

Abstract
The question of whether DNA methylation contributes to the stabilization of gene expression patterns in differentiated mammalian tissues remains controversial. Using genome-wide methylation profiling, we screened 3757 gene promoters for changes in methylation during postnatal liver development to test the hypothesis that developmental changes in methylation and expression are temporally correlated. We identified 31 genes that gained methylation and 111 that lost methylation from embryonic day 17.5 to postnatal day 21. Promoters undergoing methylation changes in postnatal liver tended not to be associated with CpG islands. At most genes studied, developmental changes in promoter methylation were associated with expression changes, suggesting both that transcriptional inactivity attracts de novo methylation, and that transcriptional activity can override DNA methylation and successively induce developmental hypomethylation. These in vivo data clearly indicate a role for DNA methylation in mammalian differentiation, and provide the novel insight that critical windows in mammalian developmental epigenetics extend well beyond early embryonic development.
INTRODUCTION
Methylation of cytosines within CpG dinucleotides is an epigenetic mechanism critical for mammalian development, serving an important role in genomic imprinting, X-chromosome inactivation and silencing of retrotransposons (1,2). Due to the mitotic stability of locus-specific CpG methylation patterns, it has long been postulated that DNA methylation additionally serves the broader developmental function of stabilizing gene expression patterns in differentiated mammalian tissues (3,4). Early studies failed to detect correlations between tissue-specific gene expression and CpG methylation, causing some to reject this conjecture (5). Single-gene studies (6) and recent genome-wide analyses of DNA methylation (7–9) have, however, identified numerous genes with tissue-specific expression and hypomethylation, reinvigorating the hypothesis. A key unaddressed issue that would elucidate the specific role of DNA methylation in differentiation is the developmental dynamics of gene-specific changes in methylation and expression. Limited data in transgenic mice suggest that changes in transcription are succeeded by methylation changes which stabilize the transcriptional state (10), but in vivo data on normal mammalian development are lacking.
If DNA methylation successively stabilizes gene expression states during mammalian differentiation, developmental changes in methylation and expression should be temporally correlated. Except for studies of genomically imprinted genes (11), retrotransposons (12,13) and X-chromosome inactivation (14), few such temporal correlations have been reported. Indeed, previous analyses of developmental epigenetics have largely focused on narrow windows of early embryonic and germline development (2). The interpretation of DNA methylation changes in somatic tissues during fetal development is complicated because maturation of tissue rudiments is often associated with dramatic changes in the relative proportions of cellular subtypes. Recognizing this fact, we set out to investigate the developmental dynamics of gene-specific DNA methylation and expression in the mouse liver during late fetal and early postnatal life, a period during which the organ is undergoing extensive functional maturation (15).
The endodermal hepatoblasts that comprise the fetal liver primordium have the potential to develop into either hepatocytes or bile duct cells. In addition to hepatoblasts, the fetal mouse liver is transiently home to hematopoietic cells, which migrate to the liver around embryonic day 11 (E11), and emigrate to the bone marrow around the time of birth (16). Despite this added complexity, the mouse liver is an attractive model in which to study developmental epigenetics because the organ is fully differentiated by postnatal day 21 (P21) (17) and is relatively homogenous, consisting predominantly of hepatocytes (70% by cell number) together with endothelial, stellate and kupfer cells (18).
Since tissue-specific gene expression is often achieved without tissue-specific differences in promoter DNA methylation (5), we first screened for genes undergoing methylation changes, then assessed these for expression changes. Human studies have demonstrated that combining methylated CpG island (CGI) amplification (19) with microarray hybridization (MCAM) provides a sensitive and reliable tool for genome-wide methylation profiling (8). Application of this technique in the mouse has not been reported. In this study, we used MCAM to identify genes undergoing methylation changes during late fetal to early postnatal mouse liver development, and examined associations between methylation and expression in a subset of these. Our data show that MCAM is capable of identifying even subtle developmental changes in methylation in normal tissues. In almost every case examined, developmental changes in CpG methylation were associated with transcriptional changes in the postnatal liver. These changes were not attributable to the emigration of hematopoietic cells from the liver. Taken together, our findings support the hypothesis that CpG methylation serves to stabilize gene-specific transcriptional states during postnatal liver development.
RESULTS
Identification of DNA methylation changes in postnatal liver
We used MCAM (8) for genome-wide methylation analysis. In this method, genomic DNA is digested first with the methylation-sensitive restriction endonuclease SmaI (leaving blunt ends); methylated SmaI sites remain as templates for subsequent digestion with the methylation-insensitive isoschizomer XmaI (which leaves a 4-nucleotide overhang). Ligation of adapters to these ‘sticky ends’, followed by whole-genome PCR, results in preferential amplification of SmaI/XmaI genomic intervals methylated at both ends. For each cohybridization, two such whole-genome PCR products were labeled differentially with Cy3 or Cy5, and cohybridized to mouse proximal promoter microarrays. The arrays contained 45–60mer oligonucleotide probes covering from −1.0 to +0.3 kb relative to the transcription start sites of 17 000 mouse transcripts defined by RefSeq (20). Bioinformatic analysis predicted that 8611 probes corresponding to 3757 unique genes on the array are potentially informative, assuming a maximal SmaI/XmaI amplicon size of 2 kb. We also annotated all the SmaI/XmaI intervals relative to CGIs and repetitive sequences. The approach yields excellent coverage of promoter-region CGIs; of the 3757 gene promoters flanked by SmaI/XmaI restriction sites, both restriction sites were within CGIs in 52% (1961), one restriction site was within a CGI in 35% (1324) and only 13% (472) were not associated with CGIs.
Many X-linked promoter CGIs are hypermethylated specifically on the inactive X chromosome in female mammals (14). Therefore, as an initial validation of MCAM in the mouse, we compared genomic methylation patterns of males and females. Genomic DNA was isolated from male and female C57BL6/J mouse liver at E17.5, and a female versus male MCAM cohybridization was performed. Signal intensity at autosomal probes showed excellent agreement between the male and female, but most probes on the X chromosome yielded 50–100-fold higher signal in the female relative to the male (Fig. 1A). Since only one of the two X chromosomes in each female cell undergoes promoter hypermethylation, and promoters on the single X chromosome in males are hypomethylated, this experiment demonstrates that even an absolute methylation difference of only 50% (female versus male) yields a robust MCAM signal.
Figure 1.

Validation of MCAM in the mouse. (A) Scatter plot of normalized signal intensity for a female versus male MCAM cohybridization. Probes not on the X chromosome show close agreement in male and female genomic DNA, whereas those on the X chromosome yield 50–100-fold higher signal in the female than in the male. (B) Manual bisulfite sequencing results at Fcgrt, comparing five E17.5 and five P21 mice. The arrows indicate CpG sites; the upper CpG site is the 5′ end of the SmaI/XmaI interval that yielded a ‘hit’ in the MCAM assay. (C) Scatter plot of P21:E17.5 methylation ratio obtained by bisulfite sequencing versus P21:E17.5 MCAM ratio. In addition to the 13 MCAM hits, three genes that did not meet the final criteria used to identify hits (Mcm2, Gzma and Tex19) are shown. Since two P21 versus E17.5 MCAM cohybridizations were performed, there are two data points for each gene validated. The points do not depart significantly from the line of identity (shown).
To detect methylation changes occurring during normal development, we isolated genomic DNA from C57BL6/J mice at E17.5 and P21 and performed two independent P21 versus E17.5 MCAM cohybridizations. Focusing on genes that showed concordant methylation changes in both cohybridizations, we identified 31 genes that gained methylation and 111 that lost methylation from E17.5 to P21 (Supplementary Material, Table S1). We used quantitative bisulfite sequencing (Fig. 1B) to verify the methylation changes in 13 of the MCAM hits. Overall, P21:E17.5 methylation ratios measured by bisulfite sequencing were highly correlated with MCAM P21:E17.5 signal ratio (R2 = 0.76), and the linear relationship between bisulfite and MCAM values did not differ significantly from the line of identity (Fig. 1C). These data clearly demonstrate that MCAM provides a reliable method by which to screen for DNA methylation changes during normal development. Further, by extending our bisulfite sequencing studies to a larger number of E17.5 and P21 mice (n = 5 at each age), we demonstrated that our approach of screening for concordant methylation changes in two independent P21 versus E17.5 MCAM cohybridizations effectively identifies developmental changes (Fig. 1B), rather than coincidental concordance of pairwise interindividual differences.
Since MCAM is based upon SmaI/XmaI digestion, it was important to determine if the developmental changes we detected are limited to the informative SmaI/XmaI sites, or in fact reflect methylation changes occurring over broad genomic regions. We performed a detailed methylation analysis on three genes that lost methylation (Azgp1, Fcgrt and Phyhd1) and three that gained methylation from E17.5 to P21 (Def6, Lingo4 and Nrbp2). Genes were selected on the basis of robust (>3-fold) methylation changes by MCAM. In most cases, both informative SmaI/XmaI sites (separated by hundreds of base pairs) and respective neighboring CpG sites showed concordant changes in methylation (Fig. 2). These data also illustrate that MCAM is capable of detecting even small methylation changes. For example, nearly the entire MCAM signal at Lingo4 is due to an increase from just 5 to 30% at one SmaI/XmaI site (Fig. 2E).
Figure 2.

Detailed analysis of DNA methylation. The upper section of each panel displays the gene region studied by bisulfite sequencing. Vertical lines indicate CpG sites, and downward arrows indicate the boundaries of each informative SmaI/XmaI interval. The lower section of each panel shows percent methylation (Mean ± SEM) versus CpG site location relative to the transcription start site (n = 5 mice per age). Shaded data points indicate the SmaI/XmaI sites that were informative by MCAM. (A) Azgp1. (B) Fcgrt. (C) Phyhd1. (D) Def6. (E) Lingo4. (F) Nrbp2.
Quantitative sequencing of post-bisulfite PCR products, while highly sensitive, provides no information on the distribution of CpG methylation on individual DNA molecules. This could be very important if, for example, during this developmental period a cell type with a methylated promoter is being replaced by one with an unmethylated promoter, or vice versa. We therefore performed bisulfite cloning and sequencing at the same six genes studied by quantitative bisulfite sequencing. Given that some of the informative SmaI/XmaI-containing regions previously evaluated were several hundred base pairs from the transcription start site we tried, when possible, to center the assays at the transcription start site of the genes. In every case, the clonal sequencing (Fig. 3) showed methylation changes that were consistent with the quantitative sequencing results. For example, while our quantitative assays at Def6 and Lingo4 were located hundreds of base pairs from the transcription start sites (Fig. 2D and E), the clonal assays closer to the transcription start sites (Fig. 3D and E) yielded similar results. Most importantly, rather than a homogeneous pattern of methylation that might be consistent with differential promoter methylation in different cell types at E17.5 and P21, clonal methylation was heterogeneous at all genes tested (Fig. 3).
Figure 3.

Clonal bisulfite sequencing results. Each panel compares bisulfite sequencing results for E17.5 and P21. The position relative to the transcription start site is indicated. Each row of circles represents a single clone; open circles represent unmethylated cytosines, and filled circles indicate methylation. In all cases, methylation patterns of individual clones were heterogeneous, and the temporal methylation changes detected in the quantitative assays were corroborated. (A) Azgp1. (B) Fcgrt. (C) Phyhd1. (D) Def6. (E) Lingo4. (F) Nrbp2.
Genomic characteristics of regions undergoing methylation change
The MCAM approach used in this study is biased toward CGIs for two reasons. First, we used a proximal promoter microarray, and over half of all promoters in the mouse genome are associated with CGIs (21). Second, the SmaI/XmaI restriction site (CCCGGG) is overrepresented in CGIs. Hence, it is not surprising that almost all (87%) of the potentially informative SmaI/XmaI intervals represented on the microarray are associated with CGIs. Compared with all the potentially informative intervals, however, genomic regions undergoing gain or loss of methylation during postnatal liver development tended not to be associated with CGIs (P < 1 × 10−10 for both comparisons); this CGI underrepresentation was strongest among genes that gained methylation (Table 1). Since flanking repetitive elements have been implicated in the de novo methylation of non-CGI promoters (5), we also analyzed whether the regions undergoing developmental changes in methylation were exceptional in their proximity to repetitive elements. Overall, 14% of the 3757 potentially informative Sma1/XmaI intervals were associated with repetitive elements, the same proportion as in the gene sets that gained or lost methylation. We examined the distributions of LINE, SINE and LTR retrotransposons, and DNA transposons in 6 kb windows centered on the transcription start sites of genes undergoing methylation changes, compared with those of 2562 genes that did not change methylation (Supplementary Material, Fig. S1). The only significant finding was that, compared with the reference set, genes that lost methylation contained fewer LTR retrotransposons downstream of the transcription start site (P = 0.0018).
Table 1.
Association of methylation changes with CGIs
| All genes analyzed (n = 3757) |
Methylation increased (n = 31) |
Methylation decreased (n = 111) |
||||
|---|---|---|---|---|---|---|
| n | % | n | % | n | % | |
| Both SmaI/XmaI sites in CGI | 1961 | 52.2 | 5 | 16.1 | 7 | 6.3 |
| One SmaI/XmaI site in CGI | 1324 | 35.2 | 5 | 16.1 | 66 | 59.5 |
| Neither SmaI/XmaI site in CGI | 472 | 12.6 | 21 | 67.7 | 38 | 34.2 |
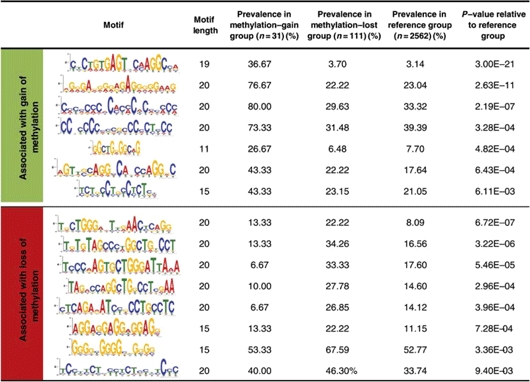
Since specific sequence motifs have been associated with tissue-specific methylation at promoter CGIs (8), we used a weight matrix finding algorithm (MEME) (22) and a motif alignment and search tool (MAST) (23) to determine if the developmental methylation changes were associated with specific sequence motifs. We analyzed 2 kb sequences centered on the transcription start sites of genes undergoing methylation changes, compared with those of 2562 genes that did not change methylation. The top 20 significantly enriched motifs in each group were identified by MEME, and MAST was then used to identify motifs significantly enriched among the genes that changed methylation, compared with the reference group. Both the gene regions that gained methylation and those that lost methylation were significantly enriched in distinct sequence motifs (Table 2). Strikingly, the enrichments were most robust in the group of promoters that gained methylation, although this group includes only 31 genes. There was no overlap between motifs enriched in the ‘gained methylation’ group and those enriched in the ‘lost methylation’ group (Table 2).
Table 2.
Sequence motifs enriched near the transcription start sites of genes that gained or lost methylation from E17.5 to P21
 |
Previous studies (24) indicate that polycomb-mediated trimethylation of histone H3 lysine 27 (H3K27me3) in embryonic stem cells may designate gene promoters for DNA methylation during later differentiation. We therefore tested whether genes that gained methylation during postnatal liver development are enriched in known polycomb targets. From three recent studies (24–26), we identified a consensus set of 1002 polycomb targets in mouse stem cells. Including only those genes present in both the list of polycomb targets and the list of genes potentially informative by MCAM, we then determined the proportion of polycomb targets among genes that gained, lost or did not change methylation (Supplementary Material, Fig. S2). Although not statistically significant, we did find a trend in the expected direction; 25.0% of the genes that gained methylation are polycomb targets, versus only 14.9% in the reference group.
Morphometric analysis of hematopoietic cell prevalence in developing liver
The fetal liver is temporarily home to a sizable population of hematopoietic cells; we therefore wished to determine if the methylation changes we found might be attributable to their emigration from the liver during late fetal development. We performed a histological evaluation to assess the prevalence of hematopoietic cells in the mouse liver from E17.5 to P21 (Fig. 4). At E17.5, hematopoietic cells were the major cell population, but by P5 their emigration from the liver was essentially complete (Fig. 4B).
Figure 4.
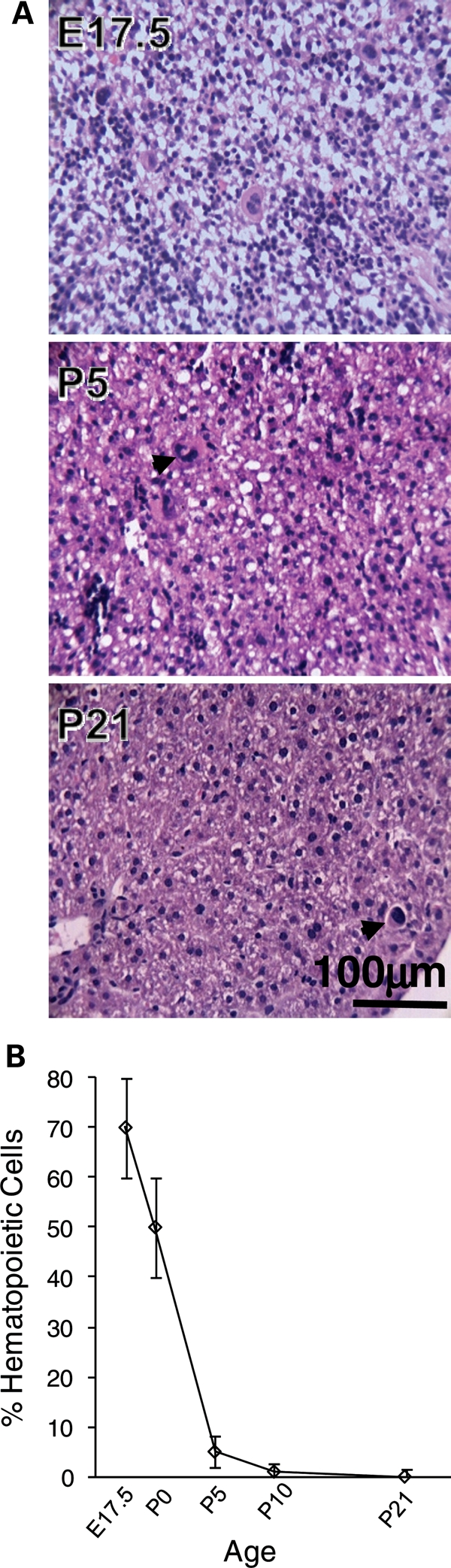
Dynamics of hematopoietic cell migration from the liver. (A) Representative high power fields of hematoxylin–eosin stained liver from E17.5, P5 and P21 mice. Note the rapid disappearance of hematopoietic cells between E17.5 and P5 (for description see Materials and Methods) including megakaryocytes (dark arrows), and the coordinate emergence of hepatocytes. Hepatocytes were the dominant cell type by P5. A single megakaryocyte was detected in one of the P21 specimens (shown). (B) Time course showing percentage hematopoietic cells versus age. Each point indicates the mean and SD of livers from 3–5 mice. Hematopoietic cells were the dominant cell type at E17.5, but almost completely gone by P5.
Temporal analyses of DNA methylation and gene expression
Having identified a set of gene promoters undergoing changes in DNA methylation from E17.5 to P21, we used real time RT–PCR to assess associated changes in gene expression. We focused on the same six genes evaluated in the detailed methylation analyses. To assess developmental dynamics of DNA methylation and gene expression, we performed quantitative measurements of both at E17.5, P0, P5, P10 and P21 (Fig. 5). All three genes that lost methylation showed coordinate increases in expression (Fig. 5A–C). In contrast, developmental increases in DNA methylation were less consistently related to expression changes. At Def6, we found the anticipated decrease in expression (Fig. 5D). At Lingo4, however, expression increased coordinately with methylation from E17.5 to P21 (Fig. 5E). Notably, at both Def6 and Lingo4 expression changed most rapidly (from P0 to P5), directly before the most rapid increase in methylation (from P5 to P10). At Nrbp2, methylation and expression appeared to be uncoupled; the transient peak in Nrbp2 expression at P0 preceded the rise in methylation from P5 to P21 (Fig. 5F). To examine whether further methylation changes occur in these genes, we measured DNA methylation in P120 mouse liver. Overall, methylation was fairly stable after P21 (Supplementary Material, Fig. S3); the largest post-weaning methylation change was at Nrbp2, which increased from 37 to 55%.
Figure 5.

Temporal analysis of DNA methylation and expression. Each panel displays both mRNA expression and percentage DNA methylation versus age. Transcript levels are expressed relative to those at P21 or E17.5 (whichever is higher) using the 2−ΔΔCt method. Points represent mean ± SEM of n = 5 mice per age for both expression and methylation. (A) Azgp1. (B) Fcgrt. (C) Phyhd1. (D) Def6. (E) Lingo4. (F) Nrbp2. At Azgp1, Fcgrt, Phyhd1 and Def6 expression is inversely correlated with methylation. At Lingo4 expression and methylation are directly correlated, while at Nrbp2 developmental changes in expression and methylation appear uncoupled.
Bioinformatic analyses of gene expression and gene ontology
If developmental changes in DNA methylation function to stabilize epigenetic states in differentiated tissues, then DNA methylation changes from E17.5 to P21 should generally predict expression changes from late fetal to adult liver. We queried a public gene expression database (www.mouseatlas.org) to assess hepatic expression changes from E18 to P84 among genes that gained or lost DNA methylation during early postnatal development. Additionally, we included a group of 2562 genes in which methylation did not change from E17.5 to P21. Genes not mapping to any expressed sequence tags at either time point were excluded. Compared with the other two classes, genes that lost methylation from E17.5 to P21 showed a greater tendency for expression to increase from late fetal to adult liver (Fig. 6A). Performing the same analysis in tissues of ectodermal (brain) and mesodermal (spleen) lineage showed no group differences (Fig. 6A), indicating that the developmental increases in expression in this group of genes are specific to liver. We also tested whether the methylation changes we identified might contribute to the maintenance of tissue-specific gene expression. Using data downloaded from a mouse gene expression atlas (http://symatlas.gnf.org), we calculated expression in liver as a Z score relative to expression in 36 other tissues (see Materials and Methods). Compared with genes that gained methylation from E17.5 to P21, those that lost methylation during this period were expressed at higher levels in adult liver relative to other tissues (Fig. 6B). Together, these data support the hypothesis that developmental changes in DNA methylation participate in the regulation of locus-specific transcriptional competence in differentiated tissues.
Figure 6.

Association of gene expression with postnatal methylation changes. (A) Box plots of P84:E18 expression ratio in liver, brain and spleen of genes that lost or gained methylation from E17.5 to P21, compared with those in which methylation did not change. Each box plot depicts the median (thick line), 25th–75th percentiles (box) and 5th–95th percentiles of the distribution (whiskers). Among genes that lost methylation in liver from E17.5 to P21, the expression increase from E18 to P84 is greater than that of the reference group, specifically in liver. (B) Distribution of expression Z score in liver versus 36 other tissues among genes that either gained or lost methylation from E17.5 to P21. Genes that lost methylation during early postnatal development are generally expressed at higher levels in liver.
We also performed a gene ontology (GO) analysis (http://babelomics.bioinfo.cipf.es/) to determine if gene regions undergoing postnatal methylation change in liver are associated with particular biological processes, molecular functions or cellular components. The reference group consisted of 3000 genes that met our criteria for hybridization signal but for which methylation was unchanged from E17.5 to P21. There were no significant GO terms among the 111 genes that lost methylation from E17.5 to P21. Among the 31 genes that gained methylation, one GO cellular component term (voltage-gated sodium channel complex) was significantly over-represented (P = 0.005). This result, however, is attributed to only two genes (Scn2b and Scn4b) among those that gained methylation (Supplementary Material, Table S1).
DISCUSSION
DNA methylation and gene expression during postnatal liver development
The hypothesis that gene-specific patterns of DNA methylation participate in the maintenance of tissue-specific gene expression in mammals, first proposed over 30 years ago (3,4), remains controversial (1,27). One outstanding issue is the elaboration of ontogenic periods during which DNA methylation is in flux. A widely held model is that developmental changes in DNA methylation occur principally in the preimplantation embryo and during primordial germ cell development (1,2,10). This view, however, is irreconcilable with the increasing number of reports demonstrating tissue-specific patterns of DNA methylation and associated gene expression (6,7,9,28). If diverse adult tissues display distinct patterns of epigenetic regulation and DNA methylation, clearly these must be established after gastrulation. But currently, little is known about the role of epigenetic mechanisms in late fetal and early postnatal development.
To our knowledge, ours is the first study to quantitatively assess temporal relationships between DNA methylation and gene expression during normal mammalian development. We show for the first time that during postnatal life developmental changes in DNA methylation—both increases and decreases—are associated with functional changes in hepatic gene expression. Given that developmental changes in gene expression can be regulated by many mechanisms, this study took the novel approach of first screening for methylation changes, and then determining if these are associated with expression changes. In all three genes studied that lost methylation from E17.5 to P21, transcript levels were inversely correlated with DNA methylation (Fig. 5A–C). Among the studied genes that gained methylation from E17.5 to P21, the association between methylation and expression was more variable. At Def6 (Fig. 5D), down-regulation of transcription was correlated with and largely preceded hypermethylation, consistent with transgenic studies (29), suggesting that transcriptional activity prevents de novo methylation. The direct relationship between developmental changes in DNA methylation and expression at Lingo4 (Fig. 5E) was unexpected. Lingo4 transcriptional activation largely preceded methylation changes. While it is generally assumed that DNA methylation correlates with transcriptional silencing, direct correlations between gene expression and methylation in non-promoter regions have been reported (30,31). We are not aware of any previous reports of promoter methylation correlating directly with transcriptional activity. Since DNA methylation can have either an attractive or repulsive effect on methylation-sensitive DNA binding proteins (10), however, there is no a priori reason to expect that promoter methylation must universally facilitate transcriptional silencing.
The DNA methylation changes we identified in the postnatal liver tended to occur at non-CGI promoters (Table 1). A previous epigenomic analysis using restriction landmark genome scanning (RLGS) in mice (9) reported that two-third of ‘tissue differentially methylated regions’ are within CGIs. This conclusion was perhaps skewed by the innate bias of RLGS toward CGIs. Indeed, a more recent RLGS study in mice (32) reported that genomic regions showing tissue-specific methylation are only rarely associated with CGIs, consistent with our findings.
The functional significance of DNA methylation at CpG-poor promoters is controversial. Our temporal analyses (Fig. 5) showed strong correlations between methylation and expression in five of six genes examined, and our bioinformatic analyses (Fig. 6) showed that, overall, the methylation changes we identified are associated with developmental changes in and tissue-specific expression. These results appear to contradict an earlier analysis using methylated DNA immunoprecipitation (MeDIP) in primary human fibroblasts, which concluded that CpG-poor promoters are predominantly hypermethylated, and that ‘repression by DNA methylation requires high CpG density’ (33). More recently, Rakyan et al. (34) used MeDIP to compare genome-wide methylation patterns across seven human tissues, and found a significant negative correlation between tissue-specific promoter methylation and gene expression, even at CpG-poor promoters. Our findings are clearly consistent with those of Rakyan et al.
We computationally identified several sequence motifs significantly enriched in promoter regions that gained or lost DNA methylation during postnatal liver development (Table 2). Interestingly, two of the motifs most highly enriched among genes that gained methylation (rows 3 and 4 in Table 2) are potential binding sites for the transcription factor Sp1 (consensus recognition sequence CCCCNCCCCNCCCC). This observation seems somewhat inconsistent with previous reports that Sp1 sites protect CGIs from methylation during embryogenesis (35). In any event, the sequence motifs identified here may be useful in future transgenic experiments aiming to characterize cis- and trans-regulatory mechanisms involved in the developmental establishment of DNA methylation.
The timing of our study overlaps with the migration of hematopoietic cells from the liver. We originally selected E17.5 as the baseline time point based on a study (36) indicating that hematopoietic cells are no longer a major population in the mouse liver at this time. When we attempted to verify this, however, we obtained results more consistent with earlier studies (17) indicating that hematopoietic cells comprise a major proportion of mouse liver cells at E17.5 (Fig. 4). Therefore, some of the methylation changes detected by MCAM may reflect the changing cellular composition of the liver from E17.5 to P21. But clearly this is not the entire story. In all six of the genes studied significant changes in methylation and expression continued after P5 (Fig. 5), by which time the hematopoietic migration was nearly complete (Fig. 4). Further, our clonal bisulfite sequencing studies (Fig. 3) show no evidence of clone-specific methylation, as might be expected if, for example, a cell type containing a methylated promoter is being replaced by one with an unmethylated promoter. Hence, in many cases the methylation changes identified here are likely to reflect developmental maturation of epigenetic regulation in hepatic cells during postnatal development.
Owing perhaps to the relatively small number of genes identified, GO analysis did not detect associations among gene methylation changes and specific developmental pathways. Nonetheless, our results do highlight several genes at which epigenetic changes may be important to liver development. Jarid2 (37), Bmp4 (38) and Onecut1 (39) encode important transcriptional regulators critical to early liver development. All of these genes were found to gain methylation in the postnatal liver, suggesting they may be epigenetically down-regulated following the completion of hepatic morphogenesis. Conversely, among the genes found to lose methylation postnatally were Fdps, which encodes an enzyme involved in cholesterol biosynthesis, and Trf, which encodes the iron transporter transferrin (40). Both are highly expressed in adult liver; their loss of methylation postnatally is therefore consistent with hepatic functional maturation.
The timing of epigenetic modifications is of central relevance to the developmental origins hypothesis, which proposes that early environmental influences on development cause permanent changes in structure and function that affect adult metabolism and disease risk. Myriad human epidemiologic data indicate that environmental factors affecting fetal and early postnatal growth play a role in such ‘developmental programming’ (41), and epigenetic mechanisms are increasingly implicated in these effects (42). Since mammalian epigenetic regulation is most labile to environment when DNA methylation is undergoing developmental establishment or maturation (43,44), elucidating a potential epigenetic basis for developmental programming will require an understanding of epigenetic changes in diverse tissues throughout mammalian development (not just in the early embryo).
Sensitivity and coverage of MCAM
Our validation data indicate that MCAM has excellent specificity and can detect even small methylation differences. Also, although MCAM is based upon SmaI/XmaI restriction sites, it usually identifies methylation changes occurring regionally, effectively increasing the method's genomic coverage.
Current genome-wide methylation analysis methods can be divided into antibody-based methods such as MeDIP (33), and methylation-sensitive restriction enzyme-based approaches (such as MCAM) [A nascent method, whole-genome bisulfite shotgun sequencing (45), has yet to be applied to an entire mammalian genome]. Although theoretically capable of providing better coverage than restriction-enzyme based approaches, MeDIP works best in genomic regions of fairly high CpG density (33). Further, even in regions of high CpG density the method appears to lack sensitivity, and apparently detects only full-scale methylation differences (i.e. 100 versus 0%) (33). This lack of sensitivity was also documented in a recent study comparing various genome-wide methylation analysis methods (46).
Methylation-sensitive restriction enzyme-based approaches include RLGS (9), microarray hybridization of labeled NotI fragments (7), and the HELP assay (47,48), which is based upon digestion of genomic DNA with the methylation-sensitive restriction enzyme HpaII, followed by ligation-mediated PCR and microarray hybridization. A recent study comparing various genome-wide methylation assays (46) demonstrated that the HELP assay suffers from low specificity. This is likely attributable to incomplete digestion of unmethylated restriction sites causing false positive hits, a problem that generally plagues digestion-based approaches. In MCAM, serial digestion of each sample with methylation sensitive and insensitive isoschizomers effectively eliminates this problem (8). Only SmaI/XmaI sites that are (i) uncleaved by SmaI and (ii) subsequently cleaved by XmaI serve as template for amplification; failure to digest cannot be misinterpreted as a methylation signal. This unique characteristic likely explains the high specificity of MCAM (Fig. 1C). Additionally, MCAM is sufficiently sensitive to detect even small methylation changes (Fig. 2). In the most striking example (Fig. 2E), we detected developmental hypermethylation at Lingo4, in which essentially the entire MCAM signal was derived from a methylation increase at the 3′ SmaI/XmaI site from 5 to 30%.
A general concern regarding restriction enzyme-based approaches for methylation profiling is their limited genomic coverage. Of the ∼21 000 000 CpG sites in the mouse genome (45), only ∼75 000 (0.4%) are within SmaI/XmaI sites that are potentially informative by MCAM. But it is misleading to say that MCAM only ‘covers’ 0.4% of the genome. Rather, since variation in DNA methylation usually occurs regionally (33), MCAM actually enables the identification of broad genomic regions of differential DNA methylation. Indeed, although the SmaI/XmaI intervals yielding MCAM ‘hits’ were several hundred base pair long, in almost every case we found concordant methylation changes at both ends, as well as at CpG sites adjacent to the informative SmaI/XmaI sites (Fig. 2). Further, even when the informative SmaI/XmaI sites were several hundred base pair away (as in Def6 and Lingo4), our clonal bisulfite sequencing studies showed similar methylation changes near the transcription start site (Fig. 3D and E). Clearly, MCAM generally identified not single CpG sites, but rather broad genomic regions undergoing methylation changes. Of the 48 657 potentially informative SmaI/XmaI intervals in the mouse genome, the proximal promoter array we used included probes within only 3757. Having demonstrated the utility of MCAM at promoter regions, custom arrays designed specifically for MCAM will include only probes within potentially informative SmaI/XmaI intervals, dramatically improving the depth and genomic coverage of the method.
Methylated CGI amplification (MCA) was originally developed to screen for aberrant CGI hypermethylation in cancer (19). When coupled with microarray hybridization, the method was dubbed ‘MCAM’ (8). Our data, however, clearly demonstrate that the genomic coverage of the method extends beyond CGIs: most of the developmental changes we identified are not at CGIs (Table 1). Hence, we suggest that future studies refer to the method as ‘Methylation-Specific Amplification and Microarray hybridization’ (MSAM).
Using a DNA methylation microarray approach, we have identified promoter regions in the mouse genome at which DNA methylation is undergoing developmental maturation in the early postnatal period. Our characterization of the temporal relationships between developmental changes in DNA methylation and expression in vivo provides support for the hypothesis that DNA methylation functions during mammalian differentiation to maintain the silence of genes whose activity is not required in specific tissues. Importantly, however, our data also show that promoter methylation does not irrevocably prevent transcriptional activation; even during postnatal development, promoter demethylation was detected and correlated with transcriptional activation. Lastly, our successful application of MCAM to detect subtle DNA methylation changes during normal development underscores the broad potential applicability of the method.
MATERIALS AND METHODS
Animals and tissue collection
C57BL6/J mice (Jackson Laboratories, Bar Harbor, ME) were used. For collection of fetal liver samples, timing of coitus was determined by observation of vaginal plugs; the morning a plug was observed was considered 0.5 days post coitum. At 17.5 days post coitum (E17.5), pregnant females were killed by CO2 asphyxiation, and fetal liver collected. Fetal sex was determined by PCR for Sry. P21 liver was collected directly after removing mice from their dams in the morning. All applicable institutional and governmental regulations concerning the ethical use of animals were followed during this research. The protocol was approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Methylated CpG island amplification and microarray hybridization
MCAM was performed as previously described (8), with the following modifications. During the ligation step, adaptors appropriate for mouse genomic DNA were used: RXMA24: 5′-AGCACTCTCCAGCCTCTCACCGAC-3′, and RXMA12: 5′-CCGGGTCGGTGA-3′. Following the amplification step, E17.5 and P21 MCA products were labeled with Cy3 and Cy5, respectively. Mouse proximal promoter microarrays were obtained from Agilent Technologies (Santa Clara, CA). Computational identification of potentially informative SmaI/XmaI genomic intervals, and annotation relative to CGIs and repetitive regions were performed as previously described, using the March 2005 (mm6) mouse genome assembly. Two P21 versus E17.5 cohybridizations were performed, and [based upon our earlier study in humans (8)] a SmaI/XmaI interval was considered a ‘hit’ if the signal ratios and intensities of all probes within the interval met the following criteria in both cohybridizations: (i) median signal ratio >2 or <0.5, (ii) median upper signal intensity >1000 and (iii) median P-value log ratio <0.0001. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (49) and are accessible through GEO Series accession number GSE15634 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE15634).
DNA methylation analysis methods
Bisulfite modification of genomic DNA was performed as previously described (50). Validation of MCAM hits was performed by direct manual sequencing and phosphorimager quantitation (50) (n = 11), or by bisulfite pyrosequencing (51) (n = 5). We compared both methods on a subset of samples and obtained identical results (data not shown). A list of genes validated, including primers used, is provided in Supplementary Material, Table S2. Whenever possible, we obtained bisulfite sequencing data on both SmaI/XmaI sites for each SmaI/XmaI interval. The temporal analyses of DNA methylation (Fig. 5) are based on % methylation values averaged over the following CpG sites (relative to transcription start site): Azgp1 -17, -7; Fcgrt -155, -146; Phyhd1 19, 21; Def6 244, 262; Lingo4 -234, -226; Nrbp2 -557, -552, -549, -542, -534. Bisulfite cloning and sequencing were performed for six genes to analyze clonal methylation. We cloned the bisulfite-PCR products into the TA vector pCR2.1 (Invitrogen), then extracted and sequenced plasmid DNA from the resulting clones, using the sequencing primer provided by the manufacturer. PCR primer sequences are described in Supplementary Material, Table S2.
Histological evaluation of liver
Flash frozen pieces of liver were fixed in 10% formaldehyde for more than 24 h, paraffin embedded, sectioned and hematoxylin–eosin stained according to standard pathological procedures. Hematopoietic cells were identified by dense pink cytoplasm and round dark nuclei in erythroid precursors and larger size of nuclei and lesser cytoplasm in granulocytic precursor cells. Megakaryocytes were easily recognizable, large cells with pink cytoplasm and large, often multilobulated nuclei. The percentage of hematopoetic cells was estimated in 10 high power fields of the sections from each animal from E17.5, P0, P5, P10, P21 (3–5 animals per age).
Gene expression measurements
Total RNA was isolated (RNA Stat-60, Tel-Test, Inc., Friendswood, TX), and reverse transcribed using random priming (M-MLV reverse transcriptase, Promega, Madison, WI). No-RT negative controls were included in all assays. For Def6 and Lingo4, real-time RT–PCR was performed using Syber Green PCR Master Mix (Applied Biosystems, Foster City, CA) according to manufacturer's instructions. Gene-specific cDNA was amplified using primers spanning an intron, and all RT–PCR products were examined by agarose gel electrophoresis to confirm single bands of the correct size. Bands resulting from amplification of contaminating DNA were never observed. The primers used are provided in Supplementary Material, Table S3. For the other genes the following TaqMan gene expression assays (Applied Biosystems) were used, according to manufacturer's instructions: Azgp1 (Mm00516330_m1), Fcgrt (Mm01205449_g1), Phyhd1 (Mm00549288_m1), Nrbp2 (Mm00522920_g1) and Beta actin (Mm00607939_s1). Expression changes were quantitated relative to β-actin as an endogenous control, using the 2−ΔΔCt method.
Bioinformatic analysis
To analyze the distribution of repetitive elements in the vicinity of genes of interest, we downloaded the gene annotation file (refGene.txt.gz) and RepeatMasker files (chr_rmsk.txt.gz) from the UCSC genome browser (mm9, NCBI Build 37). The distances of repetitive element midpoints from relevant transcription start sites were calculated. For genes with multiple transcription start sites, the one nearest the informative SmaI interval was used. To search for sequence motifs enriched in the different gene groups, we used MEME and MAST as described previously (8). We used Fatigo functional enrichment analysis (http://babelomics.bioinfo.cipf.es/) (52) to identify GOclassifications significantly over- or underrepresented among our gene lists; cited P-values were adjusted for multiple testing. To globally assess developmental changes in expression among the genes identified by MCAM, we downloaded SAGE data from the Mouse Atlas of Gene Expression (http://www.mouseatlas.org) for E18 and P84 C57BL6 mouse liver, brain and spleen. We used the Mouse Atlas DiscoverySpace platform to map expressed tags to genes, then cross-referenced these expression data to the gene lists from our MCAM experiment. To globally assess gene expression in liver relative to other tissues, we downloaded mouse expression data from the GNF SymAtlas (http://symatlas.gnf.org/SymAtlas/). For each gene identified as undergoing postnatal methylation change in liver, a Z score was calculated for expression in liver relative to other tissues: Z=((liver expression)−(average expression in other tissues))/(SD of expression in other tissues). Tissues included were brain, pituitary, eye, lymph node, trachea, uterus, ovary, adipose tissue, adrenal gland, bladder, epidermis, digits, snout, tongue, medial olfactory epithelium, prostate, vomeralnasal organ, lung, stomach, large intestine, bone marrow, bone, spleen, thymus, brown fat, heart, skeletal muscle, placenta, mammary gland, kidney, small intestine, salivary gland, thyroid, pancreas and testis.
Statistical analysis
Chi-square tests were used to perform group comparisons of the proportions of SmaI/XmaI intervals with no, 1, or both SmaI/XmaI sites within a CGI (Table 1). To analyze group differences in developmental expression changes (Fig. 6A) and liver-specific expression (Fig. 6B), values were log transformed to improve normality, then two-tailed, unpaired t-tests were performed.
Whereas MCA is molecule-specific (both SmaI/XmaI sites on a specific DNA molecule must be methylated for the SmaI/XmaI interval to be amplified), our quantitative bisulfite sequencing methods provide no molecule-specific information. We therefore created an algorithm to compare methylation ratios obtained by bisulfite sequencing with hybridization ratios obtained by MCAM. If methylation at the two sites on a single allele is uncorrelated, the product of fractional methylation at the two sites should compare best with the MCAM result. If they are highly correlated, the average methylation should compare best with the MCAM result. We assumed partial correlation, calculating the mean of the average and the product of methylation at the two SmaI/XmaI sites to obtain the final percent methylation ratios that were compared with the MCAM ratios in Figure 1C:
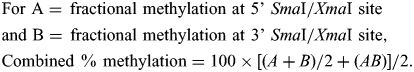 |
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by National Institutes of Health [5K01DK070007 to R.A.W.], the March of Dimes Birth Defects Foundation [#5-FY05-47 to R.A.W.] and the United States Department of Agriculture [CRIS #6250-51000-049 to R.A.W.]. L.S. is a Sidney Kimmel Foundation Scholar.
ACKNOWLEDGEMENTS
We wish to acknowledge Adam Gillum for his assistance in creating the figures.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Jaenisch R., Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 2003;33(Suppl.):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 2.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 3.Holliday R., Pugh J.E. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–232. [PubMed] [Google Scholar]
- 4.Riggs A.D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell. Genet. 1975;14:9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- 5.Walsh C.P., Bestor T.H. Cytosine methylation and mammalian development. Genes Dev. 1999;13:26–34. doi: 10.1101/gad.13.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ehrlich M. Expression of various genes is controlled by DNA methylation during mammalian development. J. Cell. Biochem. 2003;88:899–910. doi: 10.1002/jcb.10464. [DOI] [PubMed] [Google Scholar]
- 7.Ching T.T., Maunakea A.K., Jun P., Hong C., Zardo G., Pinkel D., Albertson D.G., Fridlyand J., Mao J.H., Shchors K., et al. Epigenome analyses using BAC microarrays identify evolutionary conservation of tissue-specific methylation of SHANK3. Nat. Genet. 2005;37:645–651. doi: 10.1038/ng1563. [DOI] [PubMed] [Google Scholar]
- 8.Shen L., Kondo Y., Guo Y., Zhang J., Zhang L., Ahmed S., Shu J., Chen X., Waterland R.A., Issa J.P. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007;3:2023–2036. doi: 10.1371/journal.pgen.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song F., Smith J.F., Kimura M.T., Morrow A.D., Matsuyama T., Nagase H., Held W.A. Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc. Natl. Acad. Sci. USA. 2005;102:3336–3341. doi: 10.1073/pnas.0408436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 11.Reik W., Dean W., Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 12.Lane N., Dean W., Erhardt S., Hajkova P., Surani A., Walter J., Reik W. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis. 2003;35:88–93. doi: 10.1002/gene.10168. [DOI] [PubMed] [Google Scholar]
- 13.Walsh C.P., Chaillet J.R., Bestor T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 14.Heard E. Recent advances in X-chromosome inactivation. Curr. Opin. Cell Biol. 2004;16:247–255. doi: 10.1016/j.ceb.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Greengard O. Enzymic differentiation in mammalian liver. Science. 1969;163:891–895. doi: 10.1126/science.163.3870.891. [DOI] [PubMed] [Google Scholar]
- 16.Lemaigre F., Zaret K.S. Liver development update: new embryo models, cell lineage control, and morphogenesis. Curr. Opin. Genet. Dev. 2004;14:582–590. doi: 10.1016/j.gde.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Herbst R.S., Babiss L.E. In: Mechanisms of Differentiation. Fisher P.B., editor. Vol. 1. Boca Raton: CRC Press; 1990. pp. 15–45. [Google Scholar]
- 18.Gao B., Jeong W.I., Tian Z. Liver: an organ with predominant innate immunity. Hepatology. 2008;47:729–736. doi: 10.1002/hep.22034. [DOI] [PubMed] [Google Scholar]
- 19.Toyota M., Ho C., Ahuja N., Jair K.W., Li Q., Ohe-Toyota M., Baylin S.B., Issa J.P. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res. 1999;59:2307–2312. [PubMed] [Google Scholar]
- 20.Pruitt K.D., Tatusova T., Maglott D.R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antequera F. Structure, function and evolution of CpG island promoters. Cell. Mol. Life Sci. 2003;60:1647–1658. doi: 10.1007/s00018-003-3088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bailey T.L., Williams N., Misleh C., Li W.W. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–W373. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bailey T.L., Gribskov M. Combining evidence using P-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- 24.Mohn F., Weber M., Rebhan M., Roloff T.C., Richter J., Stadler M.B., Bibel M., Schubeler D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell. 2008;30:755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 25.Boyer L.A., Plath K., Zeitlinger J., Brambrink T., Medeiros L.A., Lee T.I., Levine S.S., Wernig M., Tajonar A., Ray M.K., et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 26.Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P., et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki M.M., Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 28.Futscher B.W., Oshiro M.M., Wozniak R.J., Holtan N., Hanigan C.L., Duan H., Domann F.E. Role for DNA methylation in the control of cell type specific maspin expression. Nat. Genet. 2002;31:175–179. doi: 10.1038/ng886. [DOI] [PubMed] [Google Scholar]
- 29.Brandeis M., Frank D., Keshet I., Siegfried Z., Mendelsohn M., Nemes A., Temper V., Razin A., Cedar H. Sp1 elements protect a CpG island from de novo methylation. Nature. 1994;371:435–438. doi: 10.1038/371435a0. [DOI] [PubMed] [Google Scholar]
- 30.Hu M., Yao J., Cai L., Bachman K.E., van den Brule F., Velculescu V., Polyak K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 2005;37:899–905. doi: 10.1038/ng1596. [DOI] [PubMed] [Google Scholar]
- 31.Ladd-Acosta C., Pevsner J., Sabunciyan S., Yolken R.H., Webster M.J., Dinkins T., Callinan P.A., Fan J.B., Potash J.B., Feinberg A.P. DNA methylation signatures within the human brain. Am. J. Hum. Genet. 2007;81:1304–1315. doi: 10.1086/524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakamoto H., Suzuki M., Abe T., Hosoyama T., Himeno E., Tanaka S., Greally J.M., Hattori N., Yagi S., Shiota K. Cell type-specific methylation profiles occurring disproportionately in CpG-less regions that delineate developmental similarity. Genes Cells. 2007;12:1123–1132. doi: 10.1111/j.1365-2443.2007.01120.x. [DOI] [PubMed] [Google Scholar]
- 33.Weber M., Hellmann I., Stadler M.B., Ramos L., Paabo S., Rebhan M., Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 34.Rakyan V.K., Down T.A., Thorne N.P., Flicek P., Kulesha E., Graf S., Tomazou E.M., Backdahl L., Johnson N., Herberth M., et al. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs) Genome Res. 2008;18:1518–1529. doi: 10.1101/gr.077479.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macleod D., Charlton J., Mullins J., Bird A.P. Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 1994;8:2282–2292. doi: 10.1101/gad.8.19.2282. [DOI] [PubMed] [Google Scholar]
- 36.Chagraoui J., Lepage-Noll A., Anjo A., Uzan G., Charbord P. Fetal liver stroma consists of cells in epithelial-to-mesenchymal transition. Blood. 2003;101:2973–2982. doi: 10.1182/blood-2002-05-1341. [DOI] [PubMed] [Google Scholar]
- 37.Takeuchi T., Watanabe Y., Takano-Shimizu T., Kondo S. Roles of jumonji and jumonji family genes in chromatin regulation and development. Dev. Dyn. 2006;235:2449–2459. doi: 10.1002/dvdy.20851. [DOI] [PubMed] [Google Scholar]
- 38.Rossi J.M., Dunn N.R., Hogan B.L., Zaret K.S. Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 2001;15:1998–2009. doi: 10.1101/gad.904601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Margagliotti S., Clotman F., Pierreux C.E., Beaudry J.B., Jacquemin P., Rousseau G.G., Lemaigre F.P. The Onecut transcription factors HNF-6/OC-1 and OC-2 regulate early liver expansion by controlling hepatoblast migration. Dev. Biol. 2007;311:579–589. doi: 10.1016/j.ydbio.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 40.Kasik J.W., Rice E.J. Transferrin gene expression in maternal liver, fetal liver and placenta during pregnancy in the mouse. Placenta. 1993;14:365–371. doi: 10.1016/s0143-4004(05)80458-8. [DOI] [PubMed] [Google Scholar]
- 41.Gluckman P.D., Hanson M.A. Living with the past: evolution, development, and patterns of disease. Science. 2004;305:1733–1736. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- 42.Jirtle R.L., Skinner M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waterland R.A., Jirtle R.L. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weaver I.C., Cervoni N., Champagne F.A., D'Alessio A.C., Sharma S., Seckl J.R., Dymov S., Szyf M., Meaney M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 45.Meissner A., Mikkelsen T.S., Gu H., Wernig M., Hanna J., Sivachenko A., Zhang X., Bernstein B.E., Nusbaum C., Jaffe D.B., et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008 doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irizarry R.A., Ladd-Acosta C., Carvalho B., Wu H., Brandenburg S.A., Jeddeloh J.A., Wen B., Feinberg A.P. Comprehensive high-throughput arrays for relative methylation (CHARM) Genome Res. 2008;18:780–790. doi: 10.1101/gr.7301508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khulan B., Thompson R.F., Ye K., Fazzari M.J., Suzuki M., Stasiek E., Figueroa M.E., Glass J.L., Chen Q., Montagna C., et al. Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res. 2006;16:1046–1055. doi: 10.1101/gr.5273806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mill J., Tang T., Kaminsky Z., Khare T., Yazdanpanah S., Bouchard L., Jia P., Assadzadeh A., Flanagan J., Schumacher A., et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waterland R.A., Lin J.R., Smith C.A., Jirtle R.L. Post-weaning diet affects genomic imprinting at the insulin-like growth factor 2 (Igf2) locus. Hum. Mol. Genet. 2006;15:705–716. doi: 10.1093/hmg/ddi484. [DOI] [PubMed] [Google Scholar]
- 51.Shen L., Guo Y., Chen X., Ahmed S., Issa J.P. Optimizing annealing temperature overcomes bias in bisulfite PCR methylation analysis. Biotechniques. 2007;42:48, 50, 52. doi: 10.2144/000112312. passim. [DOI] [PubMed] [Google Scholar]
- 52.Al-Shahrour F., Minguez P., Tarraga J., Montaner D., Alloza E., Vaquerizas J.M., Conde L., Blaschke C., Vera J., Dopazo J. BABELOMICS: a systems biology perspective in the functional annotation of genome-scale experiments. Nucleic Acids Res. 2006;34:W472–W476. doi: 10.1093/nar/gkl172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.