

Abstract
Despite the pressing need for an AIDS vaccine, the determinants of protective immunity to HIV remain concealed within the complexity of adaptive immune responses. We dissected immunodominant virus-specific CD8+ T cell populations in Mamu-A*01+ rhesus macaques with primary SIV infection to elucidate the hallmarks of effective immunity at the level of individual constituent clonotypes, which were identified according to the expression of distinct T cell receptors (TCRs). The number of public clonotypes, defined as those that expressed identical TCR β-chain amino acid sequences and recurred in multiple individuals, contained within the acute phase CD8+ T cell population specific for the biologically constrained Gag CM9 (CTPYDINQM; residues 181–189) epitope correlated negatively with the virus load set point. This independent molecular signature of protection was confirmed in a prospective vaccine trial, in which clonotype engagement was governed by the nature of the antigen rather than the context of exposure and public clonotype usage was associated with enhanced recognition of epitope variants. Thus, the pattern of antigen-specific clonotype recruitment within a protective CD8+ T cell population is a prognostic indicator of vaccine efficacy and biological outcome in an AIDS virus infection.
The global HIV pandemic demands an effective vaccine. However, while immunogenic vectors enter advanced clinical trials, the parameters on which to base measurements of efficacious immunity in a prospective manner remain unclear. Indeed, the recent Merck STEP trial failure has exposed our rudimentary understanding of protective determinants within the adaptive T cell response to HIV (1–3). It is established that specific CD8+ T cell immunity suppresses HIV replication in vivo and that certain patterns with respect to antigen targeting and MHC class I restriction are consistently associated with low levels of virus load (4–6). However, simple quantitative correlates, at least in peripheral blood, have proved elusive (7, 8). This paradox is exemplified by the SIV model, in which CD8+ T cell responses to the structurally constrained Gag CM9 epitope restricted by Mamu-A*01 are protective yet insufficient in terms of magnitude alone to define outcome (9, 10).
In the absence of consistent numerical correlates of immune control, recent observational studies have focused on functional profiling in attempts to identify the properties that demarcate effective HIV-specific CD8+ T cell responses (11–15). Indeed, a broad consensus indicates that polyfunctionality within pathogen-specific T cell populations, which is related to the sensitivity of antigen recognition among other parameters, correlates with improved outcome measures (3, 16). However, the qualitative properties of CD8+ T cell populations are clearly affected by viral replication, and the extent to which such functional associates reflect deterministic attributes remains uncertain. Similarly, phenotypic analyses of HIV-specific CD8+ T cell populations have yet to provide definitive indicators of immune control (3). At a more fundamental level, a given cognate T cell response is defined by the nature of its constituent clonotypes, which are defined on the basis of their expressed TCRs and can be considered the elemental units of any antigen-specific T cell population. Thus, the primary interface between the virus and adaptive T cell immunity occurs at the level of TCR-mediated recognition of peptide-MHC antigen; these signal transduction events, in turn, dictate the ontogeny and biological characteristics of individual cognate T cells in vivo. Given the seminal importance of clonotype-dependent TCR-mediated recognition events, it is not unreasonable to propose that the potential efficacy of a composite virus-specific CD8+ T cell population might depend on the idiosyncrasies with which individual cognate TCRs engage the targeted viral antigen.
In a previous study, we examined the clonotypic composition of immunodominant CD8+ T cell populations in acute SIV infection to illuminate the role of TCR usage in the process of mutational immune escape (17). In the present study, we conducted a detailed prospective study of vaccine-induced SIV-specific CD8+ T cell responses to the same immunodominant epitopes to establish whether the mobilized antigen-specific TCR repertoire can influence virologic outcome.
RESULTS
Disparate outcomes after vaccination and challenge despite induction of potent SIV-specific CD8+ T cell responses
Eight Mamu-A*01+/B*17− rhesus macaques were vaccinated with SIVmac239-derived immunogens using a DNA prime/adenovirus serotype 5 (Ad5) boost regimen and subsequently exposed to repeated intrarectal challenges with homologous virus until infection occurred (Fig. 1 A). Longitudinal CD4+ T cell trajectories and plasma virus load (pVL) dynamics are summarized for all participant macaques in Fig. 1 B. Consistent with a previous report that indicated a central role for vaccine-induced CD8+ T cell responses in the outcome of subsequent infection (9), the vaccinated macaques in this study developed lower peak and postprimary set point pVL levels and maintained memory CD4+ T cells to a greater extent compared with control nonvaccinated macaques infected under identical conditions (18). Furthermore, although potent SIV-specific CD8+ T cell responses were induced by vaccination in all cases, virus-specific humoral responses were not a confounding factor; no env gene-derived products were incorporated in the vaccine formulations, and neutralizing antibodies were detected after infection only in the vaccinee with the least favorable course (r97111; Fig. 1 B). However, no significant correlations were observed between the magnitude of the vaccine-induced CD8+ T cell response to SIV and the degree of virologic control after infection; this held for the classically immunodominant Mamu-A*01–restricted CD8+ T cell responses specific for the Tat SL8 (STPESANL; residues 28–35) and Gag CM9 (CTPYDINQM; residues 181–189) epitopes, as well as for subdominant responses induced to other viral products (18). Notably, these latter CD8+ T cell responses conferred no additional protection in the acute and early chronic phases of SIV infection when compared with Gag vaccination alone using the same platform (9, 18). Thus, this cohort encapsulates the conundrum that while the presence of a response is clearly beneficial, the precise attributes of the induced CD8+ T cell populations that confer protection remain obscure.
Figure 1.

Vaccination regimen and biological outcomes after challenge. (A) DNA plasmids encoding SIVmac239-derived genes were administered i.m. together with adjuvant at weeks 0, 4, and 8, followed at week 24 by Ad5 vectors encoding the genes described in Materials and methods. Repeated weekly intrarectal challenges with SIVmac239 were commenced at week 50 and continued until infection occurred, as determined by pVL measurements 7 d later. (B) Longitudinal pVL measurements (top) and peripheral blood CD4+ T cell counts (bottom) are shown for each macaque over a 500-d time period from infection; the symbol color code for each macaque is shown. (C) The frequency of CM9 and SL8 pMamu-A*01 tetramer+ CD8+ T cells is shown for each macaque at week 2 after vaccination with Ad5 and at week 4 after infection; the color code corresponds to that in B.
Vaccine- and infection-induced SIV epitope–specific CD8+ T cell populations exhibit similar clonotypic profiles
The observation that disparate outcomes were not dependent on simple quantitative measures of response magnitude alone prompted us to seek qualitative determinants of CD8+ T cell–mediated immune protection in this cohort. Given that downstream cellular effector functions originate from the initial interaction between TCR and cognate peptide–MHC class I antigen, we first examined the clonotypic architecture of immunodominant SIV-specific CD8+ T cell populations. In each macaque, the dominant response 2 wk after boosting with Ad5 vectors (the postvaccination [PV] time point) was directed against the CM9 epitope (Fig. 1 C); this is consistent with antigen targeting patterns observed in Mamu-A*01+ macaques during primary SIV infection but with substantial skewing away from the typically codominant SL8 epitope (19). After infection, the vaccinated macaques mounted massive anamnestic CD8+ T cell responses to the SL8 and CM9 epitopes that together comprised >50% of the total acute phase SIV-specific CD8+ T cell population (18). Cognate SL8- and CM9-specific CD8+ T cells were visualized directly ex vivo with fluorochrome-conjugated pMamu-A*01 tetrameric complexes and sorted viably by flow cytometry at both the PV time point and 4 wk after infection (the postinfection [PI] time point); an anchored template-switch RT-PCR that reproducibly generates an accurate hierarchical representation of TCRB gene expression was then used to ascertain the clonotypic composition of each sorted population (17). The amino acid sequences of all expressed TCRB CDR3s at both time points are shown for the SL8-specific clonotypes in Fig. S1 and for the CM9-specific clonotypes in Fig. 2. Strikingly, both vaccine-induced antigen-specific CD8+ T cell populations in all eight macaques exhibited clonotypic profiles similar to those observed in a previous study of acute SIV infection (17). In particular, the SL8-specific clonotypes expressed predominantly TCRBV14 and TCRBJ1.5 in association with a restricted CDR3 motif characterized by the presence of an arginine residue at position 6 (Fig. S1), whereas the CM9-specific clonotypes exhibited greater CDR3 diversity and a preference for TCRBV13 (Fig. 2). Furthermore, CD8+ T cell populations of both specificities elicited by vaccination exhibited the phenomenon of TCRB “sharing” between different macaques (Fig. S1 and Fig. 2); all common TCRB CDR3 sequences were encoded with distinct interindividual nucleotide profiles and restricted to the relevant antigen specificity across the entire dataset (Table S1 and Table I). Such public clonotypes (20) are defined in this study on the basis of CDR3 and TCRBJ identity because of minor 5′ regional variations between detected TCRBV13 sequences (Table S2) and were more prevalent in the motif-dominated SL8-specific CD8+ T cell populations, which were generally more polyclonal (median = 16.5 clonotypes; range = 10–38 clonotypes) than the corresponding CM9-specific populations (median = 8 clonotypes; range = 2–16 clonotypes). Overall, these characteristics are remarkably analogous to those observed in primary SIV infection (17), thereby indicating that the intrinsic nature of cognate antigen rather than the context of presentation is a primary determinant of clonotype recruitment in the periphery.
Figure 2.

Clonotypic architecture of CM9-specific CD8+ T cell populations. TCRB CDR3 amino acid sequences, TCRBV and TCRBJ usage, and the relative frequency of CD8+ T cell clonotypes specific for the CM9 epitope are shown for all eight macaques at week 2 after vaccination with Ad5 (left) and at week 4 after infection with SIVmac239 (right). The number of challenges required for infection to occur is indicated in each case underneath the individual macaque identification codes. Data are separated according to macaques with the lowest (A) and highest (B) set point pVL values. Colored boxes in the frequency column (percentages) indicate clonotypes within individual macaques that have the same TCRB CDR3 amino acid sequence at both time points shown; colored boxes in the CDR3 sequence column indicate public clonotypes. Public clonotypes were defined on the basis of TCRB amino acid sequences that were present in more than one macaque; TCRB sequences reported both in this study and in a previous study were included in the analysis (reference 17). The corresponding TCRB CDR3 nucleotide sequences are shown in Table I.
Table I.
CM9-specific public clonotype TCRB CDR3 nucleotide alignments
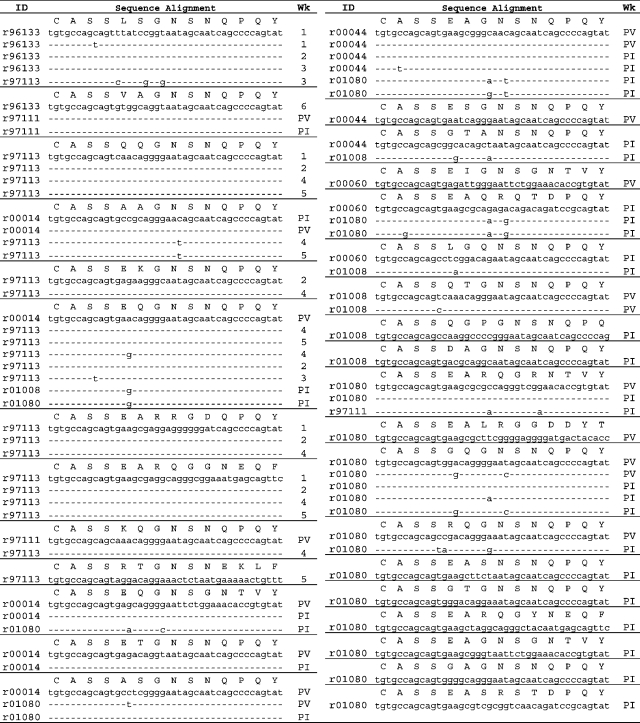 |
CDR3 codon usage of CM9-specific CD8+ T cell public clonotypes. Each section displays a distinct TCRB CDR3 amino acid sequence, and the nucleotide alignments for all corresponding clonotypes in each macaque. Public clonotypes were defined on the basis of TCRB amino acid sequences that were present in more than one macaque; previously reported TCRB sequences were included in this analysis (reference 17). 1–6: time points from PV for macaques r97113 and r96133 (1 is PV, and 2–6 are subsequent blood draws as shown in Fig. 3).
Vaccine-induced SIV epitope–specific CD8+ T cell clonotypes are maintained and expanded during acute SIV infection
The CD8+ T cell responses mounted after SIVmac239 infection were substantially greater in magnitude and mobilized with faster kinetics compared with the corresponding epitope-specific responses in nonvaccinated controls (18). Within these anamnestic responses, which were generally expanded compared with those induced by vaccination (Fig. 1 C), a notable degree of clonotypic preservation was apparent for both SL8-specific (Fig. S1) and CM9-specific (Fig. 2) CD8+ T cell populations. Indeed, conservation of clonotype usage was apparent in longitudinal follow-up studies of macaques r96133 and r97113 beyond week 4 after infection (Fig. 3 and not depicted). This is consistent with a previous study in humans, in which vaccine-induced clonotypes specific for the HLA B*2705–restricted HIV-1 p24 Gag KK10 epitope (KRWIILGLNK; residues 263–272) were well represented after infection (21). However, there was also a high degree of clonotype turnover between the PV and PI time points, with evident recruitment of new clonotypes and loss of vaccine-primed clonotypes in both the SL8- and CM9-specific CD8+ T cell populations (Fig. S1 and Fig. 2); similar shifts in clonotypic composition were observed in a recent study of vaccinated pigtail macaques (22) and tend to characterize the natural evolution of immunodominant CD8+ T cell responses in both HIV and SIV infection (12, 17). Overall, the immunodominant primary SIV-specific CD8+ T cell populations characterized in the present cohort exhibited significantly less diversity at the clonotypic level than the corresponding repertoires in nonvaccinated macaques at a similar time point after infection (P = 0.03 for SL8/TL8, and P = 0.008 for CM9 using the Mann-Whitney U test comparing Simpson's diversity indices with standardization for clone sample size) (23). Although we cannot unequivocally attribute repertoire narrowing to antigen preexposure during immunization, it is salutary to anticipate that such reductions in clonotypic diversity might adversely complicate vaccination by facilitating viral escape (17, 24).
Figure 3.

Longitudinal analysis of clonotypic structure in macaques r97113 and r96133. (A, top) pVL (SIV RNA copies/ml) and CD4+ T cell count (cells/µl) in macaque r97113 at the indicated time points; (bottom) number and relative frequency of individual T cell clonotypes, with monoclonal populations defining the narrowest clonotypic structure, specific for CM9 (red bars) and SL8 (blue bars). (B) The usage of public (dark shading) and private (light shading) clonotypes in the CD8+ T cell populations specific for CM9 (red; left) and SL8 (blue; right) in macaque r97113 at the indicated time points; the number of public clonotypes over total clonotypes is shown at each time point. (C) Experiments performed as in A for macaque r96133. (D) Experiments performed as in B for macaque r96133.
Public clonotype usage in primary Gag CM9–specific CD8+ T cell populations predicts the outcome of SIV infection
Evidence from vaccination studies indicates a protective role for the Mamu-A*01–restricted CM9-specific CD8+ T cell response (9); this is consistent with experiments demonstrating that strict biological constraints operate in this region of the virus and limit the potential for escape through antigenic mutation (25, 26). We therefore hypothesized that the clonotypic constitution of CM9-specific CD8+ T cell populations could determine virologic outcome in macaques infected with SIV. Initially, we explored this possibility retrospectively using data reported in a previous study of 12 rhesus macaques acutely infected with SIVmac251 (17). Among several candidate measures of clonal composition at week 5 after infection, the best predictor of postprimary set point pVL proved to be the number of public clonotypes within the CM9-specific CD8+ T cell population (P = 0.0006 and r2 = 0.71; Fig. 4 A and not depicted); this parameter is solely a measure of interindividual clonotype sharing within the antigen-specific repertoire and is distinct from both clonal diversity and TCR sequence. Multivariate linear regressions were also considered using stepwise model building to test for the influence of other potentially predictive parameters; such variables included absolute CD4+ and CD8+ T cell counts, mean log10 pVL at time points before week 6, and additional measures of clonality such as the total number of clonotypes and the proportion of the response that comprised public clonotypes. None of these covariates lead to a model that was significantly more predictive of postprimary set point pVL (i.e., mean log10 pVL from week 6 to 16) than the simple model based on the number of public clonotypes (unpublished data). Similarly, the inclusion of these other variables did not substantially change estimates of the relationship between the number of public clonotypes and postprimary set point pVL. Furthermore, the results were unaffected by small changes in parameterization of the response variable, such as modeling the mean log10 pVL or using the area under the curve from week 10 to 16, or by altering the definition of public clonotype at the amino acid level as follows: (a) matching TCRBV, TCRB CDR3, and TCRBJ; (b) matching TCRB CDR3 and TCRBJ; and (c) matching TCRB CDR3 alone. Subsequently, we tested the hypothesis generated from this analysis using the dataset described in this study; indeed, the rationale for the current study was based on the initial finding, and this second stage was conducted prospectively with investigator blinding. There was a significant inverse relationship between the number of public clonotypes at week 2 after vaccination and the mean log10 pVL from week 6 to 16 (Pearson: r = −0.81 and P = 0.014; Spearman: r = −0.82 and P = 0.015). The linear regression parameter (slope = −0.55) indicates that the estimated effect of each public clonotype was a mean reduction in postprimary pVL of 0.55 log10 RNA copies/ml during this period (Fig. 4 B). Notably, no such correlation with outcome was observed for clonal diversity per se (Pearson: P = 0.11) despite a strong association between this parameter and the number of public clonotypes (Pearson: r = 0.85 and P = 0.007; diversity of clonotypes with standardization for clone sample size) (23). There was no significant relationship between the number of public clonotypes at week 4 after infection and mean log10 pVL from week 6 to 16 (Pearson: P = 0.92). As described, no other measures of clonality were significant predictors of postprimary set point pVL, either alone or as covariates (unpublished data); similarly, we could find no clonotypic correlates of either central memory CD4+ T cell preservation (18, 27, 28) or the number of challenges required for infection to occur. Finally, to validate the association further, the original cohort was revisited with public clonotypes assigned according to the same definition but compiled using the entire dataset from all 20 macaques. This expanded range for the number of public clonotypes exerted only a minimal influence on a simple linear regression performed against mean log10 pVL from week 6 to 16 (P = 0.007 and r2 = 0.54). In all analyses, results using the computed area under the pVL trajectory curve from week 6 to 16 were consistent with those derived using the mean pVL for the same period. No significant correlations were observed for parallel analyses of SL8/TL8-specific CD8+ T cell clonotypes in either dataset (unpublished data); these negative findings act as an important control and are not unexpected given the propensity with which the infecting virus mutates at this site with minimal biological constraints to evade immune recognition and curtail the efficacy of the cognate response (17, 18, 29–31). Thus, although SL8/TL8-specific CD8+ T cell populations are typically polyclonal in acute SIV infection and exhibit a substantial degree of TCR sharing (Fig. S1 and Fig. 3), they are neither protective nor clonotypically diverse (17, 30). Instead, the monomorphic nature of SL8/TL8-specific TCRs, which presumably require a TCRB CDR3 motif to engage a relatively featureless antigen regardless of interindividual sharing (32), predisposes to viral escape at the level of TCR recognition (17); this contrasts with the diverse range of clonotypes that can be mobilized in response to the CM9 epitope, and in the face of such considerations, the clonotypic composition of the SL8/TL8-specific response with respect to the degree of TCR sharing becomes largely irrelevant. Public clonotype usage can therefore be associated with either favorable or unfavorable biological outcomes according to the nature of the targeted epitope and the characteristics of the available cognate repertoire. Consequently, it is important to emphasize that the predictive value of “publicity” within epitope-specific CD8+ T cell responses is context dependent.
Figure 4.

The number of public clonotypes in primary Gag CM9–specific CD8+ T cell populations predicts virologic outcome in SIV infection. Simple linear regressions predicting mean log10 pVL from week 6 to 16 after infection by the number of public clonotypes present in the CM9-specific CD8+ T cell population at week 5 after infection without prior vaccination in a previously reported cohort of 12 macaques (A; reference 17) and at week 2 after vaccination in the cohort of macaques described in this study (B). In A, the relationship remained highly significant with minimal change in the coefficient when the most influential data point was discarded from the analysis; the analysis presented in B was performed to confirm the relationship observed in A.
Public clonotype usage does not skew the phenotypic properties or functional capabilities of composite antigen-specific CD8+ T cell populations
To investigate the mechanistic basis for the observed clonotypic association with virologic outcome, we attempted to discern the biological properties conferred by public TCR usage that distinguish such T cells from those with identical antigen specificity derived from the private repertoire. Initially, we reasoned that differential TCR/pMamu-A*01 engagement might trigger subtly different cellular outputs in the composite antigen-specific T cell populations. Indeed, recent studies linking the functional attributes of HIV-specific CD8+ T cells to pVL in infected individuals provide a potential precedent for such a link (3, 11, 12, 14). We therefore assessed the functional output of CM9-specific CD8+ T cell populations in macaques r96133 and r97113 at the prechallenge time point, thereby excluding any confounding impact of replicating SIV on these cells. These macaques were selected on the basis that they epitomize the association expounded in the present study; specifically, despite similar levels of clonal diversity, r97113 had a favorable outcome after infection and a CD8+ T cell response to CM9 that was predominantly public, whereas the opposite applied to r96133 (Figs. 2 and 3). In each case, both IFN-γ and TNF-α production were triggered in response to antigen with almost identical peptide sensitivities for the former effector function (Fig. 5, A–C). Screening for additional soluble factors released on antigen encounter yielded similarly uniform results overall, although PBMCs from r96133 failed to produce significant levels of MIP-1α and were comparatively meager with respect to lymphotoxin A secretion (Fig. 5 B). Furthermore, these vaccine-induced CD8+ T cell populations exhibited indistinguishable effector memory (CD28−CD45RA+CD95+) phenotypic profiles (Fig. 5 D); interestingly, this suggests that the impaired maturational profile characteristic of HIV-specific CD8+ T cells in infected individuals arises as a consequence of virus exposure (33, 34). Thus, consistent with the findings of a recent mouse study (35), no major disparities were apparent in the interactions of these constitutively distinct CM9-specific CD8+ T cell populations with cognate wild-type antigen. Functional and phenotypic homogeneity irrespective of clonotypic composition was also observed in multiparametric analyses of CM9-specific CD8+ T cell populations at the PI time point (n = 6; Fig. S2 and not depicted). Collectively, these data suggest that such approaches are insufficient in isolation to monitor the efficacy of T cell responses with respect to biological outcome.
Figure 5.

Functionality, antigen sensitivity, and immunophenotype of CM9-specific CD8+ T cell populations. Data from the prechallenge time point are shown for macaques r96133 (A, C, and D, left; B, red) and r97113 (A, C, and D, right; B, green). (A) Intracellular production of IFN-γ and TNF-α after direct ex vivo stimulation of PBMCs with CM9 peptide at a concentration of 2 µg/ml. Plots are gated on CD3+CD8+ cells. Corresponding background values in the absence of cognate peptide, expressed with respect to the total CD8+ memory T cell pool, were 0.05 and 0.02% for IFN-γ, 0.46 and 0.22% for TNF-α, and 0.15 and 0% for IFN-γ/TNF-α, respectively, for macaques r96133 and r97113. (B) PBMCs were stimulated with CM9 peptide at a concentration of 2 µg/ml and assayed for production of the indicated soluble factors by cytokine bead array. Results are shown with background subtracted. (C) PBMCs were stimulated with CM9 peptide at the concentrations indicated in an IFN-γ ELISPOT assay. Results are shown with background subtracted. (D) Phenotypic profiles, as defined by expression of CD28 and CD45RA, of CM9 pMamu-A*01 tetramer+ CD8+ T cells. Plots are gated on CD3+CD8+CD95+ cells; the CM9 pMamu-A*01 tetramer+ events are shown as red dots superimposed on a density plot showing the phenotype of all gated cells. Similar phenotypic profiles were observed in the functionally responsive cells identified in A (not depicted). MFI, mean fluorescence intensity; SFC, spot-forming cell.
Antigen-specific CD8+ T cell populations comprising multiple public clonotypes exhibit promiscuous variant antigen recognition properties
Given the absence of obvious functional or phenotypic differentiators between public and private CM9-specific responses with respect to wild-type antigen, we reasoned that differential recognition properties might be apparent in response to variant antigens. Cross-reactivity is an intrinsic property of the T cell recognition system (36) and has potentially important biological consequences (37, 38). Faced with a persistent, genetically unstable pathogen, antigen-specific CD8+ T cell populations with enhanced variant recognition capabilities might enable more efficacious immune control (39); such cross-reactivity could be achieved either by clonotypic diversity (17, 40–42) or by the incorporation of highly promiscuous clonotypes within the composite population (43–45). We tested this possibility in macaques r96133 and r97113, selected on the basis of a robust and consistent distinction in terms of public clonotype usage (Figs. 2 and 3), using IFN-γ ELISPOT analysis of PBMCs from the prechallenge time point (Fig. 6); importantly, the discrete but related parameter of activation threshold per se was not a confounder in these experiments because recognition sensitivities for wild-type antigen were almost identical (Fig. 5 C). All possible amino acid substitutions were tested at positions 1, 7, and 8 in the CM9 peptide; these are potential TCR contact residues that have previously been shown by alanine scanning experiments to be the only intraepitopic sites tolerant of variability within the confines of viral fitness, although replication competence is affected to a substantial extent in the case of P7 mutations (26). Notably, a greater degree of recognition flexibility was apparent in the public clonotype–dominated CM9-specific CD8+ T cell population from macaque r97113 (Fig. 6). Similar results were observed in additional macaques with diametrically opposed patterns of CM9-specific clonotype usage (Fig. S3 and not depicted); interestingly, some public clonotype–rich CM9-specific CD8+ T cell populations exhibited enhanced recognition of variants relative to wild-type antigen. The extent to which individual variants were recognized by public versus private clonotypes could not be assessed in these experiments due to limiting cell numbers. However, CD8+ T cells specific for the CM9 variants L1 and M8 sorted from macaque r97113 with pMamu-A*01 tetrameric complexes refolded around the relevant peptides demonstrated almost identical clonotypic compositions to the corresponding wild-type CM9-specific population; these data exclude the possibility that the observed cross-reactivity was mediated by clonotypically distinct CD8+ T cell populations (unpublished data). Thus, public clonotype usage is associated with enhanced cross-reactivity to potentially relevant antigenic variants.
Figure 6.

Sensitivity of CM9-specific CD8+ T cell populations to epitope variants. PBMCs from macaques r97113 (top) and r96133 (bottom) at the prechallenge time point were stimulated in IFN-γ ELISPOT assays with CM9 wild-type peptide or individual peptides containing each possible amino acid substitution at positions P1, P7, and P8; mutations at these positions can be incorporated either with minimal impact on viral fitness (P1 and P8) or without disabling the virus entirely (P7; reference 26). Each peptide was assayed at a concentration of 10−7 M, selected on the basis of the titrations shown in Fig. 5 C; broader screening was precluded by limiting cell numbers. The CM9 sequence is shown at the top and the substituted amino acid in each position is shown on the left of each panel. Data are color coded to indicate the size of the response to the variant epitope as a percentage of the response to the CM9 wild-type peptide.
DISCUSSION
In the present study, we conducted a detailed analysis of antigen-specific CD8+ T cell clonotype usage in relation to virologic outcome in acute SIV infection. A strong immunological correlate of protection was identified, in which set point pVL was negatively associated with the number of public clonotypes present within the initially mobilized Gag CM9–specific CD8+ T cell population. Subsequently, we tested this association prospectively in a vaccine trial. Again, the number of public clonotypes in the vaccine-induced CM9-specific CD8+ T cell population correlated inversely with virologic outcome after SIV challenge. Compared with private clonotype–dominated CM9-specific CD8+ T cell responses, populations with substantial public clonotype representation exhibited greater levels of cross-reactivity to epitope variants at virologically permissive residues despite comparable activation thresholds and equivalent clonal diversity; these data establish the testable hypothesis that public clonotypes are intrinsically more promiscuous than private clonotypes. In contrast, phenotypic profiles and functional properties elicited in response to wild-type antigen were largely similar regardless of clonotypic architecture, although it remains possible that important differences might emerge from more extensive analyses. Thus, the nature of the initial interactions between virus and emergent antigen-specific CD8+ T cell clonotypes is an independent predictor of outcome in SIV infection.
Mechanistically, the observed association between public clonotype usage and enhanced cross-reactivity profiles is somewhat at odds with the finding that the contingent protective effect is apparent immediately after infection. The CM9 epitope is generally stable throughout the early periods of infection, with escape mutants appearing late and variably; indeed, no CM9 mutations were detected within the course of this study (macaques r00060 and r01008; unpublished data). How can we explain this apparent discrepancy? One possibility is that emerging escape mutations below the level of detection or at anatomically disparate sites are quelled more effectively by public clonotypes and that these imperceptible events favorably influence other facets of host immunity and, hence, the subsequent outcome of infection. Alternatively, public clonotype usage might be linked to a distinct determinant of virologic control, with enhanced variant recognition simply representing an epiphenomenon. For example, the kinetics with which cognate T cells are mobilized from the naive pool in response to incoming virus could dramatically modify the massive pathological damage that can occur during acute SIV infection (46), and thereby associate better downstream virologic outcomes with more pronounced public clonotype content if the latter are preferentially recruited into earlier and more robust CM9-specific CD8+ T cell populations. Mechanistically, such a selective priming bias could occur as a function of precursor frequency, and it does appear that public clonotypes are generated more often as a consequence of convergent recombination (20, 47–49). Indeed, it is tempting to speculate that this process establishes an “immunological homunculus” (50) within the naive T cell pool comprising highly prevalent public T cell clonotypes with a proclivity for cross-reactivity that can expand rapidly in response to a substantial number of antigenic assaults and thereby provide an early protection system against many pathogens. This scenario is consistent with several observations. First, it could explain the classical early immunodominance of the SL8/TL8 epitope in Mamu-A*01+ macaques during acute SIV infection (19); SL8/TL8-specific CD8+ T cell populations are dominated by public clonotypes bearing a TCRB CDR3 codon-degenerate motif (17), and a high presursor frequency of such TCRs generated through convergent recombination could fuel primary responses to this antigen. Indeed, the SL8-specific response is markedly less dominant after infection in the presence of vaccine-induced CD8+ T cell populations specific for other epitopes (Fig. 1) (18), further supporting the notion that TCR repertoire skewing underlies this pattern of immunodominance. Second, the associations between public clonotype usage and virologic outcome were only apparent at the initial point of antigen contact (the PV time point in the vaccination study and week 5 after infection in the unmanipulated acute SIV cohort); although the publicity of individual responses was maintained to some extent throughout the period of study (Fig. 3), these correlations were lost at later time points (the PI time point and week 12 after infection, respectively). Thus, consistent with an effect mediated by the predominance of cognate public clonotypes within the naive T cell pool, it seems that the nature of the primary interactions is determinant and that the initially relevant clonal topography is obscured with subsequent adaptation to persistent antigen (51). Third, recruitment of all cognate clonotypes, both public and private, during the vaccination process is likely to mask any advantage conferred by high precursor frequencies in the naive T cell pool; these considerations could explain the lack of association between public clonotype usage and virologic outcome at the PI time point, and the weaker correlation at the PV time point with respect to the acute infection cohort. Overall, these considerations invoke subtle differences in the nature of the primary CD8+ T cell response as profound determinants of outcome (52). However, ongoing recruitment of antigen-specific CD8+ T cells from a munificent naive pool may continue to sustain a more effective response throughout the course of infection (53); in this regard, the correlation between clonotypic diversity and the number of public clonotypes is notable despite the fact that the latter parameter in isolation did not achieve significance with respect to measures of disease progression in the present study.
In summary, these data provide compelling evidence that the fundamental nature of the initial encounter between a constrained viral antigen and the adaptive cellular immune system dictates subsequent biological outcome. The potential for vaccine-based initiatives to alter such interactions with beneficial effect will likely depend on the balance between the extent to which differential public clonotype usage is predetermined by the composition of naive pool (54), or perhaps even the heterologous memory pool (37), versus stochastic elements in the initial antigen-specific selection process. Further studies are necessary to clarify the associations described in this paper, especially with respect to mechanistic determinants at the clonal level and quantitative relationships at the sites of active viral replication. However, the observation that clonotypic parameters can predict virologic outcome provides ample reason to consider such effects in the design and monitoring of vaccine efficacy trials.
MATERIALS AND METHODS
Vaccination and infection procedures.
All animals in this study were colony-bred Mamu-A*01+ Indian rhesus macaques (Macaca mulatta). Vaccination studies were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin and were conducted in full accordance with established regulations and guidelines using a DNA prime/adenovirus boost platform, as previously described (55). In brief, 5 mg of a VIR vector DNA construct containing codon-optimized SIVmac239 gag, tat, rev, or mutated nef genes was added to 7.5 mg CRL1005 (CytRx) mixed with benzalkonium chloride (Ruger). Eight Mamu-B*17− macaques were vaccinated intramuscularly (i.m.) with 0.5 ml of each DNA preparation at weeks 0, 4, and 8. At week 24, recombinant E1-deleted replication-incompetent Ad5 vectors containing the same individual SIVmac239 genes were injected i.m. at a dose of 1011 recombinant viral particles. All vaccines were administered at four separate sites, one for each SIVmac239 gene insert (18). Intrarectal challenges with SIVmac239 (300 TCID50) were started at week 50 (56); infection occurred after two to seven weekly challenges (18). Infection studies with SIVmac251 were previously described (17). The Mamu-A*01–restricted Tat epitope (residues 28–35) differs at position 1 between SIVmac239 (SL8) and SIVmac251 (TL8); the corresponding Gag epitope (residues 181–189) is identical in both strains (CM9).
Virus load quantification.
pVL was determined for all SIVmac239 studies using previously published protocols (57, 58) with minor modifications (18).
IFN-γ ELISPOT assays.
Direct ex vivo production of IFN-γ in response to stimulation of PBMCs with the concentrations of wild-type CM9 peptide or variants thereof indicated in the figures was assessed in ELISPOT assays, as previously described (59, 60). All conditions were tested in duplicate. Spot imaging and quantification were automated with an ImmunoSpot Series 3B Analyzer (CTL Analyzers LLC). Background counts in the absence of exogenous peptide were subtracted from experimental counts; for comparative analyses, results were calculated as the percentage of spots induced by variant peptide with respect to wild type.
Cytokine bead arrays.
Direct ex vivo production of multiple soluble factors in response to stimulation with wild-type CM9 peptide at a concentration of 2 µg/ml was assayed using the Cytokine Bead Array Flex Set (BD) containing mAbs and detection reagents specific for 25 different cytokines, each of which was shown in preliminary experiments to cross react with rhesus macaque cytokines. Unstimulated samples were used in parallel to determine background levels of cytokine secretion resulting from in vivo activation; these values were subtracted from the data shown in Fig. 5 B.
Flow cytometry and cell sorting.
Soluble fluorochrome-labeled pMamu-A*01 tetramers were produced and used as previously described (17, 61). For functional analyses of CD8+ T cell populations, 2 × 106 PBMCs were stimulated with cognate peptide at a final concentration of 2 µg/ml in R10 medium (RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum, 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 mM l-glutamine) for 6 h in the presence of 5 µg/ml anti-CD28–Alexa Fluor 680 mAb, 10 µg/ml anti-CD49d mAb, and 10 µg/ml brefeldin A; for each experimental condition, parallel negative controls without cognate peptide were included. Subsequent staining procedures to exclude aberrant binding events, identify surface markers, and detect intracellular cytokine production or granzyme B content were conducted as previously described with minor modifications (62). For phenotypic analyses of pMamu-A*01–allophycocyanin (APC) tetramer–labeled cells, the following directly conjugated mAbs were used: anti-CD3–Cy7APC, anti-CD8–Pacific blue, anti-CD28–FITC, anti-CD45RA–Texas red–PE, and anti-CD95–Cy5PE. At least 106 events were acquired on an LSR II flow cytometer (BD) equipped for the detection of 17 fluorescent parameters, and data were analyzed with FlowJo software (Tree Star, Inc.). In all cases, electronic compensation was performed with antibody-capture beads (BD) stained separately with individual mAbs present in the experimental samples. Flow cytometric cell sorting was conducted with a modified FACSAria (BD) at 70 PSI; postsort purity was consistently >98%.
Clonotype analysis.
Tetramer-labeled CD8+ T cells were sorted viably into 1.5-ml microtubes containing 100 µl RNAlater (Applied Biosystems); for the CM9-specific populations in this study, the median number of sorted cells was 5,000 (range = 740–10,000). All expressed TCRB gene products were amplified without bias using an anchored template-switch RT-PCR (17, 41); amplicons were subcloned, sampled, sequenced, and analyzed as previously described (17, 63). For consistency with previous studies, Arden's nomenclature is retained throughout (64); macaque TCRBV and TCRBJ sequences were assigned according to the closest human equivalent (17).
Statistics.
The presented analysis comprised hypothesis generation and confirmation as two distinct steps. First, the relationship between several candidate measures of SIV antigen-specific CD8+ T cell repertoires at week 5 after infection and mean postprimary pVL from week 6 to 16 was explored using simple linear regressions in an analysis restricted to data derived from a previous study of SIVmac251 infection (17). Due to heteroskedasticity, a logarithmic scale was used for pVL data. Variables considered included the total number of unique clonotypes (diversity), the total number of public clonotypes, and the percentage of captured clonotypes that were public; these parameters were assessed both as absolute values and in relation to response magnitude. The number of public clonotypes at week 5 after infection emerged as the strongest predictor of mean pVL during the later interval. Further multivariate linear regression models were considered to examine the independence of this predictive variable, to assess whether predictions could be significantly improved with combinations of variables, and to determine the impact of additional variables on the observed relationships. Second, an independent dataset was collected from the cohort of eight macaques as described in this study; this information was used to test the hypothesis generated in the first step. Simple linear regressions were used as the primary models to relate the number of public clonotypes, identified both within this cohort and in relation to the first cohort, at both week 2 after vaccination and week 4 after infection to mean log10 pVL from week 6 to 16 after infection. As the time points at which pVL was measured were not quite evenly spaced, we also computed the area under the curve from week 6 to 16 and repeated the linear regressions with this variable. For thoroughness, the alternative measures described previously were also examined in the second cohort. In a final step, we returned to the first cohort of macaques with the expanded compilation of public clonotypes based on the incorporation of data from the second cohort to confirm that the observed relationships were maintained.
Online supplemental material.
Fig. S1 shows the clonotypic architecture of SL8-specific CD8+ T cell populations at week 2 after vaccination with Ad5 and at week 4 after infection with SIVmac239; the corresponding nucleotide sequences are displayed in Table S1. Fig. S2 presents a functional analysis of CM9-specific CD8+ T cell populations from six macaques in the acute phase after infection; based on the simultaneous and independent measurement of five separate effector functions directly ex vivo in a flow cytometric panel comprising 13 distinct parameters, no significant differences were observed in the functional profile of CM9-specific CD8+ T cells irrespective of virologic outcome. Fig. S3 shows the antigen sensitivity and cross-reactivity profiles of CM9-specific CD8+ T cell populations from four macaques at week 2 after vaccination with Ad5; consistent with the data shown in Fig. 6, CM9-specific CD8+ T cell populations that were enriched for public clonotypes exhibited greater levels of cross-reactivity in response to both natural and selected monosubstituted peptide epitope variants. Table S2 displays the 3′ sequences of expressed TCRBV13 family genes. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20081127/DC1.
Acknowledgments
This work was supported by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health, and by the Australian Research Council. D.A. Price is a Medical Research Council (UK) Senior Clinical Fellow. M.P. Davenport is a Sylvia and Charles Viertel Senior Medical Research Fellow.
The authors have no conflicting financial interests.
Footnotes
Abbreviations used: Ad5, adenovirus serotype 5; i.m., intramuscularly; PI, postinfection; PV, postvaccination; pVL, plasma virus load.
References
- Sekaly R.P. 2008. The failed HIV Merck vaccine study: a step back or a launching point for future vaccine development? J. Exp. Med. 205:7–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins D.I., Burton D.R., Kallas E.G., Moore J.P., Koff W.C. 2008. Nonhuman primate models and the failure of the Merck HIV-1 vaccine in humans.Nat. Med. 14:617–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appay V., Douek D.C., Price D.A. 2008. CD8+ T cell efficacy in vaccination and disease.Nat. Med. 14:623–628 [DOI] [PubMed] [Google Scholar]
- Sewell A.K., Price D.A., Oxenius A., Kelleher A.D., Phillips R.E. 2000. Cytotoxic T lymphocyte responses to human immunodeficiency virus: control and escape.Stem Cells. 18:230–244 [DOI] [PubMed] [Google Scholar]
- Scherer A., Frater J., Oxenius A., Agudelo J., Price D.A., Gunthard H.F., Barnardo M., Perrin L., Hirschel B., Phillips R.E., McLean A.R. 2004. Quantifiable cytotoxic T lymphocyte responses and HLA-related risk of progression to AIDS.Proc. Natl. Acad. Sci. USA. 101:12266–12270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulder P.J., Watkins D.I. 2008. Impact of MHC class I diversity on immune control of immunodeficiency virus replication.Nat. Rev. Immunol. 8:619–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts M.R., Ambrozak D.R., Douek D.C., Bonhoeffer S., Brenchley J.M., Casazza J.P., Koup R.A., Picker L.J. 2001. Analysis of total human immunodeficiency virus (HIV)-specific CD4(+) and CD8(+) T-cell responses: relationship to viral load in untreated HIV infection.J. Virol. 75:11983–11991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addo M.M., Yu X.G., Rathod A., Cohen D., Eldridge R.L., Strick D., Johnston M.N., Corcoran C., Wurcel A.G., Fitzpatrick C.A., et al. 2003. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load.J. Virol. 77:2081–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimiro D.R., Wang F., Schleif W.A., Liang X., Zhang Z.Q., Tobery T.W., Davies M.E., McDermott A.B., O'Connor D.H., Fridman A., et al. 2005. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with DNA and recombinant adenoviral vaccine vectors expressing Gag.J. Virol. 79:15547–15555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniuszko M., Bogdan D., Pal R., Venzon D., Stevceva L., Nacsa J., Tryniszewska E., Edghill-Smith Y., Wolinsky S.M., Franchini G. 2005. Correlation between viral RNA levels but not immune responses in plasma and tissues of macaques with long-standing SIVmac251 infection.Virology. 333:159–168 [DOI] [PubMed] [Google Scholar]
- Betts M.R., Nason M.C., West S.M., De Rosa S.C., Migueles S.A., Abraham J., Lederman M.M., Benito J.M., Goepfert P.A., Connors M., et al. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells.Blood. 107:4781–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida J.R., Price D.A., Papagno L., Arkoub Z.A., Sauce D., Bornstein E., Asher T.E., Samri A., Schnuriger A., Theodorou I., et al. 2007. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover.J. Exp. Med. 204:2473–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichterfeld M., Yu X.G., Mui S.K., Williams K.L., Trocha A., Brockman M.A., Allgaier R.L., Waring M.T., Koibuchi T., Johnston M.N., et al. 2007. Selective depletion of high-avidity human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T cells after early HIV-1 infection.J. Virol. 81:4199–4214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daucher M., Price D.A., Brenchley J.M., Lamoreaux L., Metcalf J.A., Rehm C., Nies-Kraske E., Urban E., Yoder C., Rock D., et al. 2008. Virological outcome after structured interruption of antiretroviral therapy for human immunodeficiency virus infection is associated with the functional profile of virus-specific CD8+ T cells.J. Virol. 82:4102–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehr M., Cahenzli J., Haas A., Price D.A., Gostick E., Huber M., Karrer U., Oxenius A. 2008. Emergence of polyfunctional CD8+ T cells after prolonged suppression of human immunodeficiency virus replication by antiretroviral therapy.J. Virol. 82:3391–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seder R.A., Darrah P.A., Roederer M. 2008. T-cell quality in memory and protection: implications for vaccine design.Nat. Rev. Immunol. 8:247–258 [DOI] [PubMed] [Google Scholar]
- Price D.A., West S.M., Betts M.R., Ruff L.E., Brenchley J.M., Ambrozak D.R., Edghill-Smith Y., Kuroda M.J., Bogdan D., Kunstman K., et al. 2004. T cell receptor recognition motifs govern immune escape patterns in acute SIV infection.Immunity. 21:793–803 [DOI] [PubMed] [Google Scholar]
- Wilson N.A., Reed J., Napoe G.S., Piaskowski S., Szymanski A., Furlott J., Gonzalez E.J., Yant L.J., Maness N.J., May G.E., et al. 2006. Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239.J. Virol. 80:5875–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothe B.R., Horton H., Carter D.K., Allen T.M., Liebl M.E., Skinner P., Vogel T.U., Fuenger S., Vielhuber K., Rehrauer W., et al. 2002. Dominance of CD8 responses specific for epitopes bound by a single major histocompatibility complex class I molecule during the acute phase of viral infection.J. Virol. 76:875–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi V., Price D.A., Douek D.C., Davenport M.P. 2008. The molecular basis for public T-cell responses? Nat. Rev. Immunol. 8:231–238 [DOI] [PubMed] [Google Scholar]
- Betts M.R., Exley B., Price D.A., Bansal A., Camacho Z.T., Teaberry V., West S.M., Ambrozak D.R., Tomaras G., Roederer M., et al. 2005. Characterization of functional and phenotypic changes in anti-Gag vaccine-induced T cell responses and their role in protection after HIV-1 infection.Proc. Natl. Acad. Sci. USA. 102:4512–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.Z., Asher T.E., Venturi V., Davenport M.P., Douek D.C., Price D.A., Kent S.J. 2008. Limited maintenance of vaccine-induced simian immunodeficiency virus-specific CD8 T-cell receptor clonotypes after virus challenge.J. Virol. 82:7357–7368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi V., Kedzierska K., Turner S.J., Doherty P.C., Davenport M.P. 2007. Methods for comparing the diversity of samples of the T cell receptor repertoire.J. Immunol. Methods. 321:182–195 [DOI] [PubMed] [Google Scholar]
- Meyer-Olson D., Shoukry N.H., Brady K.W., Kim H., Olson D.P., Hartman K., Shintani A.K., Walker C.M., Kalams S.A. 2004. Limited T cell receptor diversity of HCV-specific T cell responses is associated with CTL escape.J. Exp. Med. 200:307–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyerl F.W., Barouch D.H., Yeh W.W., Bazick H.S., Kunstman J., Kunstman K.J., Wolinsky S.M., Letvin N.L. 2003. Simian-human immunodeficiency virus escape from cytotoxic T-lymphocyte recognition at a structurally constrained epitope.J. Virol. 77:12572–12578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyerl F.W., Bazick H.S., Newberg M.H., Barouch D.H., Sodroski J., Letvin N.L. 2004. Fitness costs limit viral escape from cytotoxic T lymphocytes at a structurally constrained epitope.J. Virol. 78:13901–13910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattapallil J.J., Douek D.C., Buckler-White A., Montefiori D., Letvin N.L., Nabel G.J., Roederer M. 2006. Vaccination preserves CD4 memory T cells during acute simian immunodeficiency virus challenge.J. Exp. Med. 203:1533–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letvin N.L., Mascola J.R., Sun Y., Gorgone D.A., Buzby A.P., Xu L., Yang Z.Y., Chakrabarti B., Rao S.S., Schmitz J.E., et al. 2006. Preserved CD4+ central memory T cells and survival in vaccinated SIV-challenged monkeys.Science. 312:1530–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen T.M., O'Connor D.H., Jing P., Dzuris J.L., Mothe B.R., Vogel T.U., Dunphy E., Liebl M.E., Emerson C., Wilson N., et al. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia.Nature. 407:386–390 [DOI] [PubMed] [Google Scholar]
- Allen T.M., Mortara L., Mothe B.R., Liebl M., Jing P., Calore B., Piekarczyk M., Ruddersdorf R., O'Connor D.H., Wang X., et al. 2002. Tat-vaccinated macaques do not control simian immunodeficiency virus SIVmac239 replication.J. Virol. 76:4108–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich T.C., Dodds E.J., Yant L.J., Vojnov L., Rudersdorf R., Cullen C., Evans D.T., Desrosiers R.C., Mothe B.R., Sidney J., et al. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo.Nat. Med. 10:275–281 [DOI] [PubMed] [Google Scholar]
- Chu F., Lou Z., Chen Y.W., Liu Y., Gao B., Zong L., Khan A.H., Bell J.I., Rao Z., Gao G.F. 2007. First glimpse of the peptide presentation by rhesus macaque MHC class I: crystal structures of Mamu-A*01 complexed with two immunogenic SIV epitopes and insights into CTL escape.J. Immunol. 178:944–952 [DOI] [PubMed] [Google Scholar]
- Champagne P., Ogg G.S., King A.S., Knabenhans C., Ellefsen K., Nobile M., Appay V., Rizzardi G.P., Fleury S., Lipp M., et al. 2001. Skewed maturation of memory HIV-specific CD8 T lymphocytes.Nature. 410:106–111 [DOI] [PubMed] [Google Scholar]
- Mueller Y.M., De Rosa S.C., Hutton J.A., Witek J., Roederer M., Altman J.D., Katsikis P.D. 2001. Increased CD95/Fas-induced apoptosis of HIV-specific CD8(+) T cells.Immunity. 15:871–882 [DOI] [PubMed] [Google Scholar]
- La Gruta N.L., Thomas P.G., Webb A.I., Dunstone M.A., Cukalac T., Doherty P.C., Purcell A.W., Rossjohn J., Turner S.J. 2008. Epitope-specific TCRbeta repertoire diversity imparts no functional advantage on the CD8+ T cell response to cognate viral peptides.Proc. Natl. Acad. Sci. USA. 105:2034–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason D. 1998. A very high level of crossreactivity is an essential feature of the T-cell receptor.Immunol. Today. 19:395–404 [DOI] [PubMed] [Google Scholar]
- Selin L.K., Cornberg M., Brehm M.A., Kim S.K., Calcagno C., Ghersi D., Puzone R., Celada F., Welsh R.M. 2004. CD8 memory T cells: cross-reactivity and heterologous immunity.Semin. Immunol. 16:335–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley J.M., Ruff L.E., Casazza J.P., Koup R.A., Price D.A., Douek D.C. 2006. Preferential infection shortens the life span of human immunodeficiency virus-specific CD4+ T cells in vivo.J. Virol. 80:6801–6809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport M.P., Price D.A., McMichael A.J. 2007. The T cell repertoire in infection and vaccination: implications for control of persistent viruses.Curr. Opin. Immunol. 19:294–300 [DOI] [PubMed] [Google Scholar]
- Charini W.A., Kuroda M.J., Schmitz J.E., Beaudry K.R., Lin W., Lifton M.A., Krivulka G.R., Necker A., Letvin N.L. 2001. Clonally diverse CTL response to a dominant viral epitope recognizes potential epitope variants.J. Immunol. 167:4996–5003 [DOI] [PubMed] [Google Scholar]
- Douek D.C., Betts M.R., Brenchley J.M., Hill B.J., Ambrozak D.R., Ngai K.L., Karandikar N.J., Casazza J.P., Koup R.A. 2002. A novel approach to the analysis of specificity, clonality, and frequency of HIV-specific T cell responses reveals a potential mechanism for control of viral escape.J. Immunol. 168:3099–3104 [DOI] [PubMed] [Google Scholar]
- Meyer-Olson D., Brady K.W., Bartman M.T., O'Sullivan K.M., Simons B.C., Conrad J.A., Duncan C.B., Lorey S., Siddique A., Draenert R., et al. 2006. Fluctuations of functionally distinct CD8+ T-cell clonotypes demonstrate flexibility of the HIV-specific TCR repertoire.Blood. 107:2373–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser J.B., Darnault C., Gregoire C., Mosser T., Mazza G., Kearney A., van der Merwe P.A., Fontecilla-Camps J.C., Housset D., Malissen B. 2003. CDR3 loop flexibility contributes to the degeneracy of TCR recognition.Nat. Immunol. 4:241–247 [DOI] [PubMed] [Google Scholar]
- Turnbull E.L., Lopes A.R., Jones N.A., Cornforth D., Newton P., Aldam D., Pellegrino P., Turner J., Williams I., Wilson C.M., et al. 2006. HIV-1 epitope-specific CD8+ T cell responses strongly associated with delayed disease progression cross-recognize epitope variants efficiently.J. Immunol. 176:6130–6146 [DOI] [PubMed] [Google Scholar]
- Dong T., Stewart-Jones G., Chen N., Easterbrook P., Xu X., Papagno L., Appay V., Weekes M., Conlon C., Spina C., et al. 2004. HIV-specific cytotoxic T cells from long-term survivors select a unique T cell receptor.J. Exp. Med. 200:1547–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley J.M., Price D.A., Douek D.C. 2006. HIV disease: fallout from a mucosal catastrophe? Nat. Immunol. 7:235–239 [DOI] [PubMed] [Google Scholar]
- Venturi V., Kedzierska K., Price D.A., Doherty P.C., Douek D.C., Turner S.J., Davenport M.P. 2006. Sharing of T cell receptors in antigen-specific responses is driven by convergent recombination.Proc. Natl. Acad. Sci. USA. 103:18691–18696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturi V., Chin H.Y., Price D.A., Douek D.C., Davenport M.P. 2008. The role of production frequency in the sharing of simian immunodeficiency virus-specific CD8+ TCRs between macaques.J. Immunol. 181:2597–2609 [DOI] [PubMed] [Google Scholar]
- Venturi V., Chin H.Y., Asher T.E., Ladell K., Scheinberg P., Bornstein E., van Bockel D., Kelleher A.D., Douek D.C., Price D.A., Davenport M.P. 2008. TCR beta-chain sharing in human CD8+ T cell responses to cytomegalovirus and EBV.J. Immunol. 181:7853–7862 [DOI] [PubMed] [Google Scholar]
- Cohen I.R. 1992. The cognitive paradigm and the immunological homunculus.Immunol. Today. 13:490–494 [DOI] [PubMed] [Google Scholar]
- Davenport M.P., Fazou C., McMichael A.J., Callan M.F. 2002. Clonal selection, clonal senescence, and clonal succession: the evolution of the T cell response to infection with a persistent virus.J. Immunol. 168:3309–3317 [DOI] [PubMed] [Google Scholar]
- Pantaleo G., Demarest J.F., Schacker T., Vaccarezza M., Cohen O.J., Daucher M., Graziosi C., Schnittman S.S., Quinn T.C., Shaw G.M., et al. 1997. The qualitative nature of the primary immune response to HIV infection is a prognosticator of disease progression independent of the initial level of plasma viremia.Proc. Natl. Acad. Sci. USA. 94:254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezys V., Masopust D., Kemball C.C., Barber D.L., O'Mara L.A., Larsen C.P., Pearson T.C., Ahmed R., Lukacher A.E. 2006. Continuous recruitment of naive T cells contributes to heterogeneity of antiviral CD8 T cells during persistent infection.J. Exp. Med. 203:2263–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows S.R., Silins S.L., Moss D.J., Khanna R., Misko I.S., Argaet V.P. 1995. T cell receptor repertoire for a viral epitope in humans is diversified by tolerance to a background major histocompatibility complex antigen.J. Exp. Med. 182:1703–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiver J.W., Fu T.M., Chen L., Casimiro D.R., Davies M.E., Evans R.K., Zhang Z.Q., Simon A.J., Trigona W.L., Dubey S.A., et al. 2002. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity.Nature. 415:331–335 [DOI] [PubMed] [Google Scholar]
- McDermott A.B., Mitchen J., Piaskowski S., De Souza I., Yant L.J., Stephany J., Furlott J., Watkins D.I. 2004. Repeated low-dose mucosal simian immunodeficiency virus SIVmac239 challenge results in the same viral and immunological kinetics as high-dose challenge: a model for the evaluation of vaccine efficacy in nonhuman primates.J. Virol. 78:3140–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifson J.D., Rossio J.L., Arnaout R., Li L., Parks T.L., Schneider D.K., Kiser R.F., Coalter V.J., Walsh G., Imming R.J., et al. 2000. Containment of simian immunodeficiency virus infection: cellular immune responses and protection from rechallenge following transient postinoculation antiretroviral treatment.J. Virol. 74:2584–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifson J.D., Rossio J.L., Piatak M., Jr., Parks T., Li L., Kiser R., Coalter V., Fisher B., Flynn B.M., Czajak S., et al. 2001. Role of CD8(+) lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment.J. Virol. 75:10187–10199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen T.M., Mothe B.R., Sidney J., Jing P., Dzuris J.L., Liebl M.E., Vogel T.U., O'Connor D.H., Wang X., Wussow M.C., et al. 2001. CD8(+) lymphocytes from simian immunodeficiency virus-infected rhesus macaques recognize 14 different epitopes bound by the major histocompatibility complex class I molecule mamu-A*01: implications for vaccine design and testing.J. Virol. 75:738–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.K., Stewart-Jones G., Dong T., Harlos K., Di Gleria K., Dorrell L., Douek D.C., van der Merwe P.A., Jones E.Y., McMichael A.J. 2004. T cell cross-reactivity and conformational changes during TCR engagement.J. Exp. Med. 200:1455–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M.J., Schmitz J.E., Barouch D.H., Craiu A., Allen T.M., Sette A., Watkins D.I., Forman M.A., Letvin N.L. 1998. Analysis of Gag-specific cytotoxic T lymphocytes in simian immunodeficiency virus–infected rhesus monkeys by cell staining with a tetrameric major histocompatibility complex class I–peptide complex.J. Exp. Med. 187:1373–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille-Reece U., Flynn B.J., Lore K., Koup R.A., Kedl R.M., Mattapallil J.J., Weiss W.R., Roederer M., Seder R.A. 2005. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates.Proc. Natl. Acad. Sci. USA. 102:15190–15194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price D.A., Brenchley J.M., Ruff L.E., Betts M.R., Hill B.J., Roederer M., Koup R.A., Migueles S.A., Gostick E., Wooldridge L., et al. 2005. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses.J. Exp. Med. 202:1349–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden B., Clark S.P., Kabelitz D., Mak T.W. 1995. Human T-cell receptor variable gene segment families.Immunogenetics. 42:455–500 [DOI] [PubMed] [Google Scholar]