


Abstract
Transcriptional regulation is mediated by the collective binding of proteins called transcription factors to cis-regulatory elements. A handful of factors are known to function at particular distances from the transcription start site, although the extent to which this occurs is not well understood. Spatial dependencies can also exist between pairs of binding motifs, facilitating factor-pair interactions. We sought to determine to what extent spatial preferences measured at high-scale resolution could be utilized to predict cis-regulatory elements as well as motif-pairs binding interacting proteins. We introduce the ‘motif positional function’ model which predicts spatial biases using regression analysis, differentiating noise from true position-specific overrepresentation at single-nucleotide resolution. Our method predicts 48 consensus motifs exhibiting positional enrichment within human promoters, including fourteen motifs without known binding partners. We then extend the model to analyze distance preferences between pairs of motifs. We find that motif-pairs binding interacting factors often co-occur preferentially at multiple distances, with intervals between preferred distances often corresponding to the turn of the DNA double-helix. This offers a novel means by which to predict sequence elements with a collective role in gene regulation.
INTRODUCTION
Transcriptional initiation is a major point of control for gene expression (1–3), and considerable effort has been devoted to deciphering the code by which transcriptional regulation occurs. Although this aspect of the genotype–phenotype connection is central to many fundamental biological processes, our understanding of how this mechanism operates at the molecular level is far from complete.
Understanding transcriptional regulation requires knowledge about the individual cis-regulatory elements that affect gene expression. Several methods have been proposed to predict individual transcription factor-binding sites by detecting statistically overrepresented motifs within the promoter (4–15). In the absence of functional data, however, overrepresentation of DNA sequence elements is not a sufficient criterion for functionality for two basic reasons. First, binding sites are generally short (5–10 bp), which means that many instances are present by chance rather than for functional reasons. Second, many motifs occur at increased frequency as a result of mutational bias or dinucleotide fluctuations near the start of transcription. The widespread overrepresentation of these motifs frequently dominates the subtle indicators of regulatory function, thus limiting the efficacy of these approaches.
Several recent studies have used spatial preferences as a criterion to predict cis-regulatory elements (16–20). With one notable exception (21), most previous studies have taken the ‘sliding window’ approach, which measures position-specific overrepresentation within several independent windows of pre-determined width (e.g. 20–25 bp). Although low-resolution approaches have been useful, an inherent limitation is that larger windows fail to recover positional enrichment at very precise locations, while smaller windows overlook general trends and are susceptible to random noise. The sliding window approach cannot operate at high resolution while simultaneously detecting broadly distributed signals, which limits the sensitivity of these methods. A second difficulty not addressed by previous studies is that the dinucleotide composition fluctuates dramatically near the start of transcription (19). This can greatly affect the frequency of motif occurrence in a position-specific manner, and raises the concern that some of the motifs predicted to exhibit positional specificity are not true cis-regulatory elements.
As a result of the limitations described above, relatively little information exists about positional biases in regulatory sequences. Important questions include whether such biases are common, how they are distributed around genes, which transcription factors (TFs) are involved, and whether they are associated with functional classes of genes. In this study, we report that a large number of likely regulatory elements exhibit position-specific overrepresentation relative to the transcription start site (TSS). The model presented here predicts regulatory elements by measuring positional enrichment at single base pair resolution while considering the data collectively; this approach allows us to detect both broad and narrow ranges of positional enrichment. By using regression analysis and a likelihood ratio test, the model can differentiate noise in the data from true position-specific overrepresentation. The method also accounts for position-specific dinucleotide fluctuations that exist within the promoter by incorporating a non-uniformly distributed background (null) model based upon the dinucleotide composition across the regulatory region. We show that this method can be used to predict novel potential cis-regulatory elements exhibiting previously unrecognized instances of positional biases on a genome-wide scale.
Since the model provides a general measure for spatial preferences, it is not limited to individual motif prediction but can also be extended to predict pairs of motifs binding interacting TFs using biases in separation distances. Transcription is not driven by individual proteins working in isolation, but is instead produced by cooperative interactions between multiple protein factors (3,22–24). Previous studies have shown that mutual relationships exist between various motifs, such as paired co-occurrences and relative orientations to the TSS (25–28). Such relationships have been effectively utilized in a variety of applications, such as the study of condition-specific and time-dependent gene expression patterns, gene network analyses, and promoter region detection (25,29–31). However, such studies are frequently limited to analyzing sequence element relationships between either known binding site motifs or those predicted using standard motif overrepresentation methods. The study presented here effectively circumvents this limitation by extending our model to analyze spatial relationships comprehensively across all motif-pairs. This allows us to predict pairs of sequence elements that are putatively bound by interacting TFs de novo, without any prior knowledge about the sequences of the predicted motifs. We find that binding sites of putatively interacting TFs frequently co-occur preferentially at multiple distances, with the interval between preferred distances corresponding approximately to the number of nucleotides in one turn of the DNA double-helix. This suggests a tendency for certain factor-pair interactions to occur in a particular orientation relative to the turn of the DNA molecule. We use the periodic phasing of inter-motif distance preferences to predict motif-pairs bound by interacting proteins, predicting functional binding site relationships between both known and novel sequence elements.
METHODS
The MPF model
Our goal is to predict regulatory motifs using spatial enrichment as an indicator of functionality. The method is an application of non-linear regression: given a set of observed motif occurrences within a set of promoter sequences, we collectively estimate the underlying frequency of occurrence according to location. Spatial biases are modeled using a continuous function which we denote as a ‘motif-positional function’ (MPF). The current study defines two types of MPFs. The first, denoted as a ‘‘motif locational function’ (MLF), measures position-specific overrepresentation of a given motif in reference to a landmark such as the TSS. The second model, denoted as a ‘motif-relational function’ (MRF), measures spatial preferences between pairs of motifs.
For a given motif w, its MLF gw(x) represents the underlying probability of occurrence according to its position x. Suppose our data set s consists of N sequences each of length L: s = {s1,s2…,sN}, where si = si(1)…si(L). We consider these sequences to be the observed outcome of an underlying biological process, and define a random variable S analogous to a single sequence in s, where S = S(1)…S(L). We then define a random variable Uk( j ) to be the k-mer starting at position j in S: Uk(j) = S(J)S(J + 1)…S(J + k − 1). Our model then defines the MLF gw(x) to be
| 1 |
where x represents the position of the motif, t represents the position of the TSS, and lw represents the length of w. Note that the position x is given relative to t, and thus the location of the TSS is given by x = 0. The value of gw(x) represents, for any individual position x, the underlying probability of occurrence of w at this precise location. The values of this function are not normalized across the values of x, and therefore the sum of the values do not, in general, equal 1 across the promoter.
In contrast to an MLF, an MRF provides a measure of inter-motif distance preferences between two motifs. For any pair of motifs w and ν, we define an MRF fw|ν(x) to be the frequency of w to occur exactly x bp from ν. Thus we set:
| 2 |
We note that the position of ν, given by i, defines the position x = 0. The function fw|ν(x) is independent of i; i.e. MRFs are defined as a conditional, rather than joint, probability.
Both MLFs and MRFs are modeled as the sum of a ‘background function’, C(x), and a ‘signal function’, H(x). Thus, for any MPF y(x) (i.e. y(x) can represent either g(x) or f(x) as defined above), we have
| 3 |
The background function C(x) represents the background frequency of the motif, namely, the frequency without explicit positional bias. This function is allowed to fluctuate according to the dinucleotide makeup of the promoter, and is modeled as a polynomial (see below). In contrast, the signal function H(x) incorporates possible spatial bias(es) into the model. The signal function of an MLF is modeled as a single unnormalized Gaussian term times a coefficient a:
| 4 |
Thus, H(x) is designed to incorporate a ‘peak’ into the function y(x). The parameters a, μ, and σ are free parameters, where a and μ give the height and location of the peak, respectively, while σ reflects the width of the peak.
Spatial biases are modeled using non-zero values for H(x), while the model where H(x) = 0 (i.e. a = 0) assumes no positional bias. Spatial preferences are predicted by fitting each of these models to the data and comparing log-likelihoods using a likelihood ratio test (see the ‘Model selection’ section below).
The signal function of an MRF is extended from that of the MLF model in order to incorporate multiple peaks into the model, as we have found that many motif-pairs co-occur preferentially at multiple distances. The signal function of each MRF is therefore modeled using a linear combination of unnormalized Gaussian terms:
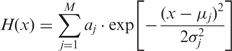 |
5 |
where M represents the number of Gaussian terms. This model is similar to that of an MLF, as MRFs for which H(x) = 0 (i.e. M = 0) assume no spatial preferences. In contrast, motif-pairs exhibiting spatial preferences are modeled using one or more Gaussian terms (M > 0). For pairs of motifs exhibiting spatial preferences, the value for M reflects the number of inter-motif distances at which a pair of motifs tend to co-occur preferentially.
Background functions
The background function C(x) represents the background frequency at position x (i.e. the frequency of occurrence without explicit spatial bias). C(x) is estimated using a ‘prototype background function’, c(x), which represents the expected frequency of occurrence. This expected frequency is determined according to the dinucleotide composition at each position within the promoters. We distinguish between the background frequency C(x) and the ‘expected’ frequency of occurrence c(x), since many motifs are either over- or under-represented with respect to their dinucleotide makeup.
For MLFs, we model the underlying expected frequency using a polynomial function:
| 6 |
This function is obtained by conducting linear regression on the set of data points 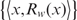 , where Rw(x) represents the expected frequency of occurrence at position x. The value Rw(x) is determined independently at each nucleotide site according to the observed dinuculeotide frequencies at that particular location. The function cw(x) then gives the underlying probability of occurrence after fitting a polynomial to this data set. The degree of this polynomial K is unique to each motif, and reflects the expected amount of fluctuation in occurrence frequency according to changes in dinucleotide content across the promoters.
, where Rw(x) represents the expected frequency of occurrence at position x. The value Rw(x) is determined independently at each nucleotide site according to the observed dinuculeotide frequencies at that particular location. The function cw(x) then gives the underlying probability of occurrence after fitting a polynomial to this data set. The degree of this polynomial K is unique to each motif, and reflects the expected amount of fluctuation in occurrence frequency according to changes in dinucleotide content across the promoters.
Formally, Rw(x) for a motif w of length lw, i.e. w = w(1)…w(lw), is given by a position-specific 1st order Markov-dependency model as described in Karlin et al. (32). The expected frequency Rw(x) of w at position x is given by
| 7 |
where Rw(i)w(i + 1)(x) gives the observed frequency of the dinucleotide w(i )w(i + 1) at position x; Rw(i)(x) represents the analogous mono-nucleotide frequency.
Since the expected frequency of many motifs differs from the actual frequency of occurrence, the background frequency Cw(x) of a k-mer w is allowed to deviate from cw(x). Namely, we allow for uniformly distributed over- and under-representation. We therefore model Cw(x) as
| 8 |
where b and d are free parameters. Thus, the background model is allowed to ‘shift’ and ‘stretch’ vertically using parameters b and d, respectively; this allows for uniformly distributed differences in the expected and observed occurrence frequencies.
The prototype background function cw|ν(x) for an MRF is a simple extension of that of the MLF model. In this case, cw|ν(x) represents the expected probability for motif w to occur x bp away from motif ν. This expected frequency is estimated according to the background functions of each individual motif. We assume the two motifs will occur randomly with respect to each other, and estimate cw|ν(x) using the conditional probability provided by Equation 2:
| 9 |
The C(·) functions on the right-most part of the equation represent those of the estimated background functions for each corresponding motif. The background function Cw|ν(x) of the MRF is then derived similarly to that of an MLF; i.e, 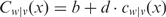 .
.
Parameter estimation and statistical significance determination
As noted briefly above, parameter estimates (i.e. b, d, a, μ, σ) are obtained using likelihood-maximization. For the model assuming no positional specificity (i.e. H(x) = 0), only the parameters b and d must be estimated. In this case, since both b and d are linear parameters within the model, the likelihood can be maximized directly using linear regression. For models incorporating positional specificity, however, the parameters must be obtained through non-linear regression analysis. This is done by optimizing the log-likelihood L(D; θy) of the data D given the model y, where θy is the parameter vector of model y. Here, the data set D is given by: 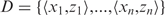 , where zi represents the number of motif occurrences at position xi. As each MPF y(x) represents the probability of motif occurrence at x, the log-likelihood of a single data point
, where zi represents the number of motif occurrences at position xi. As each MPF y(x) represents the probability of motif occurrence at x, the log-likelihood of a single data point 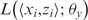 reflects the outcome of multiple Bernoulli trials with a ‘success’ being an occurrence of the motif at position xi. Thus, the log-likelihood of this data point is given by the binomial distribution:
reflects the outcome of multiple Bernoulli trials with a ‘success’ being an occurrence of the motif at position xi. Thus, the log-likelihood of this data point is given by the binomial distribution:
| 10 |
where Ni is the number of ‘trials’; i.e. the maximum possible value for zi. The total log-likelihood L(D;θy) of the data is given by the sum of the log-likelihoods across all data points 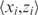 . This value is maximized using an iterative method called ‘Broyden's method’ (33) given an initial parameter estimate θ0; interested readers are referred to (33–36). Several initial parameter vectors are used during each MPF estimation; the final parameter estimates are taken to be those producing the highest log-likelihood. The method by which the initial parameter vectors are determined as well as the high level of robustness for the parameter estimates is discussed in Supplementary Data S1.
. This value is maximized using an iterative method called ‘Broyden's method’ (33) given an initial parameter estimate θ0; interested readers are referred to (33–36). Several initial parameter vectors are used during each MPF estimation; the final parameter estimates are taken to be those producing the highest log-likelihood. The method by which the initial parameter vectors are determined as well as the high level of robustness for the parameter estimates is discussed in Supplementary Data S1.
MODEL SELECTION
Model selection for any given MPF involves determining both the degree K of the prototype background function as well as the number of Gaussian terms within the signal function (either 0 or 1 for MLFs, or any non-negative number M for MRFs). These are determined in the same manner; namely, we use a likelihood ratio test (F-test) to compare the log-likelihoods of the data given two possible models. To determine the presence or absence of positional enrichment using the MLF method, we compare the log-likelihood derived from the (null) model, where H(x) is identically zero [i.e. a = 0 in Equation 4], to that of the (alternative) model where H(x) takes on non-zero values (a ≠ 0). Model selection involves comparing the log-likelihoods L(D; θyA) and L(D; θy0), where the MPF yA allows for positional specificity, while its nested null model y0 assumes no positional enrichment. The ‘scaled deviance’ Z(θyA,θy0) given by
| 11 |
follows a x2 distribution with  degrees of freedom (35); see Supplementary Data S2 for discussion.
degrees of freedom (35); see Supplementary Data S2 for discussion.
Our final statistic is
| 12 |
where n is the number of data points and model S is the ‘saturated model’, i.e. the model optimizing the log-likelihood at each data point without limits on the number of parameters. The value F follows the F-distribution with 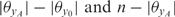 degrees of freedom (35); P-values reflecting the significance of spatial enrichment are derived using this statistic.
degrees of freedom (35); P-values reflecting the significance of spatial enrichment are derived using this statistic.
The number of Gaussian terms M within an MRF are determined similarly. However, as the number of Gaussian terms can be larger than 1, the value of M is determined in an iterative fashion. Namely, we begin by comparing the model where M = 0 to the model where M = 1 and conduct the F-test in a similar manner as described above. Note that this is equivalent to the single model comparison for an MLF, as we are determining the presence or absence of a single Gaussian term. For motif-pairs producing significant P-values, we proceed to increment the value of M (i.e. comparing models for which M = 1 to that where M = 2, then M = 2 versus M = 3, etc.) until the P-value produced from the F-test is no longer significant. The final value of M is taken to be the last value of M that has produced a significant P-value.
Determining the order K of the polynomial c(x) is also determined using an F-test in an incremental fashion. As the function c(x) is derived using linear regression, the F statistic is obtained by comparing the sum of the squares for each of two (consecutive) models. Namely, if SSy represents the sum of the squares produced from fitting model y to the data points, the F statistic is given by
| 13 |
where the degree of polynomial yA is KA, and the degree of the nested polynomial y0 is KA − 1. This value also follows the F-distribution with 1 and n − KA − 1 degrees of freedom (35). In a manner similar to the estimation of M, we increase the value of K incrementally until the comparison no longer produces a significant P-value; the final value of K is then taken to be the last value of K that produces a significant P-value.
Motif clustering procedure (MLFs)
For MLF clustering analyses, 6-mer motifs are clustered for redundancy according to both sequence similarity as well as the position and width of enrichment. Clustering is conducted by considering motifs in rank order; at each step, an individual 6-mer motif is either placed in an existing cluster or else a new cluster is created. 6-mers matching at five of the six sites (i.e. containing only one mismatch, or no mismatches with a single bp offset) are clustered if their signal functions are similar according to their KL divergence (37). The KL divergence between two signal functions is calculated by converting each function into a discrete probability distribution p(x) across each position within the promoter:
| 14 |
where the values for x are shifted according to any offset between the two motif sequences. Values of H(x) are buffered by a minimum value of 1e–45 to prevent extreme KL divergence values (i.e. 0 or infinity). The KL divergence V for two distributions, pw1(x) and pw2(x), is calculated to be
| 15 |
The V-value threshold was set to 0.2 during our analysis; motif-pairs with similar sequences were clustered if their KL divergence fell below this threshold.
Consensus sequence determination and known cis-regulatory motif comparisons
Motif clusters are condensed into a single consensus sequence according to the criteria derived from (38) and (39). Namely, each aligned site is assigned a single residue consensus if it comprises 50% of the aligned k-mers and occurs at least twice as frequently as every other nucleotide type. Double nucleotide degeneracy is applied to sites for which the two residues comprise 75% of the cases, with neither residue matching the criteria for a single site consensus. Sites not matching the criteria for either single or double nucleotide degeneracy are considered completely degenerate; triple degeneracy is not considered. During our analyses, comparisons to known regulatory elements in TRANSFAC v11.3 (39) were conducted using STAMP (40); only binding motifs found in humans were considered.
Data preparation
DNA sequences used during our analyses were taken from the UCSC Table Browser (http://genome.ucsc.edu) (41). Human analyses were conducted using the promoter sequences from the hg18, Build 36.1 assembly (42); mouse promoter data was taken from the mm9, NCBI Build 37 (43). Both data sets contained sequences comprising 500-bp upstream and 100-bp downstream of a known TSS in RefSeq (44,45). Sequence-pairs with at least 500 matching sites were filtered from the data sets. Genes without 5′ UTR annotations were excluded in order to eliminate TSS annotations caused by incomplete mRNA transcripts. The final data sets comprised a total of 20 609 non-redundant human promoters and 18 354 mouse promoters.
Program availability
The analyses presented here were conducted using the ‘Functional Region Evaluation Engine’ (FREE). Implementation of the program involves an initial formatting step; any number of subsequent analyses can be conducted after the formatting step is completed. The executable file is freely available at (http://www.biology.duke.edu/wraylab/); instructions for usage as well as an overview of user-defined parameters are included.
RESULTS
Motif Locational Functions (MLFs) provide a measure of positional enrichment
The MLF model presented here provides a measure of the amount of position-specific enrichment for any given motif. In contrast to previous models, our methodology is conducted at single-site resolution using regression analysis, allowing the data to be considered collectively across each position. MLFs are modeled using a continuous function g(x), whose values represent the underlying probability of occurrence according to position x. This function is given as the sum of the background frequency C(x) and a contribution of position-specific overrepresentation H(x); thus g(x) = C(x) + H(x). Positional bias, represented by H(x), is modeled using a Gaussian term, incorporating a ‘peak’ into the MLF (Equation 4). The mean (μ) of the Gaussian term represents the central location of enrichment, while the standard deviation (σ) reflects the ‘width’ of this enrichment. This model is illustrated in Figure 1a which shows the MLFs of the TBP and SP1-binding motifs (TATA-box and GC box). These motifs are found overrepresented 30 and 65 bp prior to the TSS, respectively (μ = −29.6 and μ = −65.3). The TATA-box is found overrepresented only at a few sites within the promoter, thus producing a small σ-value (σ = 1.9), while the GC box is enriched across a much broader range upstream of the promoter (σ = 52.2).
Figure 1.

Raw data and MLFs of four example motifs. x-axis values denote the position within the promoter, where x = 0 represents the location of the TSS. The y-axis represents the frequency of occurrence. Solid plots represent the resulting MLFs (g(x)), long dashes show the background frequency of occurrence C(x), and short dashes indicate the expected frequencies derived from dinucleotide composition (c(x)). (a) MLFs of the SP1 (gray) and TBP (black)-binding sites; significant amounts of positional enrichment are predicted for both. (b) Expected frequencies of the TBP-binding site. Each data point is derived according to the dinucleotide composition at each position. Note that c(x) was designed to not incorporate sharp increases observed in the dinucleotide data, as such rises are often a byproduct of the overrepresentation of the motif itself. (c) Two motifs (GGGCGC, gray; TGCTTC, black) without positional enrichment. Note that without positional enrichment (H(x) = 0), the MLF g(x) is the same as the background frequency C(x). A comparison between C(x) and c(x) illustrates the ability of the background model to account for uniformly distributed over- and under-representation with respect to the expected frequency (according to dinucleotide composition). The gray plot shows a high amount of fluctuation in the promoter, although this is attributed to the dinucleotide makeup of the promoter [note c(x)]; this motif is therefore not predicted to exhibit positional enrichment.
For a given motif, we predict spatial preferences using a likelihood ratio test (F-test). Namely, we compare the model assuming no positional specificity to that allowing for spatial enrichment. The former is modeled by setting H(x) to zero, and thus the underlying frequency of occurrence g(x) simply equals the background frequency C(x). We compare this model to the one where H(x) takes on non-zero values, allowing for position-specific overrepresentation. Comparing the log-likelihoods given each of these two models produces a P-value reflecting the significance of positional enrichment. Positional specificity is then predicted for motifs for which this P-value falls below a given threshold.
Our model accounts for fluctuations in dinucleotide frequencies across the promoter by allowing the values of the background frequency C(x) to vary according to position. For instance, as GC content rises near to the start of transcription, the background frequency C(x) of GC-rich motifs likewise increases close to the TSS. In order to determine the background frequency, we first estimate each motif's expected frequency across the sequences. This expected frequency is denoted as c(x). The values of c(x) are allowed to vary by position according to the dinucleotide makeup of the regulatory region, thus accounting for position-specific changes in dinucleotide composition. However, we distinguish between the ‘background’ and ‘expected’ frequency of occurrence [given by C(x) and c(x), respectively], as many motifs are either over- or under-represented with respect to their dinucleotide composition. Thus, the background frequency C(x) allows for uniformly distributed over- or under-representation. Both c(x) and C(x) are important components of our model. Namely, we must allow for differences between the expected and observed frequency of occurrence while still incorporating dinucleotide fluctuations into the background frequency of occurrence. Thus, although the background frequency is allowed to deviate from the expected frequency, C(x) is restrained to mimic the ‘shape’ of c(x) in order to preserve the expected fluctuations according to dinucleotide composition. For instance, for motifs whose frequencies are expected to vary according to position, rises and drops in C(x) are restricted to conform to those of c(x). In contrast, motifs expected to occur at a constant frequency across the region [i.e. the values of c(x) are uniform across all positions] likewise have a constant value for C(x).
We model c(x) in a continuous fashion (Equation 6). This function is determined by conducting linear regression on the set of data points representing the expected frequency of occurrence at each site. Fitting the function c(x) to these data points then gives the underlying (expected) frequency of occurrence. An example is illustrated in Figure 1b, which shows the raw data and resulting function c(x) for the TATA-box. We note that sharp rises in the observed dinucleotide frequencies at a particular location are not directly incorporated into c(x), but instead remain outlier points after fitting this function to the data. This is an important aspect of our model, as overrepresentation of a motif can itself cause rises in dinucleotide frequency. Incorporation of such rises into c(x) would therefore obscure the distinction between the signal and the background frequency at this location; this is discussed later in the Results section.
The ability of the background model to incorporate uniformly distributed over- and under-representation is illustrated in Figure 1c. Here, we show the MLFs for two motifs that do not exhibit positional enrichment. We note that although the gray plot fluctuates according to position, this would be expected according to the dinucleotide frequencies within the promoter [note the fluctuations in c(x)]. Thus, this motif is not predicted to exhibit biologically relevant positional specificity using our model.
The MLF method predicts position-specific overrepresentation for many motifs within human promoters
In order to determine which motifs exhibit positional enrichment within human promoters, we analyzed spatial enrichment for all 6-mers on a set of non-redundant RefSeq human promoters (44,45) collected from the UCSC Genome Browser (http://genome.ucsc.edu) (41,42). The data set consisted of 20 609 sequences, each comprising the region 500-bp upstream and 100-bp downstream of a known TSS. As expected, the vast majority of the motifs did not exhibit positional enrichment within the promoter data set. However, a few motifs showed highly significant position-specific overrepresentation, with 106 6-mers exhibiting spatial bias at a significance level of P < 1e–25.
To compare these results to those of a control data set, we repeated the analysis on a set of intergenic sequences, each comprising the 600-bp interval starting 2-kb upstream of a known TSS. Very few motifs were predicted to exhibit positional enrichment in this control data set, with less than 1% producing P-values under 1e–5. We then tested our model on two types of simulated data sets. The first was generated by considering the observed mono-nucleotide frequencies at each site, while the second was produced using dinucleotide frequencies at each position. One hundred data sets of both the mono- and di-nucleotide simulations were generated, with each individual data set comprising the same number of sequences as the human promoter data. For each type of simulation, we scanned for positional enrichment across all 100 data sets, recording the most significant P-value for each individual 6-mer. This P-value, denoted as Psim, was thus unique to each 6-mer. Results of these analyses, as well as those for the human promoter analysis, are shown in Figure 2. Significantly more predictions were made during the human promoter analysis than either of the simulation analyses. We also note that the dinucleotide-generated data sets produced more predictions than those produced using only mono-nucleotide frequencies, suggesting that our use of a dinucleotide-based background model leads to a more conservative significance criteria.
Figure 2.

Results from the comprehensive MLF analysis. Histograms show the cumulative number of 6-mers with positional enrichment according to their P-values in human promoters. The plots give the number of 6-mers producing Psim-values under the given thresholds during simulation analyses, where Psim represents the most significant P-value for each individual 6-mer across 100 simulated data sets. Solid plots refer to simulations conducted according to the dinucleotide frequencies across each position within the promoter, while the dotted lines represent those generated using mono-nucleotide frequencies. P-value thresholds above 1e–20 are shown in (a), while the contrast between the results of the human and simulated analyses for which P < 1e – 20 is illustrated in (b). Note that the dinucleotide-generated simulated data sets produced a significantly larger number of predictions than the mono-nucleotide-generated simulated data, while the real human promoters produce more predictions than either of the control data sets.
We used the results of both the intergenic sequence analysis as well as those of the simulated data sets to set the prediction criteria for positional specificity within human promoters. The lowest P-value produced from the intergenic sequence analysis was found slightly under 1e–15; motifs above this threshold were excluded from the list of predictions. The remaining motifs producing P-values under their Psim-value times a stringent multiple hypothesis correction factor of 1e–5 were then predicted to exhibit positional enrichment in the human RefSeq promoters. Thus, the prediction criteria (p < Psim × 1e–5) was unique to each 6-mer, subjecting motifs with lower P-values within the simulated data sets to a more stringent threshold.
The final list of predictions contained 166 6-mer motifs, representing 4% of the total number of possible 6-mers. Despite our stringent prediction criteria, the majority of the top-ranked motifs from the RefSeq promoter analysis were not filtered due to the simulation analysis. Out of the 50 top-ranked motifs exhibiting spatial bias within the real promoters, only one motif did not pass the P-value threshold determined as above.
Motif clustering predicts locational overrepresentation for both known and putatively novel cis-regulatory elements
We used the list of positionally enriched 6-mers to generate consensus motifs with degenerate sites and flexible lengths. Motifs were clustered computationally according to sequence similarity as well as the location and width of their positional enrichment. We then condensed each cluster into a single consensus sequence, generating a total of 48 consensus motifs exhibiting position-specific enrichment within human promoters. In order to test whether the predicted motifs overlapped with known regulatory elements, we compared our results to known TF-binding sites in the TRANSFAC database (39) using STAMP (40). Thirty-four of the motif clusters matched known cis-regulatory elements, comprising a total of twenty known binding sites within TRANSFAC as well as the Inr sequence element (Table 1). Several of the motifs predicted were previously known to exhibit position-specific over-representation, including the TBP, SP1, NFY, CREB, ETS, NRF1 and MYC factor-binding sites (19,46–48). We also predicted several additional motifs whose positional enrichment had not been previously documented, including fourteen novel regulatory motif candidates, denoted as d1-d14. The location of enrichment for each of the predicted motif clusters is illustrated in Figure 3. Most of these motifs were found to be enriched close to the TSS, although a few were found farther upstream of the promoter. Motifs enriched far from the promoter were frequently found to be overrepresented over a large range of the regulatory region as shown in Table 1. This is to be expected, as it is unlikely that a regulatory element enriched far from the promoter would be constrained to a highly specific location. We note the precision of the method to predict related clusters at the same location, such as the TBP-binding site as well as the Inr sequence clusters.
Table 1.
Positionally enriched motifs in human promoters
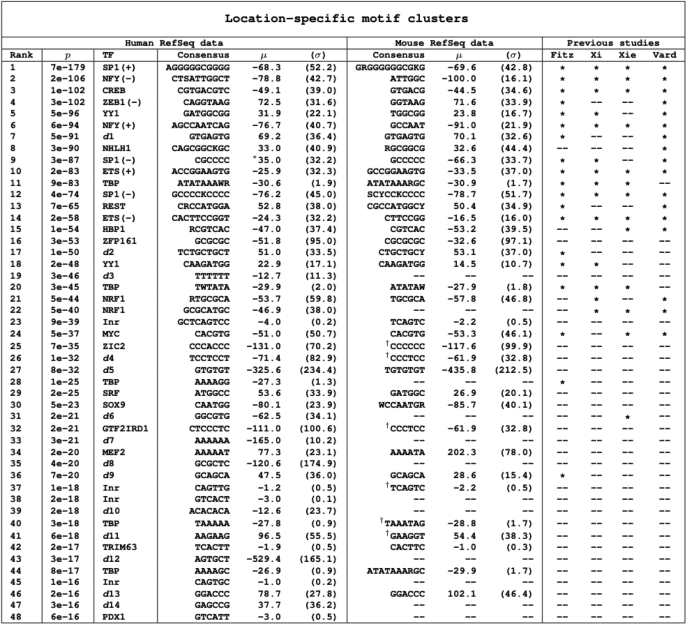 |
The location (μ) and width (σ) of enrichment are given to the right of each cluster. P-values given on the left pertain to the most significant 6-mer within the cluster. The third column shows factor names-binding to the known regulatory elements in TRANSFAC (39); putatively novel motifs are labeled d1–d14. Motifs found spatially enriched in mouse promoters are given to the right of the human analysis results. The right columns show comparisons to previous studies using the ‘sliding window method’ (18,19,47,48). Asterisks denote matches to non-redundant consensus motifs produced by these studies after k-mer clustering; only motifs predicted to be enriched at approximately the same location were considered matches. All sequence matches to TRANSFAC, mouse motif predictions, and those of previous studies were conducted using STAMP (40) (E-value threshold: 1e–6).
†Denotes a weak match.
Figure 3.

Location of enrichment for the 48 MLF motif predictions within human promoters.
Figure 4 shows the MLFs for six motifs with positional enrichment within the promoter. The MLFs of the GC-rich NHLH1 and ZFP161-binding sites are shown at the top of the figure. We note that the rise of GC content centers directly across the TSS, as indicated by the simulated data plots. However, the positional enrichment for each motif is found at other locations (μ = +33 and −51, respectively), indicating that the positional bias of these motifs is not due to dinucleotide fluctuations within the promoter. The putatively novel d3 motif comprises a homopolymeric thymine tract. Such poly(T) sequences are known to alter DNA conformation, thereby affecting transcriptional regulation by displacing the nucleosome from the DNA molecule (49–51). Similarly, the novel d10 motif, comprising a CA-dinucleotide repeat, promotes left-handed Z-DNA conformations (52–54). The positional biases of these motifs may therefore reflect a functional role for each motif at these locations. The MLFs of the novel reverse complement motifs d2 and d9 are shown at the bottom of Figure 4; each orientation of this putatively novel regulatory element show positional enrichment at the same location downstream of the TSS.
Figure 4.

MLFs of six motifs exhibiting positional enrichment. Top: MLFs for the GC-rich NHLH1-binding site and the ZFP161-binding site. Each plot shows results for both human (black) and simulated (gray) data sets. Dashed lines denote the background functions C(x). Middle: MLFs of the novel poly(T) 5-mer d3 and (AC)3 motif d10. Bottom: MLFs for the GCT-repeat motif d2 and its reverse complement d9. Each shows significant amounts of positional enrichment ∼50 bp after the TSS.
Many positionally biased motifs are shared between human and mouse
We tested whether spatially biased motifs found in human promoters would also show positional enrichment within mouse promoters. We conducted a second comprehensive MLF analysis using a sequence data set of 18 354 non-redundant mouse promoters in RefSeq (43–45). We then compared the motif predictions between the two species according to sequence similarity as well as the location of positional overrepresentation.
Our analysis predicted a total of 49 consensus motifs to exhibit positional enrichment within mouse promoters (Supplementary Data S4). Comparisons of these results to those of the human promoter analysis showed a very significant amount of overlap between motif predictions across the two species. We found that 36 (75%) of the motif clusters identified in the human data set matched spatially biased motifs detected within the mouse promoters (Table 1). Such a significant overlap provides confidence in our new motif predictions, as these motifs were predicted during independent analyses using data from two highly diverged species. In addition, the location of enrichment for our motif predictions was often found to be highly conserved between the two lineages. Many motifs with well-documented positional enrichment were found overrepresented at very similar locations across the two species, particularly the TBP, SP1, NRF1 and CREB-binding sites. This was also found to be the case for many of our novel motif predictions. For instance, the novel d1 and d2 motifs exhibited overrepresentation peaks whose position differed by only 1 bp across the two lineages.
Study comparisons highlight differences in methodologies to previous studies
Several previous studies have analyzed spatial preferences of potential regulatory motifs within the promoter (17–21,47,48). Most previous analyses, with one exception (21), have used the ‘sliding window’ approach. In this approach, the promoter region is divided into several discrete bins of pre-determined width (e.g. 20–25 bp), and positional bias is then predicted by comparing the number of motif occurrences in each window to a background frequency of occurrence. A previous study conducted by FitzGerald et al. (19) used the sliding window approach, considering motif occurrences within separate windows of 20 bp. FitzGerald et al. predicted a total of 156 8-mers to exhibit positional bias prior to clustering. A direct comparison of our results to those of FitzGerald et al. showed that 97% of the 8-mers predicted by FitzGerald et al. matched one of our predicted 6-mers (Table 2). We also found that 85% of our individual 6-mers matched a 8-mer prediction made by FitzGerald et al. However, the vast majority of these matches were to redundant motifs that had been grouped according to sequence similarity during the clustering analysis. There were also cases in which distinct 6-mers found within different cluster groups matched a single 8-mer predicted by FitzGerald et al. For instance, one of the G-rich 8-mers predicted by FitzGerald et al. matched eight of our predicted 6-mers, although this group of 6-mers included representatives from three different motif clusters. These 6-mers clearly represented distinct regulatory elements, as their enrichment was found at significantly different locations within the regulatory region.
Table 2.
Between-studies comparisons of positionally enriched kmer predictions
| Fitz | Thara | Vard | MPF | ||
|---|---|---|---|---|---|
| Positionally enriched motifs: study comparisons | |||||
| Predictions | Number | 156 | 1226 | 168 | 166 |
| kmer length | 8 | 8 | 7 | 6 | |
| GC content | Fraction | 63% | 69% | 60% | 60% |
| GC rich | Number | 28 | 387 | 19 | 48 |
| Expected | 6 | 43 | 11 | 18 | |
| (Numb/Exp) | 4.7 | 9.0 | 1.7 | 2.7 | |
| AT rich | Number | 3 | 39 | 0 | 16 |
| Expected | 6 | 43 | 11 | 18 | |
| (Numb/Exp) | 0.5 | 0.9 | 0.0 | 0.9 | |
| FitzGerald et al. | Matches | — | 521 | 60 | 141 |
| Fraction | 42% | 36% | 85% | ||
| Tharakaraman et al. | Matches | 149 | — | 101 | 156 |
| Fraction | 95% | 60% | 94% | ||
| Vardhanabhuti et al. | Matches | 125 | 507 | — | 103 |
| Fraction | 80% | 41% | 62% | ||
| MPF | Matches | 151 | 1004 | 84 | — |
| Fraction | 97% | 82% | 50% | ||
Spatially enriched k-mers are compared between studies conducted by FitzGerald et al. (19), Tharakaraman et al. (21) and Vardhanabhuti et al. (18) as well as the MPF model. The total number of (unclustered) k-mer predictions are shown in the top row. The number of GC and AT rich motif predictions (those composed of G/C or A/T consensus sites at all but one site) are shown below, along with the expected number and the ratio of actual/expected. Here, the ‘expected’ number of predictions was determined by assuming a random collection of k-mers identical in size to the set of predictions. Bottom rows show the amount of overlap between predictions across the four studies. Overlapping predictions were determined by considering all consensus sites of the predicted motifs, allowing for any offset such that at most one consensus site of the smaller motif was not aligned to the larger k-mer. For instance, 149 (95%) of the 156 motif predictions made by FitzGerald et al. matched a prediction made by Tharakaraman et al. Note that the number of matches is not symmetrical, since a single k-mer may match more than one other motif prediction.
Thus, we looked to compare the non-redundant consensus sequences produced by both studies after clustering. Motif clustering conducted by FitzGerald et al. resulted in nine non-redundant motif clusters. Eight of these clusters overlapped with one of our consensus motif cluster predictions, while our model attributed the putative spatial bias of the remaining cluster to dinucleotide fluctuations within the promoter. In contrast, less than half of our consensus sequences were detected by FitzGerald et al. Table 1 contains comparisons between our regulatory motif predictions to those of FitzGerald et al. as well as three other studies providing non-redundant motifs with spatial enrichment (18,47,48). We found that many of our motifs predicted with wider ranges of positional enrichment could not be detected using the sliding window approach. Our approach was also found to increase sensitivity to spatial biases occurring at very precise locations. For instance, FitzGerald et al., in addition to the three other studies included in Table 1, could not easily detect the well-known Inr sequence element. The Inr sequence has been previously characterized by the consensus motif YYAnWYY (55). This element is known to function specifically at a single nucleotide site at the start of transcription (55,56), and therefore it is difficult to detect using low resolution approaches. Out of 156 8-mer predictions made by FitzGerald et al. none included the YYAnW 5-mer with enrichment at the TSS. In contrast, our model identified seven 5-mers matching this consensus with significant enrichment at the start of transcription (P < 1e–15). The most common version of this motif was TCAGT, which was found overrepresented at the TSS more than seven and a half times over the background frequency (P = 6e–48). Despite the highly significant amount of positional overrepresentation exhibited by this motif, none of the studies using the sliding window approach detected any motifs containing this 5-mer (18,19,47,48). Figure 5 shows the occurrence data of this motif using 20 bp windows and using single-site resolution; we note the significant decrease of the signal when considering the data using windows of 20 bp.
Figure 5.

Occurrence frequency of the functional Inr sequence 5-mer TCAGT. The contrast is shown between occurrence data using (a) 20 bp windows and (b) single-site resolution.
Tharakaraman et al. (21,57) also scanned for positional biases within human promoters. However, their methodology allowed for varying window sizes, improving sensitivity of spatial enrichment considerably. Tharakaraman et al. predicted 1226 unclustered 8-mers to exhibit positional enrichment within the promoter. Despite such a large number of predictions made by Tharakaraman et al., we found that 82% of their predicted motifs overlapped with our results (Table 2). However, their model assumed a uniform background frequency of occurrence across the promoter. Since GC mono- and di-nucleotide composition rises substantially near the start of transcription (19), about a third of the 8-mers predicted by Tharakaraman et al. were highly GC-rich, containing at least seven out of eight G/C consensus sites. This is nine times more than what would be expected from a random selection of 8-mers. In contrast, the number of GC rich motifs predicted during our analysis is only 2.7 times higher than would be expected by chance. As many GC-rich motifs do play functional roles in gene regulation, we looked to determine whether these GC-rich 8-mers do, in fact, comprise true regulatory elements. To assess the validity of these GC-rich predictions, we compared the predictions made by Tharakaraman et al. to known protein-binding sites found in humans. We found that, among the GC-rich predictions overlapping our results, over half matched human-binding elements found in the TRANSFAC database. This represents a significant enrichment of known regulatory elements, as only about a third of all GC-rich 8-mers match human binding sites in TRANSFAC. However, among the GC-rich predictions that did not overlap our results, only 19% matched known human binding sites. This is significantly less than would be expected by chance given a random selection of 8-mers. Although this evidence is not necessarily conclusive, we would still expect some amount of enrichment for known regulatory elements in this list of predictions. Thus, it is likely that a number of these predictions are simply the result of the rise of GC content near the TSS, rather than true regulatory elements.
In contrast to the analysis of Tharakaraman et al., Vardhanabhuti et al. (18) controlled for changes in basepair composition across the promoter. In this analysis, the observed number of occurrences of a given motif was compared to an expected number of occurrences in each window of 20 bp. The expected frequency was estimated separately within each individual window by considering occurrence data of other motifs with identical basepair composition. That is, occurrence data was obtained for motifs whose columns were ‘permuted’ from the original motif, thus conserving base composition. The observed occurrences of these permuted motifs were then used to determine the expected frequency of occurrence in each individual window; both the ‘observed’ and ‘expected’ frequencies were thus unique to each window. Vardhanabhuti et al. first scanned for positional biases using known TF-binding sites in TRANSFAC, and subsequent analyses predicted spatial overrepresentation across all (novel) 7-mer motifs filtered for known binding sites in TRANSFAC. Although these latter predictions were presented as novel motifs, we found that a third of these 7-mers matched known regulatory elements. For example, the second and sixth highest-ranking motifs (ATTGGCT and AGCCAAT) match the NFY-binding site, each with a STAMP E-value under E < 1e–7 (Supplementary Data S3).
Between-studies comparisons showed consistently less overlap between the results of Vardhanabhuti et al. and those of other studies, including the one presented here (Table 2). It is likely that these differences can be explained by the methodology used to estimate the background frequency of occurrences. For instance, the occurrence frequency of a motif rich in a single nucleotide type will not be significantly different after permuting its columns, as the motif consensus itself will not be changed considerably. In particular, mono-nucleotide repeats are impossible to detect. As a result, sensitivity to many biologically relevant signals is decreased significantly. Vardhanabhuti et al. note within their study that their methodology predicts enrichment of the well-known TBP-binding element (TATA-box) at a location that differs from where it is known to function. This motif was predicted by Vardhanabhuti et al. to be enriched 45 bp prior to the TSS, although it is known to function at a very specific location 30-bp upstream of the TSS (55,56,58–60). The authors attribute this discrepancy to an increase of A/T nucleotide composition at this location, increasing the ‘expected’ number of occurrences within this window and therefore decreasing the observed/expected ratio. However, the increase of A/T nucleotide composition at this location is simply a result of the overrepresentation of the A/T rich TBP-binding site itself. This raises the concern that correcting for basepair composition in a position-specific manner can cause failure to detect real biological signals, as the signal itself can be incorporated into the background (expected) frequency. The method presented here effectively circumvents this problem, as the background frequency is modeled in a continuous fashion. Significant changes in the expected frequency caused by real biological signals remain outlier points after fitting the background model to the data (Figure 1b). We note that in the case of the TATA-box, the MLF model predicted enrichment at the correct location 30 bp prior to the TSS at a high level of confidence.
Motif relational functions (MRFs) detect spatial biases between motif-pairs
Transcription is driven by multiple regulatory elements acting in coordination, and knowledge of regulatory element interactions is essential to understand the mechanisms driving gene regulation. Since protein-protein interactions are inherently structure-specific, it is logical to expect that regulatory motifs binding interacting TF proteins preferentially co-occur non-randomly with respect to each other. Since the MPF model is designed to detect spatial biases of motif occurrence, we expanded the previous model in order to predict motif-pairs binding interacting TFs de novo according to their spatial relationships. The extended model measures inter-motif distance preferences between pairs of motifs; we denote this measure as a ‘motif relational function’ (MRF).
The MRF model represents a simple extension of the previous MLF model. In this new model, we are able to determine multiple instances of spatial biases for each individual motif-pair. This aspect of the model was designed to capture the phasing of inter-motif distance preferences, which would be expected according to the results of previous studies. Periodic distributions have been associated with DNA sequence features attributed to the structural conformations of the nucleosome (61), and TF-pair interactions are often known to occur at phased intervals around the histone complex or the winding of the DNA double-helix (3,62,63). This scheme is shown in Figure 6, which illustrates a potential preference for protein–protein interactions to occur in a specific orientation in relation to the turn of the double-helix. As explained in the methods section, the number of overrepresentation peaks (i.e. individual instances of spatial preferences) is not pre-defined but is instead estimated separately for each individual motif-pair. Specifically, the model is designed to detect each instance of spatial bias on an individual basis, continuing to add overrepresentation peaks until all statistically significant peaks have been incorporated into the model.
Figure 6.

Functional motif-pair inter-relationships. Proteins must often be positioned in a particular orientation with respect to the DNA molecule to induce potential interactions (3,62,63). Interactions between protein A and protein B occur when the latter is positioned at B1. The same interaction frequently occurs one turn of the double-helix away from B1 (i.e. at B2), since the orientation of protein B is consistent relative to the turn of the DNA molecule. However, the interaction cannot occur when protein B is at B3 due to its inconsistent orientation. The distance between factors A and B is determined by the size of the proteins and is unique between different TF-pairs. In contrast, phasing intervals (i.e. the distance between B1 and B2) remains relatively consistent, as they correspond approximately to the number of nucleotides in a turn of the DNA double-helix.
Figure 7 shows two MRFs which were both generated by motif-pairs that bind TFs with known interactions (64,65), namely, the NFY-NFY and NFY-SP1-binding motif pairs. Motif-pairs were often found to co-occur preferentially at multiple distances, with intervals separating preferred distances corresponding approximately to the turn of the DNA double-helix.
Figure 7.

MRFs of two motif-pairs-binding interacting TFs (64,65). A known occurrence of the reverse-strand NFY-binding site defines the position x = 0; x-axis values denote the position of the (a) plus-strand NFY-binding site and the (b) minus-strand SP1-binding site. y-axis values show the frequency of occurrence of these partner motifs. Each motif-pair exhibits more than one preferred distance between motifs, with intervals between peaks being ∼8–10 bp. This is consistent with the scheme illustrated in Figure 6, where the location of the peaks represents the positions of B1 and B2, and the position x = 0 corresponds to the position of factor A.
In our particular application of the MRF method, we used the phasing intervals of inter-motif distance preferences as a criterion for predicting motifs with a cooperative role in transcriptional regulation. Namely, we predicted pairwise interactions between putative regulatory motifs by scanning for consistency in the intervals between overrepresentation peaks. In order to quantify the phasing of inter-motif distance preferences, we defined a ‘peak-separation value’, xsep, to be the distance between any pair of overrepresentation peaks within the same motif-pair MRF. Thus, xsep = | μi − μi | for any two peaks i and j within the same MRF (Equation 5). We controlled for peaks potentially representing random outlier points by filtering peaks corresponding to a single-site location. We also filtered double motif occurrences separated by less than 20 bp in order to remove spurious peak phasing caused by repeat sequences and same-sequence dyads.
Our comprehensive MRF analysis was conducted across all possible pairwise combination of 5-mer motifs. Enumerating across all MRFs containing at least two overrepresentation peaks resulted in 619 MRFs exhibiting xsep-values ranging between 7.5–9.5 bp (Figure 8). This is more than twice the expected number (i.e. the average number of MRFs producing xsep-values within any other 2-bp range). While these values do not correspond precisely to the number of nucleotides in a turn of the double-helix (∼10.5 bp), it is possible that this deviation can be explained by distortions of the DNA caused by protein binding, or by other similar mechanisms.
Figure 8.

MRF peak separation distributions. The distribution of peak separation values (xsep-values) shown were produced by enumerating all peak-pairs across all possible 5-mer motif-pairs. Peak separation values are defined as xsep = | μi − μj | between overrepresentation peaks i and j within the same MRF; this value is analogous to the distance between B1 and B2 in Figure 6. Note the strong concentration of xsep-values close to ∼8–9 bp.
Given these trends, we hypothesized that motifs producing consistent xsep-values corresponding to the turn of the DNA double-helix would act as protein-binding sites. We chose to use a stringent criterion for motif-pair predictions by specifically focusing on motifs exhibiting significant concentrations of xsep-values, whose consistency was unlikely to be due to chance. Thus, we calculated the distribution of peak phasing intervals for each individual 5-mer motif across all possible motif partners, testing for significant concentrations within their xsep-value concentrations. Namely, at each iteration, we chose an individual 5-mer to be the fixed motif, and xsep-values were then determined across all MRFs produced from this fixed motif and one of the possible variable motif partners. xsep-values were then accumulated across all variable motif partners; fixed motifs producing a significant xsep-value concentration within one of the 2-bp windows centered at xsep ≈ 8,9,10 were predicted to be functional.
After correcting for multiple hypothesis testing, thirteen 5-mers were found to have significant xsep concentrations within one of these regions (P < 1e–5). Clustering these 5-mers according to sequence similarity and their corresponding xsep distributions produced nine consensus motifs (Table 3). Six of the nine consensus sequences matched known TF-binding sites in TRANSFAC, namely the NRF1, NFY, EVI1, and MADS-box protein family-binding sites. The NFY and MADS-box protein family-binding motifs were predicted on both strands, while the NRF1-binding sequence was palindromic. Three additional motifs, denoted as y1-y3, did not match any known binding sequences in TRANSFAC, and therefore represent novel cis-regulatory element candidates. Figure 9 shows the xsep-value distribution for the highest-ranking 5-mer in four of the predicted motif clusters. These include the reverse-strand MADS-box protein family-binding site, the NRF1-binding site, the novel y1 motif, and the reverse-strand NFY-binding site. Note the highly significant concentration of xsep-values around ∼8–10 bp for each motif.
Table 3.
Motifs exhibiting consistent phasing intervals between preferred inter-motif distances
| Rank | Consensus | TF | xsep | P | Partners |
|---|---|---|---|---|---|
| Motifs with consistent phasing intervals | |||||
| 1 | TTTGTA | y1 | 9 | 7e–26 | 19 |
| 2 | ATTTTT | MADS (−) | 8 | 3e–21 | 24 |
| 3 | AAAAAT | MADS (+) | 8 | 4e–19 | 16 |
| 4 | GCATGC | NRF1 | 9 | 7e–19 | 23 |
| 5 | ATTGC | y2 | 8 | 3e–12 | 8 |
| 6 | TCTTG | EV11 | 9 | le–11 | 7 |
| 7 | GAGCT | y3 | 10 | 2e–10 | 7 |
| 8 | ATTGG | NFY(−) | 10 | 4e–8 | 5 |
| 9 | CCAAT | NFY(+) | 10 | le–7 | 3 |
Phasing intervals (xsep) were considered across all MRFs produced by the (fixed) motifs shown above and one of the possible 5-mer partners. xsep-values denote the interval between distance preferences (peaks) within the same MRF for a pair of motifs (xsep = |μi − μj |). xsep-value concentrations were determined across all 2-bp intervals centered around 8–10 bp. P-values (fifth column) correspond to the significance of this concentration for the top-ranking 5-mer in each cluster. Tfs binding to known motifs in TRANSFAC (39) are shown in the third column [STAMP (40) E-value threshold: 1e–5]; novel regulatory element predictions are labeled y1–y3. The number of predicted partner clusters is given in the right column.
Figure 9.

MRF peak separation concentrations. Peak separation distributions are shown for four motifs with significant xsep-value concentrations. Each panel shows the xsep distributions for the most significant 5-mer of the reverse-strand MADS-box family-binding motif, the NRF1-binding motif, the reverse-strand NFY-binding motif and the novel regulatory element prediction y1.
Periodic phasing of inter-motif distance preferences detects known and novel regulatory element relationships
We extended the analysis in order to predict binding site partners for each of the motifs exhibiting significant xsep-value concentrations. Each fixed motif predicted during the previous MRF analysis was paired with multiple partner motifs by considering each individual fixed/partner motif-pair MRF. A 5-mer was predicted to pair with the fixed motif if the motif-pair produced phased distance preferences corresponding to the fixed motif's xsep-value concentration. The predicted partner motifs were then clustered according to sequence similarity as well as the location of their overrepresentation peaks. This procedure produced a total of 112 motif partner clusters pairing with one of the nine fixed motifs predicted in the previous analysis.
Partner motif predictions for the NFY and MADS-box protein family-binding sites are given in Tables 4 and 5, respectively. Only a few motif clusters were predicted to pair with the NFY-binding motifs; each of the partner clusters corresponded to either the NFY or the SP1-binding sequences. Both factors are known to have direct interactions with NFY (64,65).
Table 4.
Motif partners for the NFY-binding element
| TF partner | Forward strand | Reverse strand |
|---|---|---|
| NFY-binding site partners | ||
| NFY (+) | CCAAT | GCCAATC |
| (−) | GATTGGC CGATT | ATTGG |
| SP1 (+) | GGCGG | GGGCGG |
| (−) | CCGCC | |
Table 5.
MADS-box family-binding site partners
| Forward strand: AAAAAT |
Reverse strand: ATTTTT |
||||||
|---|---|---|---|---|---|---|---|
| Consensus | TF | RC | Consensus | TF | RC | ||
| MADS-box motif partners | |||||||
| 1 | AGACC | 1 | GAACTCCT | NR1I2 | |||
| 2 | CAGCTAC | TOPORS | 2 | AGCCT | Rl | ||
| 3 | AGGCTG | rl | 3 | AGTGC | HMX3 | R8 | |
| 4 | CAGCC | 4 | ATCCG | ||||
| 5 | CCTGTA | AR | 5 | ATGTT | |||
| 6 | CGCCA | E2F1 | 6 | CACCA | NFY (*) | R7 | |
| 7 | CTACTC | 7 | CCACG | ATF6 (*) | |||
| 8 | GCTGAG | NFE2 | r2 | 8 | CCCAA | IKZF1 | |
| 9 | GAACC | 9 | CCTCC | MAZ | R3 | ||
| 10 | GGCAGG | TCF3 (*) | 10 | CCTGA | |||
| 11 | GGAGG | MAZ | r3 | 11 | TCGAAC | XBP1 | |
| 12 | GGTTG | r4 | 12 | GCTGGGACA | PITX2 | ||
| 13 | TGTAATCCCA | CEBP (*) | r5 | 13 | GATCC | ||
| 14 | GTGGC | r6 | 14 | GGATTACA | CEBP (*) | R5 | |
| 15 | TGGTG | NFY (*) | r7 | 15 | GCCAC | R6 | |
| 16 | GCACT | HMX3 | r8 | I6 | GGGTTT | TERF2IP | |
| 17 | TCAAG | NKX2 | |||||
| 18 | TGACC | ESR1 | |||||
| 19 | TGATC | ||||||
| 20 | AGCCA | PCBP2 | |||||
| 21 | CAACC | R4 | |||||
| 22 | CTCGG | ZNF569 | |||||
| 23 | TCAGC | NFE2 | R2 | ||||
| 24 | TGCCT | ||||||
TFs binding to the known cis-regulatory elements in TRANSFAC (39) are shown in the third columns. Binding factors with known direct interactions to SRF, a MADS-box family member, are labeled with asterisks. Reverse complements across opposing strands of the MADS-box fixed motif, as determined by STAMP (E-value threshold: 1e–5), are labeled r1–r8 and R1–R8 (e.g. r1 is the reverse complement of R1, etc.).
The MADS-box protein family-binding sites were predicted to pair with more partner motifs than the NFY-binding element. Sixteen and 24 partner clusters are predicted to pair with the forward- and reverse-strand MADS-box motif, respectively. A total of 24 (60%) of the partner clusters were found to match known TF-binding sequences in TRANSFAC, comprising a total of 19 known regulatory elements. Several of these regulatory elements bind proteins with direct interactions to SRF, a member of the MADS-box protein family. These include binding motifs of TCF3 (66), CEBP (67), NFY (68) and ATF6 (69). We found that partner motifs pairing with the MADS-box-binding site were frequently predicted in both orientations. There were eight reverse complement matches between partner motifs pairing across opposing strands of the MADS-box-binding motif, comprising half of the motif predictions pairing with the forward-strand MADS-box parent motif (Table 5). We found that the mutual directionality of the fixed/partner motif-pairs was highly conserved, with each separate strand of the MADS-box motif pairing with only a single strand of the partner motif. There were no same-strand matches between partner motifs across opposing strands of the parent MADS-box-binding element; i.e. individual strands of the partner motifs were not found to pair to both orientations of the MADS-box-binding motif.
DISCUSSION
Measuring spatial overrepresentation at high-scale resolution provides a powerful, but not yet frequently applied, approach to predicting functional motifs on a genome-wide scale. The MPF model provides a general measure of position-specific enrichment for a motif at single-site precision in relation to a given reference point. The methodology is distinct from previous methods that use general overrepresentation as a criterion for regulatory function. Although such motif finders have been useful for certain applications, assessments of these methods have shown that the efficacy of such approaches is somewhat limited (70). The method presented here is designed to predict either individual regulatory elements or functional motif-pair relationships using spatial biases, rather than the overall frequency of occurrence, as a criteria for functionality. The MLF model shows that many known regulatory elements exhibit strong locational preferences relative to the TSS. The MRF model predicts motif-pair relationships de novo by analyzing inter-motif distance preferences without prior knowledge about the corresponding motif sequences. Our implementation of this model predicts cis-regulatory elements and their pairwise interactions using periodically phased distance preferences between pairs of motifs.
The MLF method predicts positional enrichment for 48 consensus motifs
The comprehensive MLF analysis predicted 48 consensus motifs to exhibit positional enrichment within the promoter (Table 1). Thirty-four (71%) of these consensus sequences matched previously documented cis-regulatory elements, comprising a total of 22 known regulatory elements. The remaining 14 comprise putatively novel cis-regulatory element predictions without known binding partners.
While most previous studies predict spatial preferences by counting motif occurrences within multiple independent windows of ∼20–25 bp (18–20,47,48), the MLF model considers the data collectively at single-site resolution, estimating the underlying frequency of occurrence according to position. Although prior studies have shown positional enrichment of motifs, the increased spatial resolution of our approach provides evidence for finer-scale functional constraints on the position of motifs. Inspection of the results of FitzGerald et al. (19) and Vardhanabhuti et al. (18) showed that the most common Inr 5-mer consensus (TCAGT) did not appear in the predictions of either study. Occurrences of this motif are constrained to a single nucleotide site, and thus the positional specificity of this motif could not be detected using larger 20-bp windows. Previous methodologies cannot operate at single-site resolution without a loss of sensitivity to spatial preferences occurring across larger ranges of the regulatory region. We also note that several of our motif predictions not predicted by these previous methods were found overrepresented across wide ranges of the upstream flanking region. For instance, the ZFP161-binding motif shows clear enrichment across a range of ∼200 bp (Figure 4); this consensus motif had not previously been detected using the sliding window method. We show that considering the data collectively leads to a significant increase of sensitivity, and allows us to detect positional biases occurring either across broad portions of the promoter or constrained to a single nucleotide site.
The model presented here accounts for dinucleotide fluctuations found in and around the promoter. We show from the results of two simulation analyses that changes of basepair composition within the promoter can produce spurious motif predictions due to the rise of GC content near the TSS. This effect was found to be more prominent in simulated data sets conserving dinucleotide frequencies at each site than those generated using only mono-nucleotide basepair composition. Comparisons to previous studies suggest that models not accounting for nucleotide composition are sensitive to the significant rise of GC content near the TSS. Methods assuming a uniform background frequency are prone to predict GC-rich motifs which are likely to reflect only changes in basepair composition, rather than motif functionality. However, the cross-studies comparisons also show that accounting for fluctuations in mono- and di-nucleotide frequencies must be conducted collectively across all positions, rather than independently at each location. For instance, Vardhanabhuti et al. (18) estimated the expected frequency of occurrence separately at each location within the promoter. However, overrepresentation of a positionally enriched motif can itself increase the basepair composition at the same location. Estimating the expected frequency within each separate window therefore incorporates such increases in basepair composition into the background frequency, although this rise in the expected frequency is simply a direct byproduct of the biologically relevant overrepresentation. As discussed by the authors of this study, their method was unable to predict positional specificity of the TATA-box at its correct functional location (48,58–60); it is highly likely that many other existing signals went undetected using this model. In contrast, the model presented here estimates the background frequency of occurrence collectively in a continuous fashion. In this way, increases of the expected frequency resulting from true biological signals remain outlier points after fitting the expected frequency to the data, and they are therefore not directly incorporated into the background frequency of occurrence (Figure 1b). We note that in the case of the TATA-box, our model predicts enrichment at its correct location 30 bp prior to the TSS at a high level of significance. Thus, we find that considering the data collectively is essential in order to control for dinucleotide fluctuations within the promoter.
The results from our analyses indicate that many regulatory motifs exhibit positional enrichment in or near the promoter. It is likely that these motifs may perform specific functions at their respective locations of enrichment. We note that some of the known motifs are also known to function at a diverse number of locations, and thus functional occurrences of these motifs are not restricted to occur within their location of enrichment. For instance, functional occurrences of the SRF-binding motif are not limited to its predicted location of enrichment (71). However, SRF binding is known to be facilitated by the YY1 factor, with each protein occupying the same binding location on the DNA, albeit on opposite strands of the molecule (72,73). As YY1 has been shown to function at specific locations within the promoter (48), it is not surprising that the SRF-binding motif also exhibits positional enrichment near that of YY1. Thus, although functional occurrences of the SRF-binding element occur at other locations, it is likely that SRF may play a unique role within its location of enrichment in concert with the YY1 factor. It is possible that other factors also known to bind at a diverse number of locations may also perform specific functional roles at which their binding motifs preferentially occur, and future studies may elucidate the biological relevance regarding the roles of these motifs within their area of enrichment.
Our predictions also include 14 motifs (d1–d14) that do not match any known human TF-binding motifs in TRANSFAC. Although these motifs do not match any known TF-binding sequences, some have been reported to play alternative roles in gene regulation. For instance, the novel d5 and d10 motifs consist of (GT)n and (CA)n dinucleotide repeats, respectively. These sequence elements are known to promote left-handed Z-DNA structures that affect DNA supercoiling, and subsequently transcription (52–54). Similarly, the d3 motif consists of a homopolymeric thymine [poly(T)] tract. Such poly(T) tracts are known to alter the conformation of the DNA molecule, thereby disrupting nucleosome positioning (51). Both d3 and d10 show a significant overrepresentation peak centering near the start of the transcription bubble (74). A recent study conducted by Kaplan et al. (75) has shown that nucleosome occupancy is significantly decreased at this location. It is therefore likely that occurrences of these motifs in this region of the promoter may make the area more accessible to the RNA-polymerase machinery. Thus, while many of our novel regulatory element predictions are likely to function as protein-binding sites, others may affect transcriptional regulation through alternative mechanisms.
Applying our method to a set of mouse promoters showed that the majority of the motifs exhibiting spatial preferences in humans also show positional enrichment in mouse. Thirty-six (75%) of the motif clusters were shared across the two species, and the location of positional enrichment was frequently found to be highly conserved between the two lineages. Supplementary Data S4 provides results from a second mouse promoter analysis conducted on a smaller, high-quality data set provided by the RIKEN Institute. This analysis was conducted on 1354 mouse promoters generated using CAGE-tag data (56,76), a significant decrease in the number of sequences used during our previous analyses. Thirty-one motifs predicted during the human promoter analysis matched predictions from the RIKEN data set, representing a significant overlap between results produced from the two data sets. However, the positional bias of some infrequently occurring motifs could not be detected using the smaller RIKEN data set, including several regulatory elements whose positional bias is well-documented, such as the CREB, MYC, NRF1 and ETS-binding sites (19,47,48). Thus, we note that it is usually preferable to use larger data sets than smaller ones in order to minimize the amount of random noise relative to the background frequency. This increases sensitivity to the spatial bias of infrequently occurring motifs, and it also eliminates spurious peaks caused by random outlier points. We have found that our approach is not greatly affected by noise when applied to large data sets, and that the MLF method is efficient at detecting instances of spatial bias when using an adequate number of sequences.
MRF peak separation analyses predict individual and putatively interacting cis-regulatory elements
The MRF model presented here analyzes inter-motif distance preferences between pairs of sequence elements. Our application of this method predicted cis-regulatory element-pairs that putatively bind interacting TFs by considering periodically phased spatial preferences. We found that pairs of TF-binding sequences with collective roles in transcription often show elevated frequencies of co-occurrence at multiple separation distances. The model presented here explicitly uses multi-modal characteristics of inter-motif distance frequencies, and is therefore inherently different from previous uni-modal models, which have generally relied on the sliding window method or maximum-distance approaches (16–18,31,77–79). We have found that individual instances of spatial preferences are generally constrained to widths of only ∼2–3 bp, and that single overrepresentation peaks often exhibit only minimal amounts of significance. However, despite the subtlety of each individual instance of spatial preference between motif-pairs, overall trends in the phasing intervals between preferred inter-motif distances are highly significant. Our model was intentionally designed to account for spurious overrepresentation peaks by considering peak separation distances collectively across a comprehensive list of all pairs of 5-mers. We explicitly focused on motifs with distinguishing phasing intervals between preferred distances, and whose consistency was unlikely to be due to chance.
We have found that for many motif-pairs exhibiting multiple preferred separation distances, the intervals between preferred inter-motif distances are often found near the range of ∼8–9 bp. This corresponds to approximately 2 bp less than the number of nucleotides in one turn of the DNA double-helix (∼10.5 bp). This finding may reflect a structural requirement for interacting TF proteins to be positioned in a particular orientation relative to the turn of the DNA molecule, albeit at a distortion of the helical structure upon protein binding. The deviation from the expected number of 10.5 bp is worth noting, particularly due to the robustness of the signal occurring across such a large number of motif-pairs. There are some possible explanations available from the literature. Structural analyses have shown that protein binding, and the binding of multi-protein complexes in particular, distort the conformation of the DNA (80–83), thus affecting the helical characteristics of the DNA. An alternative, although not necessarily exclusive, interpretation is that the occurring protein-protein interactions may either be stabilized by alterations of the DNA molecule or require them for collective binding. Selective binding of proteins to DNA involves not only sequence-specific elements within the DNA, but also topological characteristics of the DNA molecule (84–86); this is known to be particularly true during the recruitment of multiple interacting proteins to the DNA (85,86). Thus, although the biological explanation of the observed pattern remains unclear, these results are not inconsistent with our current knowledge of protein–protein and protein–DNA interactions.
Nine motif clusters were found to exhibit highly consistent phasing intervals around 8–10 bp. Six of these consensus motifs matched known binding sites in TRANSFAC, including the binding motifs for the NFY, NRF1, EVI1 and the MADS-box protein family. We subsequently used our phasing criteria to predict multiple partner motifs for each of these nine motifs, resulting in 112 motif-pairs predicted to bind interacting TFs (Supplementary Data S5). We have illustrated several examples of our motif-pair predictions in Tables 4 and 5, which show the partner motifs pairing with the NFY-binding element and the MADS-box protein family-binding sequence. All partners predicted to pair with the NFY-binding motif correspond to either the NFY or SP1-binding elements; both NFY–NFY and NFY–SP1 factor-pair interactions are documented in the literature (64,65). The MADS-box family consensus sequences predicted during the analysis bind both the myocyte enhancer factor 2 (MEF2) and the serum response factor (SRF) (87). These two factors are known to be involved in complex extra-cellular signaling pathways, playing multiple roles involving cell differentiation and development (88–90). Both MEF2 and SRF regulate gene expression through the recruitment of multiple accessory co-factors whose presence or absence within the complex cause differential expression of their target genes (66,67,86), and therefore we would expect a large number of partner motifs to pair with their binding elements. Many predicted partner motifs were predicted on both orientations, with eight reverse complement-pairs occurring across opposing strands of the MADS-box-binding motif. The mutual directionality of the MADS-box/partner motif-pairs was found to be highly conserved, with each individual strand of the partner motif pairing with one, but not both, orientations of the MADS-box-binding motif. The majority of partner motifs pairing with the MADS-box-binding element match known TF-binding sites. In addition, 12 motif partners bind proteins known to be involved in either signal transduction pathways or developmental processes. Three such factors belong to the homeobox family, whose members play a crucial role in early development (91–93). Many of the remaining partner motifs may play unknown functional roles in concert with one of the MADS-box protein factors.
We note that the MRF method comprises a general tool that can be used to analyze spatial preferences, and that our present analysis represents only one of the many possible applications of the model. Expanding our knowledge about the nature of collective protein binding is crucial to further the understanding of gene regulation, and future studies are necessary to demonstrate the full efficacy of the method presented here.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences; National Institutes of Health [grant numbers HG004065 and 5P50-GM-081883]. Funding for open access charge: National Institutes of Health grant number HG004065 and 5P50-GM-081883.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Sayan Mukherjee, Jeff Thorne, the Wray lab and STU labs for their comments.
REFERENCES
- 1.Pedersen AG, Baldi P, Chauvin Y, Brunak S. The biology of eukaryotic promoter prediction – a review. Comput. Chem. 1999;23:191–207. doi: 10.1016/s0097-8485(99)00015-7. [DOI] [PubMed] [Google Scholar]
- 2.Latchman DS. Eukaryotic transcription factors. Biochem. J. 1990;270:281–289. doi: 10.1042/bj2700281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV, Romano LA. The evolution of transcriptional regulation in eukaryotes. Mol. Biol. Evol. 2003;20:1377–1419. doi: 10.1093/molbev/msg140. [DOI] [PubMed] [Google Scholar]
- 4.Lawrence CE, Reilly AA. An expectation maximization (EM) algorithm for the identification and characterization of common sites in unaligned biopolymer sequences. Proteins. 1990;7:41–51. doi: 10.1002/prot.340070105. [DOI] [PubMed] [Google Scholar]
- 5.Bailey TL, Elkan C. Unsupervised learning of multiple motifs biopolymers using expectation maximization. Machine Learning. 1995;21:51–80. [Google Scholar]
- 6.Roth FP, Hughes JD, Estep PW, Church GM. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat. Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- 7.van Helden J, Andre B, Collado-Vides J. Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J. Mol. Biol. 1998;281:827–842. doi: 10.1006/jmbi.1998.1947. [DOI] [PubMed] [Google Scholar]
- 8.van Helden J, Rios AF, Collado-Vides J. Discovering regulatory elements in non-coding sequences by analysis of spaced dyads. Nucleic Acids Res. 2000;28:1808–1818. doi: 10.1093/nar/28.8.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thijs G, Lescot M, Marchal K, Rombauts S, De Moor B, Rouze P, Moreau Y. A higher-order background model improves the detection of promoter regulatory elements by Gibbs sampling. Bioinformatics. 2001;17:1113–1122. doi: 10.1093/bioinformatics/17.12.1113. [DOI] [PubMed] [Google Scholar]
- 10.Sinha S, Tompa M. YMF: A program for discovery of novel transcription factor binding sites by statistical overrepresentation. Nucleic Acids Res. 2003;31:3586–3588. doi: 10.1093/nar/gkg618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavesi G, Mereghetti P, Mauri G, Pesole G. Weeder Web: discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res. 2004;32:W199–W203. doi: 10.1093/nar/gkh465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bussemaker HJ, Li H, Siggia ED. Building a dictionary for genomes: identification of presumptive regulatory sites by statistical analysis. Proc. Natl Acad. Sci. USA. 2000;97:10096–10100. doi: 10.1073/pnas.180265397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bussemaker HJ, Li H, Siggia ED. Regulatory element detection using correlation with expression. Nat. Genet. 2001;27:167–171. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- 14.Down TA, Bergman CM, Su J, Hubbard TJ. Large-scale discovery of promoter motifs in Drosophila melanogaster. PLoS Comput. Biol. 2007;3:e7. doi: 10.1371/journal.pcbi.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keles S, van der Laan M, Eisen MB. Identification of regulatory elements using a feature selection method. Bioinformatics. 2002;18:1167–1175. doi: 10.1093/bioinformatics/18.9.1167. [DOI] [PubMed] [Google Scholar]
- 16.Hannenhalli S, Levy S. Predicting transcription factor synergism. Nucleic Acids Res. 2002;30:4278–4284. doi: 10.1093/nar/gkf535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Hannenhalli S. A mammalian promoter model links cis elements to genetic networks. Biochem. Biophys. Res. Commun. 2006;347:166–177. doi: 10.1016/j.bbrc.2006.06.062. [DOI] [PubMed] [Google Scholar]
- 18.Vardhanabhuti S, Wang J, Hannenhalli S. Position and distance specificity are important determinants of cis-regulatory motifs in addition to evolutionary conservation. Nucleic Acids Res. 2007;35:3203–3213. doi: 10.1093/nar/gkm201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.FitzGerald PC, Shlyakhtenko A, Mir AA, Vinson C. Clustering of DNA sequences in human promoters. Genome Res. 2004;14:1562–1574. doi: 10.1101/gr.1953904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.FitzGerald PC, Sturgill D, Shyakhtenko A, Oliver B, Vinson C. Comparative genomics of Drosophila and human core promoters. Genome Biol. 2006;7:R53. doi: 10.1186/gb-2006-7-7-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tharakaraman K, Bodenreider O, Landsman D, Spouge JL, Marino-Ramirez L. The biological function of some human transcription factor binding motifs varies with position relative to the transcription start site. Nucleic Acids Res. 2008;36:2777–2786. doi: 10.1093/nar/gkn137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beachy PA, Varkey J, Young KE, von Kessler DP, Sun BI, Ekker SC. Cooperative binding of an Ultrabithorax homeodomain protein to nearby and distant DNA sites. Mol. Cell Biol. 1993;13:6941–6956. doi: 10.1128/mcb.13.11.6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biggin MD, McGinnis W. Regulation of segmentation and segmental identity by Drosophila homeoproteins: the role of DNA binding in functional activity and specificity. Development. 1997;124:4425–4433. doi: 10.1242/dev.124.22.4425. [DOI] [PubMed] [Google Scholar]
- 24.Elkon R, Linhart C, Sharan R, Shamir R, Shiloh Y. Genome-wide in silico identification of transcriptional regulators controlling the cell cycle in human cells. Genome Res. 2003;13:773–780. doi: 10.1101/gr.947203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pilpel Y, Sudarsanam P, Church GM. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat. Genet. 2001;29:153–159. doi: 10.1038/ng724. [DOI] [PubMed] [Google Scholar]
- 26.Keles S, van der Laan MJ, Vulpe C. Regulatory motif finding by logic regression. Bioinformatics. 2004;20:2799–2811. doi: 10.1093/bioinformatics/bth333. [DOI] [PubMed] [Google Scholar]
- 27.Senger K, Armstrong GW, Rowell WJ, Kwan JM, Markstein M, Levine M. Immunity regulatory DNAs share common organizational features in Drosophila. Mol. Cell. 2004;13:19–32. doi: 10.1016/s1097-2765(03)00500-8. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Cheng AS, Jin VX, Paik HH, Fan M, Li X, Zhang W, Robarge J, Balch C, Davuluri RV, et al. A mixture model-based discriminate analysis for identifying ordered transcription factor binding site pairs in gene promoters directly regulated by estrogen receptor-alpha. Bioinformatics. 2006;22:2210–2216. doi: 10.1093/bioinformatics/btl329. [DOI] [PubMed] [Google Scholar]
- 29.Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat. Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- 30.Ma X-T, Tang H-X. Predicting polymerase II core promoters by cooperating transcription factor binding sites in eukaryotic genes. Acta Biochimica et Biophysica Sinica. 2004;36:250–258. doi: 10.1093/abbs/36.4.250. [DOI] [PubMed] [Google Scholar]
- 31.Beer MA, Tavazoie S. Predicting gene expression from sequence. Cell. 2004;117:185–198. doi: 10.1016/s0092-8674(04)00304-6. [DOI] [PubMed] [Google Scholar]
- 32.Karlin S, Burge C, Campbell AM. Statistical analyses of counts and distributions of restriction sites in DNA sequences. Nucleic Acids Res. 1992;20:1363–1370. doi: 10.1093/nar/20.6.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broyden C. A class of methods for solving nonlinear simulationeous equations. Mathematic Comput. 1965;19:577–593. [Google Scholar]
- 34.Press WH, Teukolsky SA, Vetterling WT, Flannery BP. Numerical Recipes in C: The Art of Scientific Computing. 2nd. New York: Cambridge University Press; 2002. [Google Scholar]
- 35.Davison AC. Statistical Models. New York: Cambridge University Press; 2003. [Google Scholar]
- 36.Burden RL, Douglas JF. Numerical Analysis. 7th. Pacific Grove, CA: Wadsworth Group; 2001. [Google Scholar]
- 37.Kullback S, Leiber RA. On information theory and sufficiency. Ann. Mathematical Stat. 1951;22:79–86. [Google Scholar]
- 38.Cavener DR. Comparison of the consensus sequence flanking translational start sites in Drosophila and vertebrates. Nucleic Acids Res. 1987;15:1353–1361. doi: 10.1093/nar/15.4.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahony S, Benos PV. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007;35:W253–W258. doi: 10.1093/nar/gkm272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 43.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 44.Maglott DR, Katz KS, Sicotte H, Pruitt KD. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 2000;28:126–128. doi: 10.1093/nar/28.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pruitt KD, Maglott DR. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 2001;29:137–140. doi: 10.1093/nar/29.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bucher P. Weight matrix descriptions of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. J. Mol. Biol. 1990;212:563–578. doi: 10.1016/0022-2836(90)90223-9. [DOI] [PubMed] [Google Scholar]
- 47.Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xi H, Yu Y, Fu Y, Foley J, Halees A, Weng Z. Analysis of overrepresented motifs in human core promoters reveals dual regulatory roles of YY1. Genome Res. 2007;17:798–806. doi: 10.1101/gr.5754707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crothers DM, Haran TE, Nadeau JG. Intrinsically bent DNA. J. Biol. Chem. 1990;265:7093–7096. [PubMed] [Google Scholar]
- 50.Hizver J, Rozenberg H, Frolow F, Rabinovich D, Shakked Z. DNA bending by an adenine—thymine tract and its role in gene regulation. Proc. Natl Acad. Sci. USA. 2001;98:8490–8495. doi: 10.1073/pnas.151247298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Segal E, Widom J. Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr. Opin. Struct. Biol. 2009;19:65–71. doi: 10.1016/j.sbi.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rich A, Nordheim A, Wang AH. The chemistry and biology of left-handed Z-DNA. Annu. Rev. Biochem. 1984;53:791–846. doi: 10.1146/annurev.bi.53.070184.004043. [DOI] [PubMed] [Google Scholar]
- 53.Naylor LH, Clark EM. d(TG)n.d(CA)n sequences upstream of the rat prolactin gene form Z-DNA and inhibit gene transcription. Nucleic Acids Res. 1990;18:1595–1601. doi: 10.1093/nar/18.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rothenburg S, Koch-Nolte F, Rich A, Haag F. A polymorphic dinucleotide repeat in the rat nucleolin gene forms Z-DNA and inhibits promoter activity. Proc. Natl Acad. Sci. USA. 2001;98:8985–8990. doi: 10.1073/pnas.121176998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang C, Bolotin E, Jiang T, Sladek FM, Martinez E. Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene. 2007;389:52–65. doi: 10.1016/j.gene.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ponjavic J, Lenhard B, Kai C, Kawai J, Carninci P, Hayashizaki Y, Sandelin A. Transcriptional and structural impact of TATA-initiation site spacing in mammalian core promoters. Genome Biol. 2006;7:R78. doi: 10.1186/gb-2006-7-8-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tharakaraman K, Marino-Ramirez L, Sheetlin S, Landsman D, Spouge JL. Alignments anchored on genomic landmarks can aid in the identification of regulatory elements. Bioinformatics. 2005;21(Suppl. 1):i440–i448. doi: 10.1093/bioinformatics/bti1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aso T, Conaway JW, Conaway RC. Role of core promoter structure in assembly of the RNA polymerase II preinitiation complex. A common pathway for formation of preinitiation intermediates at many TATA and TATA-less promoters. J. Biol. Chem. 1994;269:26575–26583. [PubMed] [Google Scholar]
- 59.Kaufmann J, Smale ST. Direct recognition of initiator elements by a component of the transcription factor IID complex. Genes Dev. 1994;8:821–829. doi: 10.1101/gad.8.7.821. [DOI] [PubMed] [Google Scholar]
- 60.Martinez E, Chiang CM, Ge H, Roeder RG. TATA-binding protein-associated factor(s) in TFIID function through the initiator to direct basal transcription from a TATA-less class II promoter. EMBO J. 1994;13:3115–3126. doi: 10.1002/j.1460-2075.1994.tb06610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ioshikhes I, Trifonov EN, Zhang MQ. Periodical distribution of transcription factor sites in promoter regions and connection with chromatin structure. Proc. Natl Acad. Sci. USA. 1999;96:2891–2895. doi: 10.1073/pnas.96.6.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lewin B. Genes VII. Oxford: Oxford University Press; 2000. [Google Scholar]
- 63.White RJ. Gene Transcription: Mechanisms and Control. Malden, Mass: Blackwell Science; 2001. [Google Scholar]
- 64.Liberati C, di Silvio A, Ottolenghi S, Mantovani R. NF-Y binding to twin CCAAT boxes: role of Q-rich domains and histone fold helices. J. Mol. Biol. 1999;285:1441–1455. doi: 10.1006/jmbi.1998.2384. [DOI] [PubMed] [Google Scholar]
- 65.Benfante R, Antonini RA, Vaccari M, Flora A, Chen F, Clementi F, Fornasari D. The expression of the human neuronal alpha3 Na+,K+-ATPase subunit gene is regulated by the activity of the Sp1 and NF-Y transcription factors. Biochem. J. 2005;386:63–72. doi: 10.1042/BJ20041294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Groisman R, Masutani H, Leibovitch MP, Robin P, Soudant I, Trouche D, Harel-Bellan A. Physical interaction between the mitogen-responsive serum response factor and myogenic basic-helix-loop-helix proteins. J. Biol. Chem. 1996;271:5258–5264. doi: 10.1074/jbc.271.9.5258. [DOI] [PubMed] [Google Scholar]
- 67.Hanlon M, Sturgill TW, Sealy L. ERK2- and p90(Rsk2)-dependent pathways regulate the CCAAT/enhancer-binding protein-beta interaction with serum response factor. J. Biol. Chem. 2001;276:38449–38456. doi: 10.1074/jbc.M102165200. [DOI] [PubMed] [Google Scholar]
- 68.Yamada K, Osawa H, Granner DK. Identification of proteins that interact with NF-YA. FEBS Lett. 1999;460:41–45. doi: 10.1016/s0014-5793(99)01311-3. [DOI] [PubMed] [Google Scholar]
- 69.Zhu C, Johansen FE, Prywes R. Interaction of ATF6 and serum response factor. Mol. Cell Biol. 1997;17:4957–4966. doi: 10.1128/mcb.17.9.4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tompa M, Li N, Bailey TL, Church GM, De Moor B, Eskin E, Favorov AV, Frith MC, Fu Y, Kent WJ, et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005;23:137–144. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- 71.Cooper SJ, Trinklein ND, Nguyen L, Myers RM. Serum response factor binding sites differ in three human cell types. Genome Res. 2007;17:136–144. doi: 10.1101/gr.5875007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Natesan S, Gilman M. YY1 facilitates the association of serum response factor with the c-fos serum response element. Mol. Cell Biol. 1995;15:5975–5982. doi: 10.1128/mcb.15.11.5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen CY, Schwartz RJ. Recruitment of the tinman homolog Nkx-2.5 by serum response factor activates cardiac alpha-actin gene transcription. Mol. Cell Biol. 1996;16:6372–6384. doi: 10.1128/mcb.16.11.6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pal M, Ponticelli AS, Luse DS. The role of the transcription bubble and TFIIB in promoter clearance by RNA polymerase II. Mol. Cell. 2005;19:101–110. doi: 10.1016/j.molcel.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 75.Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 77.Hughes JD, Estep PW, Tavazoie S, Church GM. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- 78.Aerts S, Van Loo P, Thijs G, Moreau Y, De Moor B. Computational detection of cis-regulatory modules. Bioinformatics. 2003;19(Suppl. 2):ii5–ii14. doi: 10.1093/bioinformatics/btg1052. [DOI] [PubMed] [Google Scholar]
- 79.Segal E, Sharan R. A discriminative model for identifying spatial cis-regulatory modules. J. Comput. Biol. 2005;12:822–834. doi: 10.1089/cmb.2005.12.822. [DOI] [PubMed] [Google Scholar]
- 80.Tomovic A, Oakeley EJ. Computational structural analysis: multiple proteins bound to DNA. PLoS ONE. 2008;3:e3243. doi: 10.1371/journal.pone.0003243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jones S, van Heyningen P, Berman HM, Thornton JM. Protein-DNA interactions: a structural analysis. J. Mol. Biol. 1999;287:877–896. doi: 10.1006/jmbi.1999.2659. [DOI] [PubMed] [Google Scholar]
- 82.Prabakaran P, Siebers JG, Ahmad S, Gromiha MM, Singarayan MG, Sarai A. Classification of protein-DNA complexes based on structural descriptors. Structure. 2006;14:1355–1367. doi: 10.1016/j.str.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 83.Pan Y, Nussinov R. p53-Induced DNA bending: the interplay between p53-DNA and p53-p53 interactions. J. Phys. Chem. B. 2008;112:6716–6724. doi: 10.1021/jp800680w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harrington RE. DNA curving and bending in protein-DNA recognition. Mol. Microbiol. 1992;6:2549–2555. doi: 10.1111/j.1365-2958.1992.tb01431.x. [DOI] [PubMed] [Google Scholar]
- 85.Kerppola TK. Transcriptional cooperativity: bending over backwards and doing the flip. Structure. 1998;6:549–554. doi: 10.1016/s0969-2126(98)00057-4. [DOI] [PubMed] [Google Scholar]
- 86.West AG, Sharrocks AD. MADS-box transcription factors adopt alternative mechanisms for bending DNA. J. Mol. Biol. 1999;286:1311–1323. doi: 10.1006/jmbi.1999.2576. [DOI] [PubMed] [Google Scholar]
- 87.Huang K, Louis JM, Donaldson L, Lim FL, Sharrocks AD, Clore GM. Solution structure of the MEF2A-DNA complex: structural basis for the modulation of DNA bending and specificity by MADS-box transcription factors. EMBO J. 2000;19:2615–2628. doi: 10.1093/emboj/19.11.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miano JM, Long X, Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am. J. Physiol. Cell Physiol. 2007;292:C70–C81. doi: 10.1152/ajpcell.00386.2006. [DOI] [PubMed] [Google Scholar]
- 89.Chai J, Tarnawski AS. Serum response factor: discovery, biochemistry, biological roles and implications for tissue injury healing. J. Physiol. Pharmacol. 2002;53:147–157. [PubMed] [Google Scholar]
- 90.Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- 91.Whitelaw E. The role of DNA-binding proteins in differentiation and transformation. J. Cell Sci. 1989;94(Pt 2):169–173. doi: 10.1242/jcs.94.2.169. [DOI] [PubMed] [Google Scholar]
- 92.Lonai P, Orr-Urtreger A. Homeogenes in mammalian development and the evolution of the cranium and central nervous system. FASEB J. 1990;4:1436–1443. doi: 10.1096/fasebj.4.5.1968407. [DOI] [PubMed] [Google Scholar]
- 93.Nunes FD, de Almeida FC, Tucci R, de Sousa SC. Homeobox genes: a molecular link between development and cancer. Pesqui Odontol Bras. 2003;17:94–98. doi: 10.1590/s1517-74912003000100018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.