parenthesis.
Within MtmOIV, FAD can potentially adopt one of two structural conformations (IN or OUT) based on the solvent accessibility of the moiety and its orientation toward the putative substrate binding pocket. In contrast to the current literature, the FAD cofactor appears to be stabilized in the IN conformation. Thirteen direct hydrogen bonds could be identified within the binding pocket stabilizing the FAD moiety and promoting solvent inaccessibility to the FAD molecule (Figure 2B and 2D). R52A is involved in a hydrophobic interaction and a hydrogen bond interaction (3.07 Å) with the isoalloxazine ring of FAD. These interactions place R52 in a reasonable position to perform its chemistry. With RebC, it was recently demonstrated that FAD adopts an IN conformation when substrate is bound or FAD is reduced and remains in an OUT conformation in substrate free and non-reduced FAD states (63). Although a secondary structure of MtmOIV in the substrate bound state is needed to confirm the orientation of FAD, from this single state structure, FAD appears to be in the IN conformation.
In the structural alignment, the elements are numbered the same as PgaE for reference (Figure 3). MtmOIV is a known dimer (Figure 4) in solution (37) and the dimer interface can be observed within the crystal structure, stretching both along the FAD binding domain (involving residues R345, L348, N349, R351, A352, L354, A355, L356, R358, D360, E361, Q362, H363, T364, P366, L367, F370, E373, L374, T377, E379, Y383, F384, M387) and the middle domain (involving residues T183, E184, T186, V187, T232, D274). Using the PISA Server (64), the buried surface area between the dimers was calculated to be ∼1200 Å2, which is ∼5.7% of total surface area for each monomer.
Figure 3.
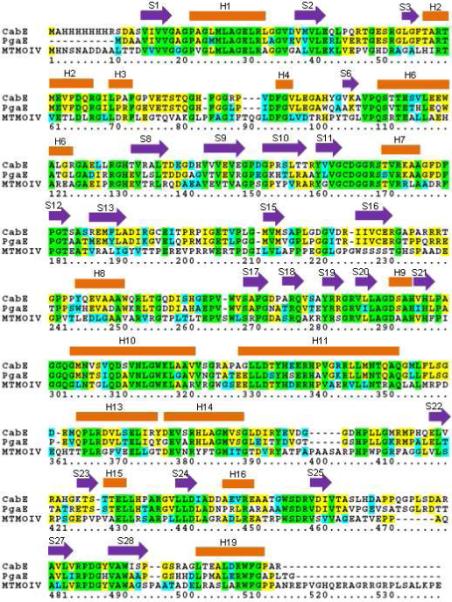
Sequence alignment of MtmOIV, PgaE, and CabE. Green shading indicates residues that are completely conserved, yellow indicates partially conserved, and cyan indicates similar residues. Secondary α-helix structures are labeled as orange blocks above the residues, and β-sheets with purple arrows. Structural labels have the same numbering as the PgaE structure (44).
Figure 4.

Biological subunit of MtmOIV as a dimer, which has ∼1200 Å2 of buried surface area (5.7% of total surface area for each monomer) which is mediated along the FAD binding domain and the middle domain.
Co-Factor binding
To further probe the FAD binding site, interactions with residues F180 and F272 were tested by mutation of these residues to alanine. Both phenylalanine residues seem involved only in more or less distant hydrophobic and/or steric interactions with the aromatic rings of FAD. F272 appears to interact with the isoalloxazine moiety of FAD, and F180 rather distantly with FAD’s adenine moiety. Thus, both mutations were designed to decrease FAD binding, but not to completely inactivate the enzyme (Figure 5A). Unfortunately, FAD binding was not maintained with these mutations and activity was almost completely lost and turnover was so limited that kinetic and catalytic parameters were not able to be solved within the substrate concentration range tested, up to 200 μM (Table 2). Kinetic activity was compared to the native enzyme by monitoring the oxidation of the NADPH cofactor in the presence of FAD and O2.
Figure 5.

A. Residues F180 (green), close to the adenine moiety of FAD, and F272 (green), close to the isoalloxazine moiety of FAD (gray stick with N atoms = blue, O-atoms = red and P atoms = orange), were selected for mutagenesis to support the FAD binding area in MtmOIV (gold/blue ribbon). B. Residues L107, R204 and F89 (all in green) were chosen to support their involvement in the substrate (premithramycin B, gray stick with O-atoms = red) binding. The isoalloxazine moiety of FAD (gray stick with N-atoms = blue, O-atoms = red, P-atoms = orange) and the enzyme show in gold/blue ribbon. The results (Table 2) show that residues F89 and R204 possibly constrict the premithramycin B binding site, while the L107A mutation reduces substrate binding, and also FAD binding.
Substrate binding
The substrate binding site for MtmOIV was partially identified by a low resolution (3.6 Å) crystal structure from an MtmOIV-premithramycin B co-crystallization experiment (data not shown). While difference density, presumably for premithramycin B, was observed in the co-crystal structure when compared to the native structure, the density was poor and not large enough to accommodate the entire ligand, possibly due to disorder in the structure of the compound, low resolution of the crystal structure, and/or low occupancy in the crystal structure (Figure 6). Since we could not determine the interactions between MtmOIV and premithramycin B directly from the crystal structure, we used the partial information gathered from the difference density in the co-crystal structure, along with what is already known about FAD-dependent enzymes, to estimate a starting location for modeling premithramycin B into the MtmOIV crystal structure using the program Autodock 4 (61). An Autogrid template was centered along the region of observed difference density in our crystal structure and the cluster of conformations with the lowest docking energy was analyzed to determine the best docking solution.
Figure 6.
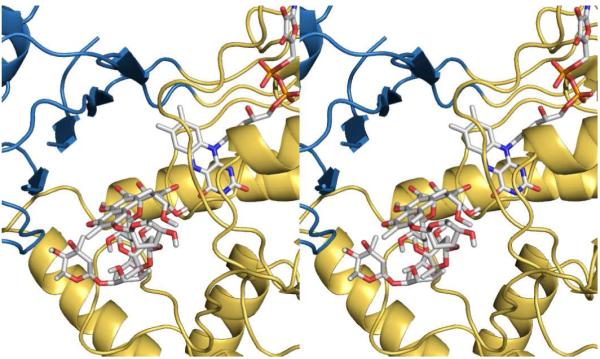
Stereoimage of the premithramycin B (gray stick) binding pocket with FAD (gray stick), based on the found density from premithramycin B/MtmOIV co-crystallization and initial Autodock computational studies, prior to refinement (for the Autodock-refined results, see Figs. 5B and 7). MtmOIV is shown as gold (FAD binding domain) and blue (middle domain) ribbon.
The estimated free energy of binding for premithramycin B was -3.57 kcal/mol, which is within acceptable limits (65). Other Autodock model data statistics: estimated Ki =s 2.41 mM, final intermolecular energy = - 6.04 kcal/mol, vdW + Hbond + desolv energy = - 5.88 kcal/mol, electrostatic energy = - 0.17 kcal/mol, final total internal energy = - 6.24 kcal/mol, torsional free energy = + 5.76 kcal/mol, unbound system’s energy = - 2.95 kcal/mol. The modeled substrate sits in the putative active site cleft (Figures 5B and 7A), and suggests a ∼ 5.3 Å distance between carbon C1 of premithramycin B and carbon C4a of FAD, to which the hydroperoxy group is known to be bound (Figure 2A and Figure 7B). This distance agrees well with reports of other FAD-dependent enzymes, which typically ranges between 4.5-5.5 Å (66). It is known that the FAD cofactor is essential for the enzyme function within the GR2 subclass (43) of FAD-dependent enzymes and that FAD’s role is to transfer an oxygen molecule from O2 to the premithramycin B, subsequently releasing H2O (36). Therefore, the FAD binding site must be in close proximity to the enzyme’s active site for the oxygen transfer, which is consistent with the observations of our modeling studies.
Figure 7.

A. Premithramycin B (green stick) binding pocket with FAD (gray stick). MtmOIV is shown in transparent gray surface representation. B. The dotted line represents the measured distance of 5.30 Å from the C4a carbon of the flavin ring of FAD to C1 of premithramycin B.
To gather supporting evidence for the suggested substrate binding site, mutants F89A, L107A and R204A were generated. Figure 5B shows these three residues to be possibly involved in substrate binding through interactions with the tetracyclic aglycone (L107) and the mycarose moiety (sugar E) of the trisaccharide chain (F89 and R204), respectively, of premithramycin B. The latter two of these three selected residues seem distant from the FAD binding site, and although an effect on the FAD-binding is possible, the mutations should more directly affect substrate binding.
Steady State Kinetics
Enzymatic turnover was correlated with the consumption of NADPH and calculated by measuring the decrease in absorption at 340 nm (ε340 = 6220 m-1 cm-1). Kinetic parameters were determined for each of the enzymes. The Km for the wild type MtmOIV was similar to previous reports at 31μm (31, 36). In contrast, the Vmax (966 nmol min-1 mg-1) of MtmOIV was significantly increased and is attributed to higher concentrations of NADPH in the assay, 0.25mm compared with 0.1mm. Kinetic analysis of the FAD and substrate binding site mutants confirm an important dependency on FAD binding. Of the six mutant enzymes, three (L107A, F180A, F272A) were extremely deficient in FAD binding with undetectable levels of FAD as measured by absorbance at 450 nm. The FAD deficiency translated into enzymatic inefficiency. Each of these enzymes had impaired kinetics with NADPH consumption curves lacking any initial fast phase. Even at substrate concentrations 3-fold > Km (102 μM premithramycin B), these three enzymes showed marginally detectable activity.
Two substrate binding site mutants (F89A and R204A) and a mutant (R52A) aimed at destabilizing an intermediate of the BVMO reaction mechanism maintained varying amounts of FAD binding (Table 2). From the purification experiments, it seems that MtmOIV wild type binds approximately 1 FAD cofactor per 2 enzyme monomers, and F89A has similar FAD content while R204A binds approximately half the FAD of wild type and F89A. R52A has 8-fold lower FAD content than the wild type enzyme and also shows the greatest reduction in activity of the three mutants. R52A activity is reduced by >10-fold (comparison of kcat/Km values between the two enzymes). The increase in Km (R52A: 79 μm) from wild type suggests an additional consequence of the mutation aside from FAD content reduction. Inefficiency at stabilizing a reaction intermediate or a reduced affinity for the initial substrate could explain the altered kinetics of R52A. Clearly, the mutation negatively affects the kinetics of MtmOIV.
Both F89A and R204A mutants maintain high FAD content compared to the other mutants and the activity of these two enzymes is increased from wild type with kcat/Km values of 47 and 41 s-1 mm-1, respectively, compared to 35 s-1 mm-1 of wild type. Each enzyme accomplishes this increase through a distinct mechanism. While F89A has a comparable Km to wild type and a significantly increased kcat value and R204A has a comparable kcat but a decreased Km effectively heightening the activity of each mutant in respect to wild type enzyme.
In addition to NADPH, NADH is also a functional coenzyme in the MtmOIV reaction mechanism; however the exact kinetic parameters had not yet been defined (36). Within error, we found a similar Km in the presence of 0.25mm NADH but kcat was decreased limiting the kcat/Km value to about 70% of the wild type enzyme with NADPH as the cofactor.
DISCUSSION
The structure of the BVMO MtmOIV has been determined revealing many similarities to other characterized FAD-dependent enzymes. The structure contains an FAD molecule non-covalently bound adjacent to a large substrate binding cavity which is able to perfectly accommodate the substrate premithramycin B. Bordering this cavity is MtmOIV’s active site including the flavin ring of the FAD cofactor, which is required for catalysis.
MtmOIV does have a novel structure for a BVMO compared to the only so far published structures of BVMOs, namely phenylacetone monooxygenase (PAMO) from the thermophilic bacterium Thermobifida fusca (PDB code 1W4X) (33, 67) and 3,6-diketocamphane monooxygenase from Pseudomonas putida (by comparison with the shown structure, which is not deposited in the PDB database) (32). PAMO has significant differences compared to MtmOIV with an RMSD of 5.78 Å for only 282 aligned residues. The differences may be explained by the enzyme substrates which are vastly different in size, although the native functionality of phenylacetone monooxygenase in Thermobifida fusca is unknown. However, there are also similarities regarding the active site. Most important, MtmOIV possesses, like PAMO, an arginine residue (Arg-52) that is located above the flavin ring (Figure 8A), suited to stabilize both the negatively charged peroxyflavin and Criegee intermediates involved in the BV-reaction mechanism (1, 2, 33). However, while the corresponding R337 of PAMO is located on the re side of the flavin ring (33), R52 of MtmOIV lays on the si side (Figure 8A).
Figure 8.

A. Left: View of the catalytically important arginine-52 (R52, green stick, N = blue, O = red) residue of MtmOIV above the flavin ring of FAD (gray stick, N = blue, O = red, P = orange). The measured distance (dotted line) between the N-atoms of the guanidine residue of R52 and the flavin ring surface is ∼ 6.2 Å in the shown conformation. The remainder of the enzyme (ribbon) is depicted in green.
B. Below: Suggested Baeyer-Villiger reaction mechanism of the MtmOIV reaction, involving the peroxyflavin and Criegee intermediates. R = deoxysugar chains
Nevertheless, the overall mechanism of the initial MtmOIV reaction (for the likely sequence of events, see Figure 8B) should be similar as described for PAMO (33), and R52 should play a similar important role for MtmOIV as R337 for PAMO. Thus, R52 mutagenesis was expected to significantly decrease enzyme turnover by interfering with crucial interactions between the enzyme and reaction intermediates. Indeed, mutant enzyme R52A’s efficiency (13% of the native enzyme, see Table 2) decreased significantly, but did not render the enzyme inactive, possibly indicating a compensatory mechanism of intermediate stabilization. In contrast, the analyzed R337A and R337K mutants of PAMO, while still able to form and stabilize the C-4a-peroxyflavin intermediate, were unable to convert the substrate phenylacetone (67). However, since we monitored the oxidation of NADPH, the found remaining activity of the R52A mutant might also point to a biosynthetic shunt pathway, through which NADPH is oxidized, e.g. an NADPH-triggered reduction of premithramycin B. Future studies on MtmOIV will hopefully clarify some of these mechanistic questions.
MtmOIV and PgaE are highly similar both in sequence and in structure, as seen in Figures 2C and 3. However, the reactions they catalyze are fundamentally different, although both are post-polyketide synthase tailoring enzymes. PgaE is an aromatic hydroxylase isolated from a cryptic gene cluster from Streptomyces sp. PGA64 involved in angucycline biosynthesis, catalyzing the C12-hydroxylation of UWM6 as an early post-PKS tailoring step towards gaudimycin C (Figure 9) (68), which is mechanistically an electrophilic aromatic substitution reaction, while MtmOIV is a Baeyer-Villiger monooxygenase involved in the biosynthesis of the aureolic acid class anticancer agent mithramycin, which initiates it reaction sequence through a nucleophilic attack of one of the four keto functions of premithramycin B (Figures 1, 8B) (31, 38).
Figure 9.

The PgaE reaction. PgaE is an early acting hydroxylase in the biosynthesis of angucylinone gaudimycin C, catalyzing the 12-hydroxylation of UWM6 (left) to 12-hydroxy-UWM6 (right).
The reactions between the two enzymes do share the oxidative/reductive property provided by the cofactors FAD and NADPH, but the primary reaction is different. Like PgaE, MtmOIV belongs to the para-hydroxybenzoate hydroxylase (PHBH) fold family as well as the GR2 subclass of FAD dependent enzymes, that previously contained no characterized BVMO (43, 69). Both PgaE and MtmOIV process bulky tetracyclic polyketide-derived substrates in their active site, in contrast to phenylacetone monooxygenase (PAMO) that acts on a relatively small substrate (33), however the MtmOIV substrate is much more complex, because it is highly decorated with deoxysugars, while the PgaE substrate lacks any sugar moiety.
The structure of MtmOIV presented here can now be used to guide mutations that can allow different substrates to access the active site cavity and to potentially be converted into additional active anticancer drug compounds. The generation of analogues MTM SK (40), MTM SDK (20) and demycarosyl-3D-β-d-digitoxosyl-MTM (41) (Figure 10), which all possess increased efficacy in some aspects compared to MTM, shows that an improvement of the biological activity of the natural product MTM is realistic.
Figure 10.

Chemical structures of mithramycin (MTM) analogues mithramycin SK (MTM SK)(40), mithramycin SDK (MtmSDK)(20) and 3D-demycarosyl-3D-digitoxosyl-mithramycin (Demyc-Dig-MTM)(41), all generated by combinatorial biosynthesis, which have increased efficacy compared with MTM. The structural differences compared with MTM are highlighted in blue.
While MTM SK and MTM SDK differ from MTM with respect to their 3-side chain, demycarosyl-3D-β-d-digitoxosyl-MTM is an example, in which an improvement was achieved through modification of the saccharide pattern, a strategy we want to further exploit, but which is currently hampered by substrate specificity restrictions of MtmOIV. Demycarosyl-3D-β-d-digitoxosyl-MTM was one of only three fully glycosylated MTM analogues with modified sugar moieties that could be generated through combinatorial biosynthesis involving the native MtmOIV. However, it differs from MTM only with respect to the E-sugar moiety, which is d-digitoxose instead of d-mycarose, i.e. it only lacks the 3E-CH3-residue (Fig. 10).
We hoped that the MtmOIV structure would provide clues how to reengineer the enzyme to accommodate and process premithramycin B analogues with significantly different saccharide patterns, particularly with modified E-sugar moieties, because this terminal sugar of the trisaccharide chain plays a crucial role in MTM’s interaction with the DNA (70, 71). The L107A mutant shows that a residue seemingly involved in the binding of the disaccharide chain of the substrate, Leu107, seems to be crucial to anchor both the substrate and FAD in the active site, and its mutation to alanine led to loss of FAD binding and loss of activity (Table 2). Interestingly, the here described F89A and R204A mutants showed significantly increased efficiency over the wild type enzyme, based on the kcat/Km values. Thus, both of these bulky residues may be important for the stabilization of the trisaccharide chain of substrate premithramycin B in the substrate binding pocket, interacting particularly with sugar E of this chain. These two mutants show that Phe89 and Arg204 in MtmOIV are possibly involved in substrate specificity of the enzyme binding pocket. This specificity could constrict the binding pocket, designed to house the specific substrate premithramycin B in a biological environment where many possible substrates are present, e.g. premithramycin-type intermediates of the mtm pathway that are not yet completely glycosylated (29, 72). However, such specific binding could decrease even the binding of the natural substrate, and consequently reduction of these more bulky residues to a less space-filling alanine residue may in fact relax the binding pocket to allow greater binding of the natural substrate and possible binding of non-natural substrates as well. Consequently, the in vitro kinetic assays exhibit increased turnover due to a relaxation of the spatial limitations of this binding region in mutants F89A and R204A, respectively. Our future work will further focus on using the MtmOIV crystal structure to perform systematic mutagenesis along the substrate binding cavity.
In summary, we presented the crystal structure of MtmOIV, a BVMO catalyzing the key step of the biosynthesis of the anticancer agent mithramycin. Following a useful new classification system (2), MtmOIV appears to be an example of an atypical class A flavoprotein monooxygenase catalyzing a Baeyer-Villiger oxidation. The crystal structure provides supporting evidence for a flavin-catalyzed BV-reaction involving a peroxyflavin intermediate for its nucleophilic attack, in contrast to typical class A flavoprotein monooxygenases that form a reactive hydroperoxyflavin instead, and perform electrophilic attacks. The crystal structure of the atypical BVMO MtmOIV was solved by X-ray crystallography using molecular replacement, for which we took advantage of the crystal structure data of the structurally similar monooxygenase PgaE, a hydroxylase, which is -considering the hydroxylation reaction it catalyzes- a typical class A flavoprotein requiring a hydroperoxyflavin intermediate for its electrophilic aromatic substitution reaction (Figures 1, 8B, 9). Given the low similarity of MtmOIV with other BVMOs (e.g., only 8% sequence identity with PAMO) and the fact that MtmOIV lacks the typical sequence motif of type I BVMOs and is not related to type II BVMOs (4), MtmOIV represents a new class of BVMO.
Using the structural information and substrate modeling results, MtmOIV mutants were engineered that support the conclusions drawn regarding the FAD binding, and also provide further evidence regarding residues involved in the substrate binding site, which were identified by computational substrate docking experiments. The MtmOIV crystal structure may assist combinatorial biosynthetic efforts to generate new MTM analogues with drastically altered saccharide patterns, since structure-inspired MtmOIV mutants might accommodate different premithramycin-type substrates and convert them into new mithramycin analogues with potentially increased activity or decreased toxicity.
ACKNOWLEDGEMENTS
We would like to thank the staff at the SER-CAT 22-ID Beamline at the Advanced Photon Source for their assistance in data collection. We also thank Professor David W. Rodgers and the University of Kentucky Center for Structural Biology as well as the University of Kentucky Research foundation for their support.
Footnotes
This work was supported by National Institutes of Health Grant CA 091901 to J.R. The crystal data for MtmOIV were deposited in the protein data bank (PDB), and have been assigned with the RCSB ID code rscb050786 and PDB ID code 3FMW.
- ASU
- asymmetric unit
- BVMO
- Baeyer-Villiger monooxygenase
- BV
- Baeyer-Villiger
- MOPS
- 3-(N-morpholino)propanesulfonic acid
- MTM
- mithramycin
- RMSD
- root mean square deviation
REFERENCES
- 1.Mihovilovic MD, Rudroff F, Grotzl B, Kapitan P, Snajdrova R, Rydz J, Mach R. Family clustering of Baeyer-Villiger monooxygenases based on protein sequence and stereopreference. Angew. Chem. Int. Ed. Engl. 2005;44:3609–3613. doi: 10.1002/anie.200462964. [DOI] [PubMed] [Google Scholar]
- 2.van Berkel WJH, Kamerbeek NM, Fraaije MW. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 3.Kamerbeek NM, Fraaije MW, Janssen DB. Identifying determinants of NADPH specificity in Baeyer-Villiger monooxygenases. Eur. J. Biochem. 2004;271:2107–2116. doi: 10.1111/j.1432-1033.2004.04126.x. [DOI] [PubMed] [Google Scholar]
- 4.Kamerbeek NM, Janssen DB, van Berkel WJH, Fraaije MW. Baeyer-Villiger Monooxygenases, and Emerging Family of Flavin-Dependent Biocatalysts. Adv. Synth. Catal. 2003;345:667–678. [Google Scholar]
- 5.Willetts A. Structural studies and synthetic applications of Baeyer-Villiger monooxygenases. Trends Biotechnol. 1997;15:55–62. doi: 10.1016/S0167-7799(97)84204-7. [DOI] [PubMed] [Google Scholar]
- 6.Hilker I, Gutierrez MC, Furstoss R, Ward J, Wohlgemuth R, Alphand V. Preparative scale Baeyer-Villiger biooxidation at high concentration using recombinant Escherichia coli and in situ substrate feeding and product removal process. Nat. Protoc. 2008;3:546–554. doi: 10.1038/nprot.2007.532. [DOI] [PubMed] [Google Scholar]
- 7.Reetz MT, Brunner B, Schneider T, Schulz F, Clouthier CM, Kayser MM. Directed evolution as a method to create enantioselective cyclohexanone monooxygenases for catalysis in Baeyer-Villiger reactions. Angew. Chem. Int. Ed. Engl. 2004;43:4075–4078. doi: 10.1002/anie.200460272. [DOI] [PubMed] [Google Scholar]
- 8.Ehrlich KC, Montalbano B, Boue SM, Bhatnagar D. An aflatoxin biosynthesis cluster gene encodes a novel oxidase required for conversion of versicolorin a to sterigmatocystin. Appl. Environ. Microbiol. 2005;71:8963–8965. doi: 10.1128/AEM.71.12.8963-8965.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia XY, Tian ZH, Shao L, Qu XD, Zhao QF, Tang J, Tang GL, Liu W. Genetic characterization of the chlorothricin gene cluster as a model for spirotetronate antibiotic biosynthesis. Chem. Biol. 2006;13:575–585. doi: 10.1016/j.chembiol.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Wright JLC, Hu T, McLachlan JL, Needham J, Walter JA. Biosynthesis of DTX-4: Confirmation of a Polyketide Pathway, Proof of a Baeyer-Villiger Oxidation Step, and Evidence for an Unusual Carbon Deletion Process. J. Am. Chem. Soc. 1996;118:8757–8758. [Google Scholar]
- 11.Rix U, Remsing LL, Hoffmeister D, Bechthold A, Rohr J. Urdamycin L: A novel metabolic shunt product that provides evidence for the role of the urdM gene in the urdamycin A biosynthetic pathway of Streptomyces fradiae TÜ2717. ChemBioChem. 2003;4:109–111. doi: 10.1002/cbic.200390002. [DOI] [PubMed] [Google Scholar]
- 12.Townsend CA, Christensen SB, Davis SG. Bisfuran formation in aflatoxin biosynthesis: the role of versiconal acetate. J. Am. Chem. Soc. 1982;104:6154–6155. [Google Scholar]
- 13.Kharel MK, Zhu L, Liu T, Rohr J. Multi-oxygenase complexes of the gilvocarcin and jadomycin biosyntheses. J. Am. Chem. Soc. 2007;129:3780–3781. doi: 10.1021/ja0680515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dover LG, Corsino PE, Daniels IR, Cocklin SL, Tatituri V, Besra GS, Futterer K. Crystal structure of the TetR/CamR family repressor Mycobacterium tuberculosis EthR implicated in ethionamide resistance. J. Mol. Biol. 2004;340:1095–1105. doi: 10.1016/j.jmb.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Fraaije MW, Kamerbeek NM, Heidekamp AJ, Fortin R, Janssen DB. The Prodrug Activator EtaA from Mycobacterium tuberculosis Is a Baeyer-Villiger Monooxygenase. J. Biol. Chem. 2004;279:3354–3360. doi: 10.1074/jbc.M307770200. [DOI] [PubMed] [Google Scholar]
- 16.Garbe LA, Tressl R. Enzymatic Baeyer-Villiger oxidation as the key step in decano-4-lactone and decano-5-lactone degradation by Sporobolomyces odorus. Chem. Biodivers. 2004;1:900–915. doi: 10.1002/cbdv.200490072. [DOI] [PubMed] [Google Scholar]
- 17.Koma D, Sakashita Y, Kubota K, Fujii Y, Hasumi F, Chung SY, Kubo M. Degradation pathways of cyclic alkanes in Rhodococcus sp. NDKK48. Appl. Microbiol. Biotechnol. 2004;66:92–99. doi: 10.1007/s00253-004-1623-5. [DOI] [PubMed] [Google Scholar]
- 18.Kataoka M, Honda K, Sakamoto K, Shimizu S. Microbial enzymes involved in lactone compound metabolism and their biotechnological applications. Appl. Microbiol. Biotechnol. 2007;75:257–266. doi: 10.1007/s00253-007-0896-x. [DOI] [PubMed] [Google Scholar]
- 19.Blume SW, Snyder RC, Ray R, Thomas S, Koller CA, Miller DM. Mithramycin inhibits Sp1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J. Clin. Invest. 1991;88:1613–1621. doi: 10.1172/JCI115474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Albertini V, Jain A, Vignati S, Napoli S, Rinaldi A, Kwee I, Nur-e-Alam M, Bergant J, Bertoni F, Carbone GM, Rohr J, Catapano CV. Novel GC-rich DNA-binding compound produced by a genetically engineered mutant of the mithramycin producer Streptomyces argillaceus exhibits improved transcriptional repressor activity: implications for cancer therapy. Nucl. Acids Res. 2006;34:1721–1734. doi: 10.1093/nar/gkl063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bianchi N, Rutigliano C, Passadore M, Tomassetti M, Pippo L, Mischiati C, Feriotto G, Gambari R. Targeting of the HIV-1 long terminal repeat with chromomycin potentiates the inhibitory effects of a triplex-forming oligonucleotide on Sp1-DNA interactions and in vitro transcription. Biochem. J. 1997;326:919–927. doi: 10.1042/bj3260919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Black AR, Black JD, Azizkhan-Clifford J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell. Physiol. 2001;188:143–160. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 23.Gambari R, Feriotto G, Rutigliano C, Bianchi N, Mischiati C. Biospecific interaction analysis (BIA) of low-molecular weight DNA-binding drugs. J. Pharmacol. Exp. Ther. 2000;294:370–377. [PubMed] [Google Scholar]
- 24.Kennedy BJ, Torkelson JL. Long-term follow-up of stage III testicular carcinoma treated with mithramycin (plicamycin) Med. Pediatr. Oncol. 1995;24:327–328. doi: 10.1002/mpo.2950240511. [DOI] [PubMed] [Google Scholar]
- 25.Hadjipavlou AG, Gaitanis LN, Katonis PG, Lander P. Paget’s disease of the spine and its management. Eur. Spine J. 2001;10:370–384. doi: 10.1007/s005860100329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pecherstorfer M, Brenner K, Zojer N. Current management strategies for hypercalcemia. Treat. Endocrinol. 2003;2:273–292. doi: 10.2165/00024677-200302040-00005. [DOI] [PubMed] [Google Scholar]
- 27.Ferrante RJ, Ryu H, Kubilus JK, D’Mello S, Sugars KL, Lee J, Lu P, Smith K, Browne S, Beal MF, Kristal BS, Stavrovskaya IG, Hewett S, Rubinsztein DC, Langley B, Ratan RR. Chemotherapy for the brain: the antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington’s disease. J. Neurosci. 2004;24:10335–10342. doi: 10.1523/JNEUROSCI.2599-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rohr J, Mendez C, Salas J. The Biosynthesis of Aureolic Acid Group Antibiotics. Bioorg. Chem. 1999;27:41–54. [Google Scholar]
- 29.Nur-e-Alam M, Méndez C, Salas JA, Rohr J. Elucidation of the glycosylation sequence of mithramycin biosynthesis: isolation of 3A-deolivosylpremithramycin B and its conversion to premithramycin B by glycosyltransferase MtmGII. ChemBioChem. 2005;6:632–636. doi: 10.1002/cbic.200400309. [DOI] [PubMed] [Google Scholar]
- 30.Prado L, Fernandez E, Weissbach U, Blanco G, Quiros LM, Brana AF, Mendez C, Rohr J, Salas JA. Oxidative cleavage of premithramycin B is one of the last steps in the biosynthesis of the antitumor drug mithramycin. Chem. Biol. 1999;6:19–30. doi: 10.1016/s1074-5521(99)80017-9. [DOI] [PubMed] [Google Scholar]
- 31.Gibson M, Nur-e-Alam M, Lipata F, Oliveira MA, Rohr J. Characterization of Kinetics and Products of the Baeyer-Villiger Oxygenase MtmOIV, The Key Enzyme of the Biosynthetic Pathway toward the Natural Product Anticancer Drug Mithramycin from Streptomyces argillaceus. J. Am. Chem. Soc. 2005;127:17594–17595. doi: 10.1021/ja055750t. [DOI] [PubMed] [Google Scholar]
- 32.Isupov MN, Lebedev AA. NCS-constrained exhaustive search using oligomeric models. Acta Crystallogr. D Biol. Crystallogr. 2008;64:90–98. doi: 10.1107/S0907444907053802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malito E, Alfieri A, Fraaije MW, Mattevi A. Crystal structure of a Baeyer-Villiger monooxygenase. Proc. Natl. Acad. Sci. USA. 2004;101:13157–13162. doi: 10.1073/pnas.0404538101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Beilen JB, Mourlane F, Seeger MA, Kovac J, Li Z, Smits TH, Fritsche U, Witholt B. Cloning of Baeyer-Villiger monooxygenases from Comamonas, Xanthobacter and Rhodococcus using polymerase chain reaction with highly degenerate primers. Environ. Microbiol. 2003;5:174–182. doi: 10.1046/j.1462-2920.2003.00401.x. [DOI] [PubMed] [Google Scholar]
- 35.Morii S, Sawamoto S, Yamauchi Y, Miyamoto M, Iwami M, Itagaki E. Steroid monooxygenase of Rhodococcus rhodochrous: sequencing of the genomic DNA, and hyperexpression, purification, and characterization of the recombinant enzyme. J. Biochem. 1999;126:624–631. doi: 10.1093/oxfordjournals.jbchem.a022494. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez D, Quiros LM, Braña AF, Salas JA. Purification and characterization of a monooxygenase involved in the biosynthetic pathway of the antitumor drug mithramycin. J. Bacteriol. 2003;185:3962–3965. doi: 10.1128/JB.185.13.3962-3965.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang C, Gibson M, Rohr J, Oliveira MA. Crystallization and X-ray diffraction properties of Baeyer-Villiger monooxygenase MtmOIV from the mithramycin biosynthetic pathway in Streptomyces argillaceus. Acta Cryst. 2005;F61:1023–1026. doi: 10.1107/S1744309105033221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prado L, Fernandez E, Weissbach U, Blanco G, Quiros LM, Brana AF, Méndez C, Rohr J, Salas JA. Oxidative cleavage of premithramycin B is one of the last steps in the biosynthesis of the antitumor drug mithramycin. Chem. Biol. 1999;6:19–30. doi: 10.1016/s1074-5521(99)80017-9. [DOI] [PubMed] [Google Scholar]
- 39.Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J. Inhibition of c-src transcription by mithramycin: structure-activity relationships of biosynthetically produced mithramycin analogues using the c-src promoter as target. Biochemistry. 2003;42:8313–8324. doi: 10.1021/bi034091z. [DOI] [PubMed] [Google Scholar]
- 40.Remsing LL, Gonzalez AM, Nur-e-Alam M, Fernandez-Lozano MJ, Braña AF, Rix U, Oliveira MA, Méndez C, Salas JA, Rohr J. Mithramycin SK, a novel antitumor drug with improved therapeutic index, mithramycin SA, and demycarosylmithramycin SK: three new products generated in the mithramycin producer Streptomyces argillaceus through combinatorial biosynthesis. J. Am. Chem. Soc. 2003;125:5745–5753. doi: 10.1021/ja034162h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baig I, Pérez M, Braña AF, Gomathinayagam R, Damodaran C, Salas JA, Méndez C, Rohr J. Mithramycin analogues generated by combinatorial biosynthesis show improved bioactivity. J. Nat. Prod. 2008;71:199–207. doi: 10.1021/np0705763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pérez M, Baig I, Braña AF, Salas JA, Rohr J, Méndez C. Generation of new derivatives of the antitumor antibiotic mithramycin by altering the glycosylation pattern through combinatorial biosynthesis. ChemBioChem. 2008;9:2295–2304. doi: 10.1002/cbic.200800299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dym O, Eisenberg D. Sequence-structure analysis of FAD-containing proteins. Protein Science. 2001;10:1712–1728. doi: 10.1110/ps.12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koskiniemi H, Metsa-Ketela M, Dobritzsch D, Kallio P, Korhonen H, Mantsala P, Schneider G, Niemi J. Crystal structures of two aromatic hydroxylases involved in the early tailoring steps of angucycline biosynthesis. J. Mol. Biol. 2007;372:633–648. doi: 10.1016/j.jmb.2007.06.087. [DOI] [PubMed] [Google Scholar]
- 45.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:538–557. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 46.Collaborative Computational Project, N The CCP4 suite: programs for protein crystallography. Acta Crystr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 47.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stein N. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Cryst. 2008;41:641–643. [Google Scholar]
- 49.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystr. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 52.Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delano WL. The PyMOL Molecular Graphics System. Delano Scientific; San Carlos: 2002. [Google Scholar]
- 54.Gille C, Frommel C. STRAP: editor for STRuctural Alignments of Proteins. Bioinformatics. 2001;17:377–378. doi: 10.1093/bioinformatics/17.4.377. [DOI] [PubMed] [Google Scholar]
- 55.Wang W, Malcolm BA. Two-stage polymerase chain reaction protocol allowing introduction of multiple mutations, deletions, and insertions, using QuikChange site-directed mutagenesis. Methods Mol. Biol. 2002;182:37–43. doi: 10.1385/1-59259-194-9:037. [DOI] [PubMed] [Google Scholar]
- 56.Bachmann HS, Siffert W, Frey UH. Successful amplification of extremely GC-rich promoter regions using a novel ‘slowdown PCR’ technique. Pharmacogenetics. 2003;13:759–766. doi: 10.1097/00008571-200312000-00006. [DOI] [PubMed] [Google Scholar]
- 57.Jameson GN, Jin W, Krebs C, Perreira AS, Tavares P, Liu X, Theil EC, Huynh BH. Stoichiometric production of hydrogen peroxide and parallel formation of ferric multimers through decay of the diferric-peroxo complex, the first detectable intermediate in ferritin mineralization. Biochemistry. 2002;41:13435–13443. doi: 10.1021/bi026478s. [DOI] [PubMed] [Google Scholar]
- 58.Holt A, Sharman DF, Baker GB, Palcic MM. A continuous spectrophotometric assay for monoamine oxidase and related enzymes in tissue homogenates. Anal. Biochem. 1997;244:384–392. doi: 10.1006/abio.1996.9911. [DOI] [PubMed] [Google Scholar]
- 59.Schuettelkopf AW, van Aalten DMF. PRODRG - a tool for high-throughput crystallography of protein-ligand complexes. Acta Cryst. 2004;D60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 60.van Aalten DM, Bywater R, Findlay JB, Hendlich M, Hooft RW, Vriend G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided Mol. Des. 1996;10:255–262. doi: 10.1007/BF00355047. [DOI] [PubMed] [Google Scholar]
- 61.Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J. Mol. Recognit. 1996;9:1–5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 62.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 63.Ryan KS, Chakraborty S, Howard-Jones AR, Walsh CT, Ballou DP, Drennan CL. The FAD cofactor of RebC shifts to an IN conformation upon flavin reduction. Biochemistry. 2008;47:13506–13513. doi: 10.1021/bi801229w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 65.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- 66.Ryan KS, Howard-Jones AR, Hamill MJ, Elliott SJ, Walsh CT, Drennan CL. Crystallographic trapping in the rebeccamycin biosynthetic enzyme RebC. Proc. Natl. Acad. Sci. USA. 2007;104:15311–15316. doi: 10.1073/pnas.0707190104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Torres Pazmino DE, Baas B-J, Janssen DB, Fraaije MW. Kinetic Mechanism of Phenylacetone Monooxygenase from Thermobifida fusca. Biochemistry. 2008;47:4082–4093. doi: 10.1021/bi702296k. [DOI] [PubMed] [Google Scholar]
- 68.Kallio P, Liu Z, Mantsala P, Niemi J, Metsa-Ketela M. Sequential action of two flavoenzymes, PgaE and PgaM, in angucycline biosynthesis: chemoenzymatic synthesis of gaudimycin C. Chem. Biol. 2008;15:157–166. doi: 10.1016/j.chembiol.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 69.Mattevi A. The PHBH fold: not only flavoenzymes. Biophys. Chem. 1998;70:217–222. doi: 10.1016/s0301-4622(97)00126-9. [DOI] [PubMed] [Google Scholar]
- 70.Sastry M, Fiala R, Patel DJ. Solution structure of mithramycin dimers bound to partially overlapping sites on DNA. J. Mol. Biol. 1995;251:674–689. doi: 10.1006/jmbi.1995.0464. [DOI] [PubMed] [Google Scholar]
- 71.Sastry M, Patel DJ. Solution structure of the mithramycin dimer-DNA complex. Biochemistry. 1993;32:6588–6604. doi: 10.1021/bi00077a012. [DOI] [PubMed] [Google Scholar]
- 72.Fernandez E, Weissbach U, Sanchez Reillo C, Braña AF, Méndez C, Rohr J, Salas JA. Identification of two genes from Streptomyces argillaceus encoding glycosyltransferases involved in transfer of a disaccharide during biosynthesis of the antitumor drug mithramycin. J. Bacteriol. 1998;180:4929–4937. doi: 10.1128/jb.180.18.4929-4937.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]