

Abstract
Substantial oxidative N-debenzylation reaction along with PhCH=O formation occurs from a hydroperoxo copper(II) complex which has a dibenzylamino substrate (-N(CH2Ph)2 appended as a substituent on one pyridyl group of its tripodal tetradentate TMPA {≡ TPA ≡ tris(2-pyridylmethyl)amine)} ligand framework. During the course of the (LN(CH2Ph)2)CuII(−OOH) reactivity, formation of a substrate and −OOH (an oxygen atom) derived alkoxo CuII(−OR) complex occurs. The observation that the same CuII(−OR) species occurs from CuI/PhIO chemistry suggests the possibility that a copper-oxo (cupryl) reactive intermediate forms during alkoxo species formation, and new ESI-MS data obtained provides some further support for this high-valent intermediate. Net H-atom abstraction chemistry is proposed, based on kinetic isotope effect studies provided here and that previously published for a closely related CuII(−OOH) species incorporating dimethylamine (-N(CH3)2) as the internal substrate (J. Am. Chem. Soc. 2007, 129, 6720-6721); the CuI/PhIO reactivity, with similar isotope effect results, provides further support. The reactivity of these chemical systems closely resembles proposed oxidative N-dealkylation mechanisms effected by the copper-monooxygenases dopamine β-monooxygenase (DβM) or peptidylglycine-α-hydroxylating monooxygenase (PHM).
Introduction
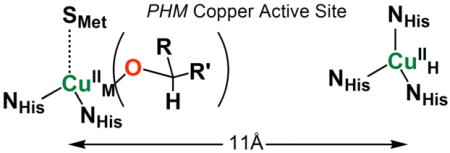
The study of copper(I)-dioxygen adducts and their (reduced) derivatives is of considerable current interest.1-8 In large part these efforts comprise the desire to elucidate fundamental aspects such as structural type along with associated spectroscopy, electronic structure, and of course reactivity patterns. Inspirations of such investigations also come from aspects of bioinorganic chemistry with interest to provide fundamental information which may be relevant to metalloenzymes with copper containing active sites where the processing of molecular oxygen occurs. Included in the latter are the copper monooxygenases peptidylglycine-α-hydroxylating monooxygenase (PHM) and dopamine β-monooxygenase (DβM), mammalian proteins serving in the biosynthesis of various neuropeptides or hormones.9-12 Figure 1 outlines the specific reactions carried out, each involving insertion of one oxygen atom (from O2) into the substrate, for DβM a net benzylic hydroxylation while for PHM hydroxylation is followed by N-dealkylation. The active sites of each enzyme are known to be very similar and each contains two copper ions. Previous biophysical studies have shown these to be relatively far apart and more recent X-ray crystallographic structures on PHM indeed show them to be this way, ∼ 11 Å apart.12 Thus, the mechanism of action focus on copper chemistry taking place at one of the two copper ions (CuM ≡ CuB) while the other (CuH ≡ CuA) serves to transfer electrons.
Figure 1.
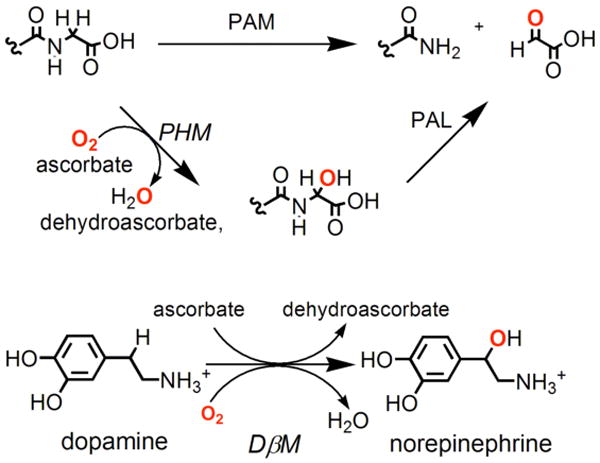
Homologus copper proteins DβM (bottom) transform dopamine to norepinephrine and PHM (top) hydroxylate the C-terminus of the peptide backbone followed by N-dealkylation by PAL.
With a history of biochemical/biophysical studies on these enzymes, and in the context of considering various copper-dioxygen derived species, either known or unknown in inorganic chemistry, a hydroperoxo-copper(II) CuII(−OOH), a superoxo-copper(II) CuII(O2•−); derived from initial CuI-O2 chemistry or high-valent cupryl [CuII-O•] have been discussed as species which might be responsible for an initial hydrogen-atom abstraction reaction.2,4,8,9,11-18 For some time a hydroperoxo complex was suggested, but more recently both experimental and computational (bio)chemistries rather suggest that the CuII(O2•−) moiety is relevant.4,9-11,13 Further, one crystal structure of PHM reveals what is best described as a CuMII(O2•−) moiety perfectly juxtaposed to the relevant substrate C–H bond. Still other theoretical treatments prefer a prior (rather then subsequent) O-O cleavage from CuII(−OOH) leading to a high-valent [Cu-O]2+ or [Cu-O]+ (equiv to CuII-O•) moieties (“cupryl”) which effects H-atom transfer.14,15 In part to obtain answers or provide insights, synthetic bioinorganic chemists have been actively pursuing the chemistry of mononuclear copper-dioxygen derived complexes, especially in reactivity toward substrates.2,19-27 Which species may effect H-atom abstractions followed by the net transfer of an oxygen atom to the substrate, i.e., monooxygenase activity, Scheme 1?
Scheme 1.
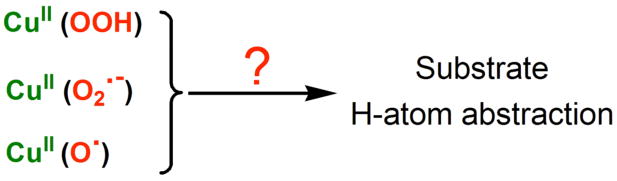
In biomimetic studies approached via coodination chemistry efforts, two types of hydroperoxide-copper species have to date been produced, μ-1,1-hydroperoxo-dicopper(II) and mononuclear complexes. We have recently demonstrated substrate C–H activation chemistries starting from well-characterized dinuclear μ-−OOH-dicopper(II) complexes, oxidative N-dealkylation or RCH2C≡N oxidative cleavage (to the RCH=O aldehyde + cyanide).28,29 Suzuki has also observed a hydrocarbon attack from a CuII2(−OH)2 + H2O2 reaction, giving a CuII-−OORligand-substrate product.30 A number of rather well characterized mononuclear CuII(−OOH) complexes have been described,31-37 however substrate oxidations has been limited to organic sulfides37 or olefins (but in low yields).32 In our own labs, we have in the last year been able to demonstrate C–H activation and thus oxygenation from CuII(−OOH) complexes, as further discussed. There have been notable advances in the generation of mononuclear CuII(O2•−) or CuIII(O22−) species,2,24,25,38 but chemistry where such entities attack substrate C–H bonds have yet to be described. There are as yet no discrete examples or direct evidence for mononuclear high-valent copper-oxo species, however see further discussion below.
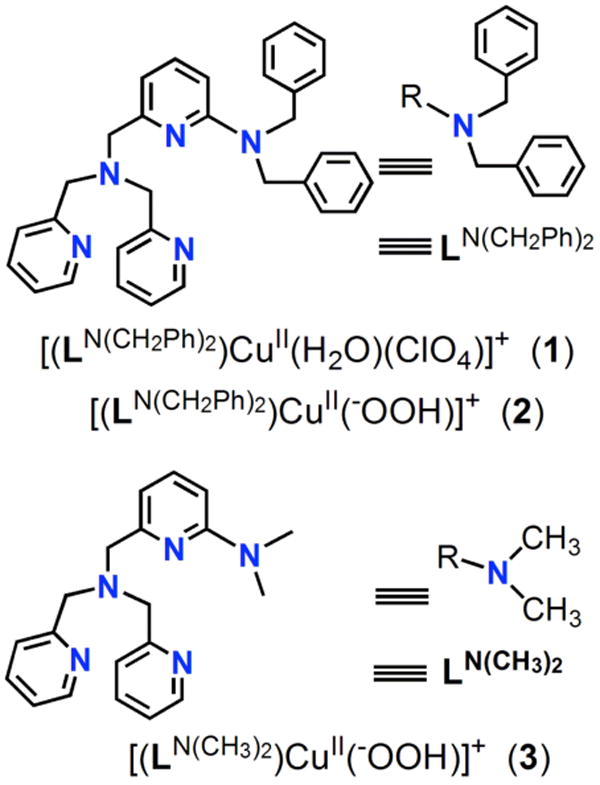
Within the context of the introduction given above, we here describe the chemistry of a new ligand LN(CH2Ph)2 and its copper complex promoted oxidative chemistry. While the main focus is on a mononuclear CuII(−OOH) species, [(LN(CH2Ph)2)CuII(−OOH)]+ (2), we complement it with (ligand)CuI-O2 reactivity and a separate attempt to interrogate the possible involvement of cupryl chemistry. The work follows our recent report where we employed a close analogue [(LN(CH3)2)CuII(−OOH)]+ (3), where both ligands are the derivatives of TMPA {≡ TPA ≡ tris(2-pyridylmethyl)amine)}, showing that the hydroperoxo group coordinated to copper(II) can effect oxidative N-dealkylation chemistry on the juxtaposed dimethylamino substrate.27 In this report the chemistry of the LN(CH2Ph)2 analogue is fully described.
Experimental Section
Materials and Methods
Unless otherwise stated all solvents and chemicals used were of commercially available analytical grade. Acetone, dietyhlether, pentane, propionitrle, methanol and tetrahydrofuran (THF) were used after passing them through a 60 cm long column of activated alumina (Innovative Technologies, Inc.) under argon. Deoxygenation of solvents was effected by either repeated freeze/pump/thaw cycles or bubbling with argon for 30 - 45 minutes. Dioxygen was dried by passing through a short column of supported P4O10 (Aquasorb, Mallinkrodt). Preparation and handling of air sensitive compounds were performed under an argon atmosphere using standard Schlenk techniques or in an MBraun Labmaster 130 inert atmosphere (< 1 ppm O2, < 1 ppm H2O) drybox filled with nitrogen. Elemental analyses were performed by Desert Analytics (Tucson, AZ). 1H-NMR and 13C-NMR spectra were measured on a Bruker 400 MHz spectrometer. Chemical shifts were reported as δ values relative to an internal standard (Me4Si) and the residual solvent proton peak. UV-vis spectra were recorded on a Hewlett-Packard Model 8453A diode array spectrophotometer equipped with two-window quartz H.S. Martin Dewar filled with cold MeOH (25 °C to −85 °C) maintained and controlled by a Neslab VLT-95 low temp circulator. Spectrophotometer cells used were made by Quark Glass with column and pressure/vacuum side stopcock and 2 mm path length. The copper complexes are ClO4− and B(C6F5)4− salt complexes unless otherwise stated. While we have experienced no problems in working with perchlorate compounds, they are potentially explosive and care must be taken not to work with large quantities. ESI mass spectra were acquired using a Finnigan LCQDeca ion-trap mass spectrometer equipped with an electrospray ionization source (Thermo Finnigan, San Jose, CA). For meta-stable species (as described below), samples were introduced from low temperature solutions with a liquid-N2 precooled plastic syringe, and quickly injected into the instrument sample port which feeds the instrument syringe pump operating at 10μL/min via a silica capillary line. The heated capillary temperature was 250 °C and the spray voltage was 5 kV. X-ray diffraction was performed at the X-ray diffraction facility at the Johns Hopkins University. The X-ray intensity data were measured on an Oxford Diffraction Xcalibur3 system equipped with a graphite monochromator and an Enhance (Mo) X-ray Source (λ = 0.71073Å) operated at 2 kW power (50 kV, 40 mA) and a CCD detector. The frames were integrated with the Oxford Diffraction CrysAlisRED software package. X-band electron paramagnetic resonance (EPR) spectra were recorded on a Bruker EMX CW-EPR spectrometer controlled with a Bruker ER 041 XG microwave bridge operating at X-band (∼9 GHz). The low temperature experiments were carried out via N2(l) finger dewar.
Synthesis of [(LN(CH2Ph)2)CuII(H2O)(ClO4)]+ (1)
The synthesis and characterization of the ligand LN(CH2Ph)2 was recently reported.39 Ligand LN(CH2Ph)2 (0.250 g, 0.515 mmol) was treated with CuII(ClO4)2·6H2O (0.191 g, 0.516 mmol) (Aldrich) in acetone (10 mL) and stirred for 25 min at RT. The complex was precipitated as greenish blue solid upon addition of Et2O (140 mL). The supernatant was decanted and the resulting crystalline solid was washed two times with Et2O and dried under vacuum to afford [(LN(CH2Ph)2)CuII(H2O)(ClO4)]ClO4·acetone (1) (0.305 g, 72% yield). Single crystals were obtained by vapor diffusion of Et2O into a solution of the complex in acetone. Anal. Calcd. [(LN(CH2Ph)2)CuII(H2O)(ClO4)]ClO4·(acetone) (1); C35H39Cl2CuN5O10: C, 51.01; H, 4.77; N, 8.50. Found: C, 50.88; H, 4.88; N, 8.37. EPR spectrum: X-band spectrometer in acetone at 77 K: g‖ = 2.252, g⊥ = 2.047, A‖ = 170 G, A⊥ = 29.5 G.
Generation of [(LN(CH2Ph)2)CuII(−OOH)]+ (2) and reactivity study
In 20 mL acetone solution, complex 1 (0.337 g, 0.409 mmol) was dissolved and was cooled to −80 °C using an acetone/dry-ice bath. Addition of Et3N (0.412 g, 4.08 mmol) and 50 wt % H2O2 (252 μL, 4.09 mmol) (Aldrich) led to the formation of green complex [(LN(CH2Ph)2)CuII(−OOH)]ClO4 (2), λ = 380 nm (ε = 1400 M-1cm-1). When an aliquot of this −80 °C green solution was injected into the mass spectrometer, a parent peak cluster at m/z 581.03 corresponding to the positive ion [(LN(CH2Ph)2)CuII(OOH)]+ was observed. When H218O2 was used for the formation of 2, the positive ion peak clusters shifted to m/z 585.09. The alkoxide product [(LN(CH2Ph)2)CuII(OOH)]+ (2) was detected with a parent peak cluster at m/z 563.05 corresponding to the positive ion [(LN(CH2Ph)(PhCHO-))CuII]+ (4). An EPR spectrum of the reaction mixture containing 4 reveals typical Cu(II) axial species supporting its mononuclear formulation. When the formation of 2 was carried out with H218O2, the positive ion peak clusters of 4 shifted to m/z 565.20. X-band EPR of 2, in acetone at 77 K: g‖ = 2.240, g⊥ = 2.041, A‖ = 180 G, A⊥ = 27 G. The hydroperoxo species [(LN(CH2Ph)2)CuII(-OOH)]ClO4 (2) was kept at −80 °C and stirred for 15 min. Upon warming, the solution was carefully concentrated to 4 mL under vacuum and the ESI-MS of the reaction solution was recorded. The resulting copper solution was washed three times (200 mL) with Et2O whereupon the Et2O solutions were combined and dried over Na2SO4. The Et2O solution was analyzed by GC and GC-MS (see below, for conditions) confirming the formation of PhCHO (authentic commercial PhCHO was also used for comparison) (yield, 43 %). When the formation of 2 was carried out with H218O2 and similar follow-up procedures were performed, ∼ 65 % 18O-atom incorporation in PhCHO occurs.
Isolation of Organic Products
The residual material after Et2O washing was treated with 250 mL saturated Na2EDTA/H2O solution and the organic part was extracted by using 80 mL CH2Cl2. The CH2Cl2/Na2EDTA/H2O extraction was repeated three times to ensure complete demetallation. The organic CH2Cl2 solution was dried over anhydrous Na2SO4, filtered, and concentrated by rotory evaporation. ESI-MS of the organic extract was recorded. Isolation and purification by column chromatography (with Al2O3, 2% MeOH + CH2Cl2) revealed unreacted ligand LN(CH2Ph)2 (0.032 g, ∼16 %) and the N-debenzylated product LNH(CH2Ph) (0.077 g, ∼48%). The products were characterized by ESI-MS, 1H-NMR and 13C-NMR spectroscopies and thin-layer chromatography (TLC). Formation of LN(CH2Ph)(COPh) and LN(CH2Ph)(COPh) (0.014 g, ∼7%; ESI-MS, 522.53, M+Na and 536.31, M+Na respectively) and LNH(COPh) (0.004 g, ∼3%) ESI-MS (410.65, M+H and 432.51, M+Na) were also observed. Several attempts for isolation of these organic products in absolute pure form were unsuccessful due to their comparable Rf values, however their formulation can be confirmed from both pre and post demetallation. We did not find any evidence for LNH2 formation.
Experiment adding only one equiv peroxide
Triethylamine, Et3N (0.041 g, 0.408 mmol) and 50 wt % H2O2 (25 μL, 0.408 mmol) was added to the 20 mL acetone solution (−80 °C) of complex 1 (0.337 g, 0.409 mmol). Following procedures similar to that described above showed the production of unreacted ligand LN(CH2Ph)2 (0.130 g, ∼65 %), LNH(CH2Ph) (0.026 g, ∼16%), LN(CH2Ph)(COPh) plus LN(CH2Ph)(COPh) (0.002 g, ∼1%). A similar reaction in methanol as solvent lead to formation of ∼14 % LNH(CH2Ph), <1% LN(CH2Ph)(COPh) and LN(CH2Ph)2 in ∼68 % yield. The starting ligand LN(CH2Ph)2 was obtained in ∼64 % yield along with ∼14% LNH(CH2Ph) and <1% LN(CH2Ph)(COPh) in propionitrile as solvent for an analogous [(LN(CH2Ph)2)CuII(H2O)(ClO4)]+ (1)/H2O2/Et3N reaction.
LNH(CH2Ph): 1H-NMR (CDCl3): δ 3.70 (s, 2H, CH2Py), 3.89 (s, 4H, 2CH2Py), 4.48 (d, 2H, CH2Ph), 5.13 (s, 1H, NH), 6.22 (d, 1H), 6.83 (d, 1H), 7.09 (t, 2H), 7.11-7.44 (m, 5H), 7.45-7.68 (m, 5H), 8.49 (d, 2H). 13C-NMR (CDCl3): δ 46.51, 60.21, 77.39, 104.74, 112.04, 121.86, 122.83, 126.79, 127.41, 128.55, 136.38, 137.89, 139.43, 143.28, 148.99, 157.76, 158.29, 159.84. ESI-MS (396.68, M+H; 418.33, M+Na). Rf = 0.23, Alumina, 2% MeOH + CH2Cl2.
Reaction of [(LN(CH2Ph)2)CuI]+ (6) with O2
The synthesis and characterization of copper(I)-complex, [(LN(CH2Ph)2)CuI]B(C6F5)4 (6) was previously reported.39 Formation of bis(μ-oxo)dicopper(III) complex, [{(LN(CH2Ph)2)CuIII}2(O2-)2](B(C6F5)4)2 (9) from O2-reactivity of 6 was confirmed by low-temperature UV-vis and rR spectroscopy. For the determination of ligand oxidation chemistry, a solution of [(LN(CH2Ph)2)CuI]+ (6) (0.125 g, 0.101 mmol) in a 25 mL Schlenk flask with 6 mL Et2O was prepared in the glovebox. Removal to the benchtop and cooling to −80 °C, followed by bubbling O2 via a long syringe needle (20 s) was carried out, and this was followed by removal of excess dioxygen via evacuation and purging with argon. After 30 mins, the solution was warmed to RT and demetallation procedures with Na2EDTA/H2O/CH2Cl2 was performed; this results in the production of dipicolylamine, (0.001 g, ∼ 4%), LNH(CH2Ph) (0.002 g, ∼ 5%) and unreacted LN(CH2Ph)2 (0.037 g, ∼ 75%).
Synthesis of [(LNH(CH2Ph))CuII(Cl)]+ (10)
Here, we followed synthetic procedures previously employed to generate crystalline copper(II)-chloride complexes via ligand-Cu(I)-CHCl3 chemistry.27,40 Isolated LNH(CH2Ph) (0.050 g, 0.126 mmol) and [CuI(CH3CN)4]ClO4 (0.042 g, 0.128 mmol) were placed in a 25 mL Schlenk flask under argon. Dioxygen-free CH3CN (2 mL) was added under argon to form a yellow solution and this was stirred for 10 min. Deoxygenated CHCl3 (0.3 mL) was then added under argon whereupon the solution ceased to look transparent, and its color turned to green. After 2 h, 20 mL of Et2O was added in order to precipitate the copper complex. X-ray quality crystals of this compound [(LNH(CH2Ph))CuII(Cl)]+ (10) were obtained after precipitation prompted by Et2O, from the reaction solution. The green precipitate was recrystallized two times from CH3CN/Et2O. Anal. Calcd. C25H29Cl2CuN5O6: C, 47.66; H, 4.64; N, 11.12. Found: C, 47.71; H, 4.34; N, 11.08. After vacuum-drying the green crystals weighed 0.054 g (68%).
Reaction of [(LN(CH2Ph)2)CuI]B(C6F5)4 (6) with PhIO
[(LN(CH2Ph)2)CuI]B(C6F5)4 (6)39 (0.018 g, 0.015 mmol) was dissolved in 18 mL CH3CN inside the drybox and was taken in a 25 mL Schlenk flask. In a separate Schlenk flask, PhIO (0.033g, 0.150 mmol) was taken with 4.0 mL CH3CN under argon and was stirred for 30 min and cooled to −40 °C. With a syringe, 1 mL of this CH3CN solution was introduced anaerobically into the −40 °C solution of 6. ESI-MS of this low temperature copper solution was recorded, showing the initial strong peak at m/z 564.25 due to high-valent [(LN(CH2Ph)2)CuIII=O]+ species formation. The decomposition of this 564 species was followed with respect to time, which leads to the alkoxo species, [(LN(CH2Ph)(PhCHO−))CuII]+ (4) (m/z = 563.18). When the formation of 4 was carried out with PhIO plus H218O, the positive ion peak clusters of 4 shifted to m/z = 565.34.
Reaction of [(LN(CH3)2)CuI]B(C6F5)4 (7)39 with PhIO
[(LN(CH3)2)CuI]B(C6F5)4 (7) (0.015 g, 0.014 mmol) was dissolved in 15 mL CH3CN inside the drybox and was taken in a 25 mL Schlenk flask. In a separate Schlenk flask, PhIO (0.031 g, 0.140 mmol) was taken with 5.0 mL CH3CN under argon and was stirred for 30 mins. With a syringe, 1 mL of this CH3CN solution (−40 °C) was introduced anaerobically into the −40 °C solution of 7. ESI-MS of the low temperature copper solution was recorded, showing the strong peak at m/z 411.18. When a similar reaction was carried out with [(LN(CH3)(CD3))CuI]+ and PhIO, the positive ion peak clusters contains dominant peak at m/z = 413.24 and 414.14 due to the generation of [(LN(CH3)(CD2O−))CuI]+ and [(LN(CD3)(CH2O−))CuI]+, respectively.
Chemistry of [(LN(CH3)2)CuII(−OOH)]+ (3) and its Reactivity Study
The synthesis, characterization and reactivity studies of copper(II)-hydroperoxo complex, [(LN(CH3)2)CuII(−OOH)]ClO4 (3) were previously reported.27 Via further examination in the present study, we were able to isolate a small amount of one decomposition product [(LN(CH3)2)CuII(HCOO−)(ClO4)CuII-(LNH(CH3))](ClO4)2 (11) with dicopper(II) centers bridged by (HCOO−) group from the reactivity of 3 in acetone. Compound 11 was characterized crystallographically and its EPR and ESI-MS characterization studies were performed (see Supporting information). We also previously described the chemistry of [(LN(CH3)(CD3))CuII(−OOH)]+ (3-CD3) 27 for which now we find that the extracted unreacted ligand has its isotope label scrambled (see discussion below).
Results and Discussion
Ligand Design and Hydroperoxo-Copper(II) Complexes
We have previously carried out and published extensive studies concerning the dioxygen reactivity of the parent ligand copper(I) complex [(TMPA)CuI(CH3CN)]+ along with a variety of analogs.41-43 Masuda and coworkers44,45 elaborated on the TMPA framework inputting potential hydrogen-bonding moieties, placing them in 6-pyridyl positions. In a series of studies, they showed that ligand-copper(I) compounds react with O2 to form binuclear copper-dioxygen adducts, formally μ-1,2-peroxodicopper(II) complexes, which do show altered UV-vis and resonance Raman (rR) spectroscopic properties due to H-bonding.44,45 Further, Masuda generated a variety of hydroperoxo-copper(II) complexes via {ligand-CuII + H2O2 + base} reactions, including those shown in Chart 2.33-36 As indicated one of these could be characterized by X-ray crystallography.33
Chart 2.
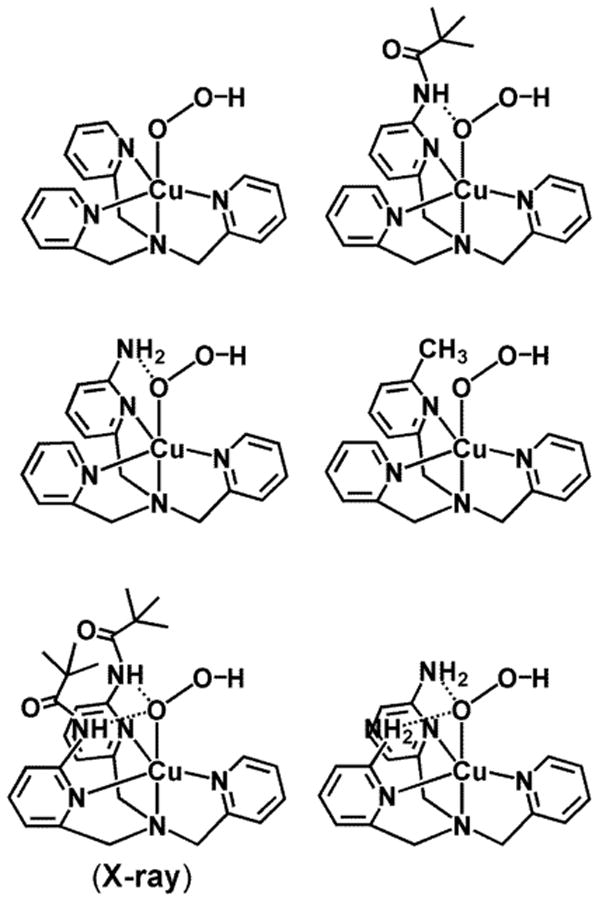
However, oxidative reactivity of these mononuclear CuII(−OOH) species (with tetradentate chelates) towards exogenous substrates was not observed. One reason for such inactivity could be that potential substrates could not achieve an appropriate proximity (either spatially or temporarily) to the CuII(−OOH) moiety. Another explanation could be that stabilization of the CuII(−OOH) unit with H-bonding comes at the cost of reactivity. In the last year, concerning hydroperoxo-Cu(II) compounds, we reported two examples where we ‘mounted’ a potentially oxidizable substrate within the ligand framework: N(CH3)2, i.e., LN(CH3)2 (Chart 1)27 or a hydrocarbon aryl group,26 thus fixed by covalent bonding to be potentially juxtaposed to a CuII(−OOH) moiety or a species derived from it via O-O cleavage. As mentioned in the Introduction, here we detail here the overall oxidative copper ion chemistry with LN(CH2Ph)2.
Chart 1.
Copper(II) Complex [(LN(CH2Ph)2)CuII(H2O)(ClO4)]ClO4 (1)
For the purpose of generating a hydroperoxo complex by starting with Cu(II) and adding H2O2, we straightforwardly synthesized complex 1 (see Experimental Section). X-ray quality crystals were obtained, and the structure is described here, Figure 2.
Figure 2.

ORTEP diagram of [(LN(CH2Ph)2)CuII(H2O)(ClO4)](ClO4) (1). Thermal ellipsoids are drawn by ORTEP and represent 50% probability surfaces. Cu(1)-O(1) = 1.972 Å; Cu(1)-N(1) = 1.989 Å; Cu(1)-N(3) = 1.997 Å; Cu(1)-N(2) = 2.010 Å; ∠ O(1)-Cu(1)-N(1) = 94.34°; ∠ O(1)-Cu(1)-N(3) = 99.06°; ∠ N(1)-Cu(1)-N(3) = 166.31°; Cu1-N4 = 2.935 Å.
The copper(II) ion is five-coordinate. The oxygen atom (O1) from a water molecule along with the alkylamino nitrogen (N2) and pyridyl groups not substituted in the 6-position (N1 and N3) comprise the basal plane of the structure found in a pyramidal geometry around copper center. One of the two perchlorate anions coordinates as the fifth axial ligands, Cu1-O2perchlorate = 2.3515(11) Å. A simple geometric analysis indicates the overall coordination geometry as a slightly distorted square pyramid (τ = 0.156, where τ = 0.00 for a perfect square pyramid and τ = 1.00 for a trigonal bipyramid).46 The copper ion (Cu1) in fact lies in the best least squares basal plane. The pyridyl substituents arm (with N4) is moved and twisted away from the copper ion (Cu…N4 = 2.9345(13) Å, most likely because of steric issues with the 6-dibenzylamino substituent.40,47 The structure is likely maintained in solution, as a typical axial EPR is observed (Figure 3a).
Figure 3.
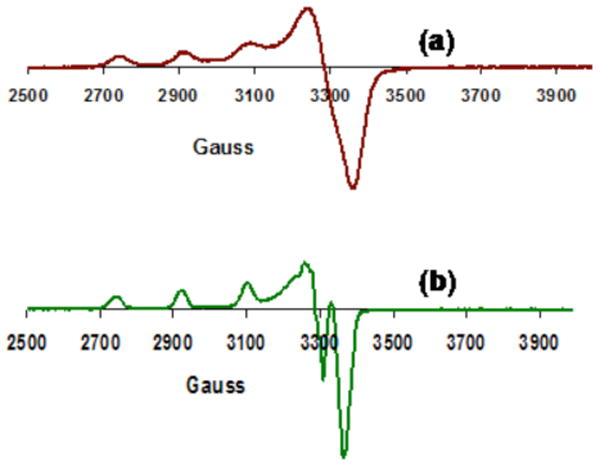
EPR (X-band, 77K, acetone) spectra of (a) [(LN(CH2Ph)2)CuII(H2O)(ClO4)](ClO4) (1), g‖ = 2.252, g⊥ = 2.047, A‖ = 170 G, A⊥ = 29.5 G. (b) [(LN(CH2Ph)2)CuII(−OOH)](ClO4) (2) generated from reaction of 1 with H2O2, g‖ = 2.240, g⊥ = 2.041, A‖ = 180 G, A⊥ = 27 G.
Hydrogen Peroxide Reactivity of [(LN(CH2Ph)2)CuII(H2O)(ClO4)]ClO4 (1)
Generation of [(LN(CH2Ph)2)CuII(−OOH)]+ (2)
The green product solution which forms following addition of 2-3 equiv H2O2/Et3N using 50 % H2O2(aq) to a greenish blue acetone solution of 1 at −80 °C is formulated as the hydroperoxide species [(LN(CH2Ph)2)CuII(−OOH)]+ (2). The band at λmax = 380 nm (ε 1400 M−1cm−1) is assignable to a −OOH –> CuII LMCT absorption based on the correspondence with a number of literature examples.27,33-37 An axial CuII EPR spectrum is also observed for 2, consistent with a single mononuclear species formulation, but which is distinguishable from 1 (Figure 3b).
Direct evidence for [(LN(CH2Ph)2)CuII(−OOH)]+ (2) comes from Electrospray Ionization Mass Spectrometry. Injection of a −80 °C acetone solutions of 2 gives a highest molecular weight parent peak cluster with m/z = 581.03 and expected 63,65 Cu pattern, corresponding to the monocation as formulated. As indicated (Figure 4a), the most prominent ESI-MS peak at m/z = 548.59 matches a complex having lost (H)OOH, [(LN(CH2Ph)2)CuII]+. When formation of 2 was instead carried out using H218O2, the positive ion peak shifts to 585.09 due to formation of [(LN(CH2Ph)2)CuII(−18O18OH)]+ (2-18O); fitting of the parent peak pattern around m/z = 585 indicates > 99 % 18-O incorporation.
Figure 4.
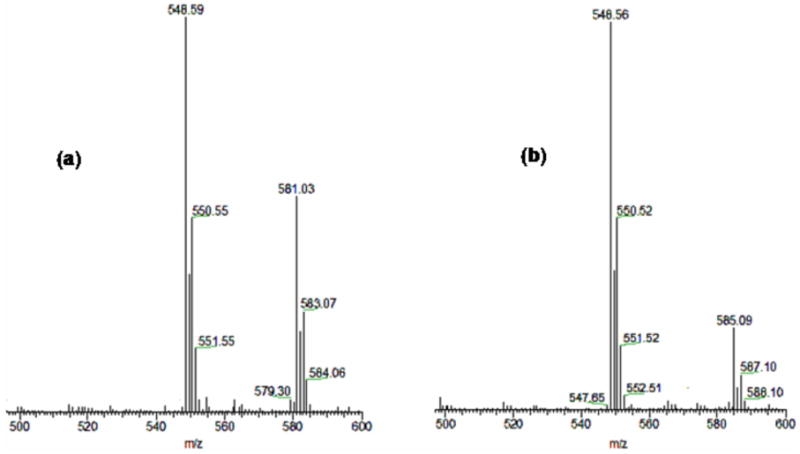
ESI-MS spectra of (a) [(LN(CH2Ph)2)CuII(−OOH)](ClO4) (2) from the reaction of 1 with H2O2 and (b) [(LN(CH2Ph)2)CuII(−18O18OH)](ClO4) (2-18O) generated by 1/H218O2.
Hydrogen peroxide or −OOH are known to attack the carbonyl group of acetone and Itoh and coworkers21 recently observed a thus-formed alkylperoxo adduct bound to copper(II) in related but tridentate ligand-copper complex chemistry. However, by ESI-MS, we see no evidence for such a hydroperoxo-acetone-copper(II) complex. Also, see further discussion below.
Ligand Oxidative Reactivity for [(LN(CH2Ph)2)CuII(−OOH)]+ (2)
Hydroperoxo-copper(II) species 2 is stable in solution at −80 °C, but warming results in a change to a darker green color. Thin-layer chromatography (TLC) and ESI-MS data obtained from the product solution which has been stripped of copper ion (by addition of Na2EDTA(aq) and extraction into CH2Cl2; see Experimental Section) shows that the formation of several new organic products has occurred. Thus, the singly N-dealkylated (i.e., N-debenzylation) compound LNH(CH2Ph) {(m/z = 396.48 (M+H), 418.49 (M+Na)}, the singly oxygenated organic compound LN(CH2Ph)(COPh) {(m/z = 500.32 (M+H), 522.35 (M+Na)}, the over-oxidized/oxygenated amide derived from benzoic acid LNH(COPh) {(m/z = 410.47 (M+H), 432.41 (M+Na)} and the doubly-oxygenated (bis-amide) parent ligand LN(COPh)2 {(m/z = 514.22 (M+H), 536.27 (M+Na)}. Unreacted ligand LN(CH2Ph)2 {(m/z = 486.36 (M+H), 418.49 (M+Na)} is also present. The absolute yields (average of three runs) are summarized in Scheme 2.
Scheme 2.
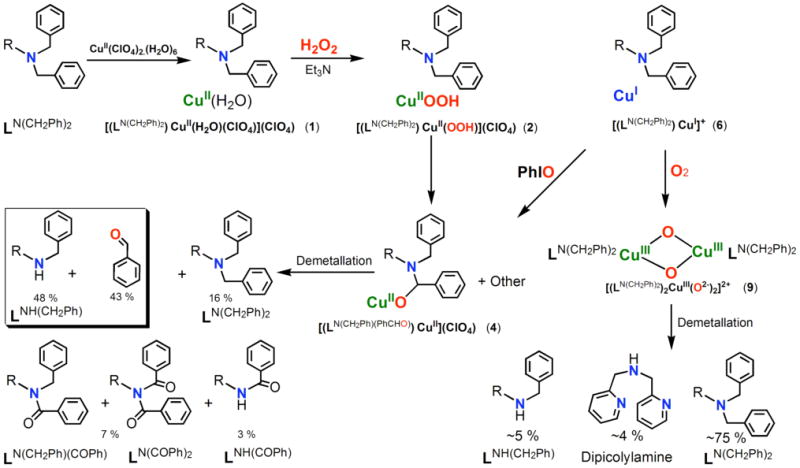
We also observed over-oxidized products, i.e., more than a single two-electron substrate oxidation (peroxide as oxidizing/oxygenating agent) in our previously published results with LN(CH3)2 and [(LN(CH3)2)CuII(−OOH)]+ (3).27 However, here, no corresponding LNH2 product derived from 2 was observed. In the present chemistry, chromatographic separation/isolation reveals that only ∼ 16 % yield of the original LN(CH2Ph)2 ligand remains after reactions. The major (∼48 %) new organic product formed is the oxidatively singly N-debenzylated compound LNH(CH2Ph) (Scheme 2). As would be expected in an N-dealkylation reaction, a corresponding aldehyde should form, and indeed we observe a ∼ 43 % yield of benzaldehyde as determined by GC and GC-MS spectroscopy. When labeled hydrogen peroxide is used in the reaction giving [(LN(CH2Ph)2)CuII(−18O18OH)]+ (2-18O), 18-O labelled PhC(18O)H is formed with ∼65% of the theoretically expected O-18 incorporated. Benzaldehyde, with its pretty exchangeable (with water) carbonyl group, is well-documented to not always retain a high degree of 18-O incorporation.48,49 As mentioned and from the yields obtained (Scheme 2), small but significant amounts of formally four or eight electron over-oxidized products form, ketones LN(CH2Ph)(COPh) plus LN(COPh)2 (7%) and LNH(COPh) (3%) (Scheme 2). These are likely to derive from the reaction of initial two-electron oxidation products with excess hydrogen peroxide (see also below).
When only one equiv H2O2/Et3N is used to generate [(LN(CH2Ph)2)CuII(−OOH)]+ (2), warming and workup leads to considerably more (∼ 65%) unreacted LN(CH2Ph)2, the yield of primary mono N-debenzylated ligand-substrate LNH(CH2Ph) drops to 16 %; only 1 % (total) of LN(CH2Ph)(COPh) plus LN(COPh)2 are obtained. No LNH(COPh) or LNH2 are observed. Thus, the major reaction product is again the two-electron (from one H2O2) oxidatively N-dealkylated ligand, LNH(CH2Ph), is formed in a “dose” dependent manner. Following our comments above, we further wanted to rule out the possible involvement of solvent to activate the hydroperoxo species. Thus, we carried out the formation of [(LN(CH2Ph)2)CuII(−OOH)]+ (2) in both propionitrile and methanol solvents, separately. In both cases, product distributions were found to be the similar as were found for acetone. We conclude that the CuII(−OOH)]+ moiety truly effects the oxidations observed.
Some further insights into the overall chemistry occurring in this system come from direct ESI-MS reaction solution, following warming to RT, but prior to extraction of the copper ion with EDTA (vide supra). The ESI-MS data (in Supporting Information) reveal the presence of several copper complexes, [(LNH(CH2Ph))Cu]+ (m/z = 457.48) and [(LNH(CH2Ph))Cu(acetone)]+ (m/z = 517.27), unreacted [(LN(CH2Ph)2)Cu]+ (m/z = 548.53). In addition, a prominent (intense) ESI-MS cluster is found at m/z = 563.05l, corresponding to alkoxide species [(LN(CH2Ph)(PhCHO-))Cu]+ (4), see Scheme 2. In a corresponding experiment with [(LN(CH2Ph)2)CuII(−18O18OH)]+ (2-18O), 18-O does appear in this alkoxide product, [(LN(CH2Ph)(PhCH(18)O-))Cu]+ (m/z = 565.19; 94 % 18-O incorporation). As will be described below, this finding is relevant to the biomimetic nature of the reaction chemistry and also provides some insight to mechanistic considerations.
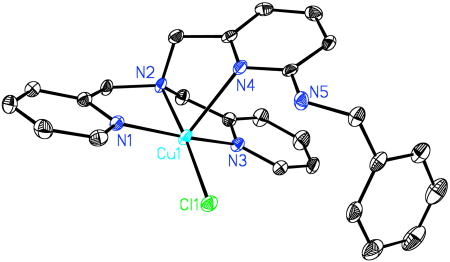
X-ray Crystal Structure of [(LNH(CH2Ph))CuII(Cl)]ClO4 (10)
We wished to further verify our major organic product assignment, and thus for LNH(CH2Ph) as isolated and purified via column chromatography, we formed a copper complex product. Under Ar, reaction of LNH(CH2Ph) and CuI(MeCN)4(ClO4) in acetonitrile followed by addition of CHCl3 leads to the chloro-copper(II) complex, [(LNH(CH2Ph))CuII(Cl)]+ (10) (see Experimental Section). An X-ray structure of the pentacoordinate complex was obtained (see diagram), and it confirms the presence and identity of LNH(CH2Ph). The full X-ray structure is presented in Supporting Information.
 |
Corresponding CuI/O2 Chemistry with LN(CH2Ph)2
While the results so far presented implicate the CuII(−OOH) moiety as being responsible for the observed reactivity (Scheme 2), as before27 we have examined the product(s) of O2 reaction with [(LN(CH2Ph)2)CuI]B(C6F5)4 (6), to determine if copper(I)-dioxygen might be responsible. This seems to not be the case, vide infra.
In the context of other works, the 6/O2 reactions result in the formation of the metastable bis(μ-oxo)dicopper(III) complex, [{(LN(CH2Ph)2)CuIII}2(O2-)2](B(C6F5)4)2 (9), which could in fact be characterized by low-temperature UV-vis and rR spectroscopy.39 Warming such solutions to RT and following Na2EDTA/H2O/CH2Cl2 demetallation procedures, the organic products and yields of these (which are low) are found to be quite different from the [(LN(CH2Ph)2)CuII(H2O)(ClO4)]+ (1)/H2O2/Et3N chemistry, Scheme 2. Some oxidative N-dealkylation product LNH(CH2Ph) is observed but only in ∼ 5% total yield. A small amount of new product, dipicolylamine (∼ 4% yield) is also observed; this must be derived from CuIII2(O2−)2 attack on the benzylic position of the substituted (i.e., with N(CH2Ph)2) pyridyl moiety, analogous to that previously seen by Suzuki and coworkers.50,51 Most of the organic recovered (∼ 75%) is unreacted starting ligand, LN(CH2Ph)2 (Scheme 2). CuI/O2 reaction would normally progress through a copper-superoxo [CuII(O2−)] initial species,6,7 thus the observed oxidative chemistry of [(LN(CH2Ph)2)CuII(H2O)(ClO4)]+ (1)/H2O2/Et3N does not proceed via a superoxo species. The lack of effective N-debenzylation reaction of LN(CH2Ph)2 by [CuIII2(O2-)2]2+ is most likely due to the axial positioning of the N,N-dibenzyl arm which prevents the oxo-atom attack. Suzuki and coworkers demonstrated such an axial ligand elongation for 6-substituted 2-pyridyl ligand arms in [{(6-Me2TPA)CuIII}2(O2-)2]2+ (X-ray, Me2TPA = (2-pyridylmethyl)bis(6-methyl-2-pyridylmethyl)amine).50
Reaction of [(L)CuI]+ with PhIO
One might anticipate that [(LN(CH2Ph)2)CuII(−OOH)]+ (2) potentially could undergo O-O cleavage chemistry giving a high-valent copper-oxo species, something which is sought after but not known (see further discussion below). There is a very broad and more established FeIII-OOR (R = H or alkyl or acyl; either heme or non-heme iron) homolytic or heterolytic O-O cleavage (bio)chemistry.52-57 In this context, iodosylbenzene or analogue donors have been widely utilized with reduced metal complexes to generate higher valent metal-oxo complexes, {i.e., PhIO + ligand-Mn+; (M = heme, non-heme Fe, Mn, …) ––> ligand-Mn+2=(O2-) + PhI}.58-61 As referred to in the Introduction, a copper-oxo moiety is seen to either be responsible for H-atom abstraction in PHM or DβM, or form later in the reaction sequence/mechanism.
In fact we were previously successful in effecting C-H activation and ligand methyl group hydroxylation leading to an alkoxo product [(TMG3trenO−)CuII]+, see diagram {TMG3tren = tris(2-(N-tetramethylguanidyl)ethyl)amine}.24
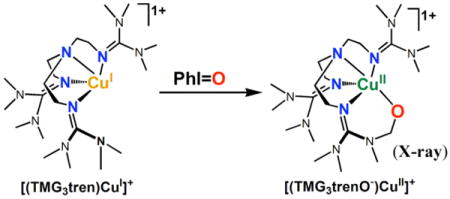 |
Most interestingly, for the present system, reaction of PhIO with the cuprous complex [(LN(CH2Ph)2)CuI]+ (6) did produce [(LN(CH2Ph)(PhCHO−))CuII]+ (4) (Scheme 2), as detected by ESI-MS (m/z = 563.18) as corroborated when employing PhI18O (m/z = 565.34 due to [(LN(CH2Ph)(PhCH(18)O−)CuII]+ (4-18O) in complementary experiments (see Supporting Information). One thus could suggest that the cupryl species [(LN(CH2Ph)2)CuII-O•]+ (8) formed during the reaction and effected the hydroxylation. Caution is required however, since it might be that an iodosylbenzene bound copper complex (e.g., [(LN(CH2Ph)2)CuI-OIPh]+) is the true oxygenating agent and that prior O-O cleavage does not occur.
Potentially exciting observations occurred when we followed the [(LN(CH2Ph)2)CuI]+ (6) / PhIO chemistry using ESI-MS as a function of time; the reaction was carried out at −40 °C and aliquot samples were injected into the mass spectrometer while initially at this temperature. Most interestingly, early on in the reaction, ∼ 1 minute after addition of PhIO to [(LN(CH2Ph)2)CuI]+ (6) (CH3CN solvent, −40 °C) the predominant higher molecular weight ESI-MS species detected occurs with strongest peak at m/z = 564.25 (Figure 5), which in fact correspond to a cupryl species, [(LN(CH2Ph)2)CuIII=O]+ (8, CuIII=O ≡ CuII-O•). This peak is only observed early on, as the product alkoxo complex [(LN(CH2Ph)(PhCHO−))CuII]+ (4) (m/z = 563.18) comes in and dominates. Notice that theoretical fitting of this early time mass spectrum best corresponds to a mixture of 70 % cupryl (8) and 30 % alkoxo product 4 (Figure 5). In the Supporting Information we show a succession of mass spectra (as a function of time), showing how the putative cupryl is there initially but disappears over ∼ 5-6 minutes; it is almost not there at all after ∼ 3 minutes. The yields of the organics from the reaction of [(LN(CH2Ph)2)Cu(I)]B(C6F5)4 (6) with PhIO could not be determined due to insufficient separation and ‘tailing’, observed in column and thin-layer chromatography.
Figure 5.
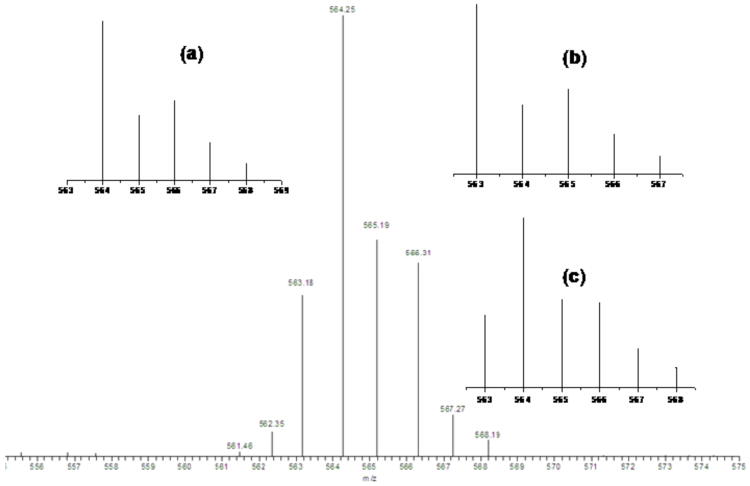
ESI-MS spectra from the reaction of [(LN(CH2Ph)2)CuI]+ (6) with PhIO suggesting the transient formation of the cupryl complex [(LN(CH2Ph)2)CuII-O•]+ (8) at m/z = 564.25 (∼70% formation) and [(LN(CH2Ph)(PhCHO−))CuII]+ (4) at m/z = 563.18 (∼30% formation). Insets showing the expected pattern for (a) 100% formation of 8 and (b) 100% formation of 4, and the best fitting to the data with (c) 70% formation of 8 plus 30% formation of 4. See text for further discussion.
Similarly we here carried out a PhIO reaction with the previously studied ligand complex with dimethylamino substituents/substrate, [(LN(CH3)2)CuI]+ (7).27,39 ESI-MS data indicate that the corresponding alokoxide product forms, i.e. [(LN(CH3)(CH2O−))CuII]+ (5) (m/z = 411.18) (see Supporting Information). This forms from [(LN(CH3)2)CuII(−OOH)]+ (3) chemistry also, as reported.27 Interestingly when the partially deuteriated ligand complex [(LN(CH3)(CD3))CuI]+ (7-CD3) was reacted with PhIO, the ESI-MS analysis leads to the conclusion that for this reaction, the kinetic isotope effect (kH/kD) is 1.9 based on the relative formation of [(LN(CD3)(CH2O−))CuII]+ (m/z = 414.14) and [(LN(CH3)(CD2O−))CuII]+ (m/z = 413.24) (Figure 6). This may be compared to the finding that kH/kD ∼ 2.2 for the [(LN(CH3)(CD3))CuII(−OOH)]+ (3) reaction.27 Considering that 1.9 and 2.2 are nearly within experimental error, we suggest that both the [(L)CuII(−OOH)]+ and [CuI]+/PhIO chemistry proceed through a common reactive intermediate, the cupryl species [(L)CuII-O•]+ (see diagram). The data are suggestive but probably not good enough to yet firmly establish cupryl chemistry.
Figure 6.
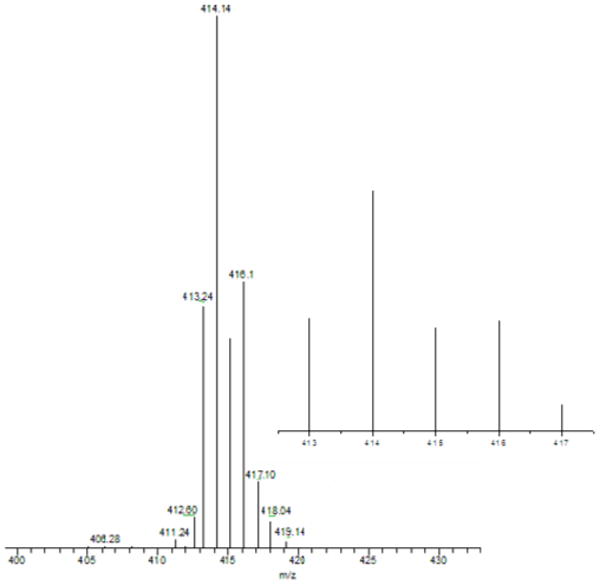
ESI-MS spectra obtained from the reaction of [(LN(CH3)(CD3))CuI]+ (7) with PhIO. The ratio of formation of [(LN(CD3)(CH2O−))CuII]+ (m/z = 414.14) versus [(LN(CH3)(CD2O−))CuII]+ (m/z = 413.24) suggest that kH/kD ∼ 1.9. The inset shows the expected (calculated) ESI-MS spectral pattern for a reaction which would lead to kH/kD ∼ 1.9.
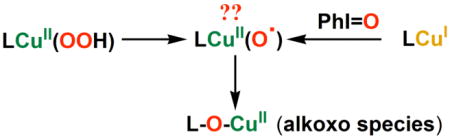 |
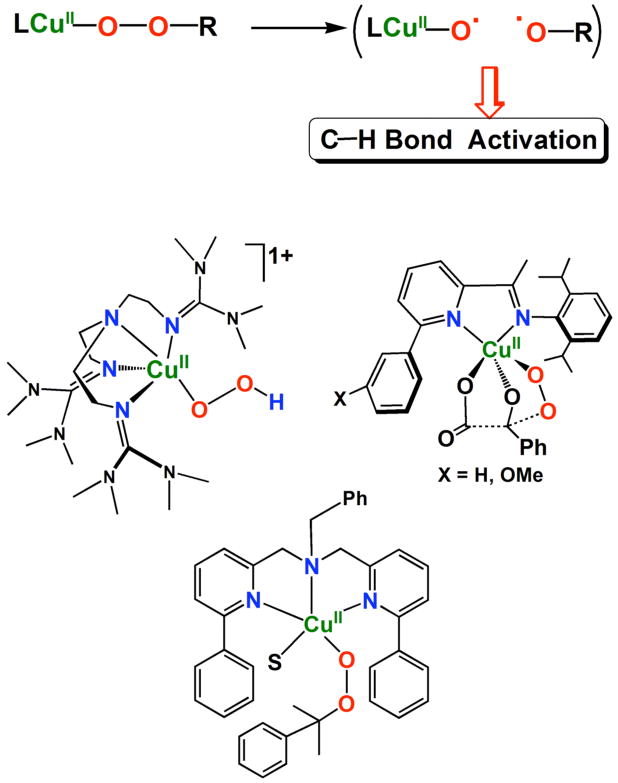
To summarize this section of discussion, we employed PhIO reactions with reduced complexes [(L)CuI]+ to probe the possibility that high-valent curpyl species are involved in the reactions of [(L)CuII(−OOH)]+, i.e., following hemolytic O-O bond cleavage. For both the ligand complexes with LN(CH2Ph)2 and LN(CH3)2, PhIO reactions do give the expected alkoxo products (Scheme 2) and the mass spectrometric data (Figure 5) appear to give direct evidence for the presence or formation of the cupryl complex for the one case, [(LN(CH2Ph)2)CuII-O•]+ (8). We do not wish to overly strongly state that we have detected the first copper cupryl. The overall reaction yields are not exceptional for the PhIO reaction chemistry, although this is probably not the ideal oxo-transfer agent. Also, while detecting the cupryl species in the ESI-MS experiments using PhIO, for some reason it was not observed when employing PhI18O; however that final product alkoxide did contain 18-O. In addition to the chemistry described here (and the initial report),27 CuII-O-OR homolytic cleavage has been suggested to lead to a CuII-O• transient reactive species which may effect substrate oxidation/oxygenation chemistry.20,22,24,62-64 Scheme 4 depicts some of the recent synthetic examples. 27.20,22
Scheme 4.
Mechanistic Discussion
Oxidative N-dealkylation is a relatively common reaction in the biotransformation of alkylamines; it is also catalyzed by cytochrome P-450 monooxygenases. Considerable experimental effort has gone into differentiating between conventional hydrogen-atom transfer (HAT) versus a sequential reaction where a single-electron transfer (SET) oxidation is followed by deprotonation.57,65-67 Oxidative N-dealkylation of the ligand bound to copper in CuIII2(O2−)2 compounds have been observed where mechanistic studies favored initial HAT.7 We also have reported on cases where a CuIII2(O2−)2 (or side-on bound peroxo-dicopper(II) species in equilibrium with this) oxidatively N-dealkylate subsititued dimethylanilines but where a shift in mechanism from ET to HAT was observed, depending on the ease of substrate one-electron oxidation.68
As discussed above for the complexes N,N-dimethyl group at the 6-position of one arm of TMPA derived ligand, observed KIE values are ∼2.2 from [(LN(CH3)(CD3))CuII(−OOH)]+ (3-CD3)27 chemistry and ∼1.9 from [(LN(CH3)(CD3))CuI]+ (7-CD3)/PhIO chemistry. These results suggest a net H-atom abstraction occurs. Organic products isolated (i.e., recovered dialkyl ligand) from [(LN(CH3)(CD3))CuII(−OOH)]+ chemistry included those due to scrambling of isotopes via radical intermediates, and LN(CH3)(CD3), LN(CH3)(CD2H), LN(CH3)(CDH2) and LN(CH3)2 were all observed. However, more studies would be required to differentiate between HAT and a SET step.
Based on the results described here, we outline our proposed mechanism of reaction for [(LN(CH2R′)2)CuII(−OOH)]+ chemistry (Scheme 5). Initial net HAT (either directly from the hydroperoxide or occurring following O-O cleavage) substrate H-atom abstraction would lead to a high-valent CuII-O• (≡ CuIII=O) and substrate imminium radical cation species; ‘rebound’ gives the alkoxide (Path A). Most often in the synthetic system and always in the enzyme, protonation (from active-site solvent) releases the organic products observed, the amine and corresponding aldehyde.
Scheme 5.
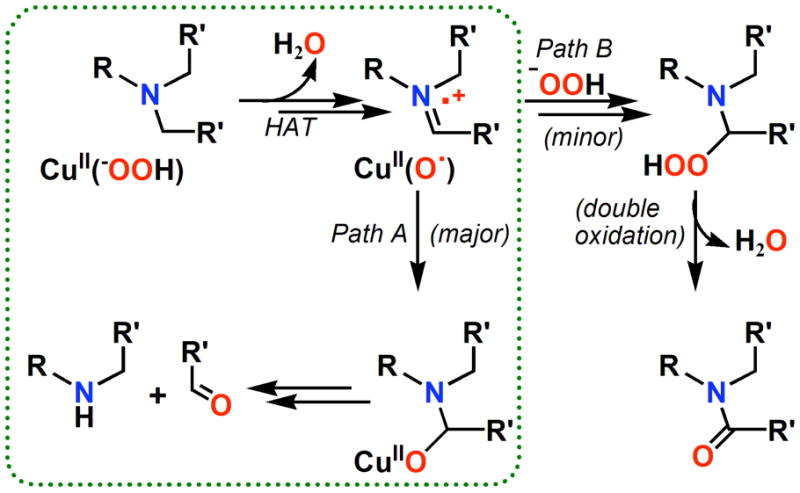
In Scheme 5, we also show (Path B) how an over-oxidized product amide would most likely form in the model synthetic system, via facile nucleophilic attack of a 2nd equivalent of hydroperoxide anion. In connection with other over-oxidation product chemistry, we recently isolated a small amount of a decomposition product, [(LN(CH3)2)CuII(HCOO−)(ClO4)CuII-(LNH(CH3))](ClO4)2 (11), for which we were able to obtain an X-ray structure (Figure 7) (See Supporting Information); this originates from previously described chemistry of [(LN(CH3)2)CuII(−OOH)]+ (3). This odd dicopper(II) complex possesses a bridging formate (HCOO−) group, wherein one of the copper ligands is LN(CH3)2, i.e., unreacted starting ligand, and the other copper ion is supported by a singly N-demethylated product LNH(CH3). An EPR spectrum of the isolated crystals then dissolved in acetone indicates the presence of two independent Cu(II) ions, with overlapping axial spectra (see Supporting Information). Further, an ESI-MS of the material contains two strong peaks; the one at m/z = 427.04 corresponds to [(LNH(CH3))CuII(HCOO−)]+ and the other at m/z = 441.04 corresponds to [(LN(CH3)2)CuII(HCOO−)]+. Generation of formate (HCOO−) can be envisaged since formaldehyde was earlier on detected as a product derived from 3. Similarly, we are able to detect a small ESI-MS peak at m/z = 579.17 from the decomposition product mixture of [(LN(CH2Ph)2)CuII(−OOH)]+ (2), which corresponds to a copper complex species containing benzoate, [(LNH(CH2Ph))CuII(PhCOO−)]+, i.e. a further oxidation product of benzaldehyde which was a primary product derived from 2. All of these over-oxidized products are obtained in very small yields. Never-the-less they are indicative of the extensive chemistry occurring in the manner proposed.
Figure 7.

ORTEP diagram of the cationic component of [(LN(CH3)2)CuII(HCOO−)(ClO4)CuII(LNH(CH3))](ClO4)2 (11). Thermal ellipsoids are drawn by ORTEP and represent 50% probability surfaces. Cu(1)-O(2) = 1.942 Å; Cu(1)-N(3) = 2.020 Å; Cu(1)-N(2) = 2.022 Å; Cu(1)-N(1) = 2.023; ∠ O(2)-Cu(1)-N(3) = 96.00°; ∠ O(2)-Cu(1)-N(2) = 175.51°; ∠ N(3)-Cu(1)-N(2) = 82.15°; ∠ O(2)-Cu(1)-N(1) = 99.39°.
PAM Chemistry Mimic
We have shown here that the hydroperoxo group reaction in [(LN(CH2Ph)2)CuII(−OOH)]+ (2) leads to the formation of oxidatively N-dealkylated LNH(CH2Ph) plus PhCH=O. The −N(CH2Ph)2 substrate (ligand substituent) resides in close proximity to the CuII(−OOH) moiety (Scheme 3, R′ = Ph) and essentially identical chemistry occurs for R′ = H.27 As described above, a copper(II)-alkoxide complex forms prior to organic product release (Scheme 3).
Scheme 3.

Thus, the present model systems (R′ = H, Ph) very closely resemble much of the proposed oxidative N-dealkylation reaction mechanism effected by PAM (Figure 1). There, an active site reactive species forms at CuM with His2Met coordination, ideally juxtaposed to the prohormone peptide substrate C–H group of the terminal glycine. Further, our cupric-alkoxide complex, formed following from [(LN(CH2Ph)2)CuII(−OOH)]+ (2) or [(LN(CH2Ph)2)CuI]+ (6)/PhIO reactivity mimics the ‘product substrate CuII-alkoxide complex’ discussed for both PHM and DβM (see diagram).9 However, as stated in the Introduction, current thinking is that a copper-superoxo complex may be the active species. Or it could be a cupryl species derived from CuII(−OOH) O-O bond cleavage. Our results, with reactivity pattern so close to that of PHM/PAM, rather favors the view that the reactive species is a hydroperoxo or cupryl group.
 |
Summary
This report summarizes our advances in oxidative N-dealkylation chemistry with hydroperoxo copper(II) complexes which have an dialkylamino substrate (-N(CH2Ph)2 or -N(CH3)2) appended as a substituent on one pyridyl group of the tripodal tetradentate TMPA ligand framework. In the previous Communication and from the present full report, we can argue that a CuII(−OOH) or species generated from it (i.e., a high-valent copper-oxo species) can initiate useful substrate hydroxylation reactions, those in fact occurring in the enzyme PAM (PHM + PAL) (Figure 1). Net substrate H-atom abstraction occurs starting with the CuII(−OOH) moiety and the likely structure of an intermediate substrate organic radical species is proposed, accounting for the products observed. The formation of a substrate and −OOH (an oxygen atom) derived alkoxo CuII(−OR) complex has been proven; the latter has also been proposed in PHM mechanisms. The fact that the alkoxo complex forms either from CuII(−OOH) or CuI/PhIO chemistry suggests the possibility that a high-valent copper-oxo reactive intermediate forms, and ESI-MS data provides some further support. Clearly, however, further studies on this or other systems need to be carried out to truly substantiate the existence of a cupryl species, and to elucidate the full chemistry of already existing CuII(−OOH) and CuII(O2−•) complexes.
Supplementary Material
Supporting Information Available: X-ray structures with selected bond lengths and angles, ESI-MS and other spectroscopic details, along with CIF files. This material is available free of charge on the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by grants from the National Institutes of Health (K.D.K., GM28962).
References
- 1.Itoh S, Fukuzumi S. Acc Chem Res. 2007;40:592–600. doi: 10.1021/ar6000395. [DOI] [PubMed] [Google Scholar]
- 2.Cramer C, Tolman W. Acc Chem Res. 2007;40:601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suzuki M. Acc Chem Res. 2007;40:609–617. doi: 10.1021/ar600048g. [DOI] [PubMed] [Google Scholar]
- 4.Itoh S. Curr Opin Chem Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 5.Hatcher L Quant, Karlin KD. J Biol Inorg Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 6.Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 7.Lewis EA, Tolman WB. Chem Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 8.Rolff M, Tuczek F. Angew Chem Int Ed. 2008;47:2344–2347. doi: 10.1002/anie.200705533. [DOI] [PubMed] [Google Scholar]
- 9.Klinman JP. J Biol Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 10.Bauman AT, Yukl ET, Alkevich K, McCormack AL, Blackburn NJ. J Biol Chem. 2006;281:4190–4198. doi: 10.1074/jbc.M511199200. [DOI] [PubMed] [Google Scholar]
- 11.Chen P, Solomon EI. Proc Nat Acad Sci, USA. 2004;101:13105–13110. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prigge ST, Eipper B, Mains R, Amzel LM. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 13.Chen P, Solomon EI. J Am Chem Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 14.Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Inorg Chem. 2006;45:3034–3041. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]
- 15.Crespo A, Marti MA, Roitberg AE, Amzel LM, Estrin DA. J Am Chem Soc. 2006;128:12817–12828. doi: 10.1021/ja062876x. [DOI] [PubMed] [Google Scholar]
- 16.Evans JP, Ahn K, Klinman JP. J Biol Chem. 2003;278:49691–49698. doi: 10.1074/jbc.M300797200. [DOI] [PubMed] [Google Scholar]
- 17.Decker A, Solomon EI. Curr Opin Chem Biol. 2005;9:152–163. doi: 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 18.Schröder D, Holthausen MC, Schwarz H. J Phys Chem B. 2004;108:14407–14416. [Google Scholar]
- 19.Fujii T, Yamaguchi S, Hirota S, Masuda H. Dalton Trans. 2008:164–170. doi: 10.1039/b712572k. [DOI] [PubMed] [Google Scholar]
- 20.Kunishita A, Ishimaru H, Nakashima S, Ogura T, Itoh S. J Am Chem Soc. 2008;130:4244–4245. doi: 10.1021/ja800443s. [DOI] [PubMed] [Google Scholar]
- 21.Kunishita A, Teraoka J, Scanlon JD, Matsumoto T, Suzuki M, Cramer CJ, Itoh S. J Am Chem Soc. 2007;129:7248–7249. doi: 10.1021/ja071623g. [DOI] [PubMed] [Google Scholar]
- 22.Hong S, Huber SM, Gagliardi L, Cramer CC, Tolman WB. J Am Chem Soc. 2007;129:14190–14192. doi: 10.1021/ja0760426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujii T, Yamaguchi S, Funahashi Y, Ozawa T, Tosha T, Kitagawa T, Masuda H. Chem Comm. 2006:4428–4430. doi: 10.1039/b609673e. [DOI] [PubMed] [Google Scholar]
- 24.Maiti D, Lee DH, Gaoutchenova K, Würtele C, Holthausen MC, Sarjeant AAN, Sundermeyer J, Schindler S, Karlin KD. Angew Chem Int Ed. 2008;47:82–85. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- 25.Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. J Am Chem Soc. 2007;129:264–265. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]
- 26.Maiti D, Lucas HR, Sarjeant AAN, Karlin KD. J Am Chem Soc. 2007;129:6998–6999. doi: 10.1021/ja071704c. [DOI] [PubMed] [Google Scholar]
- 27.Maiti D, Sarjeant AA Narducci, Karlin KD. J Am Chem Soc. 2007;129:6720–6721. doi: 10.1021/ja0719024. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Sarjeant AAN, Karlin KD. Inorg Chem. 2006;45:7160–7172. doi: 10.1021/ic060598x. [DOI] [PubMed] [Google Scholar]
- 29.Li L, Sarjeant AAN, Vance MA, Zakharov LN, Rheingold AL, Solomon EI, Karlin KD. J Am Chem Soc. 2005;127:15360–15361. doi: 10.1021/ja054948a. [DOI] [PubMed] [Google Scholar]
- 30.Itoh K, Hayashi H, Furutachi H, Matsumoto T, Nagatomo S, Tosha T, Terada S, Fujinami S, Suzuki M, Kitagawa T. J Am Chem Soc. 2005;127:5212–5223. doi: 10.1021/ja047437h. [DOI] [PubMed] [Google Scholar]
- 31.Kodera M, Kita T, Miura I, Nakayama N, Kawata T, Kano K, Hirota S. J Am Chem Soc. 2001;123:7715–7716. doi: 10.1021/ja010689n. [DOI] [PubMed] [Google Scholar]
- 32.Ohta T, Tachiyama T, Yoshizawa K, Yamabe T, Uchida T, Kitagawa T. Inorg Chem. 2000;39:4358–4369. doi: 10.1021/ic000018a. [DOI] [PubMed] [Google Scholar]
- 33.Wada A, Harata M, Hasegawa K, Jitsukawa K, Masuda H, Mukai M, Kitagawa T, Einaga H. Angew Chem Int Ed. 1998;37:798–799. doi: 10.1002/(SICI)1521-3773(19980403)37:6<798::AID-ANIE798>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 34.Yamaguchi S, Wada A, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Chem Lett. 2004;33:1556–1557. doi: 10.1021/ic0496572. [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi S, Nagatomo S, Kitagawa T, Funahashi Y, Ozawa T, Jitsukawa K, Masuda H. Inorg Chem. 2003;42:6968–6970. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]
- 36.Yamaguchi S, Masuda H. Sci Technol Adv Mat. 2005;6:34–47. [Google Scholar]
- 37.Fujii T, Naito A, Yamaguchi S, Wada A, Funahashi Y, Jitsukawa K, Nagatomo S, Kitagawa T, Masuda H. Chem Comm. 2003:2700–2701. doi: 10.1039/b308073k. [DOI] [PubMed] [Google Scholar]
- 38.Würtele C, Gaoutchenova E, Harms K, Holthausen MC, Sundermeyer J, Schindler S. Angew Chem Int Ed. 2006;45:3867–3869. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]
- 39.Maiti D, Woertink JS, Sarjeant AA Narducci, Solomon EI, Karlin KD. Inorg Chem. 2008 doi: 10.1021/ic702437c. in press. [DOI] [PubMed] [Google Scholar]
- 40.Lucchese B, Humphreys KJ, Lee DH, Incarvito CD, Sommer RD, Rheingold AL, Karlin KD. Inorg Chem. 2004;43:5987–5998. doi: 10.1021/ic0497477. [DOI] [PubMed] [Google Scholar]
- 41.Hatcher LQ, Karlin KD. Adv Inorg Chem. 2006;58:131–184. [Google Scholar]
- 42.Karlin KD, Zuberbühler AD. Formation, Structure and Reactivity of Copper Dioxygen Complexes. In: Reedijk J, Bouwman E, editors. Bioinorganic Catalysis: Second Edition, Revised and Expanded. Marcel Dekker, Inc.; New York: 1999. pp. 469–534. [Google Scholar]
- 43.Karlin KD, Kaderli S, Zuberbühler AD. Acc Chem Res. 1997;30:139–147. [Google Scholar]
- 44.Yamaguchi S, Wada A, Funahashi Y, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Eur J Inorg Chem. 2003:4378–4386. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]
- 45.Wada A, Honda Y, Yamaguchi S, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Inorg Chem. 2004;43:5725–5735. doi: 10.1021/ic0496572. [DOI] [PubMed] [Google Scholar]
- 46.Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC. J Chem Soc Dalton Trans. 1984:1349–1356. [Google Scholar]
- 47.Maiti D, Woertink JS, Vance MA, Milligan AE, Sarjeant AA Narducci, Solomon EI, Karlin KD. J Am Chem Soc. 2007;129:8882–8892. doi: 10.1021/ja071968z. [DOI] [PubMed] [Google Scholar]
- 48.Lee D, Lippard SJ. Inorg Chem. 2002;41:827–837. doi: 10.1021/ic011008s. [DOI] [PubMed] [Google Scholar]
- 49.Greenzald P, Luz Z, Samuel D. Trans Faraday Soc. 1968;64:2780–2786. [Google Scholar]
- 50.Hayashi H, Fujinami S, Nagatomo S, Ogo S, Suzuki M, Uehara A, Watanabe Y, Kitagawa T. J Am Chem Soc. 2000;122:2124–2125. [Google Scholar]
- 51.Mizuno M, Honda K, Cho J, Furutachi H, Tosha T, Matsumoto T, Fujinami S, Kitagawa T, Suzuki M. Angew Chem Int Ed. 2006;45:6911–6914. doi: 10.1002/anie.200602477. [DOI] [PubMed] [Google Scholar]
- 52.Shan X, Que JL. J Inorg Biochem. 2006;100:421–433. doi: 10.1016/j.jinorgbio.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 53.Costas M, Mehn MP, Jensen MP, Que L. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 54.Soper JD, Kryatov SV, Rybak-Akimova EV, Nocera DG. J Am Chem Soc. 2007;129:5069–5075. doi: 10.1021/ja0683032. [DOI] [PubMed] [Google Scholar]
- 55.Nam W, Han HJ, Oh SY, Lee YJ, Choi MH, Han SY, Kim C, Woo SK, Shin W. J Am Chem Soc. 2000;122:8677–8684. [Google Scholar]
- 56.Traylor TG, Traylor PS. Reaction of Dioxygen and Its Reduced Forms with Heme Proteins and Model Porphyrin Complexes. In: Valentine JS, Foote CS, Greenberg A, Liebman JF, editors. Active Oxygen: Active Oxygen in Biochemistry. Chapman & Hall; New York: 1995. pp. 84–187. [Google Scholar]
- 57.Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 58.Que L., Jr Acc Chem Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- 59.McLain JL, Lee J, Groves JT. Biomimetic Oxidations Catalyzed by Transition Metal Complexes. 2000:91–169. [Google Scholar]
- 60.Song WJ, Seo MS, George S DeBeer, Ohta T, Song R, Kang MJ, Tosha T, Kitagawa T, Solomon EI, Nam W. J Am Chem Soc. 2007;129:1268–1277. doi: 10.1021/ja066460v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qin K, Incarvito CD, Rheingold AL, Theopold KH. J Am Chem Soc. 2002;124:14008–14009. doi: 10.1021/ja028382r. [DOI] [PubMed] [Google Scholar]
- 62.Chen P, Fujisawa K, Solomon EI. J Am Chem Soc. 2000;122:10177–10193. [Google Scholar]
- 63.Comba P, Knoppe S, Martin B, Rajaraman G, Rolli C, Shapiro B, Stork T. Chem Eur J. 2008;14:344–357. doi: 10.1002/chem.200700865. [DOI] [PubMed] [Google Scholar]
- 64.Klinman JP. Chem Rev. 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 65.Ortiz de Montellano PR. Cytochrome P450 Monooxgenase. 3rd. Kluwer Academic/Plenum Publishers; New York: 2005. [Google Scholar]
- 66.Bhakta M, Hollenberg PF, Wimalasena K. Chem Comm. 2005:265–267. doi: 10.1039/b412221f. [DOI] [PubMed] [Google Scholar]
- 67.Nehru K, Seo MS, Kim J, Nam W. Inorg Chem. 2007;46:293–298. doi: 10.1021/ic0614014. [DOI] [PubMed] [Google Scholar]
- 68.Shearer J, Zhang CX, Zakharov LN, Rheingold AL, Karlin KD. J Am Chem Soc. 2005;127:5469–5483. doi: 10.1021/ja045191a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: X-ray structures with selected bond lengths and angles, ESI-MS and other spectroscopic details, along with CIF files. This material is available free of charge on the Internet at http://pubs.acs.org.