

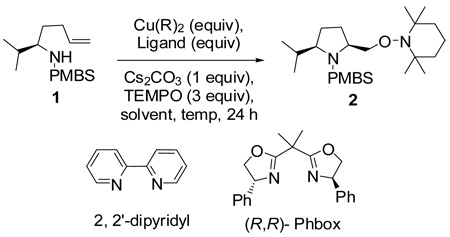
Abstract

The diastereoselectivity of the copper-promoted intramolecular aminooxygenation of various alkene substrates was investigated. α-Substituted 4-pentenyl sulfonamides favor the formation of 2,5-cis-pyrrolidines (dr >20:1) giving excellent yields which range from 76–97% while γ-substituted substrates favor the 2,3-trans pyrrolidine adducts with moderate selectivity (ca. 3:1). A substrate whose N-substituent was directly tethered to the α-carbon exclusively yielded the 2,5-trans pyrrolidine. The synthetic utility of the method was demonstrated by a short and efficient formal synthesis of (+)-monomorine.
Pyrrolidine moieties are frequently found in biologically active molecules.1 These include glycosidase inhibitors such as alexine2 and australine,3 antiviral agents such as preussin,4 antileukemia agents such as harringtonine5 and crambescidin6 and the angiotensin-converting enzyme (ACE) inhibitor ramipril.7 Due to the therapeutic importance of these pyrrolidine alkaloids, considerable effort has been devoted to the stereoselective synthesis of substituted pyrrolidines.8 In this paper, we report a copper(II) promoted diastereoselective synthesis of disubstituted pyrrolidines via an intramolecular aminooxygenation of alkenes.
The intramolecular aminooxygenation of alkenes can be catalyzed and promoted using a number of reagents and catalysts, but few result in the stereoselective synthesis of pyrrolidines.9,10,11,12 Donohoe has reported a diastereoselective osmium-catalyzed aminohydroxylation that results in the synthesis of 2,5-cis-pyrrolidines.9a However, Donohoe’s reaction requires two coordinating groups in the substrate, the sulfonamide nitrogen that forms the C-N bond, and an additional vicinal alcohol, to achieve high diastereoselectivity.
The diastereoselective copper-promoted aminooxygenation reactions reported in this paper do not require additional coordinating groups to provide high levels of 2,5-cis-pyrrolidine selectivity. Furthermore, analysis of the conformational factors that control the diastereoselectivity in these reactions led to the development of a 2,5-trans-pyrrolidine selective reaction as well (vide infra).
Recent reports from our group showed that the copper(II)-promoted synthesis of 2,5-disubstituted pyrrolidines via intramolecular alkene carboamination occur in high diastereoselectivity with predominating cis stereochemistry (Scheme 1).13a Mechanistic studies of these reactions13a revealed a pathway involving a primary carbon radical intermediate that was trapped efficiently with 2,2,6,6-tetramethylpiperidine-N-oxyl radical (TEMPO), a standard carbon radical trapping agent (Scheme 1).13b These results led us to investigate the diastereoselectivity of this net alkene aminooxygenation reaction using substrates bearing substituents α as well as γ to the sulfonamide unit.
Scheme 1.

Copper(II) promoted diastereoselective formation of 2,5-cis-pyrrolidines and TEMPO trapping of radical intermediate.
We first investigated the aminooxygenation reaction of 4-pentenyl sulfonamide 1 using catalytic amounts of copper(II) salts (Table 1).11f, 11g The use of the bisoxazoline ligand [(R, R)-Phbox] gave better conversion than the 2,2’-dipyridyl ligand under catalytic conditions using O2 (1 atm) (Table 1, entries 1 and 2). The (R, R)-Phbox ligand and substrate 1 both favor formation of the C2(S) stereocenter (the reaction is matched).11f We have previously used these conditions to catalyze the aminooxygenation reactions of slightly more reactive achiral substrates.11f,11g However, neither reaction of sulfonamide 1 went to completion. While the catalytic reaction shows promise (yield = 60%, Table 1, entry 2), its optimization is ongoing. On the other hand, we were able to rapidly identify a highly efficient and operationally simple reaction process by use of a slight excess (1.5 equiv) of a readily available and inexpensive copper(II) carboxylate, copper(II) 2-ethylhexanoate [Cu(EH)2]. Because Cu(EH)2 is neither very expensive nor toxic and since the reaction is quite operationally simple, not requiring an O2 atmosphere, we concluded that the stoichiometric reaction provides an acceptable solution until superior catalytic conditions are identified.
Table 1.
Effects of varying the amount of Cu(R)2, ligand, temperature and solvent.a
 | |||||
|---|---|---|---|---|---|
| entry | R (equiv) | ligand (equiv) | solvent | temp (°C) | yield (%)b |
| 1c | OTf (0.2) | 2,2’-dipyridyl (0.2) | xylenes | 130 | 20d |
| 2c | OTf (0.2) | (R,R)-Phbox (0.2) | xylenes | 130 | 60d |
| 3 | EH (3) | - | DMF | 160 | 80 |
| 4 | EH (1.5) | - | DMF | 160 | 40 |
| 5 | EH (1.5) | - | xylenes | 130 | 94 |
| 6e | EH(1.5) | - | xylenes | 130 | 78 |
| 7 | EH (1.5) | - | xylenes | 120 | 54d |
| 8 | EH (1.5) | - | DMF | 130 | 40d |
| 9 | EH (1.5) | - | CF3Ph | 120 | 63d |
| 10 | EH (1.0) | - | CF3Ph | 120 | 38d |
| 11 | EH (1.0) | - | xylenes | 130 | 68d |
All reactions were run in pressure tubes at 0.1 M w/r to 1.
Yield refers to amount of product isolated after purification by flash chromatography on silica gel.
The reaction was run under O2 (1 atm).
The remainder of the material is the starting olefin 1.
The reaction was run using 1.5 equiv of TEMPO. EH = 2-ethylhexanoate, PMBS = p-methoxybenzenesulfonyl
Copper(II) 2-ethylhexanoate is more reactive than many copper carboxylates owing to its high solubility in organic solvents.13a, 14 Among the solvents (DMF, xylenes, CF3Ph) and temperatures (120–160 °C) surveyed, we found that xylenes at 130 °C provided the optimal yield (94%, Table 1, entry 5). Slightly more than one equivalent of Cu(EH)2 (1.5 equiv) was also required for optimal yield, and the reaction was complete within 24 h. Lower TEMPO amounts (1.5 equiv) gave slightly lower yield (entry 6), so the reactions were run using 3 equivalents of TEMPO.
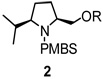
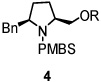
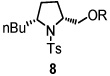
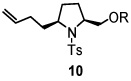
Using the optimized reaction conditions (Table 1, entry 5), the reactions of a number of α and γ-substituted 4-pentenyl sulfonamides were examined (Table 2). Similar to the diastereoselectivity results in the analogous copper(II) promoted carboamination reaction of 4-pentenyl sulfonamides (Scheme 1), substrates 1, 3, 5, 7 and 9 generated the 2,5-cis-pyrrolidines 2, 4, 6, 8 and 10 in excellent yields with >20:1 selectivity (Table 2, entries 1–5). Gratifyingly, no hydroamination side products were observed in these reactions. The crystal structure of 2 indicated cis stereochemistry with an absolute configuration of C2(S), C5(R) (see Supporting Information).15 The relative configurations of pyrrolidines 4, 6, 8 and 10 were assigned by analogy and by nOe experiments. Upon changing the nitrogen protecting group from tosyl (Ts) or p-methoxybenzenesulfonyl (PMBS) to 4-nitrophenyl sulfonyl (Ns), there was a slight decrease in the yield, but a high level of stereocontrol was still observed (Table 2, entry 6). The Ns group is usually more easy to remove.16 γ-Substituted 4-pentenyl sulfonamides 13 and 16 gave only moderate selectivity (3:1 for 13 and 2:1 for 16) favoring the trans pyrrolidine adducts (Table 2, entries 7 and 8). Little difference between the electron-donating OMe and electron-withdrawing CF3 benzyl substituents was observed. The relative configurations of the cis and trans isomers were assigned by X-ray crystallography and nOe experiments (see Supporting Information). The trans pyrrolidine is presumably favored due to equatorial placement of the γ-substituent in the cyclic transition state (see Supporting Information). It was not clear if substrate 19, with an internal disubstituted olefin, would favor aminooxygenation (20) or oxidative amination (21). In the carboamination series13a only 21 was obtained. To our delight, the aminooxygenation product 20 was isolated as the major product with >20:1 diastereoselectivity and 7:1 aminooxygenation:oxidative amination selectivity. The stereochemistry of 20 was determined by X-ray crystallography (see Supporting Information).15
Table 2.
Diastereoselective aminooxygenation of alkenes.a
| entry | substrate | product | yield (%)b (selectivity)d |
|
|---|---|---|---|---|
| 1 | 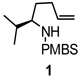 |
 |
94 (dr >20:1) |
|
| 2 | 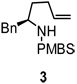 |
 |
97 (dr >20:1) |
|
| 3 | 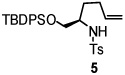 |
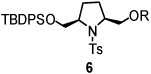 |
96 (dr >20:1) |
|
| 4 | 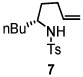 |
 |
94 (dr >20:1) |
|
| 5 | 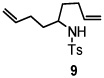 |
 |
97 (dr >20:1) |
|
| 6 | 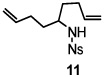 |
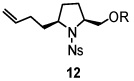 |
76 (dr >20:1) |
|
| 7 | 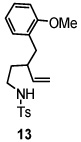 |
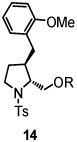 |
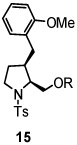 |
80c (trans: cis = 3:1) |
| 8 | 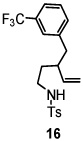 |
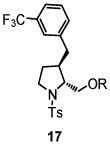 |
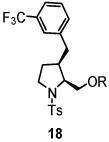 |
88c (trans:cis = 2:1) |
| 9e | 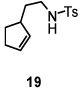 |
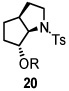 |
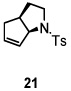 |
57f (20, dr >20:1) 20:21 = 7:1 |
R = 2,2,6,6-tetramethylpiperidine
Reaction conditions: 1.5 equiv of Cu(EH)2, 3 equiv of TEMPO, 1 equiv of Cs2CO3, xylenes (0.1 M), 130 °C, 24 h, pressure tube.
Yield refers to amount of product isolated upon purification by column chromatography on SiO2.
Yield refers to the sum of the products isolated by chromatography on SiO2.
Diastereomeric ratio was determined by analysis of the crude 1H NMR spectrum.
The reaction was run at 140 °C.
Yield refers to amount of 20 isolated.
The mechanism illustrated in Scheme 2 provides a rationalization for the high degree of cis stereoselectivity observed in the aminooxygenation reaction of the α substituted substrates. It was reasoned that this reaction underwent a mechanism similar to the copper(II) promoted intramolecular carboamination reaction.13a The first C–N bond is proposed to form in a stereoselective manner via syn aminocupration through the chair-like transition state 23 or possibly the boat-like transition state 24, generating an unstable organocopper(II) species 25. This organocopper(II) species then undergoes homolysis, forming a primary carbon radical intermediate which is trapped by the TEMPO radical,11f, 13 forming the cis aminooxygenation product 2.
Scheme 2.

Proposed mechanism for the formation of 2,5-cis-pyrrolidine.
To demonstrate the synthetic utility of the method, we performed a short and efficient formal synthesis of (+)-monomorine (28). (+)-Monomorine is an indolizidine alkaloid that is known to be a trail pheromone of a health hazard pharaoh ant Monomorium pharaonis L.17 Along with the other indolizidine alkaloids, (+)-monomorine has been the target of many organic chemists for some time. As a result, a number of different ways of synthesizing it have been reported.18 Particularly relevant to this study was the synthesis reported by Bäckvall and co-workers19 wherein they used aldehyde 27 as an intermediate in their synthesis of (+)-monomorine. Utilizing our method, aldehyde 27 was synthesized in 6 steps and 40% overall yield. Our route to 27 is 3 steps shorter than Bäckvall’s approach. The 2,5-cis-pyrrolidine 8 was formed from substrate 7 in 94% yield and high selectivity (>20:1) (Table 2, entry 4). Substrate 7 was readily synthesized from commercially available d-norleucine (see Supporting Information). The TEMPO adduct 8 was oxidized to aldehyde 27 using mCPBA in 68% yield (Scheme 3).20 We have previously also demonstrated that dissolving metal reduction can chemoselectively reveal one or both of the free amine and free alcohol functionalities.11f
Scheme 3.

Formal synthesis of (+)-monomorine.
On the basis of the proposed transition state for the α substituted 4-pentenyl sulfonamides (Scheme 2), we predicted that if the N-substituent was directly tethered to the α-carbon (e.g., substrate 29), the reaction would occur via transition state 30, which places the α-substitutent in a pseudoequatorial position, thereby favoring the formation of the 2,5-trans pyrrolidine adduct (Scheme 4). When sulfonamide 29 was subjected to the optimized reaction conditions for this copper(II) promoted aminooxygenation, it produced the trans pyrrolidine 31 in poor yield but with high diastereoselectivity (>20:1). We hypothesized that addition of excess TEMPO (3 equiv) caused the decomposition of the starting material via benzylic oxidation. When the amount of TEMPO was decreased to 1.5 equivalents, we were able to increase the yield to 59% with the same level of selectivity. The trans configuration of pyrrolidines 31 was confirmed by X-ray crystallography (Figure 1).15 We have previously demonstrated that the SO2 moiety in sultams such as 31 can be removed by dissolving metal reduction.13a, 21
Scheme 4.

Diastereoselective aminooxygenation of sulfonamide 29 forming the trans pyrrolidine 31.
Figure 1.

Crystal structure of 31.
In conclusion, we have developed a high yielding route for the synthesis of disubstituted pyrrolidines via the intramolecular copper promoted aminooxygenation of alkenes. These reactions afford the 2,3-trans-pyrrolidines in moderate selectivity and both the 2,5-cis- and 2,5-trans-pyrrolidines in excellent diastereoselectivity. The efficiency of this approach was demonstrated with the formal synthesis of (+)-monomorine. More efforts to apply this method to the total synthesis of interesting biologically active nitrogen heterocycles and further investigations into substrate scope and catalytic methods are currently underway.
Supplementary Material
Procedures and characterization data and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgment
We thank the National Institutes of Health (NIGMS) for financial support of this work (R01 GM078383). We also gratefully acknowledge Dr. Shao-Liang Zheng, Dr. Mateusz Pitak and Dr. Stephan Scheins (SUNY, Buffalo X-ray crystallography facility) for obtaining crystal structures of compounds 2, 17, 20 and 31.
References
- 1.(a) Lewis JR. Nat. Prod. Rep. 2001;18:95–128. doi: 10.1039/a909077k. [DOI] [PubMed] [Google Scholar]; (b) O’Hagan D. Nat. Prod. Rep. 2000;17:435–446. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]
- 2.(a) Nash RJ, Fellows LE, Dring JV, Fleet GWJ, Derome AE, Hamor TA, Scofield AM, Watkin DJ. Tetrahedron Lett. 1988;29:2487–2490. [Google Scholar]; (b) Scofield AM, Rossiter JT, Witham P, Kite GC, Nash RJ, Fellows LE. Phytochemistry. 1990;29:107–109. [Google Scholar]
- 3.(a) Kato A, Kano E, Adachi I, Molyneux RJ, Watson AA, Nash RJ, Fleet GWJ, Wormald MR, Kizu H, Ikeda K, Asano N. Tetrahedron: Asymmetry. 2003;14:325–331. [Google Scholar]; (b) Molyneux RJ, Benson M, Wong RY, Tropea JE, Elbein AD. J. Nat. Prod. 1988;51:1198–1206. [Google Scholar]
- 4.(a) Kinzy TG, Harger JW, Carr-Schmid A, Kwon J, Shastry M, Justice M, Dinman JD. Virology. 2002;300:60–70. doi: 10.1006/viro.2002.1567. [DOI] [PubMed] [Google Scholar]; (b) Achenbach TV, Slater PE, Brummerhop H, Bach T, Müller R. Antimicrob. Agents Chemother. 2000;44:2794–2801. doi: 10.1128/aac.44.10.2794-2801.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Powell RG, Weisleder D, Smith CR, Jr, Rohwedder WK. Tetrahedron Lett. 1970;11:815–818. doi: 10.1016/s0040-4039(01)97839-6. [DOI] [PubMed] [Google Scholar]
- 6.Chang LC, Whittaker NF, Bewley CA. J. Nat. Prod. 2003;66:1490–1494. doi: 10.1021/np030256t. [DOI] [PubMed] [Google Scholar]
- 7.Warner GT, Perry CM. Drugs. 2002;62:1381–1405. doi: 10.2165/00003495-200262090-00016. [DOI] [PubMed] [Google Scholar]
- 8.For reviews, see Wolfe JP. Eur. J. Org. Chem. 2007:571–582. Bellina F, Rossi R. Tetrahedron. 2006;62:7213–7256. Coldham I, Hufton R. Chem. Rev. 2005;105:2765–2810. doi: 10.1021/cr040004c. Mitchinson A, Nadin A. J. Chem. Soc. Perkin Trans. I. 2000:2862–2891. Pichon M, Figadère B. Tetrahedron: Asymmetry. 1996;7:927–964.
- 9.For intramolecular osmium-catalyzed aminooxygenation reactions, see Donohoe TJ, Churchill GH, Wheelhouse KMP, Glossop PA. Angew. Chem. Int. Ed. 2006;45:8025–8028. doi: 10.1002/anie.200603240. Donohoe TJ, Chughtai MJ, Klauber DJ, Griffin D, Campbell AD. J. Am. Chem. 2006;128:2514–2515. doi: 10.1021/ja057389g. Donohoe TJ, Bataille CJR, Gattrell A, Kloeges J, Rossignol E. Org. Lett. 2007;9:1725–1728. doi: 10.1021/ol070430v.
- 10.For intramolecular Pd-catalyzed aminooxygenation reactions, see Alexanian EJ, Lee C, Sorensen EJ. J. Am. Chem. Soc. 2005;127:7690–7691. doi: 10.1021/ja051406k. Szolcsanyi P, Gracza T. Chem. Commun. 2005:3948–3950. doi: 10.1039/b506731f. Desai LV, Sanford MS. Angew. Chem. Int. Ed. 2007;46:5737–5740. doi: 10.1002/anie.200701454.
- 11.For other intramolecular aminooxygenation reactions, see Noack M, Gottlich R. Chem. Commun. 2002:536–537. doi: 10.1039/b111656h. Chikkana D, Han H. Synlett. 2004:2311–2314. Correa A, Tellitu I, Dominguez E, SanMartin R. J. Org. Chem. 2006;71:8316–8319. doi: 10.1021/jo061486q. Cochran BM, Michael FE. Org. Lett. 2008;10:5039–5042. doi: 10.1021/ol8022165. Mahoney JM, Smith CR, Johnson JN. J. Am. Chem. Soc. 2005;127:1354–1355. doi: 10.1021/ja045608c. Fuller PH, Kim JW, Chemler SR. J. Am. Chem. Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m. Sherman ES, Chemler SR. Adv. Synth. Catal. 2009;351:467–471. doi: 10.1002/adsc.200800705.
- 12.For intermolecular aminohydroxylation of alkenes, see O’Brien P. Angew. Chem. Int. Ed. 1999;38:326–329. doi: 10.1002/(SICI)1521-3773(19990201)38:3<326::AID-ANIE326>3.0.CO;2-T. Bolm C, Hildebrand JP, Muniz K. In: Catalytic Asymmetric Synthesis. 2nd ed. Ojima I, editor. Wiley-VCH; 2000. pp. 412–424. Schlingloff G, Sharpless BK. In: Asymmetric Oxidation Reactions. Katsuki T, editor. Oxford University Press; 2001. pp. 104–114. Nilov D, Rieser O. Adv. Synth. Catal. 2002;344:1169. Bodkin JK, McLeod MD. J. Chem. Soc., Perkin Trans. 1. 2002:2733. Michaelis DJ, Ischay MA, Yoon TP. J. Am. Chem. Soc. 2008;130:6610–6615. doi: 10.1021/ja800495r. Michaelis DJ, Shaffer CJ, Yoon TP. J. Am. Chem. Soc. 2007;129:1866–1867. doi: 10.1021/ja067894t. Liu G, Stahl SS. J. Am. Chem. Soc. 2006;128:7179–7181. doi: 10.1021/ja061706h. Michaelis DJ, Williamson KS, Yoon TP. Tetrahedron. 2009 doi: 10.1016/j.tet.2009.03.012.
- 13.(a) Sherman ES, Fuller PH, Kasi D, Chemler SR. J. Org. Chem. 2007;72:3896–3905. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vogler T, Studer A. Synthesis. 2008:1979–1993. [Google Scholar]
- 14.(a) Fuller PH, Chemler SR. Org. Lett. 2007;9:5477–5480. doi: 10.1021/ol702401w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Antilla JC, Buchwald SL. Org. Lett. 2001;3:2077–2079. doi: 10.1021/ol0160396. [DOI] [PubMed] [Google Scholar]; (c) Baran PS, Richter PS. J. Am. Chem. Soc. 2004;126:7450–7451. doi: 10.1021/ja047874w. [DOI] [PubMed] [Google Scholar]
- 15.CCDC 696031 (2), 711960 (17), 714177 (20) and 704323 (31) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 16.Kan T, Fukuyama T. Chem. Commun. 2004:353–359. doi: 10.1039/b311203a. [DOI] [PubMed] [Google Scholar]
- 17.Ritter FJ, Rotgans IEM, Tulman E, Verweil PE, Stein F. Experientia. 1973;29:530–531. [Google Scholar]
- 18. Toyooka N, Zhou D, Nemoto H. J. Org. Chem. 2008;73:4575–4577. doi: 10.1021/jo800593n. and references therein Lesma G, Colombo A, Sacchetti A, Silvani A. J. Org. Chem. 2009;74:590–596. doi: 10.1021/jo801638u.
- 19.(a) Riesinger SW, Löfstedt J, Petterson-Fasth H, Bäckvall J. Eur. J. Org. Chem. 1999:3277–3280. [Google Scholar]; (b) Petterson-Fasth H, Riesinger SW, Bäckvall J. J. Org. Chem. 1995;60:6091–6096. [Google Scholar]
- 20.Inokuchi T, Kawafuchi H. Tetrahedron. 2004;60:11969–11975. [Google Scholar]
- 21.Evans P, McCabe T, Morgan BS, Reau S. Org. Lett. 2005;7:43–46. doi: 10.1021/ol0480123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Procedures and characterization data and NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.