

Summary
Apoptosis is a widely accepted component of the pathogenesis of Parkinson's disease (PD), a debilitating neurodegenerative disorder characterized by loss of dopaminergic neurons in the substantia nigra. However, additional death programs were implicated, and current understanding of the cycle of intracellular events that leads to the demise of these neurons is limited. Gene therapy strategies proposed to inhibit apoptosis have met with relatively limited success. Here we report that the anti-apoptotic HSV-2 gene ICP10PK protects neuronally differentiated PC12 cells from death caused by 1-methyl-4-phenylpyridinium (MPP+) (in vitro PD model) through inhibition of calpain I activation and the resulting inhibition of Bax translocation to the mitochondria, AIF release and caspase-3 activation. Neuroprotection is through ICP10PK-mediated activation of the PI3-K/Akt survival pathway and up-regulation/stabilization of the anti-apoptotic protein Bcl-2 and the cytoprotective chaperone Hsp70.
Keywords: ICP10PK, Parkinson's disease, MPP+, Programmed cell death, calpain, gene therapy
Introduction
Parkinson's disease (PD) is a debilitating neurodegenerative disorder that is characterized by selective loss of dopaminergic neurons in the substantia nigra. The intracellular cascade of events leading to neuronal death is still poorly understood, and appears to involve multiple death programs and inflammatory responses potentially related to microglial activation.1 Therapeutic strategies to inhibit apoptosis were considered, but they met with relatively limited success,2,3 and the surviving neurons did not retain normal synaptic function.4 Further confounding decisions about the appropriate choice of therapy, recent findings suggest that the death of dopaminergic neurons is not due to canonical apoptosis, but rather to caspase-independent programmed cell death (PCD)5-8 or necrosis.9 Consequently, treatment is largely symptomatic rather than preventive.10
Previous studies have shown that the herpes simplex virus type 2 (HSV-2) protein ICP10PK inhibits caspase-dependent apoptosis and protects from various neurotoxic stimuli including viral infection, treatment with a protein kinase C inhibitor, disruption of osmolar environment, loss of trophic growth support and excitotoxicity. Neuroprotection was seen in cultured cells, organotypic slices and animal models, and it was through the activation of redundant survival pathways.11-16 Most importantly, the surviving neurons retained synaptic function,11,13 suggesting that ICP10PK has strong and versatile neuroprotective potential that might be harnessed in the treatment of PD. However, the ability of ICP10PK to inhibit caspase-independent PCD is still unknown. The studies described in this report were designed to address this question, using an in vitro model of PD in which neuronally differentiated PC12 cells are treated with MPP+. We report that ICP10PK inhibits MPP+-induced PCD by inhibiting calpain I activation and AIF release, through activation of the PI3-K/Akt survival pathway. This is the first report that ICP10PK inhibits calpain I activation, and it underscores the therapeutic promise of ICP10PK in neurodegenerative diseases associated with multiple death programs.
Results
ICP10PK inhibits MPP+-induced toxicity
To examine whether ICP10PK inhibits MPP+-induced cell death, neuronally differentiated PC12 cells were treated with the ICP10PK vector ΔRR, that is neuroprotective in other toxicity paradigms,11,13-17 and exposed to MPP+ (1 mM, 24 hrs) at 24 hrs after infection. PCD was evaluated based on DNA fragmentation measured by TUNEL. The % TUNEL+ cells was calculated by counting 5 randomly selected fields, (at least 250 cells, in a 3mm2 area) and results are expressed as % TUNEL+ cells/total cells (determined by DAPI staining) ± SEM11,12,16. The vector deleted in ICP10PK (ΔPK) and cells mock-infected with PBS served as control. The % TUNEL+ cells induced by MPP+ was significantly reduced by ΔRR (p<0.001), but not ΔPK, suggesting that ICP10PK inhibits MPP+-induced toxicity (Figure 1a). However, use of ΔRR does not fully exclude the possibility that HSV-2 genes other than ICP10PK may contribute to neuroprotection. Accordingly, the second series of experiments asked whether neuroprotection also occurs in PC12 cells stably transfected with ICP10PK. We used two clonal lines, one stably transfected with ICP10PK (PC47), the other stably transfected with the ICP10 kinase-negative mutant p139™ (PC139). The establishment and properties of these cells were previously described12 and are summarized in Materials and Methods. Neuronally differentiated PC12, PC47 and PC139 cells were mock treated with PBS or treated with MPP+ (0.5, 1, or 5 mM) and examined by TUNEL 24 and 48 hours later. The % TUNEL+ cells in PC47 cultures given 5 mM MPP+ for 48 hrs (32±5%) was significantly (p<0.001) lower than in similarly treated PC12 and PC139 cultures (72±5 and 82±6%, respectively) (Figure 1b,c). Staining with antibody to tyrosine hydroxylase (TH), the rate-limiting enzyme in dopamine synthesis that is commonly used as a marker for functional dopaminergic neurons,18 indicated that 69±6% of the MPP+-treated PC47 cells were positive for both ICP10PK and TH, while the % TH+ cells was significantly lower (p<0.001) in PC12 and PC139 cultures (supplemental Figure 1). Collectively, the data indicate that ICP10PK protects from MPP+-induced toxicity independent of other viral genes, and the surviving neurons retain functionality. Stably transfected cells were used to examine the mechanism of neuroprotection.
Figure 1.

PC12 cells infected with ΔRR or stably transfected with ICP10 are protected from MPP+-induced PCD. (a) PC12 cells neuronally differentiated as described in Materials and Methods were mock-infected (PBS) or infected with ΔRR or ΔPK (0.1 pfu/cell). Twenty-four hrs later they were treated with MPP+ (1 mM; 24 hrs) and examined by TUNEL as described in Materials and Methods (***, p < 0.001 relative to similarly-treated PC12 and PC139 cells). (b) Neuronally differentiatedPC12, PC139 and PC47 cells mock-treated with PBS or treated with MPP+ (5 mM; 24 hrs) were stained by TUNEL using DAPI staining to identify cell nuclei. (c) Neuronally differentiated PC12, PC139 and PC47 cells were mock-treated with PBS or treated with MPP+ (0.5, 1 or 5 mM) for 24 or 48 hrs and examined by TUNEL (***, p < 0.001 relative to similarly treated PC12 and PC139 cells). Similar results were obtained in another clonal line of ICP10PK-transfected cells (data not shown).
ICP10PK inhibits MPP+-triggered AIF release/nuclear translocation
Both caspase-dependent19-21 and independent5-7 programs were implicated in MPP+-induced toxicity. Previous studies had shown that ICP10PK overrides caspase-dependent cascades,11-17 but its ability to inhibit caspase-independent death programs is still unknown. In a first series of experiments to address this question, neuronally differentiated PC12, PC139 and PC47 cells were treated with MPP+, exposed to the mitochondrion-selective dye MitoTracker Red 580 and stained with AlexaFluor 488-labeled AIF antibody, as described in Materials and Methods. MitoTracker staining (mitochondria) was seen in all cultures (Figure 2a). In MPP+-treated PC47 cells, AIF co-localized with MitoTracker, but co-localization was not seen in similarly treated PC12 and PC139 cells, in which AIF was intranuclear and co-localized with DAPI (Figure 2a). This is not an artifact unique to stably transfected cells, as AIF co-localized with the mitochondria in ΔRR-infected PC12 cells which are protected from PCD, but not in the unprotected ΔPK-infected cells, in which AIF was intranuclear (supplemental Figure 2).
Figure 2.
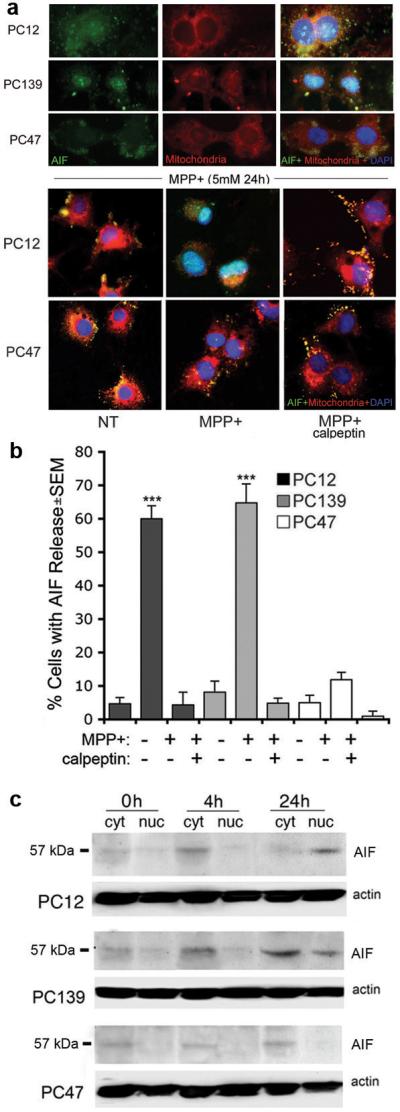
MPP+-induced AIF release/nuclear translocation is calpain-dependent and inhibited by ICP10PK. (a) Neuronally differentiated PC12, PC139, and PC47 cells were treated with MPP+ (5 mM; 24 hrs), incubated with MitoTracker Red 580, fixed and stained with AIF antibody. DAPI was used to visualize cell nuclei. AIF co-localized with DAPI in PC12 and PC139, but not PC47 cells. The experiment was repeated with cells given MPP+ together with calpeptin (50 μM) and merged images are shown for PC12 and PC47 cells. PC139 cells were similar to PC12. (b) Cells were treated as in (a) and those with AIF localization in the cytoplasm or nucleus (AIF release) were counted and the % positive cells calculated relative to the total cell number (determined by DAPI). Results are expressed as % cells with AIF release ± SEM (***, p<0.001 vs. untreated cells). (c) Cytoplasmic and nuclear fractions from neuronally differentiated PC12, PC139, and PC47 cells treated with MPP+ for 0, 4 or 24 hrs were immunoblotted with AIF antibody. The blots were stripped and re-probed with actin as loading control.
Because recent reports have associated AIF release with calpain activation7,22, we wanted to know whether ICP10PK-mediated neuroprotection is associated with calpain inhibition. Neuronally differentiated PC12, PC139, and PC47 cells were treated with MPP+ in the absence or presence of the cell-permeable calpain inhibitor calpeptin (50 μM), incubated with MitoTracker and stained with AIF antibody, as described in Materials and Methods. The results summarized in Figure 2a for PC12 and PC47 cells, indicate that calpeptin inhibited AIF translocation to the nucleus in MPP+-treated PC12 cells. Similar results were obtained for PC139 cells (data not shown), but translocation was not seen in PC47 cells, whether in the absence or presence of calpeptin (Figure 2a). The cells in which AIF did not co-localize with mitochondria (e.g. AIF was in the cytoplasm or nucleus) were counted in 5 randomly selected fields (at least 250 cells) and the % with AIF release was calculated relative to total cell numbers determined by DAPI staining. AIF release was virtually identical in MPP+-treated PC12 and PC139 cells (60±4 and 65±6%, respectively), but it was significantly lower in MPP+-treated PC47 cells (12±2%). In PC12 and PC139 cells, release was inhibited by calpeptin (4±3 and 5±2% for PC12 and PC139 cells, respectively) (Figure 2b), but calpeptin had no effect on AIF release from PC47 cells (1±1%). These percentages are not an artifact due to different levels of transgene expression, because both ICP10PK and p139™ are expressed in 96-100% of the cells12 (supplemental Fig. 1). The data indicate that MPP+-induced AIF release is calpain-dependent, and it is inhibited by ICP10PK.
To confirm that ICP10PK inhibits the MPP+-induced AIF release, cytoplasmic and nuclear fractions from neuronally differentiated PC12, PC139 and PC47 cells were obtained at 0, 4, or 24 hrs after treatment with MPP+ and immunoblotted with AIF antibody. A 57 kDa band, which is consistent with the released AIF protein,22 was barely detectable in the cytoplasmic fractions from all three cell types at 0 hrs. Its levels were significantly higher in the cytoplasmic fractions from PC12 and PC139 cells treated with MPP+ for 4 hrs, preceding its nuclear translocation, which was first seen at 24 hrs post-treatment. The levels of AIF were not increased in the cytoplasmic fractions from MPP+-treated PC47 cells and the nuclear fractions remained AIF-free (Figure 2c).
AIF is a major component of MPP+-induced PCD
MPP+ caused a significant (2.5-fold) increase in the levels of AIF, but these were reduced to the baseline level when MPP+ was given together with an AIF antisense oligonucleotide (aODN) (10 μM) (Fig. 3a). This reduction in the levels of AIF was associated with a significant (p<0.01) increase in cell survival (decreased % TUNEL+ cells) (Fig. 3c), but increased survival was not seen in cells in which MPP+ was given with the sense oligonucleotide (sODN) that did not inhibit AIF expression. The data indicate that AIF release and its nuclear translocation play a key role in MPP+-induced PCD, and they are inhibited by ICP10PK.
Figure 3.
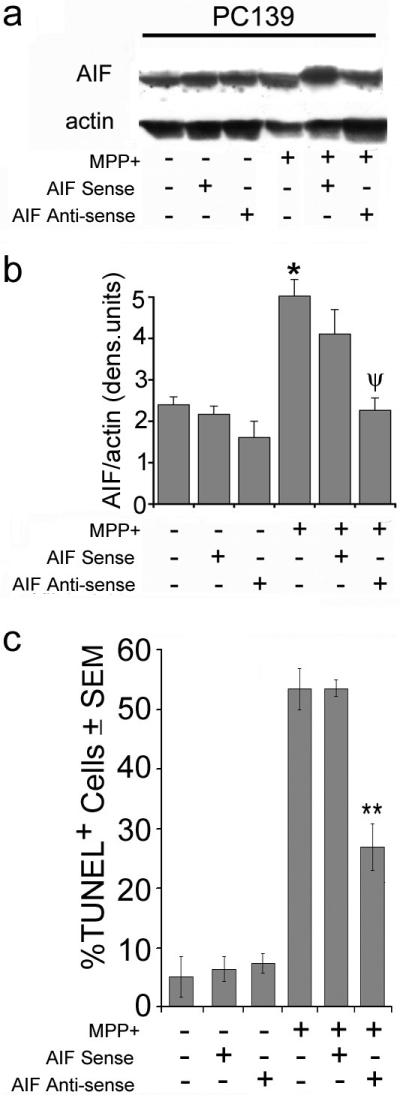
AIF plays a critical role in MPP+-induced PCD. (a) Cell extracts (50 μg protein) from PC139 cells pre-treated or not with antisense or sense AIF ODNs (24 hrs) and given MPP+ (5 mM; 24 hrs) were immunoblotted with AIF antibody. The blots were stripped and re-probed with actin antibody (loading control). (b) Data from 3 independent experiments as in (a) were quantified by densitometric scanning, as described in Materials and Methods, and the results are expressed as AIF/actin densitometric units ± SEM (*, p<0.05 vs. untreated cells; ψ, p<0.05 vs. MPP+ alone). (c) PC139 cells treated as in (a) were examined by TUNEL and the results are expressed as % TUNEL+ cells ± SEM (**, p<0.01 vs. MPP+ alone).
MPP+-induced PCD is calpain I-dependent; ICP10PK inhibits calpain I activation
Having seen that calpeptin inhibits AIF release in MPP+-treated PC12 and PC139 cells, we wanted to examine the role of calpain in ICP10PK-mediated neuroprotection. Duplicates of the cultures examined for AIF release were immunoblotted with antibody to calpain I and examined for the presence of the activated protein isoform. The ratio of the active (p76) to inactive (p80) isoforms was increased by MPP+ (2-fold) in PC12 and PC139 cells. Consistent with independent reports for calpeptin-mediated inhibition of calpain I,23,24 calpeptin restored the p76/p80 ratio in MPP+-treated PC12 and PC139 cells to that of untreated cells (data not shown), but the p76/p80 ratio was not increased in PC47 cells, whether treated or not with calpeptin (Figure 4a). In PC47 cells given MPP+ together with the PI3-K inhibitor LY294002, the p76/p80 ratio was similar to that seen in MPP+-treated PC12 and PC139 cells (Figure 4b), suggesting that ICP10PK inhibits calpain I activation through the activation of the PI3-K/Akt pathway. Calpain inhibition is a critical event in the ability of ICP10PK to inhibit MPP+-induced cell death, because the % TUNEL+ cells was reduced in PC12 and PC139 cultures treated with MPP+ in the presence of calpeptin, as compared to MPP+ treatment alone. By contrast, in PC47 cells, in which calpain I is inhibited by ICP10PK, MPP+ did not increase the % TUNEL+ cells whether in the absence or presence of calpeptin (Figure 4c).
Figure 4.

MPP+-induced PCD is calpain I-dependent and inhibited by ICP10PK. (a) Extracts of neuronally differentiated PC139 and PC47 cells untreated (NT) or treated with MPP+ (5 mM, 24 hrs) were immunoblotted with antibody that recognizes full-length (80 kDa) and cleaved/activated (76 kDa) calpain I. (b) The bands from 3 independent experiments were quantitated by densitometric scanning and the p76/p80 ratios are expressed as densitometric units ± SEM. Cells treated as in (a) but in the presence of the PI3-K inhibitor LY294002 (LY, 50 μM) were similarly analyzed. PC12 cells are similar to PC139. (c) Duplicate cultures of PC12, PC139, and PC47 cells treated with MPP+ (5 mM; 24 hrs) in the presence or absence of calpeptin (50 μM) were examined for cell death by TUNEL. Results are expressed as % TUNEL+ cells ± SEM (***, p<0.001 vs. untreated; **, p<0.01 vs. MPP+).
MPP+ triggers calpain-dependent Bax mitochondrial translocation
Because Bax was implicated in several paradigms of neuronal PCD,6,25 we wanted to know whether it is related to the ability of ICP10PK to inhibit MPP+-induced PCD. Neuronally differentiated PC12, PC139 and PC47 cells were treated, or not, with MPP+ in the presence or absence of calpeptin, incubated with MitoTracker and stained with FITC-labeled Bax antibody. The number of cells with Bax translocation (co-localization with mitochondria) was counted (5 randomly selected fields, >250 cells) and the % was calculated relative to the total cell numbers. In untreated PC12 cells, Bax was in the cytosol, but it did not co-localize with MitoTracker (Fig. 5a). Mitochondrial translocation was seen in 63±9% of the MPP+-treated PC12 cells and it was inhibited by calpeptin (reduced to 9±2%) (Figure 5a,b). Similar results were obtained for PC139 cells (58±3% and 7±1% Bax translocation for MPP+ and MPP+ with calpeptin, respectively). By contrast, in MPP+-treated PC47 cells, in which calpain I is not activated, Bax did not co-localize with MitoTracker whether in the presence or absence of calpeptin (12±2 and 10±3% in MPP+ and MPP+ with calpeptin, respectively) (Figure 5). The data indicate that MPP+ triggers calpain-dependent Bax translocation to the mitochondria and this is inhibited by ICP10PK.
Figure 5.

ICP10PK-mediated inhibition of calpain prevents Bax mitochondrial translocation. (a) MPP+-treated (5 mM, 24 hrs) PC12 and PC47 cells incubated with MitoTracker Red 580 in the absence and presence of calpeptin (50 μM) were stained with Bax antibody (green). DAPI (blue) was used to visualize the nuclei. Bax co-localizes with the mitochondria (yellow) only in MPP+-treated PC12 cells. Results for PC139 cells are similar to those obtained for PC12 cells. (b) Cells with Bax mitochondrial localization were counted and the % calculated relative to total cell numbers determined by DAPI. Results are expressed as % cells with Bax translocation ± SEM. ***, p<0.001 vs. untreated cells.
MPP+ triggers calpain-dependent caspase-3 activation
The contribution of caspase activation to the death of MPP+-treated dopaminergic neurons is still controversial.5,7 Having seen that ICP10PK inhibits calpain activation, we wanted to know whether its neuroprotective activity is caspase-independent. Neuronally differentiated PC12, PC139, and PC47 cells were treated with MPP+ or mock-treated with PBS and stained by double immunofluorescence with FITC-labeled antibody to activated caspase-3 (caspase-3p20) and AlexaFluor 546-labeled ICP10 antibody. The caspase-3p20 staining cells were counted and the % positive cells calculated as described above. Caspase-3p20 staining was seen in a minimal number (∼2%) of mock-treated PC12, PC139 and PC47 cells (Figure 6a,b). MPP+ caused a small, albeit significant (p<0.01), increase in the % staining cells in PC12 and PC139 cultures (21±4 and 25±6%, respectively). This increase was inhibited by calpeptin (5±4 and 7±2% caspase-3p20+ cells, respectively), indicating that caspase is downstream of calpain activation. MPP+ did not increase the % caspase-3p20+ cells in PC47 cultures, whether treated or not with calpeptin (1±1 and 5±3%, respectively) (Figure 6a-c). Triple staining with AlexaFluor 350-labeled ICP10 antibody, AlexaFluor 546-labeled TH antibody, and FITC-labeled caspase-3p20 antibody, confirmed that most PC47 cells were negative for caspase-3p20 while evidencing ICP10PK and TH co-localization (supplemental Figure 3). Significantly, although caspase is a relatively minor component of the death network, downstream of calpain, it contributes to the death of PC12 and PC139 cells, as evidenced by the finding that its inhibition with ZVAD-fmk caused a 25% reduction in the % TUNEL+ cells. ZVAD-fmk had no effect on PC47 cells (Figure 6d).
Figure 6.

MPP+-induced caspase-3 activation is calpain-dependent and inhibited in PC47 cells. (a) PC12, PC139, and PC47 cells were treated (24 hrs) with MPP+ (5 mM) and stained with FITC-labeled antibody to caspase-3p20 (activated caspase-3) and AlexaFluor 546-conjugated ICP10 antibody. (b) PC12 and PC47 cells treated with MPP+ as in (a) but in the absence or presence of calpeptin (50 μM) were stained with FITC-labeled-caspase-3p20 antibody. (c) Caspase-3p20+ cells in (b) were counted and the % calculated relative to the total cell number (determined by DAPI). Results are expressed as % caspase-3p20+ cells ± SEM. (**, p<0.01 vs. untreated cells). (d) PC12, PC139, and PC47 cells, untreated (NT) or treated with MPP+ (24 hrs, 5 mM) in the absence or presence of the pan-caspase inhibitor ZVAD-fmk (100 μM) were examined by TUNEL. Results are expressed as % TUNEL+ cells ± SEM. (ψ, p<0.001 vs. untreated; **, p<0.01 vs. MPP+ treatment alone).
ICP10PK activates the PI3-K/Akt pathway and up-regulates/stabilizes the anti-apoptotic protein Bcl-2
To confirm that ICP10PK inhibits calpain I through the activation of the PI3-K/Akt survival pathway, neuronally differentiated PC12, PC139, and PC47 cells were treated with MPP+ for 0, 24 or 48 hrs in the presence or absence of LY294002 and examined for Akt activation by immunoblotting with antibody specific for the phosphorylated Akt (pAkt). The blots were stripped and re-probed with antibody to total Akt, used as control. Duplicate cultures treated with MPP+ in the presence of the unrelated MEK inhibitor, U0126 (20 μM) were studied in parallel and served as control. The results were quantitated by densitometry and are expressed as pAkt/Akt ratios. Low levels of pAkt were seen in all cultures at 0 hrs. In PC12 cells, pAkt was not seen after MPP+ treatment whether it was given with or without inhibitors. Low levels of pAkt were seen in PC139 cells, but its levels were significantly higher in MPP+-treated PC47 cells, particularly at 48 hrs after treatment. pAkt was inhibited by LY294002, but not U0126 (Figure 7a), supporting the conclusion that ICP10PK activates the PI3-K/Akt pathway and activation is kinase-dependent.
Figure 7.

Akt is activated and Bcl-2 is up-regulated/stabilized in MPP+-treated PC47 cells. (a) Extracts of PC12, PC139, and PC47 cells collected at 0, 24 and 48 hrs after treatment with MPP+ (5 mM) in the absence or presence of inhibitors of PI3-K (LY294002, 50μM; LY) or MEK (U0126, 20μM; U0) were immunoblotted with antibody specific for pAkt. The blots were stripped and re-probed with antibody to total Akt. Quantitation was by densitometric scanning and the results from 3 independent experiments are expressed as pAkt/Akt ratios ± SEM (*, p<0.05 vs. 0 hrs in all cell lines). (b) Extracts of PC12, PC139, and PC47 cells collected at 0, 4, 24, and 48 hrs after treatment with MPP+ in the absence or presence of LY294002 or U0126 as in (a) were immunoblotted with antibody to Bcl-2. The blots were stripped and re-probed with antibody to actin and results from 3 independent experiments are expressed as Bcl-2/actin ratios (densitometric units ± SEM) (*, p<0.05 vs. 24 hrs NT).
Because: (i) ICP10PK up-regulates/stabilizes the anti-apoptotic protein Bcl-2 through PI3-K/Akt activation17 and (ii) dopaminergic cell lines stably expressing Bcl-2 are protected from MPP+-induced cell death,6 we wanted to know whether Bcl-2 is also involved in ICP10PK-mediated neuroprotection in our system. Extracts from neuronally differentiated PC12, PC139, and PC47 cells treated with MPP+ for 0, 4, 24 or 48 hrs were immunoblotted with Bcl-2 antibody and the blots were stripped and re-probed with actin antibody. The results were quantitated by densitometry and expressed as Bcl-2/actin ratios, as described in Materials and Methods. Bcl-2 was expressed in all the cultures at 0 hrs, but expression was significantly reduced by MPP+ in PC12 and PC139 cells by 24 hrs after treatment. By contrast, in PC47 cells, MPP+ elicited nearly a 2.5-fold increase in Bcl-2 expression, first observed at 4 hrs and still seen at 48 hrs after treatment (Figure 7b). Bcl-2 up-regulation/stabilization was through PI3-K/Akt activation, because it was significantly (p<0.05) reduced by treatment with LY294002, but not U0126 (Figure 7b).
ICP10PK up-regulates heat shock protein 70 (Hsp70)
Increased expression/stabilization of the cytoprotective chaperone Hsp70 was associated with inhibition of AIF release.26 To examine whether Hsp70 is also associated with the ICP10PK neuroprotective activity, neuronally differentiated PC12, PC139, and PC47 cells treated with MPP+ for 0, 4 or 24 hrs were immunoblotted with Hsp70 antibody. The blots were stripped and re-probed with GAPDH antibody and protein levels were quantitated by densitometric scanning, as previously described.12 Hsp70 was expressed in PC12 and PC139 cells, but expression was lost (PC12 cells) or was minimal (PC139 cells) at 24 hrs post-treatment. Expression was significantly more robust in PC47 cells and it was retained at the same levels throughout the study interval (Figure 8b). The data indicate that Hsp70 up-regulation/stabilization is associated with ICP10PK-mediated neuroprotection.
Figure 8.

Hsp-70 is up-regulated/stabilized in MPP+-treated PC47 cells. (a) Extracts of PC12, PC139, and PC47 cells collected at 0, 4, and 24 hrs after MPP+ treatment (5 mM) were immunoblotted with antibody to Hsp70. The blots were stripped and re-probed with antibody to GAPDH and the results expressed as Hsp70/GAPDH ratios (b).
Discussion
The salient feature of the data presented in this report is the finding that ICP10PK inhibits MPP+-induced PCD by blocking calpain I activation through the activation of the PI3-K/Akt survival pathway. Previous studies had shown that ICP10PK inhibits caspase-dependent apoptosis triggered by various neurotoxic stimuli,11-17,27 but this is the first report that it also inhibits calpain-dependent PCD, underscoring its ability to override multiple death programs. The following comments seem pertinent with respect to these findings.
MPP+-induced neurotoxicity is an established model of PD, also in neuronally differentiated PC12 cells,7,9 a system we used in the present studies. We confirmed cell differentiation by neurite formation, expression of the neuronal marker MAP-2 and absence of 5′-bromo-2′-deoxyuridine incorporation, as previously described.12 ICP10PK was delivered with the neuroprotective vector ΔRR11,13-17 or by stable transfection of PC12 cells.12 The ICP10PK-deleted vector (ΔPK) and cells stably transfected with the kinase-negative ICP10PK mutant p139™, served as controls. The stably transfected cells were used in order to exclude potentially confounding variables related to viral proteins other than ICP10PK; ΔRR was used in order to exclude potentially confounding variables related to clonal line selection. MPP+ caused a dose-dependent increase in the % TUNEL+ cells in PC12 cultures as well as in cultures infected with ΔPK or transfected with p139™ (PC139), but not in cultures that were infected with ΔRR or transfected with ICP10PK (PC47). The data indicate that ICP10PK protects from MPP+ toxicity independent of other viral genes and clonal cell selection, and protection is kinase-dependent.
The levels of cytoplasmic AIF were increased in PC12 and PC139, early after MPP+ treatment (4 hrs) and this was followed by its nuclear translocation, seen at 24 hrs after treatment, concomitant with DNA fragmentation (TUNEL). Analysis of AIF release on a cell population basis indicates that AIF was released in most of the PC12 and PC139 cells (60-65%), but release was not seen in PC47 cells, although ICP10PK and p139™ were expressed equally well and in virtually all the cells (96-100%). MPP+-induced AIF release from PC12 and PC139 cells is consistent with the results obtained by immunoblotting of nuclear and cytoplasmic fractions, which only detected its released 57 kDa protein form.22 We conclude that AIF plays a key role in MPP+-induced cell death, because an antisense ODN that inhibits its expression caused a significant increase in the survival of treated PC12 and PC139 cultures, while increased survival was not seen in cultures given MPP+ together with the control (sense) ODN, that does not inhibit AIF expression. Release is due to calpain activation, as evidenced by the findings that: (i) MPP+ increased the p76/p80 ratio of the calpain I forms,24,28,29 and (ii) both AIF release and PCD (TUNEL) were inhibited by the broad-spectrum calpain inhibitor calpeptin. Calpeptin inhibited the MPP+-induced increase in the p76/p80 ratio in PC12 and PC139 cells (data not shown), as also independently reported for its effect on p76/p80 in other systems.24,29 Calpain I was not activated and AIF was not released in PC47 cells in which MPP+ did not cause PCD, indicating that the neuroprotective activity of ICP10PK is through inhibition of calpain I activation. In our studies, calpeptin was used at previously established doses,30 but other calpain inhibitors were not studied, and the possible contribution of calpains other than calpain I, to MPP+-induced toxicity is still unclear. Notwithstanding, our findings are consistent with the report that calpain I truncates AIF to its 57 kDa form, thereby causing its release from the mitochondria22 and its subsequent translocation to the nucleus, where it interacts with DNA, stimulating the formation of a ‘degradosome.’5
Although one study attributed increased AIF release to up-regulation of BimEL,7 the predominant view is that upon commitment to apoptosis, the pro-apoptotic protein Bax undergoes conformational changes, oligomerizes and forms pores in the outer mitochondrial membrane that allow the release of mitochondrial factors, including AIF.31 Our data are consistent with this interpretation. They indicate that Bax co-localized with the mitochondria in a similar proportion of MPP+-treated PC12 and PC139 cells as those showing AIF release (60-65%), and co-localization was inhibited by calpeptin. Presumably, calpain I activation caused Bax to translocate to the mitochondria, where it contributed to membrane permeabilization and AIF release. Indeed, Bax did not translocate to the mitochondria in PC47 cells, in which calpain I was inhibited and AIF was not released. Interestingly, caspase-3 was activated by MPP+ in a relatively small % of PC12 and PC139 cells (23-28%) and activation was inhibited by calpeptin, indicating that caspase-3 is downstream of calpain activation. Nonetheless, the pan-caspase inhibitor ZVAD-fmk decreased the % TUNEL+ cells in MPP+-treated PC12 and PC139 cultures by an approximately similar level (25%), indicating that the contribution of caspase activation to PCD is independent of that of AIF release. MPP+-activated calpain appears to initiate both the AIF- and caspase-3-dependent death cascades, as schematically represented in Fig. 9. By inhibiting calpain activation, ICP10PK overrides the contribution of both cascades, such that neither ZVAD-fmk nor calpeptin had any effect on the % TUNEL+ cells in PC47 cultures.
Figure 9.

Schematic representation of ICP10PK-mediated neuroprotection from MPP+-induced PCD. ICP10PK activates the PI3-K/Akt pathway through Ras activation, as previously described.13,17 The activated pathway results in up-regulation/stabilization of Bcl-2. This counteracts MPP+-induced calpain I activation and subsequent mobilization of Bax to the mitochondria, AIF release and nuclear translocation, and caspase-3 activation. ICP10PK also causes up-regulation/stabilization of Hsp70, by an as yet unknown mechanism. Arrows represent activation, dotted arrows represent potentially involved factors, while reversed “T” symbols represent inhibition.
How does ICP10PK inhibit calpain activation? We have previously shown that ICP10PK activates Ras through phosphorylation (and, thereby, inhibition) of the negative Ras regulator Ras-GAP as well as binding of the GDP/GTP exchange factor SOS (complexed to the adaptor protein Grb2), thereby favoring its interaction with Ras.32,33. In turn, Ras activates the Raf-1/MEK/ERK and PI3-K/Akt pathways that override death cascades. In virus-infected hippocampal cultures, ICP10PK-mediated neuroprotection is through the Raf-1/MEK/ERK pathway,15,16 but both MEK/ERK and PI3-K/Akt are involved in the ability of ICP10PK to protect from excitotoxic injury.13,17 In PC47 cells, calpain inhibition was blocked by the PI3-K inhibitor LY294002, but not the MEK inhibitor U0126, suggesting that ICP10PK overrides the calpain cascades through the activation of the PI3-K/Akt pathway. Consistent with this interpretation, the levels of phosphorylated (activated) Akt were significantly increased in PC47 cells (susceptible to LY294002 inhibition), but not in PC12 and PC139 cells. The inhibitors used in these studies had previously been shown to block ICP10PK-mediated neuroprotection in other toxicity paradigms, and similar results were obtained for LY294002 at 100 μM, U0126 at 10 μM and the MEK inhibitor PD98059 (25-100μM) or the PI3-K inhibitor wortmannin.13,15,16,33 PI3-K/Akt activation culminated in Bcl-2 up-regulation/stabilization,17 as evidenced by: (i) significantly higher levels and prolonged duration of Bcl-2 expression in PC47 than PC12 and PC139 cells, and (ii) Bcl-2 reduction with LY294002, but not U0126. Hsp70 was also up-regulated/stabilized in MPP+-treated PC47, but not PC12 and PC139 cells, albeit by an as yet unknown mechanism. We assume that this is also through PI3-K/Akt activation, because Hsp70 was not up-regulated in cells in which ICP10PK-mediated neuroprotection is strictly MEK/ERK dependent12 and other studies had shown that its up-regulation is PI3-K/Akt-dependent.34 The mechanism of Hsp70 up-regulation/stabilization by ICP10PK is currently under investigation.
It is widely accepted that the MPP+ model reproduces many of the clinical and pathological hallmarks of PD,35,36 a widespread neurodegenerative disorder, triggered by genetic factors and environmental toxins and characterized by the loss of dopaminergic neurons in the substantia nigra. The mechanism responsible for neuronal degeneration characteristic of PD is still poorly understood and appears to involve multiple death programs. They include activation of caspases19,28,37 or calpain,7,28 mitochondrial dysfunction and increased release of pro-apoptotic factors, such as cytochrome c and AIF,5,7 as well as Bax up-regulation and its translocation to the mitochondria.25,35,37 Overexpression of Bcl-26,35,38 or Hsp709 were shown to contribute to protection. However, the exact relationship between these potentially distinct components of the death network is still unclear, presumably contributing to difficulties in identifying an effective neuroprotective strategy.10 Some studies attributed MPP+ toxicity to cytochrome c release from the mitochondria and the resulting activation of caspases-9 and -3,19,39 and showed that caspase inhibitors are neuroprotective.20 Caspase inhibitors failed to protect in other studies5,6 and a caspase-independent pathway, in which AIF was released from the mitochondria and translocated to the nucleus, was shown to cause DNA fragmentation.5,7 Dichotomy between the findings for cell death and neurite dysfunction was also reported21 and additional studies attributed neuronal death to both caspase-dependent and independent pathways,6,35,40 necrosis9 or distinct PCD pathways that involve oxidative stress,2 or autophagy.41 Calpains were also implicated in PD and MPP+-induced toxicity,8 but their contribution to cell death is still controversial. Some reports suggested that calpains act independently of the caspases in different apoptotic pathways,42 while others concluded that they cooperate.31 In the latter case, calpain activation was found to follow the activation of the caspases43 or initiate it.31 Calpain cleavage of caspases-9 and -3 was reported to attenuate or facilitate their activity during apoptosis,44,45 but more recent data suggested that calpains function in caspase-independent neuronal cell apoptosis.6,7,22,42
Our studies suggest that calpains, at least calpain I, play a determining role in the death of dopaminergic neurons, through both mitochondrial dysfunction which is characterized by AIF release, and caspase-3 activation. They also demonstrate that ICP10PK inhibits calpain activation, thereby interfering with both of these cascades. ICP10PK overrides these death programs through activation of the PI3-K/Akt survival pathway that results in the up-regulation and stabilization of Bcl-2 and Hsp70, both of which are known to protect from PD pathology.6,9 While we acknowledge that in vitro models may not be clinically relevant, our findings suggest that ICP10PK has the distinct advantage over other previously considered gene therapy strategies in that it targets those components of the death regulatory network which seem to play a decisive role in the demise of dopaminergic neurons. In this context, it may be significant to point out that the physiopathology of PD was also associated with death programs that were not studied in the present series of experiments, including growth factor depletion (viz. NGF),46 activation of the JNK pathway,35 and glial cell activation and production of pro-inflammatory cytokines (viz. TNF-α, and IL-1β),46 all of which are also inhibited by ICP10PK.11,12,14,27 ICP10PK also up-regulates XIAP,12 which was also implicated in PD therapy.35,47
We conclude that ICP10PK has a broad spectrum neuroprotective activity, which is characterized by remarkable functional redundancy associated with the activation of survival pathways that can inhibit multiple death programs and modulate neuron-glial cell cross-talk. By targeting most of the death programs implicated in PD physiopathology, ICP10PK holds great promise as a PD gene therapy strategy that warrants further investigation in in vivo models.
Materials and methods
Cells and neuronal differentiation
The establishment, properties and differentiation patterns of the PC12 cells stably transfected with ICP10PK (PC47) or its kinase-negative mutant p139™ (PC139) were previously described.12 Briefly, 96-100% of the cells express the respective transgenes. Expression is equally robust, but only ICP10 retains kinase activity. Similar expression and kinase activity were seen throughout the study interval (passages 27-48). Transgene expression did not alter NGF-induced differentiation. Thus, PC139 and PC47 cells cultured (4 or 12 days) with NGF evidenced virtually identical neurite formation as PC12 cells studied in parallel, and there was no visible difference between the 4 and 12 day cultures. Neurite formation reflects differentiation, because the microtubule-associated protein (MAP-2), which is an established marker of mature neurons,48 was only expressed in cultures grown with NGF (supplemental Figure 1). Its levels were similar in PC12, PC139 and PC47 cells. The NGF-differentiated cells were largely non-dividing, as determined by 5-bromo-2′-deoxyuridine incorporation (see supplemental Figure 1). In the present experiments, the cells were cultured (4 days) with NGF (100 ng/ml, 2.5S; Roche Molecular Bioproducts, Indianapolis, IN) and treated with MPP+ (Sigma, St. Louis, MO) used at a concentration of 5 mM unless otherwise stated. MPP+ stocks were made fresh for each experiment.
Antibodies and reagents
The generation and specificity of the rabbit polyclonal antibody to ICP10, which recognizes an epitope that is retained by both ICP10PK and p139™was described previously.12,15,16,49 The following antibodies were purchased and used according to manufacturer's instructions. Antibodies to activated caspase-3 (caspase-3p20), phosphorylated (activated) Akt (pAkt), Akt, and Bcl-2 were purchased from Cell Signaling Technology (Danvers, MA). Antibodies to AIF, TH, actin, GAPDH, Hsp70, Bax, and calpain were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Fluorescent-conjugated secondary antibodies were anti-mouse-FITC (BD Biosciences, San Jose, CA), anti-goat AlexaFluor 546, anti-rabbit-AlexaFluor 488, -AlexaFluor 546, or -AlexaFluor 350 (Invitrogen-Molecular Probes; Eugene, OR). The MEK inhibitor U0126 was purchased from Promega, and used at a final concentration of 20 μM. The PI3-K inhibitor, LY294002, and the calpain inhibitor calpeptin were purchased from Calbiochem (San Diego, CA), and both used at a final concentration of 50 μM. The mitochondrion-selective dye MitoTracker Red 580, the uptake of which is independent of mitochondrial membrane potential, was purchased from Invitrogen (Eugene, OR) and used to visualize mitochondria, as per manufacturer's instructions. Phosphorothioate antisense (TCCACACCGGAACATTTCGGCGA) and sense (TCGCCGAAATGTTCCGGTGTGGA) oligonucleotides specific for AIF were synthesized by the Biopolymer/Genomics Core Facility at the University of Maryland School of Medicine, reconstituted in sterile water, and used at a final concentration of 10 μM.
TUNEL
The In situ Cell Death Detection kit (Roche) was used according to the manufacturers' instructions. Cells were counted in five randomly chosen microscopic fields (at least 250 cells), and results are expressed as % TUNEL+ (apoptotic) cells ± SEM, as previously described.12,15,16
Immunoblotting
Cell extraction and immunoblotting was done as previously described.12,15,16 Protein concentrations were determined by the bicinchoninic assay (Pierce, Rockford, IL) and 100 μg protein samples were resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. For nuclear and cytoplasmic separation, fractions were obtained by resuspending cells in 50 mM Tris-Hcl buffer (pH 8.0) with 50 mM NaCl, 1% NP-40, 1 mM dithiothreitol (DTT) and protease inhibitors on ice, followed by centrifugation (7000 g; 1 min). The supernatant is the cytoplasmic fraction. The nuclear pellet was resuspended in the same buffer but with 450 mM NaCl (10 min, 4°C) and sonicated until in solution. In this case, 50 μg protein was loaded onto the lanes. Quantitation was by densitometric scanning with the Bio-Rad GS-700 imaging densitometer (Bio-Rad, Hercules, CA) and results are expressed as densitometric units.
Viruses
The construction and properties of the HSV-2 mutants ΔRR and ΔPK were previously described.13
Immunofluorescent staining
Cells were fixed in 4% PFA and incubated (4°C, overnight) with mouse anti-caspase-3p20 or anti-Bax antibodies, goat anti-TH, or rabbit anti-ICP10, anti-Bcl-2, or anti-AIF antibodies. They were washed in 0.1%Tween-20 in PBS, and incubated (25°C, 1 hr) with fluorescent-conjugated secondary antibodies (FITC-conjugated anti-mouse, AlexaFluor-conjugated anti-goat or AlexaFluor 488-, AlexaFluor 546-, or AlexaFluor 350-conjugated anti-rabbit). Slides were mounted in Vectashield with DAPI and visualized on a Nikon E4100 fluorescent microscope utilizing FITC (330-380nM), UV (for DAPI) (465-495nM), and Texas Red (540-580nM) cubes. Staining cells were counted in 3 randomly selected 3 mm2 fields (at least 250 cells) and the % positive cells was calculated relative to the total number of cells visualized by DAPI. Data are expressed as the mean % positive cells ± SEM. When used, MitoTracker Red 580 was added 45 min before the cells were fixed and stained with the appropriate antibody, as per manufacturer's instructions.
Statistical analyses
Analysis of variance (ANOVA) was performed with Sigma Stat version 3.1 for Windows (Systat Software, Point Richmond, CA).
Supplementary Material
Footnotes
Supplementary information is available at Gene Therapy's website.
References
- 1.Yuan H, Zheng JC, Liu P, Zhang SF, Xu JY, Bai LM. Pathogenesis of Parkinson's disease: oxidative stress, environmental impact factors and inflammatory processes. Neurosci Bull. 2007;23:125–130. doi: 10.1007/s12264-007-0018-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dawson TM, Dawson VL. Neuroprotective and neurorestorative strategies for Parkinson's disease. Nat Neurosci. 2002;5(Suppl):1058–1061. doi: 10.1038/nn941. [DOI] [PubMed] [Google Scholar]
- 3.Shastry BS. Parkinson disease: etiology, pathogenesis and future of gene therapy. Neuroscience Research. 2001;41:5–12. doi: 10.1016/s0168-0102(01)00254-1. [DOI] [PubMed] [Google Scholar]
- 4.Do Thi NA, Saillour P, Ferrero L, Paunio T, Mallet J. Does neuronal expression of GDNF effectively protect dopaminergic neurons in a rat model of Parkinson's disease? Gene Ther. 2007;14:441–450. doi: 10.1038/sj.gt.3302844. [DOI] [PubMed] [Google Scholar]
- 5.Chu CT, Zhu JH, Cao G, Signore A, Wang S, Chen J. Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J Neurochem. 2005;94:1685–1695. doi: 10.1111/j.1471-4159.2005.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi WS, Lee EH, Chung CW, Jung YK, Jin BK, Kim SU, et al. Cleavage of Bax is mediated by caspase-dependent or -independent calpain activation in dopaminergic neuronal cells: protective role of Bcl-2. J Neurochem. 2001;77:1531–1541. doi: 10.1046/j.1471-4159.2001.00368.x. [DOI] [PubMed] [Google Scholar]
- 7.Liou AK, Zhou Z, Pei W, Lim TM, Yin XM, Chen J. BimEL up-regulation potentiates AIF translocation and cell death in response to MPTP. Faseb J. 2005;19:1350–1352. doi: 10.1096/fj.04-3258fje. [DOI] [PubMed] [Google Scholar]
- 8.Crocker SJ, Smith PD, Jackson-Lewis V, Lamba WR, Hayley SP, Grimm E, et al. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson's disease. J Neurosci. 2003;23:4081–4091. doi: 10.1523/JNEUROSCI.23-10-04081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quigney DJ, Gorman AM, Samali A. Heat shock protects PC12 cells against MPP+ toxicity. Brain Res. 2003;993:133–139. doi: 10.1016/j.brainres.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Grandas F. The putative neuroprotective role of dopamine agonists in Parkinson's disease. Neurotox Res. 2000;2:205–213. doi: 10.1007/BF03033794. [DOI] [PubMed] [Google Scholar]
- 11.Golembewski EK, Wales SQ, Aurelian L, Yarowsky PJ. The HSV-2 protein ICP10PK prevents neuronal apoptosis and loss of function in an in vivo model of neurodegeneration associated with glutamate excitotoxicity. Exp Neurol. 2007;203:381–393. doi: 10.1016/j.expneurol.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wales SQ, Li B, Laing JM, Aurelian L. The herpes simplex virus type 2 gene ICP10PK protects from apoptosis caused by nerve growth factor deprivation through inhibition of caspase-3 activation and XIAP up-regulation. J Neurochem. 2007;103:365–379. doi: 10.1111/j.1471-4159.2007.04745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gober MD, Laing JM, Thompson SM, Aurelian L. The growth compromised HSV-2 mutant DeltaRR prevents kainic acid-induced apoptosis and loss of function in organotypic hippocampal cultures. Brain Res. 2006;1119:26–39. doi: 10.1016/j.brainres.2006.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laing JM, Gober MD, Golembewski EK, Thompson SM, Gyure KA, Yarowsky PJ, et al. Intranasal administration of the growth-compromised HSV-2 vector DeltaRR prevents kainate-induced seizures and neuronal loss in rats and mice. Mol Ther. 2006;13:870–881. doi: 10.1016/j.ymthe.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perkins D, Pereira EF, Aurelian L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) functions as a dominant regulator of apoptosis in hippocampal neurons involving activation of the ERK survival pathway and upregulation of the antiapoptotic protein Bag-1. J Virol. 2003;77:1292–1305. doi: 10.1128/JVI.77.2.1292-1305.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perkins D, Pereira EF, Gober M, Yarowsky PJ, Aurelian L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J Virol. 2002;76:1435–1449. doi: 10.1128/JVI.76.3.1435-1449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laing JM, Golembewski EK, Wales SQ, Liu J, Jafri MS, Yarowsky PJ, et al. Growth-compromised HSV-2 vector DeltaRR protects from N-methyl-D-aspartate-induced neuronal degeneration through redundant activation of the MEK/ERK and PI3-K/Akt survival pathways, either one of which overrides apoptotic cascades. J Neurosci Res. 2007 doi: 10.1002/jnr.21486. [DOI] [PubMed] [Google Scholar]
- 18.Hurley MJ, Mash DC, Jenner P. Markers for dopaminergic neurotransmission in the cerebellum in normal individuals and patients with Parkinson's disease examined by RT-PCR. Eur J Neurosci. 2003;18:2668–2672. doi: 10.1046/j.1460-9568.2003.02963.x. [DOI] [PubMed] [Google Scholar]
- 19.Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, et al. Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. Proc Natl Acad Sci U S A. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Kaul S, Zhang D, Anantharam V, Kanthasamy AG. Suppression of caspase-3-dependent proteolytic activation of protein kinase C delta by small interfering RNA prevents MPP+-induced dopaminergic degeneration. Mol Cell Neurosci. 2004;25:406–421. doi: 10.1016/j.mcn.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 21.Bilsland J, Roy S, Xanthoudakis S, Nicholson DW, Han Y, Grimm E, et al. Caspase inhibitors attenuate 1-methyl-4-phenylpyridinium toxicity in primary cultures of mesencephalic dopaminergic neurons. J Neurosci. 2002;22:2637–2649. doi: 10.1523/JNEUROSCI.22-07-02637.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, et al. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniel KG, Anderson JS, Zhong Q, Kazi A, Gupta P, Dou QP. Association of mitochondrial calpain activation with increased expression and autolysis of calpain small subunit in an early stage of apoptosis. Int J Mol Med. 2003;12:247–252. [PubMed] [Google Scholar]
- 24.Hayashi M, Inomata M, Saito Y, Ito H, Kawashima S. Activation of intracellular calcium-activated neutral proteinase in erythrocytes and its inhibition by exogenously added inhibitors. Biochim Biophys Acta. 1991;1094:249–256. doi: 10.1016/0167-4889(91)90083-a. [DOI] [PubMed] [Google Scholar]
- 25.Hartmann A, Michel PP, Troadec JD, Mouatt-Prigent A, Faucheux BA, Ruberg M, et al. Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson's disease? J Neurochem. 2001;76:1785–1793. doi: 10.1046/j.1471-4159.2001.00160.x. [DOI] [PubMed] [Google Scholar]
- 26.Gurbuxani S, Schmitt E, Cande C, Parcellier A, Hammann A, Daugas E, et al. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene. 2003;22:6669–6678. doi: 10.1038/sj.onc.1206794. [DOI] [PubMed] [Google Scholar]
- 27.Perkins D, Gyure KA, Pereira EF, Aurelian L. Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J Neurovirol. 2003;9:101–111. doi: 10.1080/13550280390173427. [DOI] [PubMed] [Google Scholar]
- 28.Samantaray S, Knaryan VH, Guyton MK, Matzelle DD, Ray SK, Banik NL. The parkinsonian neurotoxin rotenone activates calpain and caspase-3 leading to motoneuron degeneration in spinal cord of Lewis rats. Neuroscience. 2007;146:741–755. doi: 10.1016/j.neuroscience.2007.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schoenwaelder SM, Kulkarni S, Salem HH, Imajoh-Ohmi S, Yamao-Harigaya W, Saido TC, et al. Distinct substrate specificities and functional roles for the 78- and 76-kDa forms of mu-calpain in human platelets. J Biol Chem. 1997;272:24876–24884. doi: 10.1074/jbc.272.40.24876. [DOI] [PubMed] [Google Scholar]
- 30.Klafki H, Abramowski D, Swoboda R, Paganetti PA, Staufenbiel M. The carboxyl termini of beta-amyloid peptides 1-40 and 1-42 are generated by distinct gamma-secretase activities. J Biol Chem. 1996;271:28655–28659. doi: 10.1074/jbc.271.45.28655. [DOI] [PubMed] [Google Scholar]
- 31.Gao G, Dou QP. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome C release and apoptotic cell death. J Cell Biochem. 2000;80:53–72. doi: 10.1002/1097-4644(20010101)80:1<53::aid-jcb60>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 32.Smith CC, Luo JH, Hunter JCR, Ordonez JV, Aurelian L. The Transmembrane Domain of the Large Subunit of HSV-2 Ribonucleotide Reductase (ICP10) Is Required for Protein Kinase Activity and Transformation-Related Signaling Pathways That Result in ras Activation. Virology. 1994;200:598–612. doi: 10.1006/viro.1994.1223. [DOI] [PubMed] [Google Scholar]
- 33.Smith CC, Nelson J, Aurelian L, Gober M, Goswami BB. Ras-GAP Binding and Phosphorylation by Herpes Simplex Virus Type 2 RR1 PK (ICP10) and Activation of the Ras/MEK/MAPK Mitogenic Pathway Are Required for Timely Onset of Virus Growth. J Virol. 2000;74:10417–10429. doi: 10.1128/jvi.74.22.10417-10429.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Magrane J, Rosen KM, Smith RC, Walsh K, Gouras GK, Querfurth HW. Intraneuronal beta-amyloid expression downregulates the Akt survival pathway and blunts the stress response. J Neurosci. 2005;25:10960–10969. doi: 10.1523/JNEUROSCI.1723-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eberhardt O, Schulz JB. Apoptotic mechanisms and antiapoptotic therapy in the MPTP model of Parkinson's disease. Toxicol Lett. 2003;139:135–151. doi: 10.1016/s0378-4274(02)00428-9. [DOI] [PubMed] [Google Scholar]
- 36.Smeyne RJ, Jackson-Lewis V. The MPTP model of Parkinson's disease. Brain Res Mol Brain Res. 2005;134:57–66. doi: 10.1016/j.molbrainres.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 37.Tatton NA. Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson's disease. Exp Neurol. 2000;166:29–43. doi: 10.1006/exnr.2000.7489. [DOI] [PubMed] [Google Scholar]
- 38.Yang L, Matthews RT, Schulz JB, Klockgether T, Liao AW, Martinou J-C, et al. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyride Neurotoxicity Is Attenuated in Mice Overexpressing Bcl-2. J Neurosci. 1998;18:8145–8152. doi: 10.1523/JNEUROSCI.18-20-08145.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang D, Zhang JJ, Liu GT. The novel squamosamide derivative (compound FLZ) attenuated 1-methyl, 4-phenyl-pyridinium ion (MPP+)-induced apoptosis and alternations of related signal transduction in SH-SY5Y cells. Neuropharmacology. 2007;52:423–429. doi: 10.1016/j.neuropharm.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 40.Han BS, Hong HS, Choi WS, Markelonis GJ, Oh TH, Oh YJ. Caspase-dependent and -independent cell death pathways in primary cultures of mesencephalic dopaminergic neurons after neurotoxin treatment. J Neurosci. 2003;23:5069–5078. doi: 10.1523/JNEUROSCI.23-12-05069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shacka JJ, Roth KA, Zhang J. The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008;13:718–736. doi: 10.2741/2714. [DOI] [PubMed] [Google Scholar]
- 42.Takano J, Tomioka M, Tsubuki S, Higuchi M, Iwata N, Itohara S, et al. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J Biol Chem. 2005;280:16175–16184. doi: 10.1074/jbc.M414552200. [DOI] [PubMed] [Google Scholar]
- 43.Porn-Ares MI, Samali A, Orrenius S. Cleavage of the calpain inhibitor, calpastatin, during apoptosis. Cell Death Differ. 1998;5:1028–1033. doi: 10.1038/sj.cdd.4400424. [DOI] [PubMed] [Google Scholar]
- 44.Bizat N, Hermel JM, Humbert S, Jacquard C, Creminon C, Escartin C, et al. In vivo calpain/caspase cross-talk during 3-nitropropionic acid-induced striatal degeneration: implication of a calpain-mediated cleavage of active caspase-3. J Biol Chem. 2003;278:43245–43253. doi: 10.1074/jbc.M305057200. [DOI] [PubMed] [Google Scholar]
- 45.Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, et al. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- 46.Singh S, Dikshit M. Apoptotic neuronal death in Parkinson's disease: involvement of nitric oxide. Brain Res Rev. 2007;54:233–250. doi: 10.1016/j.brainresrev.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Eberhardt O, Coelln RV, Kugler S, Lindenau J, Rathke-Hartlieb S, Gerhardt E, et al. Protection by synergistic effects of adenovirus-mediated X-chromosome-linked inhibitor of apoptosis and glial cell line-derived neurotrophic factor gene transfer in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neurosci. 2000;20:9126–9134. doi: 10.1523/JNEUROSCI.20-24-09126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sano M, Nishiyama K, Kitajima S. A nerve growth factor-dependent protein kinase that phosphorylates microtubule-associated proteins in vitro: possible involvement of its activity in the outgrowth of neurites from PC12 cells. J Neurochem. 1990;55:427–435. doi: 10.1111/j.1471-4159.1990.tb04154.x. [DOI] [PubMed] [Google Scholar]
- 49.Luo JH, Smith CC, Kulka M, Aurelian L. A truncated protein kinase domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) expressed in Escherichia coli. J Biol Chem. 1991;266:20976–20983. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.