

Abstract
In its classical form, autophagy is a pathway by which cytoplasmic constituents, including intracellular pathogens, are sequestered in a double-membrane–bound autophagosome and delivered to the lysosome for degradation. This pathway has been linked to diverse aspects of innate and adaptive immunity, including pathogen resistance, production of type I interferon, antigen presentation, tolerance and lymphocyte development, as well as the negative regulation of cytokine signaling and inflammation. Most of these links have emerged from studies in which genes encoding molecules involved in autophagy are inactivated in immune effector cells. However, it is not yet known whether all of the critical functions of such genes in immunity represent ‘classical autophagy’ or possible as-yet-undefined autophagolysosome-independent functions of these genes. This review summarizes phenotypes that result from the inactivation of autophagy genes in the immune system and discusses the pleiotropic functions of autophagy genes in immunity.
Classical macroautophagy (called ‘autophagy’ here) involves the sequestration of cytoplasmic contents in a characteristic double-membraned vacuole, the autophagosome. Fusion of the outer autophagosomal membrane with the lysosome and the subsequent breakdown of the inner membrane results in the exposure of the sequestered cytoplasmic material to lysosomal hydrolases1,2 (Fig. 1). A related term, ‘xenophagy’, refers to the use of the autophagy pathway to digest foreign rather than self constituents3. The autophagy pathway has two main physiological functions: it rids the cell of unwanted constituents, and it recycles cytoplasmic material so that cells can maintain macromolecular synthesis and energy homeostasis during stressful conditions. These functions probably underlie the well described roles of autophagy in cell survival and in the prevention of neurodegeneration, cancer and aging and, at least in part, the protective roles of autophagy in pathogen-infected cells1,2.
Figure 1.

The autophagy pathway and its regulation. The autophagy pathway proceeds through a series of stages, including nucleation of the autophagic vesicle, elongation and closure of the autophagosome membrane to envelop cytoplasmic constituents, docking of the autophagosome with the lysosome, and degradation of the cytoplasmic material inside the autophagosome. Vesicle nucleation depends on a class III PI(3)K complex that contains various proteins (in light yellow box at right), as well as additional proteins that regulate the activity of this complex (such as rubicon, UVRAG, Ambra-1 and Bif-1). Vesicle elongation and completion involves the activity of two ubiquitin-like conjugation systems (light blue box at right). The autophagy pathway is positively regulated (green box at left) and negatively regulated (red box at left) by diverse environmental and immunological signals. TNF, tumor necrosis factor; PE, phosphatidylethanolamine; Gly, glycine; E1 and E2, ligases for ubiquitin-like conjugation systems.
The autophagy pathway involves the concerted action of evolutionarily conserved gene products involved in the initiation of autophagy, elongation and closure of the autophagosome, and lysosomal fusion (Fig. 1). Beclin 1, in complex with the class III phosphatidylinositol-3-OH kinase (PI(3)K) Vps34 and other proteins, is important for the nucleation of autophagosomes. This macromolecular complex is critical for integrating multiple signals that positively and negatively regulate autophagy. The autophagy (‘Atg’) protein Atg7 is required for two ubiquitin-like conjugation pathways that in concert result in the covalent conjugation of Atg5 to Atg12 and the conversion of LC3 (the mammalian ortholog of yeast Atg8) to its phosphatidylethanolamine-conjugated LC3-II form. The Atg5-Atg12 conjugate forms part of a large complex with Atg16L1 that is responsible for the membrane localization of the autophagic machinery and the generation of the autophagosome4. At present, there are more than 20 recognized Atg proteins1, and it is likely that more will be identified. For example, FIP200, a protein reported to interact with the kinases FAK, Pyk2 and ASK1, the tumor suppressor TSC1, the tumor suppressor p53 and tumor necrosis factor receptor–associated factor 2, has been found to be critical for the initiation of autophagy through interaction with the kinases ULK1 and ULK2 (mammalian orthologs of yeast Atg1)5. Furthermore, four independent groups have identified a candidate functional mammalian ortholog of yeast Atg14, known as either human Atg14 or Barkor, that interacts with Beclin 1 and the Vps34 class III PI(3)K complex to initiate autophagic vesicle nucleation6–9. Of the many proteins involved in autophagy, Beclin 1, Atg5, Atg7, LC3, Atg12 and Atg16L1 have been studied in one or more aspects of immunity.
The identification of this core molecular machinery has revolutionized the ability to detect and genetically manipulate the autophagy pathway. This has led to considerable advances in understanding the function of genes encoding molecules involved in autophagy (called ‘autophagy genes’ here) in immunity and other biological processes1,2. In terms of immunity, studies have shown that autophagy is regulated by pathways critical for the function and differentiation of cells of the immune system, including Toll-like receptors (TLRs), p47 GTPases, eIF2α kinases, and cytokines such as interferon-γ (IFN-γ)10–13. Furthermore, autophagy genes are important for thymic selection, lymphocyte development and survival, antigen presentation, killing of intracellular pathogens, and tissue homeostasis. There is also increasing evidence that autophagy genes may regulate innate immune signaling in a cell type–specific way.
Although some of the functions described above, such as the targeting of microbes for lysosomal degradation (xenophagy), may unequivocally reflect the involvement of classical autophagy in immunity (Fig. 2), it is not yet clear whether all functions of autophagy genes in immunity relate to the autophagy pathway itself (Fig. 3). This uncertainty arises from both the assays commonly used to detect autophagy and the lack of universal criteria for interpreting reverse-genetic studies. For example, the most commonly used autophagy assays, biochemical detection of the lipidated form of LC3 (LC3-II) and detection of the localization of LC3-II to punctate dots by light microscopy, are subject to many interpretations. Most investigators now recognize that detection of increased LC3-II can indicate either more autophagosome formation or a block in autophagosomal maturation, which necessitates the use of ancillary approaches to distinguish between these possibilities14. Perhaps less well appreciated is the idea that LC3 dots may represent the targeting of LC3 to structures other than autophagosomes, such as phagosomes, double-membraned scaffolds for the replication complexes of positive-strand RNA viruses, or even protein aggregates15–17. Similarly, LC3 may become lipidated, forming LC3-II, in the absence of autophagosome formation18,19. Thus, whereas LC3-II formation and localization of LC3 punctae are hallmark features of autophagosome formation (and sensitive parameters for the detection of autophagy), they lack complete specificity as markers of classical autophagy. The direct demonstration of a function for autophagosomes in a process is the gold standard for proving that autophagy is involved. An extensive discussion of the various assays used in autophagy research, as well as the criteria for their use and interpretation, has been provided in a consensus paper published by many of the authorities in the field14.
Figure 2.

Immunological processes involving autophagy. The colored boxes present five immunological processes that, on the basis of data available at present, involve classical autophagy. MIIC, MHC class II loading compartment.
Figure 3.
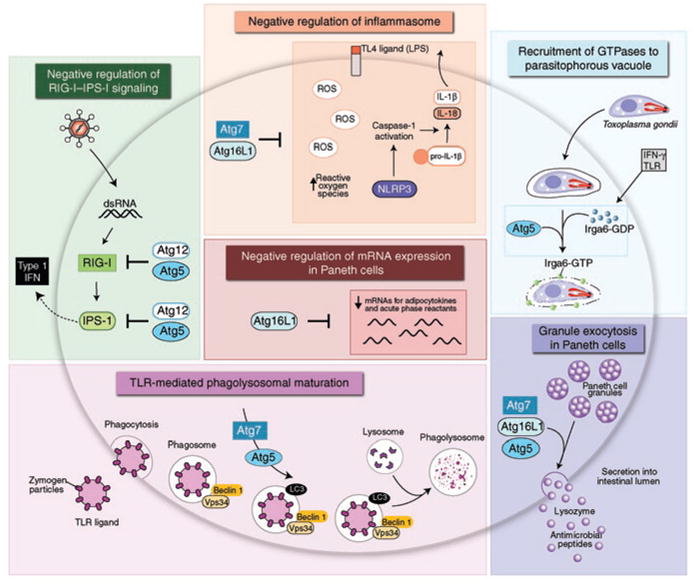
Immunological processes involving autophagy genes. The colored boxes present six immunologic processes that involve various autophagy genes but that are not yet proven to involve classical autophagy.
The genetic knockout or knockdown of core autophagy genes is an effective way to turn off the autophagy pathway, but it is often unclear whether resulting phenotypes are due to a deficiency of classical autophagy or autophagy-independent functions of the autophagy genes. As reviewed elsewhere, autophagy genes are known to have alternative functions, including those in other membrane-trafficking events, axonal elongation and cell death20,21. Furthermore, investigators often assume that phenotypes noted in autophagy gene–deficient cells or organisms are due to lack of classical autophagy without providing direct experimental proof. Notably, for many studies of the involvement of autophagy genes in immunity (Fig. 3), it is not clear how classical autophagy, involving the autophagolysosomal degradation of sequestered contents, could contribute to the described functions of autophagy genes. Thus, it is possible that autophagy genes act in other cellular processes that affect immune effector cell development and function. Here we will discuss advances related to the function of autophagy genes in the immune system.
Autophagy genes in antimicrobial defense
More than a decade ago, the first published paper on Beclin 1, a mammalian autophagy protein, demonstrated an antiviral function for exogenous neuronal expression of Beclin 1 in mice with alphavirus encephalitis22. Subsequently, endogenous autophagy genes were shown to be critical for the successful innate immune response to fungal, bacterial and viral pathogens in plants23, and viral evasion of Beclin 1 function by a herpes simplex virus neurovirulence factor was found to be essential for lethal encephalitis in mice24. These studies collectively suggested a likely function for autophagy genes in pathogen defense in vivo. In parallel, many studies demonstrated a function for autophagy in vitro in defense against invading pathogens, including group A Streptococcus, Shigella flexneri, Mycobacterium tuberculosis, Salmonella typhimurium and Toxoplasma gondii10,11,13.
Two recent studies have further confirmed an antimicrobial function for autophagy genes in host defense in vivo against intracellular pathogens and have identified previously unknown relationships among innate immune signaling, autophagy genes and potential autophagy-independent functions of autophagy genes. An innate microbial sensor, the peptidoglycan-recognition protein PRGP-LE, which recognizes bacterial diaminopimelic acid–type peptidoglycan, is crucial for autophagic control of Listeria monocytogenes infection in fly hemocytes and for host survival25. PRGP-LE-mediated resistance is associated with autophagy induction, requires the autophagy gene Atg5 and presumably involves the xenophagic degradation of bacteria (as bacteria are visualized in autophagosomes in wild-type animals). In this case, xenophagy targets cytoplasmic bacteria that have escaped from the endosome for envelopment in an autophagosome and destruction. In other cases in which a pathogen inside a vesicular structure is targeted (such as mycobacteria inside phagosomes, or salmonella inside vacuoles), the membrane dynamics involved are incompletely defined. It may be that an autophagosome can envelop the entire vesicular structure containing the pathogen or that the autophagic machinery somehow enhances phagolyosomal maturation. An alternative mechanism is discussed below for T. gondii.
Although TLR signaling has been linked to autophagy induction (discussed below), the consequences of this autophagy induction have not been studied so far; therefore, the finding of for a function for PRGP-LE in listeria infection in flies is the first to demonstrate involvement of a cytoplasmic pattern-recognition receptor in the delivery of microbes to autophagosomes for degradation and in autophagy-mediated pathogen defense. One speculation is that other pattern-recognition receptors function similarly in innate immunity to target diverse microbes or microbial products to the autophagosome. A related issue is whether autophagosomal targeting motifs, such as monoubiquitination or polyubiquitination, that can target cellular proteins and organelles to the autophagosome through the adaptor protein SQSTM1 (p62)26 also target microbial products. It will be useful to determine how the selective delivery of microbes, through cytoplasmic recognition by innate sensors and/or ubiquitination, may result in the selective removal or killing of pathogens by xenophagy, the activation of innate immune signaling and/or the generation of antigenic peptides from these pathogens for presentation to T cells.
In mice, phagocytic cell–specific deletion of Atg5 results in greater susceptibility to infection with two different types of intracellular pathogens, the bacterium L. monocytogenes and the protozoan T. gondii27. Such work has provided important confirmation that autophagy genes are involved in antimicrobial host defense in mammals. However, in contrast to studies in the fly, in which autophagy is believed to degrade bacteria that invade professional phagocytes called hemocytes, Atg5 is proposed to control T. gondii by a mechanism independent of autophagosome formation. Published work suggests that IFN-γ-mediated control of T. gondii involves the localization of immunity-related GTPases to the parasitophorous vacuole, vacuolar membrane stripping, and either the induction of green fluorescent protein–LC3–positive vesicular structures that localize together with GTPases28 or the formation of autophagic membranes near the damaged parasitophorous vacuoles29. In Atg5−/− macrophages, the GTPase IIGP1 (Irga6) fails to localize to the parasitophorous vacuole, which results in a lack of disruption of parasitophorous vacuole membranes. Thus, the autophagy machinery, or at least Atg5, may be critical in the destruction of a parasite-containing vesicular structure through the recruitment of immunity-related GTPases to the parasitophorous vacuole.
As for T. gondii–infected mouse astro-cytes28, autophagosomal membranes are not found surrounding the damaged parasitophorous vacuole in wild-type macrophages, which suggests that the GTPase-recruitment function of Atg5, rather than autophagic entrapment of T. gondii, may be the main mechanism by which Atg5 functions ‘downstream’ of IFN-γ to mediate intracellular resistance to this parasite. Studies using real-time microscopy have documented that killing of T. gondii is not consistently associated with localization of LC3 to the parasitophorous vacuole30. These data collectively provide support for a model in which an autophagy gene, Atg5, is involved in host defense independently of autophagosome formation. An important question is whether recruitment of GTPase to the parasitophorous vacuole requires autophagy proteins other than Atg5 and how Atg5, and perhaps the autophagic machinery, mediates this trafficking event.
Immunity-related GTPases and autophagy
Several different immune signals positively regulate autophagy, including PKR, TLRs, tumor necrosis factor, the CD40–CD40 ligand interaction, IFN-γ and the immunity-related GTPases10,13,27,31,32, whereas T helper type 2 cytokines (IL-4 and 1L-13) negatively regulate autophagy33. One of the most rapidly evolving areas of research centers on the relationship between autophagy and immunity-related GTPases (IRGs or p47 GTPases), a family of proteins crucial for IFN-γ-mediated resistance to intracellular pathogens34. Although only the expression of mouse p47 GTPases seems to be regulated by IFN-γ, both mouse (LRG47) and human (Irgm1) p47 GTPases are thought to be required for IFN-γ induced autophagy and antimycobacterial activity in macrophages31,32. Furthermore, delivery of T. gondii to autophagosomes in macrophages may occur in an Irgm3 (IGTP)-dependent way29. Thus, a consensus is emerging that p47 GTPases are important in the IFN-γ-dependent autophagic elimination of intracellular pathogens. The precise mechanisms by which p47 GTPases exert this effect, however, remain unknown. One theory is that their localization to pathogen-containing phagosomes or vacuoles somehow facilitates damage to the pathogen-containing compartment with subsequent targeting of pathogen to the autophagosome.
Beyond the proposed involvement of p47 GTPases in the autophagic control of intracellular pathogens, evidence suggests potentially more complex crosstalk between this family of immune effectors and autophagy. As noted above, the autophagy protein Atg5 is needed for recruitment of the GTPase Irga6 to the T. gondii parasitophorous vacuole27. Thus, autophagy proteins can function ‘upstream’ of p47 GTPases to regulate their trafficking to pathogen-containing compartments. As Irga6 that is associated with the parasitophorous vacuole is in an active GTP-bound state and Irga6 in uninfected cells is in an inactive GDP-bound state35, one obvious question is whether the effects of Atg5 on Irga6 localization occur through regulation of the GDP- or GTP-bound state of the GTPase. Another question is whether the Atg5-Atg12-Atg16L1 complex enhances the ability of Irga6, and perhaps other p47 GTPases, to form multimers on intracellular membranes. Furthermore, it is not yet known whether pathogen-containing phagosomes, in addition to parasitophorous vacuoles, may be sites of autophagy gene–dependent recruitment of p47 GTPase. Along these lines, it will be useful to determine whether Atg5 and other autophagy genes are required for the recruitment of p47 GTPases to mycobacteria-containing phagosomes.
A somewhat paradoxical function for Irgm1 in negatively regulating IFN-γ-induced autophagy has also been proposed. Mice deficient in Irgm1 have less proliferation of mature effector CD4+ T cell populations in the presence of IFN-γ36. This diminished expansion seems to be due to the induction of a caspase-independent cell death program that is blocked by PI(3)K inhibitors such as wortmanin or LY294002 or small interfering RNA specific for the autophagy gene encoding Beclin 1. Thus, Irgm1 may have dual interactions with the autophagy pathway that act in synergy to promote IFN-γ antimicrobial interactions; Irgm1 may signal autophagic sequestration of intracellular pathogens in macrophages while simultaneously limiting IFN-γ-induced autophagic death of effector T lymphocytes. That suggests the possibility of divergent functions for Irgm1 and potentially other p47 GTPases in autophagy regulation in different cell types. At present, it is unknown how Irgm1 might negatively regulate IFN-γ-induced autophagy to preserve activated CD4+ T cell populations. Furthermore, the possibility has not yet been formally excluded that the greater vacuolization in Irgm1−/− lymphocytes is a function of aberrant membrane trafficking rather than enhanced autophagic flux. As reviewed elsewhere, whether autophagy is truly involved in mediating cell death also remains controversial21.
TLRs and autophagy
TLRs and the autophagy pathway intersect at many different levels37: TLRs can regulate autophagy induction38,39, the autophagy machinery can be used to deliver viral genetic material to endosomal TLRs for efficient induction of type I interferon40, and TLRs may act in the recruitment of autophagy proteins to phagosomal membranes16. Initially it was shown that bacterial lipopolysaccharide (LPS) induces autophagy through its cognate receptor TLR4 in macrophages by a mechanism that requires the TLR adaptor TRIF but not the adaptor MyD88 and requires the ‘downstream’ molecules receptor-interacting protein 1 and p38 mitogen-activated protein kinase38. Subsequently, another group confirmed induction of autophagy by LPS-TLR4 and also reported induction of autophagy by single-stranded RNA, poly(I:C) and imiquimod through TLR3 or TLR739. TLR7-dependent induction of autophagy was found to be MyD88 dependent, and another study has proposed that in response to TLR activation, both TRIF and MyD88 trigger autophagy through a direct interaction with Beclin 1 (ref. 41). However, it is not yet known how this interaction stimulates autophagy, and further studies are needed to more fully elucidate the molecular mechanism(s) by which TLR signaling activates autophagy.
It is noteworthy that more robust TLR-dependent stimulation of autophagy in RAW264.7 mouse macrophages than in primary macrophages has been reported39 and that autophagy induction has not been reported in primary fetal liver–derived macrophages treated with LPS or several other TLR stimuli42. These primary fetal liver–derived macrophages, however, are able to mount an autophagic response to starvation or invasive bacteria, which indicates that this lack of responsiveness to TLR stimuli does not reflect a deficiency in their general ability to undergo stress-induced autophagy. These data conflict with the data outlined above showing positive regulation of autophagy by TLR signaling. One possibility is that the function of TLR signaling in autophagy induction has been examined over different time frames in different studies. Alternatively, there may be TLR-independent mechanisms for the induction of autophagy in phagocytic cells, the regulation of autophagy by TLRs may be cell type specific or the function of TLRs may be tightly regulated by the differentiation state of the macrophage. The physiological importance of TLR regulation of autophagy requires further studies in primary cells, in cells other than macrophages and dendritic cells and, perhaps most importantly, in different in vivo models of microbial infection.
Although the physiological function of TLR-mediated induction of autophagy is not yet known, it seems reasonable to speculate that it may function either in the autophagic control of intracellular pathogens and/or in other autophagy gene–dependent functions in immunity. As noted above, the autophagic control of L. monocytogenes in flies requires another type of pattern-recognition receptor, PGRP-LE25; a critical question is whether such findings represent a general paradigm that applies to other families of pattern-recognition receptors in mammalian hosts, including TLRs and Nod-like receptors. At least in vitro, LPS stimulation of macrophages (which activates TLR4) or TLR7 activation results in greater localization of M. tuberculosis in autophagosomes and lower mycobacterial survival38,39, which raises the possibility that TLR induction of autophagy may similarly participate in autophagic control of intracellular pathogens in vivo. It is also possible that TLR induction of autophagy represents a type of feed-forward mechanism for priming the autophagy machinery to deliver viral nucleic acids to endosomal TLRs to activate innate immune signaling. Furthermore, another unexplored possibility is that induction of autophagy by TLRs or pattern-recognition receptors enhances autophagy-mediated presentation of endogenous antigens to major histocompatibility complex (MHC) class II loading compartments (Fig. 2).
Studies also suggest that TLR signaling may not only induce classical autophagy but also recruit autophagy proteins to the phagosomal membrane and deliver phagocytosed material to the lysosome (Fig. 3). Engagement of TLRs during the phagocytosis of LPS-coated beads or zymosan particles induces the rapid recruitment of Beclin 1 and LC3 to phagosomal membranes in an Atg5- and Atg7-dependent way16. Furthermore, proteomic analyses of latex bead–containing phagosomes from macrophages shows enhanced recruitment of LC3 to membranes in response to autophagy inducers43. The translocation of Beclin 1 and LC3 to phagosomal membranes in macrophages treated with LPS-coated beads or zymosan particles is associated with enhanced phagosome fusion with lysosomes but not with detectable double-membraned structures characteristic of autophagosomes16. Thus, it is postulated that TLR signaling, through recruitment of autophagy proteins to the phagosome, may constitute another mechanism for expediting lysosomal microbial elimination. Similar to the failure to detect autophagosomes surrounding T. gondii parasitophorous vacuoles in some studies27,28,30, it is not yet known whether the elimination of pathogen-containing phagosomes is truly a process that requires autophagy proteins but not the formation of autophagosomes or, alternatively, whether autophagosomes are sufficiently evanescent to have escaped detection in ultrastructural studies so far. Nevertheless, the findings obtained with T. gondii and with TLR signaling, phagocytosis and autophagy proteins do raise the possibility that components of the autophagy machinery may be involved in membrane-mediated events that do not involve the classical delivery of cytoplasmic constituents or pathogens to the lysosome (Fig. 3).
Autophagy genes and cytokine secretion
The first connection between autophagy and innate immune signaling emerged after the demonstration that Atg5 is essential for the production of type I interferon in plasmacytoid dendritic cells infected with vesicular stomatitis virus by a mechanism presumed to involve autophagy-mediated delivery of viral genetic material to endosomal TLRs40 (Fig. 2). Somewhat paradoxically, several studies have shown that absent or hypomorphic expression of autophagy genes in certain cell types can result in enhanced production of type I interferon or other cytokines, including proinflammatory molecules such as IL-1β and IL-18, or adipocytokines, such as leptin and adiponectin42,44–46 (Fig. 3). As discussed below, enhanced secretion of such molecules may be involved in certain aspects of Crohn’s disease in mice deficient in the expression of the autophagy gene Atg16l1. Thus, the autophagic machinery may serve a dual function in innate immune signaling, acting not only to stimulate antiviral type I interferon responses in dendritic cells but also to ensure homeostatic balance by preventing excess innate immune activation in other cell types.
Two distinct models have been proposed to explain the higher production of type I interferon in autophagy gene–deficient fibroblasts and primary macrophages (Fig. 3). Enhanced production of type I interferon in Atg5−/− and Atg7−/− mouse embryonic fibroblasts in response to infection with vesicular stomatitis virus or double-stranded RNA has been reported44. In this model, the Atg5-Atg12 conjugate can bind to cytoplasmic viral sensors, the retinoic acid–inducible helicases RIG-1 and Mda5 and their adaptor protein IPS-1, to suppress the activity of such helicases in stimulating the production of type I interferon. Thus, it is possible that the autophagic machinery may suppress innate immune signaling by direct inhibitory interactions with these helicases and their adaptor proteins. Of note, however, a completely different, but not mutually exclusive, mechanism for the higher production of type I interferon in Atg5-deficient mouse embryonic fibroblasts and primary macrophages has also been proposed45. Dysfunctional mitochondria have been shown to accumulate in the absence of Atg5, leading to higher concentrations of IPS-1 and a reactive oxygen species–dependent increase in retinoic acid–inducible helicase signaling and production of type I interferon in response to infection with vesicular stomatitis virus (Fig. 3). Therefore, it is also possible that the homeostatic functions of constitutive autophagy, especially mitochondrial turnover and quality control, indirectly regulate the production of type I interferon.
It will be useful to determine whether this enhanced response to viral infection is present in cells deficient in autophagy genes that act in other steps of the autophagy pathway and whether the underlying mechanism reflects a deficiency of the cellular process of autophagy or alternative, as-yet-undefined functions of autophagy proteins in regulating interferon signaling. Because Atg5-deficient plasmacytoid dendritic cells with impaired type I interferon responses are more sensitive to infection with vesicular stomatitis virus and Atg5-deficient mouse embryonic fibroblasts and primary macrophages with enhanced type I interferon responses are more resistant to such infection, another critical question is how potential cell type–specific differences in autophagy gene–dependent regulation of type I interferon signaling may interact in vivo to determine the fate of viral infection. It is not yet clear whether the autophagic machinery truly has a direct immunosuppressive function that increases susceptibility to viral infection by inhibiting cytokine release or if it merely functions as a negative feedback mechanism that provides a brake on unneeded innate antiviral signaling.
Another study has shown an important function for the autophagy gene Atg16l1 in regulating the secretion of proinflammatory cytokines (Figs. 3,4). The Nod-like receptor protein cyropryrin (also called NALP or NLRP3) forms a complex known as the ‘inflammasome’, which contains the adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase-activating and caspase-recruitment domain) and caspase 1 and is responsible for the processing of pro-IL-1β to its mature, secreted form47,48. Macrophages from mice lacking Atg16l1 produce more of the proinflammatory cytokines IL-1β and IL-18 after endotoxin stimulation of TLR4 (ref. 42). Similarly, mouse chimeras engrafted with Atg16l1−/− fetal liver hematopoietic progenitors have higher serum concentrations of IL-1β and IL-18 after treatment with dextran sodium sulfate; this enhanced cytokine secretion probably contributes to pathology, as treatment with antibody to IL-1β and antibody to IL-18 results in lower susceptibility of Atg16l1−/− chimeric mice to dextran sodium sulfate–induced colitis.
Figure 4.

Possible mechanisms by which alterations in autophagy may be involved in the pathogenesis of human Crohn’s disease. The colored boxes present four potential mechanisms by which the mutation of an autophagy gene could contribute to Crohn’s disease, proposed on the basis of studies of model organisms in which various autophagy genes have been mutated in various cell types (boxes at bottom). So far, only ATG16L1 and IRGM1 have been linked to susceptibility to Crohn’s disease. ROS, reactive oxygen species.
The precise mechanisms responsible for the enhanced secretion of IL-1β and IL-18 by Atg16l1-deficient cells are not yet clear, but TRIF-dependent activation of caspase 1 and consequent enhanced processing of IL-1β in Atg16l1−/− macrophages has been noted. In addition, similar to results obtained with virus-infected Atg5-deficient cells45, higher production of reactive oxygen species in LPS-stimulated Atg16l1−/− macrophages has been found42. Thus, a gain of function in cytokine release may be obtained with deficiency in both Atg5 and Atg16l1, and in both cases it may result directly from effects of the autophagy machinery on signaling pathways that regulate cytokine production and/or indirectly from effects of autophagy on the cellular redox state.
Another study has emphasized the involvement of Atg16L1 in regulating cytokine expression in a specialized intestinal epithelial cell that is important in innate immunity, the Paneth cell (Figs. 3,4). Substantially enhanced gene expression of acute-phase reactants, PPAR signaling molecules and adipocytokines such as leptin and adiponectin was observed after transcriptional profiling by microarray of Paneth cell RNA from mice with a hypomorphic Atg16l1 allele46. Similar alterations have not been found in thymocytes from these mice, which suggests that Atg16l1 deficiency may upregulate cytokine production in a cell type–specific way. Furthermore, this study has emphasized the potential importance of the autophagic machinery in Paneth cells, as specialized cells of the innate immune system, in regulating the expression of molecules with a broad range of systemic pro- and anti-inflammatory effects. The mechanism by which deficiency in an autophagy gene results in these transcriptional changes in Paneth cells remains unknown. It is also unknown whether the polymorphism in human ATG16L1 that is associated with Crohn’s disease (ATG16L1 T300A) results in similar changes and, if so, whether such changes contribute to the pathophysiology of Crohn’s diseases and/or other types of inflammatory diseases.
Autophagy, antigen presentation and thymic selection
There is growing evidence that autophagy may be involved in the MHC class II presentation of certain endogenously synthesized peptides10,49 (Fig. 2). The involvement of autophagy in MHC class I–restricted antigen presentation remains more speculative50; one paper has suggested involvement of Atg5 in MHC class I antigen presentation51. Although it is clear that autophagosomes can deliver peptides to MHC class II loading compartments for presentation to CD4+ T cells52,53, it is not yet known whether this represents a major pathway for antigen presentation during the generation of adaptive immune responses in physiological conditions in primary cells or in vivo. It will therefore be necessary to assess the involvement of autophagy in the function of professional antigen-presenting cells during immune responses in vivo. Regardless of the results, the specific targeting of antigens to autophagosomes by fusion with the LC3 autophagy protein may represent an effective vaccine strategy for enhancing CD4+ T cell responses; for example, a 20-fold enhancement of MHC class II presentation to CD4+ T cell clones has been noted when influenza virus matrix protein is targeted to autophagosomes by fusion with LC3 (ref. 53).
One notable physiological context in which autophagy pathway–dependent endogenous loading of MHC class II may be important is in shaping the T cell repertoire during thymic selection. Thymic epithelial cells that function in positive and negative selection show constitutive autophagy, as demonstrated by the presence of Atg5-dependent green fluorescent protein–LC3 dots54,55. Normal thymic T cell selection requires Atg5 expression in the stromal cells in thymic allografts55. Experiments with thymic transplantation have shown that the selection of certain MHC class II–dependent TCRs but not MHC class I–dependent TCRs requires Atg5. Furthermore, mice with Atg5−/− thymic implants develop autoreactive CD4+ T cells, as measured both by the proportion of CD62Llo peripheral cells and the development of inflammatory infiltrates in many organs, including the intestine. This last observation supports speculation that abnormal negative selection might explain the association between a polymorphism in the autophagy protein Atg16L1 and susceptibility to Crohn’s disease (discussed below; Fig. 4). The development of autoreactive CD4+ T cells in mice with Atg5−/− thymic implants is thought to reflect a function of Atg5 and, presumably, the autophagy pathway in controlling the repertoire of self peptides presented by thymic epithelial cells that are responsible for normal thymic selection and the generation of T cell tolerance. However, autophagy genes other than Atg5 have not yet been analyzed in similar studies, and thus the function of the cellular process of autophagy in thymic selection remains to be defined. It will be useful to determine whether the mechanism responsible for the function of Atg5 in thymic selection reflects the generation of peptides or a function for nutritional, survival or transcriptional responses regulated by Atg5 or other autophagy genes.
Another important question is whether autophagy, in either the antigen-presenting cell or the cell donating antigen, is involved in efficient antigen cross-presentation. Indeed, one study has shown that small interfering RNA–mediated knockdown of either Beclin 1 or Atg12 in antigen-donor tumor cells results in less cross-presentation56. The authors postulate a mechanism involving direct involvement of the autophagosome as a carrier of protein antigens from tumor cells. More studies are needed to confirm this mechanism, as well as explore its potential clinical implications for cancer vaccines.
An additional potential link between autophagy and cross-presentation has emerged from the observations that autophagy is required for the removal of apoptotic corpses in mouse embryoid body and chick retinal development57,58. In these settings, autophagy is required for dying cells to have sufficient energy to generate the engulfment signals necessary for the clearance of such corpses by phagocytes. Therefore, one interesting, as-yet-unexplored theory is that autophagy in dying cells may also be required for the generation of signals that induce efficient uptake of cell corpses for subsequent cross-presentation. This hypothesis is potentially important for understanding and enhancing the immunogenicity of cell-based tumor vaccines. The function of autophagy in the clearance of dead cells during normal tissue turnover might also contribute to peripheral tolerance.
Autophagy genes and lymphocytes
Lymphocytes undergo extensive rearrangement of cytoplasm and organelles during the selection, expansion and contraction of antigen-specific clones and frequently confront life-or-death decisions during their development and activation. Therefore, it is not unexpected that autophagy might have critical cell-intrinsic functions in lymphocyte biology. Indeed, several studies have shown that deletion of autophagy genes in T lymphocytes and B lymphocytes alters lymphocyte development, survival and/or function (Fig. 2).
The thymus is a site of constitutive autophagy54,55, and the expression of Beclin 1 is regulated during T cell and B cell development and T cell activation59, which provides indirect evidence of potential links between autophagy and lymphocyte development. Of note, loss of Atg5 or Atg7 impairs the survival and proliferation of mature T lymphocytes in vivo60–62, and Atg5 is required in B lymphocytes for the survival of developing pre–B cells in the bone marrow and of mature B-1a cells in the periphery63. On the basis of studies of lethally irradiated mice reconstituted with Atg5−/− fetal liver progenitors, it seems that macrophages, plasmacytoid dendritic cells, neutrophils, erythrocytes and immature T cells develop and survive normally in the absence of Atg5. Therefore, a critical question is how deletion of Atg5 and Atg7 confers lymphocyte-specific and lymphocyte lineage–specific (for example, T cell and B-1a cell but not B2 B cell) effects on development and survival.
It is not yet completely clear whether the effects of the deletion of Atg5 and Atg7 on lymphocyte development are mediated through autophagy (as proposed in Fig. 2) or other effects of these genes; studies of mice with mutations in genes whose products act at other stages in the autophagy pathway may be helpful in elucidating this. One mechanism postulated for the involvement of Atg7 and Atg5 in the survival of mature T cells involves a classical autophagy function in the clearance of mitochondria and consequent prevention of the accumulation of reactive oxygen species and imbalance of the expression of pro- and antiapoptotic proteins60,62. Notably, the developmental pattern of Beclin 1 in B lymphocytes, similar to that of the antiapoptotic protein Bcl-2, shows downregulation at the transition from pro–B cell to pre–B cell58. Thus, it will be useful to determine whether suppression of autophagy, in addition to apoptosis, is necessary for the physiological cell death that occurs at this stage in B lymphocyte development and whether null mutations in autophagy genes result in excessive cell death that is incompatible with efficient development of pre–B cells in the bone marrow.
A controversial area is whether autophagy is exclusively a prosurvival pathway in lymphocytes or whether it also can be used as a death pathway. As noted before, deletion of Atg5 or Atg7 results in the impaired survival of certain populations of B cells or T cells60–63, which suggests a major function in lymphocyte survival. However, as discussed earlier, it has been postulated that IFN-γ may induce Irgm1-regulated autophagic cell death in CD4+ T cells36. In addition, knockdown of the gene encoding Beclin 1 or Atg7 results in less death of CD4+ T cells induced by growth-factor withdrawal64, and autophagy genes are required for the death of bystander CD4+ T cells triggered by the human immunodeficiency virus envelope protein65. Thus, a consensus is growing that autophagy may mediate activation-induced death of CD4+ T cells.
However, several caveats must be considered when determining whether autophagy is truly a cause of cell death (death by autophagy) rather than simply being associated with cell death (death with autophagy)21. An example of some of the complexities in determining the contribution of autophagy to T cell death is provided by a study examining the inter-relationships among signaling by the death domain protein FADD, autophagy and the death of primary CD8+ T cells66. Transgenic expression of dominant negative FADD in CD8+ T cells leads to enhanced autophagy and cell death in response to mitogenic signals, which can be prevented by either targeting of the autophagy pathway (as with pharmacological inhibitors, dominant negative Vps34 or short hairpin RNA specific for Atg7) or by use of the necropoptosis inhibitor Nec-1, which blocks the kinase RIPK1. Thus, FADD signaling may somehow dampen the autophagy response to prevent RIPK1-dependent necroptotic cell death, but it is as yet unclear whether autophagy is a true cell death pathway for proliferating CD8+ T cells or whether the autophagic machinery functions in signaling necroptosis. If autophagy does prove to be a cell death pathway for T cells, one possible explanation for the apparently conflicting observations is that basal autophagy is required for T cell survival, whereas unrestrained autophagy, which may occur in response to IFN-γ stimulation or compromise of FADD signaling, may be deleterious.
The stress-activated signaling molecules Jnk1 and Jnk2 may also be involved in the dual integration of autophagy signaling with T cell differentiation, activation and function. Jnk1 and Jnk2 both serve important but nonredundant functions in CD4+ and CD8+ T cells67–69. Notably, several studies have indicated that Jnk kinases are important in autophagic signaling. Although one study has reported more green fluorescent protein–LC3 dots in T cells lacking Jnk1 or Jnk2 (ref. 64), another study has found that Jnk1 positively regulates autophagy by a mechanism that involves phosphorylation of Bcl-2 and disruption of the Bcl-2–Beclin 1 complex, but Jnk2 does not70. The last study showed that Jnk2-deficient mouse embryonic fibroblasts have enhanced Jnk1 signaling and hyper-active autophagy70. Therefore, it is plausible that the lack of autophagy in Jnk1-deficient mice (or perhaps even enhanced autophagy in Jnk2-deficient mice) may contribute to alterations in T cell differentiation. Further assessment of the function of Jnk signaling in the regulation of autophagy in T cells and the effects of Jnk signaling–mediated autophagy regulation in T cell differentiation is clearly needed.
Autophagy genes and Crohn’s disease
One of the most exciting recent developments in the field of autophagy genes and immunity was catalyzed by genome-wide association studies identifying strong links between polymorphisms in two human autophagy genes, IRGM (called ‘IRGM1’ here) and ATG16L1, and susceptibility to inflammatory bowel disease71,72 (Fig. 4). A polymorphism in IRGM1 is associated with both Crohn’s disease and ulcerative colitis; this association is due to the absence of specific single-nucleotide polymorphisms associated with a 2.7-kilobase deletion upstream of the IRGM1 transcriptional start site71,73,74. This raises the possibility that Irgm1-mediated regulation of autophagy may contribute to susceptibility to inflammatory bowel disease; however, so far there are no studies that mechanistically link Irgm1 to features of inflammatory bowel disease in animal models, nor is it known whether the IRGM1 risk allele affects autophagy regulation.
The Crohn’s disease–associated ATG16L1 risk allele encodes a protein with a threonine-to-alanine substitution (T300A) in the carboxy-terminal domain containing tryptophan–aspartic acid dipeptide (WD) repeats. Although this domain of Atg16L1 is not conserved in yeast, is not required for the formation of multimeric complexes with Atg5-Atg12 and is not required for starvation-induced autophagy, the Atg16L1 T300A mutant protein may have lower stability and may be defective in localizing the autophagic machinery to intracellular bacteria75,76. Most notably, two studies of mice with genetically engineered mutations in Atg16l1 have identified critically important functions of this autophagy protein in innate immunity42,46.
Mice with deletion of the coiled-coil domain of Atg16L1, which results in the lack of a functional protein, have been generated42. As reported for null mutations of Atg5 and Atg7, these mice die in the first day of life, presumably because of starvation or a bioenergetic crisis during the postnatal period. As noted above, studies with macrophages from these mice have shown enhanced cytokine responsiveness after stimulation with LPS (TLR4) or stimulation of other TLRs, and chimeric mice engrafted with Atg16l1−/− hematopoietic progenitors from fetal liver have increased mortality, weight loss, colitis and serum concentrations of IL-1β and IL-18 in response to dextran sodium sulfate. This Atg16L1-dependent regulation of endotoxin-induced activation of inflammasomes demonstrates a previously unknown function for a component of the autophagic machinery in controlling inflammatory immune responses. A key question is whether the Atg16L1 T300A–encoding risk allele of ATG16L1 results in similar hyperexpression of proinflammatory genes and thus represents a potential mechanism by which this variant protein increases the risk for Crohn’s disease and potentially other inflammatory disorders (Fig. 4). It will also be useful to determine if this gain-of-function increase in cytokine release occurs with deficiency of additional autophagy proteins.
Viable mice have been bred that are hypomorphic for Atg16L1 expression by means of intronic insertion of a gene-trap vector, which has allowed direct assessment of the effects of lower Atg16L1 expression on intestinal pathology in adult mice46 (Fig. 4). As function is not completely abolished (but may be lower because of altered stability) by the T300A substitution of Atg16L1, this model of lowered mouse Atg16L1 protein expression may be relevant to humans carrying the allele encoding Atg16L1 T300A. Notably, in addition to the transcriptional changes in adipocytokines and other inflammatory regulators in Paneth cells described above, prominent histological abnormalities in Paneth cells of Atg16L1-hypomorphic mice have been reported that resemble changes in resected ileal tissue from patients with Crohn’s disease who are homozygous for the allele encoding Atg16L1 T300A. In both settings, Paneth cells show morphological defects in secretory granules and granule exocytosis, and in the Atg16L1-hypomorphic mice, deficient Paneth cell secretion of the antibacterial protein lysozyme has been noted in the small intestine.
As Paneth cells function as a barrier to bacterial invasion (in part by secreting granule contents that contain antimicrobial peptides) and as regulators of intestinal inflammation77,78, the findings reported above suggest that defects in Atgl6L1 function in Paneth cells may contribute to the pathogenesis of Crohn’s disease. Studies of mice with intestinal epithelial cell–specific deletion of other autophagy genes, including Atg5 or Atg7, have demonstrated similar abnormalities in Paneth cells46,79. Thus, several components of the autophagic machinery that act at the membrane expansion-completion stage (such as Atg16L1, Atg5 and Atg7) are involved in the maintenance of Paneth cell function. At present, it is not yet known whether this function of Atg16L1, Atg5 and Atg7 reflects involvement of classical autophagy in Paneth cell function or perhaps an as-yet-uncharacterized function for the autophagy protein–conjugation machinery in granule exocytosis (Fig. 3).
A model is thus emerging in which dysregulation of autophagy gene function may contribute to the pathogenesis of Crohn’s disease or other types of intestinal inflammatory disorders by several possible routes (Fig. 4). Lack of Atg5 in the thymus results in autoreactive CD4+ T cells and intestinal inflammatory infiltrates55; lack of Atg16l1 in macrophages results in enhanced endotoxin-induced inflammatory signaling42, and lower expression of Atg16l1 in Paneth cells results in transcriptional alterations in molecules that regulate inflammation and in defects in granule exocytosis46. More speculatively, defects in other autophagy gene–dependent processes, such as the maintenance of normal numbers of B-1a cells that make natural antibody to bacterial carbohydrate antigens62 or the survival of T cells60–62, might be involved in the pathogenesis of Crohn’s disease. Although much more work is needed to elucidate the precise effect of polymorphisms in IRGM1 and ATG16L1 on the pathogenesis of Crohn’s disease, these observations are beginning to delineate previously unknown functions for autophagy genes in the regulation of innate immunity.
Therapeutic implications
The many functions of autophagy and autophagy genes in immunity provide both opportunities and risks for manipulating autophagy therapeutically. For example, there is the opportunity to enhance CD4+ T cell–dependent vaccines by targeting antigens to the autophagic pathway. A notable idea that has arisen from the finding that autophagy is important in host resistance to viral, bacterial and parasitic infection is that pharmacological enhancers of autophagy may function as broad-spectrum antimicrobial agents. Furthermore, enhancing autophagy might ameliorate Crohn’s disease if agents that overcome deficiencies in the function of Atg16L1 or Irgm1 can be identified. The potential beneficial effects of autophagy in immunity must be taken into consideration in the clinical development of agents for other diseases, such as cancer, that are targeted at autophagy inhibition. Although inhibition of autophagy-dependent cell survival may be beneficial in cancer therapy, the adverse potential immunological effects of such approaches include diminished negative selection of thymocytes, with consequent autoimmunity, enhanced secretion of proinflammatory cytokines by hyper-responsive innate immune cells, greater susceptibility to infection with intracellular pathogens and lower survival of B-1a cells and T cells. Perhaps additional understanding of the molecular basis of the diverse functions of autophagy genes in immunity will open new doors for rational intervention in a variety of diseases without untoward immunological consequences.
Conclusion
Studies of autophagy genes and immunity have identified important potential functions for autophagy and unexpected functions for autophagy proteins in the regulation of innate immunity and inflammation. Autophagy proteins are involved in immunity-related processes that may not involve the formation of autophagosomes, such as the recruitment of immunity-related GTPases to pathogen-containing compartments, TLR-mediated phagolysosomal maturation, the regulation of RNA helicases and the inflammasome, and the exocytosis of Paneth cell granules (Fig. 3). Converging evidence suggests that autophagy proteins, although critical for normal immune responses, may also function to prevent immunological hyper-responsiveness in certain cell types, such as macrophages, fibroblasts and Paneth cells; this may at least in part underlie the associations between polymorphisms in autophagy genes and Crohn’s disease. Another emerging theme is that deletion of autophagy genes can have different effects in different immune cell populations, which may reflect an effect of the milieu of differentiated immune cells on the function of autophagy genes and/or cell type–specific differences in the complex interaction between autophagy proteins and the specialized functions of various cells of the immune system. Distinguishing between the effects of autophagy proteins that are mediated by classical autophagy and those that are perhaps mediated by other non–autophagosome-dependent mechanisms may help elucidate how proteins of a primordial stress-response pathway may have functionally diversified to shape the immunological responses of complex higher eukaryotic organisms.
Acknowledgments
We thank A. Diehl for medical illustration. Supported by the US National Institutes of Health (R01 CA074730, R01 CA096511, R01 AI054483, R01 AI065982 and U54 AI057160 to H.W.V., and R01 AI151267 and R01 CA109618 to B.L.), the Howard Hughes Medical Institute (B.L.) and the Ellison Medical Foundation (B.L.).
Footnotes
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5. Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem. 2002;277:18619–18625. doi: 10.1074/jbc.M111889200. [DOI] [PubMed] [Google Scholar]
- 5.Hara T, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Q, et al. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 2008;105:19211–19216. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong Y, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1–phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsunaga K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 10.Munz C. Enhancing immunity through autophagy. Annu Rev Immunol. 2009;27:423–449. doi: 10.1146/annurev.immunol.021908.132537. [DOI] [PubMed] [Google Scholar]
- 11.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27:11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orvedahl A, Levine B. Eating the enemy within: autophagy in infectious diseases. Cell Death Differ. 2009;16:57–69. doi: 10.1038/cdd.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3:323–328. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- 16.Sanjuan MA, et al. Toll-like receptor signaling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 17.Jackson WT, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:861–871. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki K, Noda T, Ohsumi Y. Interrelationships among Atg proteins during autophagy in Saccharomyces cerevisiae. Yeast. 2004;21:1057–1065. doi: 10.1002/yea.1152. [DOI] [PubMed] [Google Scholar]
- 19.Matsui Y, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 20.Codogno P, Meijer AJ. Atg5: more than an autophagy factor. Nat Cell Biol. 2006;8:1045–1047. doi: 10.1038/ncb1006-1045. [DOI] [PubMed] [Google Scholar]
- 21.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang XH, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, et al. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121:567–577. doi: 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Orvedahl A, et al. HSV-1 ICP34.5 Confers neurovirulence by targeting the beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Yano T, et al. Autophagic control of listeria through intracellular innate immune recognition in Drosophila. Nat Immunol. 2008;9:908–916. doi: 10.1038/ni.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci USA. 2008;105:20567–20574. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Z, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martens S, et al. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 2005;1:e24. doi: 10.1371/journal.ppat.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ling YM, et al. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao YO, Khaminets A, Hunn JP, Howard JC. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNγ–inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog. 2009;5:e1000288. doi: 10.1371/journal.ppat.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gutierrez MG, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 32.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 33.Harris J, et al. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. 2007;27:505–517. doi: 10.1016/j.immuni.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 34.Taylor GA, Feng CG, Sher A. p47 GTPases: regulators of immunity to intracellular pathogens. Nat Rev Immunol. 2004;4:100–109. doi: 10.1038/nri1270. [DOI] [PubMed] [Google Scholar]
- 35.Papic N, Hunn JP, Pawlowski N, Zerrahn J, Howard JC. Inactive and active states of the interferon-inducible resistance GTPase, Irga6, in vivo. J Biol Chem. 2008;283:32143–32151. doi: 10.1074/jbc.M804846200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng CG, et al. The immunity-related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon-γ-induced cell death. Nat Immunol. 2008;9:1279–1287. doi: 10.1038/ni.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delgado M, et al. Autophagy and pattern recognition receptors in innate immunity. Immunol Rev. 2009;227:189–202. doi: 10.1111/j.1600-065X.2008.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu Y, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 41.Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem. 2008;283:33175–33182. doi: 10.1074/jbc.M804478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 43.Shui W, et al. Membrane proteomics of phagosomes suggests a connection to autophagy. Proc Natl Acad Sci USA. 2008;105:16952–16957. doi: 10.1073/pnas.0809218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jounai N, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA. 2007;104:14050–14055. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tal MC, et al. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci USA. 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–622. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 48.Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 49.Lunemann JD, Munz C. Autophagy in CD4+ T-cell immunity and tolerance. Cell Death Differ. 2009;16:79–86. doi: 10.1038/cdd.2008.113. [DOI] [PubMed] [Google Scholar]
- 50.Vyas JM, van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. 2008;8:607–618. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.English D, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol. 2009;10:480–487. doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paludan C, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 53.Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008;455:396–400. doi: 10.1038/nature07208. [DOI] [PubMed] [Google Scholar]
- 56.Li Y, et al. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008;68:6889–6895. doi: 10.1158/0008-5472.CAN-08-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qu X, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 58.Mellen MA, de la Rosa EJ, Boya P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ. 2008;15:1279–1290. doi: 10.1038/cdd.2008.40. [DOI] [PubMed] [Google Scholar]
- 59.Arsov I, et al. BAC-mediated transgenic expression of fluorescent autophagic protein Beclin 1 reveals a role for Beclin 1 in lymphocyte development. Cell Death Differ. 2008;15:1385–1395. doi: 10.1038/cdd.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stephenson LM, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy. doi: 10.4161/auto.5.5.8133. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182:4046–4055. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- 63.Miller BC, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2007;4:309–314. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- 64.Li C, et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol. 2006;177:5163–5168. doi: 10.4049/jimmunol.177.8.5163. [DOI] [PubMed] [Google Scholar]
- 65.Espert L, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006;116:2161–2172. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bell BD, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci USA. 2008;105:16677–16682. doi: 10.1073/pnas.0808597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arbour N, et al. c-Jun NH2-terminal kinase (JNK)1 and JNK2 signaling pathways have divergent roles in CD8+ T cell-mediated antiviral immunity. J Exp Med. 2002;195:801–810. doi: 10.1084/jem.20011481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conze D, et al. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8+ T cell activation. J Exp Med. 2002;195:811–823. doi: 10.1084/jem.20011508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dong C, et al. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 70.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phospho-rylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barrett JC, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Massey DC, Parkes M. Genome-wide association scanning highlights two autophagy genes, ATG16L1 and IRGM, as being significantly associated with Crohn’s disease. Autophagy. 2007;3:649–651. doi: 10.4161/auto.5075. [DOI] [PubMed] [Google Scholar]
- 73.The wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCarroll SA, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rioux JD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS ONE. 2008;3:e3391. doi: 10.1371/journal.pone.0003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci USA. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaser A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cadwell K, Patel KK, Komatsu M, Virgin HW, Stappenbeck TS. A common role for Atg16L1, Atg5, and Atg7 in small intestinal Paneth cells and Crohn’s disease. Autophagy. 2008;5:250–252. doi: 10.4161/auto.5.2.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]