

Abstract
Structural and biochemical characterization of protein kinases that confer oncogene addiction and harbor a large number of disease-associated mutations, including RET and MET kinases, have provided insights into molecular mechanisms associated with the protein kinase activation in human cancer. In this article, structural modeling, molecular dynamics, and free energy simulations of a structurally conserved mutational hotspot, shared by M918T in RET and M1250T in MET kinases, are undertaken to quantify the molecular mechanism of activation and the functional role of cancer mutations in altering protein kinase structure, dynamics, and stability. The mechanistic basis of the activating RET and MET cancer mutations may be driven by an appreciable free energy destabilization of the inactive kinase state in the mutational forms. According to our results, the locally enhanced mobility of the cancer mutants and a higher conformational entropy are counterbalanced by a larger enthalpy loss and result in the decreased thermodynamic stability. The computed protein stability differences between the wild-type and cancer kinase mutants are consistent with circular dichroism spectroscopy and differential scanning calorimetry experiments. These results support the molecular mechanism of activation, which causes a detrimental imbalance in the dynamic equilibrium shifted toward the active form of the enzyme. Furthermore, computer simulations of the inhibitor binding with the oncogenic and drug-resistant RET mutations have also provided a plausible molecular rationale for the observed differences in the inhibition profiles, which is consistent with the experimental data. Finally, structural mapping of RET and MET cancer mutations and the computed protein stability changes suggest a similar mechanism of activation, whereby the cancer mutations which display the higher oncogenic activity tend to have the greatest destabilization effect on the inactive kinase structure.
Introduction
A central goal of cancer research involves the discovery and functional characterization of the mutated genes that drive tumorigenesis (1,2). The Cancer Genome Atlas and related DNA sequencing initiatives have motivated sequencing studies of tumors and analysis of the genomic basis of tumorigenesis (3–8). The most common protein families implicated in cancer are protein kinases, which are signaling switches regulating the activity of their substrates by adding phosphate groups to them, and there are >500 encoded in the human genome (9–13). A landmark for understanding the molecular basis of kinase function was the elucidation of the crystal structure of protein kinase A (14,15). Since this discovery, the crystal structures of nearly 70 different protein kinases have been determined. Although the kinase catalytic domain is highly conserved, protein kinase crystal structures have revealed considerable structural differences between the closely related active and highly specific inactive forms of kinases (16–18). The dynamic interconversion between distinct inactive and active protein states is a structural hallmark of the kinase domain, which is critical for its normal function.
Cancer genome resequencing efforts have illuminated the role of kinase addiction in a variety of human cancers and have classified tumor-associated somatic mutations according to their involvement in tumorigenesis (19–22). The dominant oncogenes that confer the oncogene addiction effect include ABL, epidermal growth factor receptor (EGFR), VEGFR, BRAF, MET, FGFR3, ALK, RET, and Aurora kinases (23). A comprehensive computational analysis of the distribution of nonsynonymous coding SNP and disease-causing nonsynonymous coding SNPs within the protein kinase gene family has shown that somatic mutations occurring at structurally conserved kinase positions may be statistically enriched in cancers and form mutational hotspots that promote the tumorigenic activity of multiple protein kinases (24–26). Recent advances in understanding genomic and molecular signatures of cancer-causing mutations in protein kinases have facilitated molecular studies of the mutation-dependent activation process and have identified somatic mutations linked to nonsmall cell lung carcinoma within the EGFR tyrosine kinase gene (27–30). Structural determinations of the EGFR (31–34) and ABL cancer mutants (35,36) in complexes with various cancer drugs have provided a molecular rationale of the kinase activation mechanism, revealing structural divergence of the kinases in response to activating mutations with different degrees of sensitivity. Computational studies have begun to investigate a molecular basis of protein kinase function and the structural effects of activating mutations, which may ultimately control the activity signatures of cancer drugs and determine the scope of drug resistance mutations (37–48).
According to the KinMutbase (49,50), there are >35 unique missense MET mutations and 127 missense RET kinase mutations. A large number of inactivating and activating mutations in the human RET tyrosine kinase domain can cause different disorders, including Hirschprung disease and the thyroid gland cancers (51,52). Molecular mechanisms of RET activation in endocrine tumors are largely associated with the transforming ability of specific RET mutations (53–56). Most notably, it was discovered that >95% of multiple endocrine neoplasia type 2 (MEN 2B) cancers arise from M918T mutation, which can lead to a unique pattern of RET tyrosine phosphorylation and downstream signaling. Moreover, functional and thermodynamic analysis have demonstrated that the M918T mutation can cause a local conformational change in the RET kinase that partially releases autoinhibition, resulting in the decreased thermal stability and the increased structural flexibility of the RET mutant (54). The experimental data have shown that structurally conserved mutants M918T in RET (53,54) and M1250T in MET kinases (57–61) are associated with the mechanism of oncogenic activation and display the highest transforming potential, leading to uncontrolled cell proliferation and tumorigenesis.
The biochemical and structural analysis of the wild-type (WT) RET and M918T mutant have recently identified distinct, yet complementary effects of cancer mutations on the RET kinase function, including the increasing kinase activity, a partial release of the kinase autoinhibition, and ligand-independent phosphorylation of RET receptors (54). Structural and biochemical characterization of the human WT RET kinase dimer has been reported in both nonphosphorylated and phosphorylated forms (62). These crystal structures adopt the same active kinase conformation, independent of phosphorylation status, which only modestly affected the level of its catalytic activity (62). Importantly, in the absence of activation, RET kinase monomers may also adopt a closed, autoinhibited inactive conformation, in which the A-loop blocks access to the substrate binding pocket (54). There are other relevant examples of tyrosine kinases (EGFR, ACK1) with active conformations in their nonphosphorylated forms, which may also have a structurally different inactive conformational state (63). Of particular importance is the recent discovery that the cancer drug Sorafenib can act not only as a highly potent inhibitor of BRAF (60), but also inhibit the inactive form of the WT RET kinase and the gatekeeper RET mutations (64). The recently solved crystal structures of the autoinhibited form for the WT MET kinase (65) have suggested that activating cancer mutations may act through weakening the interactions, which stabilize the inactive state of the kinase, but may have a negligible effect on stabilization of the active form.
Some tyrosine kinase inhibitors, including the pyrazolopyrimidines PP1 and PP2 and the 4-anilinoquinazoline ZD6474 can exhibit a strong activity toward the RET kinase and the oncogenic RET kinase mutants, and thereby may be used to treat specifically RET-associated cancers (66–69). In particular, ZD6474, a low molecular weight tyrosine kinase inhibitor, blocks the enzymatic activity of RET kinase and various RET cancer mutants, including M918T, at the inhibitory concentration of 100 nM, and results in the efficient inhibition of their tumorigenic potential (68,67). Moreover, most of the RET mutants, including M918T, E768D, L790F, Y791F, S891A, and A883F, have shown a sensitivity profile to ZD6474 which is very similar to that of WT RET. In contrast, mutations substituting gate-keeper V804 either to leucine or to methionine (V804L and V804M) can render the RET kinase which is resistant to ZD6474 with only a modest inhibition effect observed at 5.0 μM (67). However, substitution of V804 with a smaller glycine residue results in the RET kinase, which is even more susceptible to inhibition (IC50 = 20 nM) than the WT RET kinase (IC50 = 100 nM) (67). The crystal structures of RET kinase complexes bound to two inhibitors, the pyrazolopyrimidine PP1 and the clinically important 4-anilinoquinazoline ZD6474, have unveiled a similar binding mode of these inhibitors, suggesting a molecular rationale for the drug resistance against specific RET mutants (62).
In this study, molecular dynamics (MD) simulations and free energy stability analysis of the structurally conserved mutations in the RET and MET kinases are undertaken from a biophysical perspective to investigate the role of these mutations in the molecular mechanism of activation. Moreover, we have also performed MD simulations and docking and binding free energy evaluations of the inhibitor binding with the oncogenic and drug-resistant RET mutants. Combined with a structural mapping of the RET and MET cancer mutations and the computed protein stability changes, the results of this study offer a plausible common mechanism, according to which activating cancer mutations may trigger a partial destabilization of the inactive kinase, while contributing to the enhanced stabilization of the active kinase form.
Methods
Structural modeling
Structural modeling of M918T RET and M1250T MET mutations has been carried out using MODELLER (70,71) with a subsequent refinement of side chains by the SCRWL3 program (72). The initial M918T RET model in the active state was obtained using crystal structures of nonphosphorylated (Protein Data Base (PDB) code 2IVS) WT RET kinase in the active state. The initial structural models of the WT RET and M918T RET mutant in the inactive, autoinhibited form were constructed using the crystal structures of the closest homologous kinase, FGFR1, in the inactive state (PDB entries 1FGI, 1FGK) as structural templates. The resulting models were compared and found to be essentially identical to a validated theoretical model of inactive human RET (PDB code 1XPD). Structural model of the M1250T MET kinase mutant in the inactive state was obtained based on the crystal structure of the unphosporylated, autoinhibited form of the WT MET kinase (PDB code 2G15); the inactive WT FLT3 (PDB code 1RJB), and the inactive WT KIT (PDB entry 1T45). Structural models of all mutants were built in MODELLER with a flexible sphere of 5 Å around each mutated residue. In the final protocol, we gradually increased the radius of this sphere in 5 Å steps until the radius reached the 25 Å value (i.e., all residues falling within this range were treated as flexible). A hybrid protocol involving 5000 steps of a conjugate-gradient minimization, followed by MD with simulated annealing (SA) refinement, was repeated 20 times to generate 100 initial models for each cancer mutant in this study. In the optimization stage, we initially used a conjugate-gradient minimization to remove unfavorable contacts and to optimize geometry. MD simulations were then run at increasing temperature values from 150 K to 1500 K, followed by simulated annealing and sampling at temperatures of 1500 K, 1000 K, 800 K, 600 K, 500 K, 400 K, 320 K, and 300 K, respectively. The models were generated using 20 iterations of the MD/SA procedure and the predicted structural model was chosen out of the 100 models as scored by the MODELLER default scoring function. These models were then relaxed using 1-ns MD simulations with NAMD 2.6 software package and the CHARMM22 force field (73) to ensure local refinement of the environment surrounding the mutant residue (all residues within 5 Å distance), while keeping the rest of the protein rigid.
Molecular dynamics simulations
All MD simulations of the RET and MET kinase forms were done for 20 ns with the explicit solvent using NAMD 2.6 with the CHARMM22 force field (73). The VMD program was used for the preparation and analysis phases of simulations (74,75). The psfgen utility in the VMD software was employed to generate a protein structure file for MD simulations with the CHARMM 27 force field. The kinase structures were solvated in a large box of water with 15 Å buffering distance. Assuming normal charge states of ionizable groups corresponding to pH 7, sodium Na+ and chloride Cl− counterions at physiological concentration of 0.15 mol/L were added to achieve charge neutrality and to mimic a realistic biological environment more closely. All Na+ and Cl− ions were placed >8 Å away from any protein atoms and from each other. The structure of RET kinase is unresolved for flexible residues 828–843, which were modeled and incorporated into the final structure using MODELLER (70,71) with an additional refinement of side chains by the SCRWL3 program (72) and minimization. During simulations with the inactive (PDB entry 1XPD) and active forms of WT RET (PDB entry 2IVS), the total number of simulated atoms in the system is 59,310 including 4908 protein atoms; 18,117 water molecules, and 22 sodium Na+ and 27 chloride Cl− counterions. In simulations with the M918T RET mutant, the total number of simulated atoms in the system is the same (59,310), including 4905 protein atoms (because of the effect of the mutated residue); 18,118 water molecules; and 22 sodium Na+ and 27 chloride Cl− counterions, respectively. During MD simulations with the WT MET and M1250T MET mutant (PDB entry 2G15), the total number of simulated atoms in the system is also the same (59,783), including 4826 protein atoms for the MET WT (4823 for the MET M1250T mutant); 18,302 water molecules for the MET WT (18,303 for the MET M1250T mutant); and 51 sodium Na+ and 25 chloride Cl− counterions.
The system was subjected to initial minimization for 10,000 steps keeping protein atoms fixed (minimization of water molecules) followed by the another 10,000 steps of minimization keeping only the protein backbone fixed so as to allow protein side chains to relax. This was followed by 10,000 steps of minimization to allow the entire system to relax freely. The equilibration and simulation protocol was carried out in steps by gradually increasing the system temperature in steps of 20 K starting from 10 K until 310 K, and at each step 20 ps equilibration was run keeping a restraint of 10 Kcal mol−1 Å−2 on protein α-carbons. Thereafter the system was equilibrated for 300 ps at 310 K (NVT) and then for a further 300 ps at 310 K using the Langevin piston (NPT) to maintain the pressure. At the final stage of equilibration, the restraints were completely removed and the entire system was equilibrated for an additional 2-ns time period. After the equilibration phase had been completed, MD simulations in NPT ensemble were run using the equilibrated structure for 20 ns of the production period, keeping the temperature at 310 K and pressure at 1 bar using the Langevin piston coupling algorithm. The integration time step of the simulations was set to 2.0 fs, the SHAKE algorithm was used to constrain the lengths of all chemical bonds involving hydrogen atoms at their equilibrium values, and the water geometry was restrained as rigid by using the SETTLE algorithm. Nonbonded van der Waals interactions were treated by using a switching function at 10 Å and reaching zero at 12 Å distance. The particle-mesh Ewald algorithm (76), as implemented in NAMD, was used to compute long-range electrostatic forces.
Molecular docking simulations
We have modeled binding of ZD6474 inhibitor with the WT RET, M918T RET, V804M, and V804G RET mutants using 1000 snapshots from the MD trajectory of the ZD6474 inhibitor bound with the WT RET, followed by 1), modeling these mutations into each of the 1000 snapshots and subsequent 10,000 steps of energy minimization; and 2), inhibitor docking with 1000 conformations of the WT and RET mutants. Docking simulations are performed using a combination of the evolutionary search algorithm and the knowledge-based energy model, which have been extensively documented in our earlier studies (77). In brief, the inhibitor conformations and orientations are sampled in a parallelepiped that encompasses the binding site obtained from the crystallographic structure of the RET kinase complexes with a large 20.0 Å cushion added to every side of this box surrounding the interface. The protein structure is held fixed in its minimized and equilibrated conformation, while rigid body degrees of freedom and the peptide rotatable angles are treated as independent variables. The initial inhibitor conformations are generated by randomizing the encoded vector, where the center of mass of the ligand is restricted to the rectangular parallelepiped that encompasses the RET crystal structures. The three rigid-body rotational degrees of freedom as well as the torsional angles for all rotatable bonds are uniformly initialized between 0 and 360°.
Free energy simulations
Free energy calculations of the protein kinase stabilities and binding free energy computations are done using the molecular mechanics AMBER force field (78) and the solvation energy term based on the continuum generalized Born and solvent-accessible surface area (GB/SA) solvation model (79,80). The length of MD simulations and the size of the investigated molecules are usually hampered by the necessity of including thousands of explicit solvent molecules. The continuum dielectric methods evaluate only the solute electrostatics and consequently reduce the number of interactions with respect to explicit solvent methods. These methods have proven to be reliable and able to provide crucial information for various biomolecules (79,80). The generalized Born (GB) theory is an approximation of the Poisson equation for continuum electrostatic solvation energy. It involves accurate evaluation of Born radii, which characterize the average spherical distances of each atom to the solvent boundary. Consequently, the GB energy expression can reliably reproduce the Poisson energy at a significantly lower computational cost (79,80). The details of the MM-GBSA model typically used in free energy simulations of biological systems have been extensively documented in our earlier studies (46,47,81–83). Here, we outline the major components of the computational model that are relevant in the context of this study. The total free energy is given as
| (1) |
where Gsolvation is the solvation free energy, and Egas is the molecular mechanical energy of the molecule summing up the electrostatic Eele interactions, van der Waals contributions Evdw, and the internal strain energy Eint. TSsolute is the sum of TStrans, TSrot, and TSvib, which are the translational, rotational, and vibrational entropy contributions, respectively. The vibrational entropy term is determined using the NMODE module of the normal mode analysis involving calculation and diagonalization of a mass-weighted second derivative matrix.
The vibrational entropy corrections for studied WT and cancer kinase mutants were calculated and averaged for 1000 snapshots, selected at 20-ps intervals along the 20-ns MD trajectories for each of these systems. It is worth stressing that because normal mode analysis scales quadratically with the size of the structure, these computations continue to be rather computationally intensive and are frequently omitted in calculations of large biological systems. In our study, we have systematically applied normal mode analysis for all studied RET and MET kinases to determine the entropy contribution to the free energy and accurately assess differences in the protein stability between WT and mutant kinases.
The electrostatic contribution to the solvation free energy Ggbele involves using the GB equation to estimate the electrostatic contributions to the solvation free energy and typically leads to a small error compared to the Poisson-Boltzmann approach (79,80). The nonpolar contribution to the solvation free energy, Ggbnp, was determined using the linear combinations-of-pairwise overlaps method where the hydrophobic contribution to the solvation free energy is determined by the solvent-accessible-surface-area (SASA) dependent term. The total solvation free energy Ggbtot is the sum of the nonpolar Ggbnp and polar Ggbele contributions. The Geletot is the total electrostatic energy and is the sum of the electrostatic solvation free energy and molecular mechanics electrostatic energy.
Using the MM-GBSA module in AMBER 8.0, we have evaluated protein stability differences between 1), WT RET kinase and the M918T mutant and 2), WT MET and M1250T MET in both inactive and active states of the enzyme. A practical implementation of this approach involved computation of the energy contributions for each of the selected 1000 snapshots at 20-ps intervals along the 20-ns MD trajectories for each of these systems, followed by averaging to obtain the total free energy values. The trajectories obtained from the earlier 20 ns MD runs were converted into individual PDB files in VMD. These 1000 coordinate frames were then converted into AMBER-compatible files using utilities from the multiscale modeling tools for structural biology tool set (http://www.mmtsb.org/). The names of phosphorylated residues of MET and RET were modified to load all the required libraries and parameters for phosphorylated residues. The AMBER parameters and coordinate files were then generated using the Leap module in AMBER and subsequently used in the MM-GBSA calculation.
The binding free energy of the inhibitor-kinase complexes is given by the expression
| (2) |
Binding free energy evaluations can be performed using either separate trajectories of the complex, protein, and ligand (separate trajectory protocol) or from a trajectory of the complex (single trajectory protocol) (84). We have used a single trajectory protocol based on 1000 snapshots of the inhibitor complexes with the WT RET and RET mutants. This approach is less computationally intensive and may also be less sensitive because of cancellation of the intramolecular energies, caused by neglecting the effects of structural adaptation during binding.
Protein stability calculations
To quantify the destabilization effect of a number of known RET and MET cancer mutations in the inactive, autoinhibited kinase form, we computed the protein stability change upon these mutations using three different approaches with the increasing level of complexity: 1), Cologne University Protein Stability Analysis Tool (CUPSAT) approach for the prediction and analysis of protein stability changes upon point mutations (85,86); 2), FOLDx approach, which allows the calculation of the free energy of a macromolecule based on its high-resolution three-dimensional structure (87); and 3), MM-GBSA free energy calculations (79,80). In the CUPSAT approach, coarse-grained atom potentials and torsion angle potentials are used to predict protein stability upon point mutations (85). FOLDx analysis of protein stability is based on the empirical force field, which was developed for the rapid evaluation of the effect of mutations on the stability, folding, and dynamics of proteins and nucleic acids (88). The free energy of folding is evaluated in this approach from the difference in Gibbs free energy between the crystal structure of the protein and a hypothetical unfolded reference state of which no structural details are known.
The protein stability changes for a spectrum of studied RET and MET mutants are also evaluated using a single trajectory MM-GBSA approach, in which 1000 snapshots were extracted from 20-ns MD trajectories of the WT RET and WT MET in the autoinhibited, inactive state. The RET and MET mutations were modeled into each of the 1000 snapshots of the WT trajectories, followed by a subsequent 10,000 steps of energy minimization. The protein stability differences are approximated based on the total free energy difference between the WT and mutant kinase conformations:
| (3) |
Results and Discussion
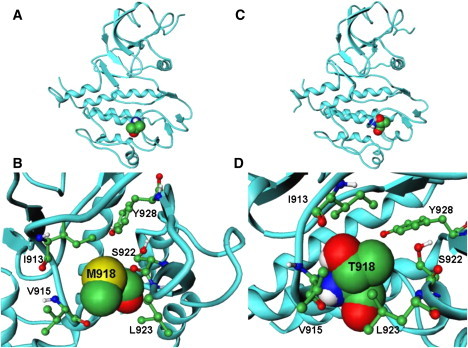
Evolutionary and structurally conserved M918 RET (Fig. 1, A and B) and M1250 MET (Fig. 1, C and D) residues are localized in the substrate binding C-lobe of the kinase core. Somatic mutations occurring at this structurally conserved kinase position are implicated in the tumorigenic activity of multiple protein kinases, including M918T RET and M1250T MET. Moreover, this mutational hotspot is also shared in ALK1 (M-R), MET (M-T), RET (M-T), and TGFbR2 (M-V). Three of the four mutations are transitions from methionine to either arginine or threonine, indicating that these mutations may disrupt the hydrophobic binding pocket by introducing polar amino acids. This methionine is highly conserved and specific for receptor tyrosine kinases, while in cytosolic tyrosine kinases, including SRC, ABL and LCK, this residue is converted to threonine. M918T in RET and M1250T in MET are situated within the WMxxEx motif, also known as the P+1 loop (Fig. 1). This loop is a small motif, which is immediately C-terminal of the A-loop and plays an important role in recognizing the residues flanking the target tyrosine in the substrate. Our study is designed to quantify the role of this mutational hotspot in altering structure and dynamics of RET and MET kinases and assess their effect on the activation mechanism.
Figure 1.
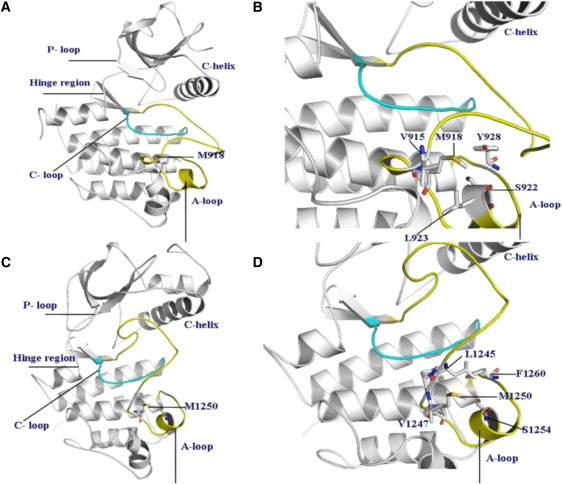
(A) The crystal structure of the wild-type RET kinase in the active form (PDB entry 2IVS). (B) A closeup of structural environment near M918T mutation in the RET kinase. (C) The crystal structure of the wild-type MET kinase in the inactive, autoinhibited form (PDB entry 2G15). (D) A closeup of structural environment near M1250T mutation in the MET kinase.
Analysis of the cancer mutation effects in the inactive RET kinase: molecular dynamics and protein stability
We have performed 20-ns MD simulations for the WT RET and tumorigenic M918T RET mutant (Fig. 1, A and B), which were independently launched from both inactive and active kinase structures. Structural and energetic analysis of these trajectories allows us to investigate the effect of the cancer mutation on protein structure and dynamics at atomic details. To provide a computational framework for a comparative structural and mechanistic analysis of WT RET and M918T RET, it is desirable to have crystal structures of these proteins in both active and inactive forms. While the existing crystal structures of both phosphorylated and nonphosphorylated RET have the same active conformation (62), it has been established that in the absence of activation, RET kinase monomers may adopt a closed, autoinhibited inactive conformation (54). Consequently, to simulate dynamics of the RET kinase in the inactive, autoinhibited form, we have built a homology model of the inactive RET using the crystal structures of the closest homologous kinase FGFR1 in the inactive form (PDB entries 1FGI and 1FGK) as structural templates. It is worth noting that the generated refined RET model is very similar to the previously validated and PDB-deposited modeled structure of the inactive human RET kinase (PDB code 1XPD).
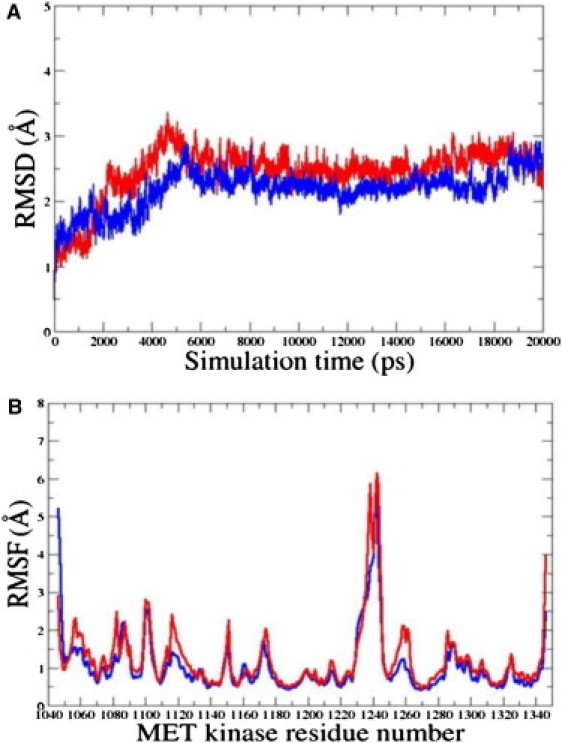
Examination of the MD trajectories in the inactive state highlights some important similarities and differences in the dynamical behavior of the WT RET and M918T RET mutant. Indeed, while WT RET tends to rapidly reach a steady equilibrium, the root mean-square deviation (RMSD) fluctuations for the M918T mutant were noticeably larger, reaching an equilibrium plateau after a considerably longer simulation period (Fig. 2 A). The equilibrium structures of the WT RET fluctuated within ∼2.0 Å from the crystal structure, and 3.0 Å for the M918T RET mutant (Fig. 2 A). To assess the effect of M918T RET mutation on the protein kinase structure and dynamics, we have also computed the root mean-square fluctuation (RMSF) values, i.e., the average residue fluctuations of the backbone residues. In agreement with the structural factors, the regions of larger thermal fluctuations and the increased protein flexibility include segments of N-terminal, C-helix, and A-loop (Fig. 2 B). MD simulations in the inactive, autoinhibited RET conformation have also shown that larger fluctuations in the M918T mutant coexist with the globally similar dynamic profiles of the WT RET and M918T mutant. Furthermore, MD simulations have pointed to the locally increased conformational mobility of the mutant structure near the mutational site at the P+1 pocket and A-loop (Fig. 2 B).
Figure 2.
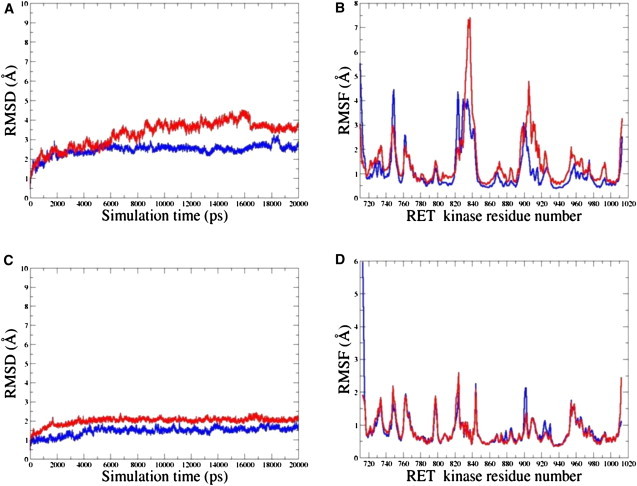
The RMSD values for Cα atoms from 20-ns simulations with the inactive RET kinase (A) and the active RET kinase. (C) The RMSF values of the RET kinase residues (using original numbering in the PDB entries 1XPD and 2IVS) from 20-ns MD simulations with the inactive RET kinase (B) and the active RET kinase (D). For all panels, time evolution of the WT RET is shown in blue; time evolution of the M918T RET mutant is shown in red.
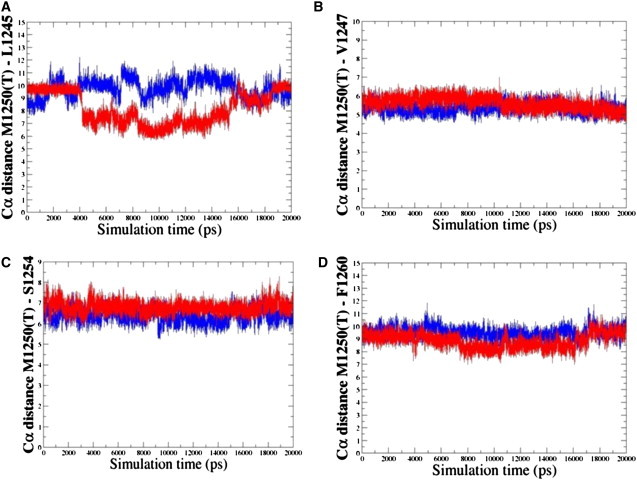
The residues comprising the P+1 substrate binding pocket in RET are highly conserved and include I913, P914, V915, M918, S922, L923, and Y928 (Fig. 1, A and B). In the autoinhibited form of WT RET, M918 interacts with residues in the activation (I913) and P+1 (P914) loops and with neighboring residues S922, L923, and Y928, which in turn interact with P+1 and catalytic loop residues, forming a closed, autoinhibited structure. In the course of simulations, the hydrophobic contacts formed by the side chain of M918 in the P+1 pocket with the I913, V915, S922, L923, and Y928 residues are maintained, indicating their importance in controlling the inactive conformation of the WT RET kinase. Mutation to a smaller hydrophilic threonine may elicit a protein response with the altered shape of the P+1 site and weakened packing interactions, particularly a loss of favorable intramolecular contacts formed by T918 with the I913 and Y928 residues (Fig. 3). Nevertheless, the mutated T918 continues to interact with V915, S922, and further strengthens interactions with L923 (Fig. 3). Structural effects of M918T are manifested through the enhanced local flexibility, which is reflected in the increased distances with the Y928 residue and a concomitant enlargement of the P+1 binding site (Fig. 4). At the same time, mutated T918 residue maintains favorable contacts with V915 and improves interactions with L923, which collectively amount to a subtly reshaped P+1 pocket. These results suggest that a locally more mobile conformation of the T918 RET mutant as compared to the WT RET, along with a subtly reshaped P+1 recognition pocket, may be functionally linked with the experimentally observed subversion in substrate specificity (89,90). Upon activation of WT RET interactions between M918 and some of these residues (S922) are reduced in strength, but are maintained with the V915, L923, and Y928 residues. The mutated T918 residue in the inactive state retains some of the normal interactions of M918 in autoinhibited WT RET, but can also acquire some of the interactions present in the active WT RET.
Figure 3.
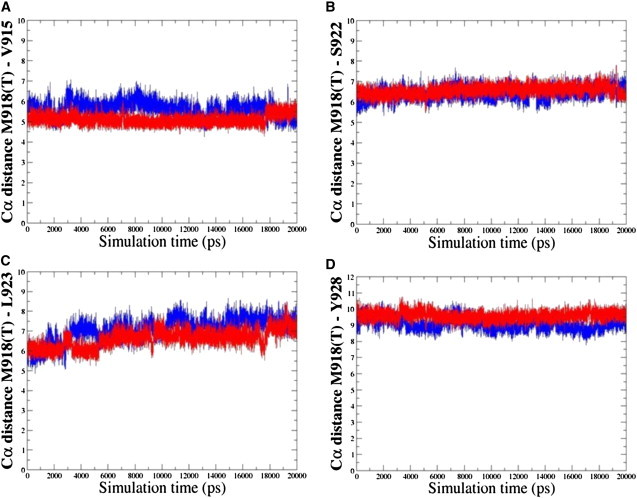
Analysis of MD simulations with the inactive RET kinase. Time evolution history of the distances between Ca atoms of the residues in the local structural environment of the mutational site. Time evolution of the distances between Ca atoms of the M918T RET and V915 RET (A), M918T RET and S922 RET (B), M918T RET and L923 RET (C), and M918T RET and Y928 RET (D). Time evolution of the distances for the M918 WT RET is shown in blue; time evolution of the distances for the T918 RET mutant is shown in red.
Figure 4.
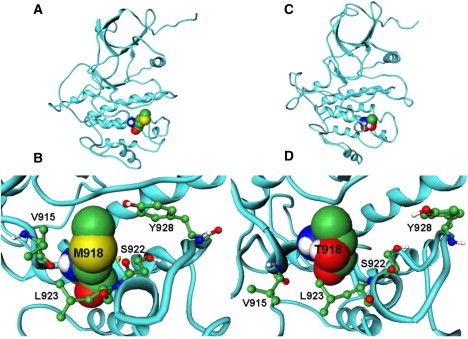
(A) The average structure of the inactive form of the WT RET kinase with M918 residue shown in CPK model. (B) A closeup of structural packing near the mutational site in the inactive WT RET kinase. (C) The average structure of the inactive form of the M918T RET kinase mutant with T918 residue shown in CPK model. (D) A closeup of structural packing near the mutational site in the inactive T918 RET kinase.
Using MM-GBSA analysis of the MD trajectories for the WT RET and M918T RET mutant, we have evaluated protein stability changes upon mutation (Tables 1 and 2). This analysis demonstrates that the higher conformational mobility and a greater entropy component of the free energy in the M918T mutant is largely offset by the loss in the enthalpy contribution. As a result, the increased protein flexibility in the inactive state of M918T RET can lead to the decreased protein stability of the mutant kinase relative to the WT RET (Tables 1 and 2). The enthalpy loss in the M918T RET mutant is determined by the less favorable molecular mechanical energy of the molecule Egas, which cannot be offset by a more favorable total solvation free energy Ggbtot (Tables 1 and 2). In fact, the energy loss is detected for all components of the molecular mechanical energy Egas, including Eele, Evdw, and Eint contributions (Table 1). These results are consistent with the functional and thermodynamic analysis, which compared the overall thermal stability and structural flexibility of the autoinhibited forms of the WT RET and M918T mutant (54). According to these experiments, in the inactive kinase state, the M918T RET mutant has a lower melting temperature and is more conformationally flexible than the WT enzyme.
Table 1.
The energy components of the MM-GBSA calculations of the protein kinase stability
| Kinase | RET inactive |
RET active |
MET inactive |
|||
|---|---|---|---|---|---|---|
| WT | M918T | WT | M918T | WT | M1250T | |
| Eele | −10074.36 (111.75) | −9678.37 (103.89) | −9012.11 (100.03) | −9185.27 (76.77) | −9056.93 (62.83) | −8970.41 (81.8) |
| Evdw | −1693.18 (20.46) | −1634.10 (20.63) | −1699.82 (11.17) | −1706.34 (15.21) | −1665.84 (12.17) | −1677.04 (16.09) |
| Eint | 3202.34 (14.10) | 3184.81 (10.81) | 3010.75 (18.41) | 3001.72 (10.7) | 3227.72 (12.56) | 3223.23 (11.97) |
| Egas | −8565.20 (108.70) | −8127.66 (108.34) | −7701.19 (97.6) | −7889.89 (75.18) | −7494.96 (66.74) | −7424.22 (83.67) |
| Ggbub | 106.80 (2.49) | 117.46 (3.36) | 96.97 (1.57) | 95.60 (2.13) | 112.69 (1.85) | 113.30 (1.18) |
| Ggbele | −4212.55 (98.71) | −4617.55 (102.96) | −4182.58 (92.63) | −4018.33 (70.19) | −3857.12 (58.72) | −3898.15 (71.24) |
| Ggbtot | −4105.75 (97.68) | −4500.09 (101.82) | −4085.61 (91.63) | −3922.73 (69.89) | −3744.43 (57.86) | −3784.86 (70.32) |
| Geletot | −14,286.92 (20.12) | −14,295.92 (12.70) | −13,194.69 (17.63) | −13,203.61 (13.69) | −12,913.95 (18.42) | −1168.56 (18.92) |
| Egas+Ggbtot | −12,670.95 (19.37) | −12,627.75 (23.98) | −11,768.80 (21.76) | −11,812.62 (20.17) | −11,239.39 (18.42) | −11,209.08 (20.98) |
| TStrans | 17.14 (0.00) | 17.14 (0.00) | 17.08 (0.00) | 17.08 (0.00) | 17.12 (0.0) | 17.12 (0.0) |
| TSrot | 17.78 (0.01) | 17.84 (0.01) | 17.70 (0.01) | 17.68 (0.01) | 17.83 (0.01) | 17.82 (0.01) |
| TSvib | 3496.14 (12.84) | 3524.26 (11.27) | 3311.5 (10.52) | 3294.59 (11.85) | 3475.16 (10.23) | 3488.04 (9.63) |
| TStotal | 3531.06 (12.85) | 3559.23 (11.27) | 3346.28 (10.52) | 3329.36 (11.85) | 3510.11 (10.24) | 3522.98 (9.63) |
All energies are in Kcal/mol. The contributions of the free energies are defined in Materials and Methods. The values in parentheses represent the standard deviation values.
Table 2.
The differences in the protein stabilities between WT and mutant RET and MET kinases
| Kinase | RET active GWT-GMUT | RET active GWT-GMUT | MET inactive GWT-GMUT |
|---|---|---|---|
| ΔEele | −359.99 | 173.16 | −86.42 |
| ΔEvdw | −59.08 | 6.52 | 11.2 |
| ΔEint | 17.53 | 9.03 | 4.49 |
| ΔEgas | −437.54 | 188.70 | −70.74 |
| ΔGgbub | −10.66 | 1.37 | −0.61 |
| ΔGgbele | 405.0 | −164.25 | 41.03 |
| ΔGgbtot | 394.34 | −162.88 | 40.43 |
| ΔEgas+ΔGgbtot | −43.20 | 25.82 | −30.31 |
| ΔTStotal | −28.17 | 16.92 | −12.87 |
| ΔGtotal | −15.03 | 8.90 | −17.44 |
All energies are in Kcal/mol. The contributions of the free energy are defined in Materials and Methods.
It is worth stressing that there are many biological systems and numerous examples where there is no a simple inverse relationship between decreased molecular fluctuations and increased stability. In general, coupling between rigid and flexible regions of a protein and correlation of various motions may lead to both increases and decreases in thermodynamic stability. There are many protein systems with both high thermal stability and flexibility (91) as well as examples of proteins where enhanced thermal stability can be achieved without increased conformational rigidity (92). Some mutations may disrupt long-range coupled regions of structural communication, while other mutations may result in local changes, depending on structural interactions near sites of substitution. In this study, the impact of the cancer mutation manifests in the overall loss of the RET kinase stability in the inactive state, caused by local structural changes, where a moderate gain in the conformational entropy is accompanied by a larger enthalpy loss and the decreased thermodynamic stability (Table 1).
Analysis of the cancer mutation effects in the active RET kinase: molecular dynamics and protein stability
MD trajectories initiated from the crystal structure of the active RET enzyme rapidly reach equilibrium, exhibiting small 1.0 Å deviations in the RMSD values for WT and 1.5–2.0 Å for the mutant (Fig. 2 C). Thermal fluctuations of the WT RET and M918T RET in the active state are largely conserved, including similar conformational flexibility of the functionally important regions near the mutational site, including the P+1 pocket and the adjacent segment of the A-loop (Fig. 5). There are no appreciable changes in the shape of the P+1 pocket in the active form of M918T RET as compared to the WT RET (Fig. 5). As a result, all favorable interactions formed in the P+1 pocket of M918T RET are preserved and even further strengthened in the active state. We have found that the M918T mutation may further stabilize the active RET conformation, leading to the increased thermodynamic stability (Table 1). A rather moderate loss in the entropy TSsolute of the active M918T RET and an unfavorable change in the total solvation energy Ggbtot are fully compensated by considerably more favorable intramolecular interactions reflected in the predicted Egas values (Tables 1 and 2).
Figure 5.
(A) The average structure of the ACTIVE form of WT RET kinase with M918 residue shown in CPK model. (B) A closeup of structural packing near the mutational site in the ACTIVE WT RET kinase. (C) The average structure of the ACTIVE form of M918T RET kinase mutant with T918 residue shown in CPK model. (D) A closeup of structural packing near the mutational site in the ACTIVE T918 RET kinase.
These results are fully consistent with circular dichroism spectroscopy experiments, in which no significant variations are seen in the secondary or tertiary structure of the M918T RET mutant (54). Interestingly, it was also proposed that the M918T mutation may affect the interlobe flexibility of the kinase domain and weaken the trans-inhibitory dimer interaction, which may favor the active state of the enzyme (62). Hence, the results of simulations suggest that a molecular basis of mutations that can contribute to the activation potential of RET kinase may be manifested through increased local mobility at functionally critical regions and reduction in the tight interactions seen in the autoinhibited form of WT RET.
We have also found that RET kinase may be less stable in the active form of the enzyme than in the inactive state. Our analysis suggests the loss of protein stability in the active form of RET as compared to the autoinhibited inactive form for both WT and mutant (Table 1). These results are consistent with the experimentally observed lower melting temperatures of the active RET form (54), which are also seen in the thermodynamic effects of the release of autoinhibition in other kinases. In addition, these data are consistent with the earlier experimental studies (93,94), which have discovered the decrease in the level of stabilization of the IRK kinase, as a result of autophosphorylation and respective conformational transition to the active form of the enzyme (93,94).
Overall, the results quantify the hypothesis that the M918T RET mutant may be more stable in the active form of the enzyme, which could provide the thermodynamic basis for diverting the normal equilibrium toward an excessively stable active kinase form for the cancer mutant. This effect may contribute to the high oncogenic activity and profound transforming potential of the M918T mutation. Together, these data suggest that changes in the intramolecular interactions may play a significant role in modulating functional effects of the M918T mutation in RET kinase. Additionally, the predicted structural effect of the M918T mutation on protein kinase dynamics and stability is supported by isothermal titration and differential scanning calorimetry experiments, which have revealed that WT RET and M918T RET kinases have very similar melting temperatures and consequently similarly stable structures in the active form (54).
Molecular dynamics and protein stability analysis of cancer mutation effects in the MET kinase
MD simulations of the MET kinase and M1250T mutant are based on the unphosporylated, autoinhibited crystal structure of the enzyme in the inactive form (95). These simulations provide further insight into structural implications of a cancer mutational hotspot on protein dynamics and activation mechanism. Examination of the MD trajectories shows more significant differences in flexibility between WT and mutant, as evidenced by larger RMSD values for the M1250T mutant reaching 2.0–2.2 Å as compared to 1.0 Å for the WT (Fig. 6 A). Moreover, larger protein fluctuations in the M1250T mutant include regions of N-terminal, C-helix, A-loop, and P+1 pocket (Fig. 6 B). The P+1 binding pocket in MET is formed by the L1245, P1246, V1247, M1250, S1254, L1255, and F1260 residues (Figs. 7 and 8). Analysis of the evolutionary and structurally conserved P+1 binding pocket reveals only very minor differences between RET and MET kinases, namely I913 RET corresponds to L1245 MET and Y928 RET is converted into F1260 MET. The local structural environment of conserved M1250 in the MET kinase is formed by a similar network of favorable packing interactions with neighboring V1247, S1254, L1245, and F1260 residues. Importantly, in the M1250T mutant a number of these favorable interactions are weakened, most notably reflected in the increased contact distances with the S1254 (Fig. 7 C) and smaller changes in the hydrophobic contacts with V1247 and F1260 residues (Fig. 7, B and D). Moreover, this mutation seems to elicit a conformational change in the L1245 side chain that may partially disturb the optimal arrangement between the L1245 and V1247 residues from the A-loop and P+1 loop, respectively (Fig. 8), which partly control the accessibility of the substrate-binding pocket. MD simulations have revealed that the M1250T substitution in MET may lead to the greater local mobility near mutational site and an expansion of the P+1 pocket as compared to the WT MET kinase structure (Fig. 8). As a result, M1250T mutant may destabilize structurally rigid, inactive MET conformation through a partial displacement of the A-loop from the autoinhibitory position (Fig. 8). A concomitant loosening of the interactions between residues in the activation and P+1 loops can lead to the eventual release of the inactive autoinhibitory conformation of the enzyme and the increased accessibility of the substrate binding pocket.
Figure 6.
(A) The RMSD values for Cα atoms from 20-ns MD simulations with the inactive MET kinase. Time evolution of the WT is shown in blue; time evolution of the M1250T mutant is shown in red. (B) The RMSF values of the RET kinase residues (using original numbering in the PDB entry 2G15) from 20-ns MD simulations with the inactive MET kinase. Time evolution of the WT is shown in blue; time evolution of the M1250T mutant is shown in red.
Figure 7.
Analysis of MD simulations with the inactive MET kinase. Time evolution of the distances between Ca atoms of the residues in the local structural environment of the mutational site. Time evolution of the distances between Ca atoms of the M1250T MET and L1245 MET (A), M1250T MET and V1257 MET (B), M1250T MET and S1254 MET (C), and M1250T MET and F1260 MET (D). Time evolution of the distances for the M1250 WT MET is shown in blue; time evolution of the distances for the T1250 MET mutant is shown in red.
Figure 8.
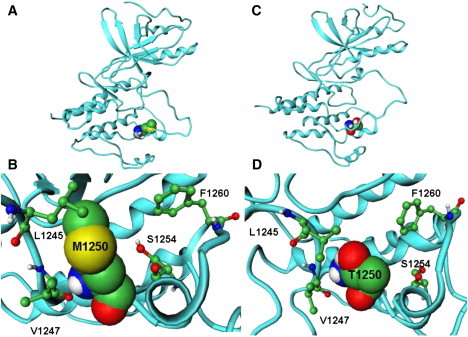
(A) The average structure of the inactive form of WT MET kinase with M1250 residue shown in CPK model. (B) A closeup of structural packing near the mutational site in the inactive WT MET kinase. (C) The average structure of the inactive form of M1250T MET kinase mutant with T1250 residue shown in CPK model. (D) A closeup of structural packing near the mutational site in the inactive T1250 MET kinase.
Hence, we have predicted that the mechanistic basis of the activating M1250T MET mutation may be driven by an appreciable free energy destabilization of the inactive kinase state in the mutational form. It appears that the higher conformational mobility of M1250T in MET, as compared to the WT MET, is largely canceled out by detrimental changes in the intramolecular interactions and the overall enthalpy loss (Table 1). These results support the molecular mechanism of activation, which causes a detrimental imbalance in the dynamic equilibrium shifted toward the active form of the enzyme. This effect may be an important contributing factor of the adverse character and transforming properties of the M1250T MET mutation.
It is worth noting at this point that while MD simulations reveal functionally relevant conformational motions and flexibility of the A-loop, computationally feasible timescales of simulations are not sufficient to observe reproducible and reversible transitions between the closed and opened enzyme forms. Our attempts to considerably extend the timescale based on MD simulations with the crystal structure of the active RET kinase and the crystal structure of the inactive MET kinase suggest that direct simulations of such large conformational changes may be still beyond our current capabilities and require alternative approaches such as targeted molecular dynamics (40,42,96).
Docking and binding simulations of the RET kinase inhibitor with the cancer mutants
The recently solved crystal structures and thermodynamic data of the WT RET (PDB entries 2IVS and 2IVT) and RET complexes with ZD6474 (PDB entry 2IVU) (62) have provided a basis for molecular docking and binding free energy simulations. We have employed the results of MD simulations performed for the WT RET and M918T mutant, and followed with MD simulations of the RET complex with ZD6474 to investigate molecular determinants mediating binding and resistance of oncogenic forms of the RET kinase with the 4-anilinoquinazoline ZD6474 inhibitor (Fig. 9). The binding free energy analysis of the V804G and V804M mutations was carried out using the results of MD simulations of the WT RET, followed by introducing the respective mutation to the 1000 representative RET conformations and docking of the inhibitor to the obtained conformational ensemble of RET mutants.
Figure 9.
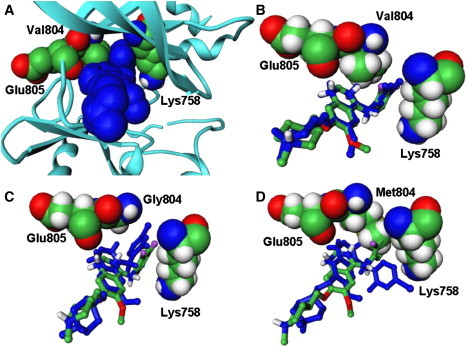
The predicted binding mode of the ZD6474 inhibitor (in blue CPK model) with the WT RET Binding site residues are shown in CPK models. (A) Superposition of the predicted binding mode (in blue stick) with the crystallographic conformation of the ZD6474 inhibitor from the WT complex(default colors, stick model) in the WT RET (B); V804G RET mutant (C); and V804M RET mutant (D).
In agreement with the experimental data, binding simulations of the RET kinase complexes have predicted the binding mode of the ZD6474 inhibitor with the WT RET, which is virtually identical to the crystallographic conformation (Fig. 9, A and B). The analysis of the crystal structure and the predicted inhibitor conformation shows a virtually identical position of the bromofluorophenyl group of ZD6474, which occupies a hydrophobic cavity near the back of the ATP site in the optimal proximity of V804 (Fig. 9 B). Although the inhibitor does make hydrogen bonds with Val-804, the size of the side chain at this position is important in controlling access to this small pocket. Indeed, simulations with the V804G RET mutant suggest a more tightly bound conformation of the inhibitor, which optimizes interactions of the bromofluorophenyl inhibitor group in the created cavity and improves the overall fit of the inhibitor in the binding site (Fig. 9 C). In contrast, Val-804 mutations to the larger Leu and Met residues can severely interfere with the binding of the inhibitor and render resistance to ZD6474. The inhibitor may compromise the favorable extended structure, being forced to adopt an intrinsically strained conformation to avoid direct van der Waals clashes with the V804M residue (Fig. 9 D). These changes may only partially improve the intermolecular inhibitor interactions with the V804M and packing against the Lys-858 side chain, while leading to overall dramatic loss in the binding affinity due to combined loss of the favorable intramolecular and intermolecular contacts (Fig. 10).
Figure 10.
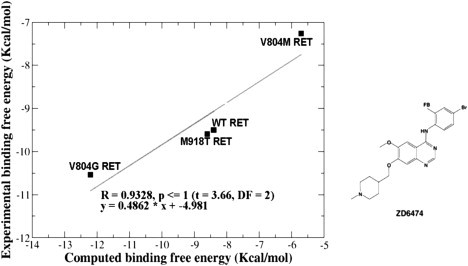
The correlation between the computed and experimental binding free energies of the ZD6474 inhibitor with WT RET, M918T, V804G, and V804M RET mutants.
The predicted binding free energies of the ZD6474 complexes with the WT RET and mutants agree with the biochemical data (66–69). A considerable improvement in the binding affinity of ZD6474 with the V804G RET mutant, as compared to the WT RET, is in full accordance with the experimentally observed inhibition values. In a sharp contrast, docking simulations with the conformations of the V804M RET mutant lead to significant changes in the binding mode of the inhibitor, which encounters severe steric clashes with the larger hydrophobic residue at the gate-keeper position and abrogates efficient inhibitor binding (Fig. 9 D). Hence, docking and binding free-energy evaluations of the inhibitor binding with the oncogenic and drug-resistant RET mutants reproduce the experimental data and provide a structural rationale of the drug inhibition profile, which is consistent with the experimental data.
Structural mapping of RET and MET cancer mutations: modeling protein stability effects
Mapping of disease-linked mutations on the dynamical profile of the WT RET and MET kinases has allowed computational predictions of protein stability changes in a number of important activating mutation sites. In the CUPSAT approach (85), coarse-grained statistical potentials, which are used to predict protein stability changes caused by mutations, may primarily reflect the enthalpy contribution to the free energy. Despite a rather simplistic model, the CUPSAT-based protein stability assessment has captured highly oncogenic RET and MET mutations as the mutations which display a larger destabilization effect on the inactive kinase structure (Figs. 11 A and 12 A). Another approach for the protein stability analysis, FOLDx (87), which provides an estimate of the differences in the empirical folding free energy, has shown a similar trend in the protein stabilities for both RET (Fig. 11 B) and MET mutants (Fig. 12 B). Modeling protein stability effects using the MM-GBSA approach (79,80) can arguably provide a more detailed, quantitative evaluation of the thermostability and complement the results obtained from knowledge-based, empirical models. Importantly, all three approaches have revealed a consistent trend and support a mechanism of activation, whereby the mutations that display the higher oncogenic activity tend to have the greatest destabilization effect on the inactive kinase structure.
Figure 11.
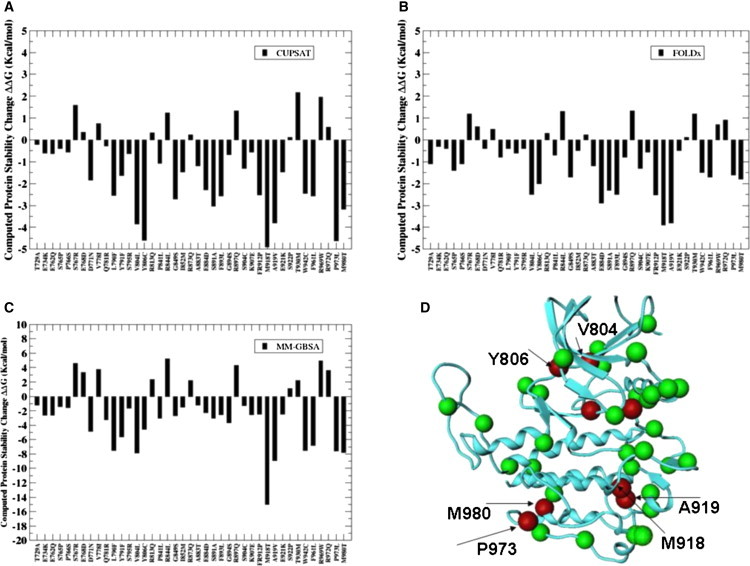
Structural mapping of cancer mutations and modeling protein stability effects in the RET kinase. Protein stability differences between the WT RET and RET mutants using CUPSAT (A), FOLDx (B), and MM-GBSA (C). Mapping of cancer mutations into the structure of the RET kinase (D). Negative values of protein stability changes correspond to destabilizing mutations. Mutational sites are in green CPK Ca models. Mutations with the largest destabilization effect are shown in red.
Figure 12.
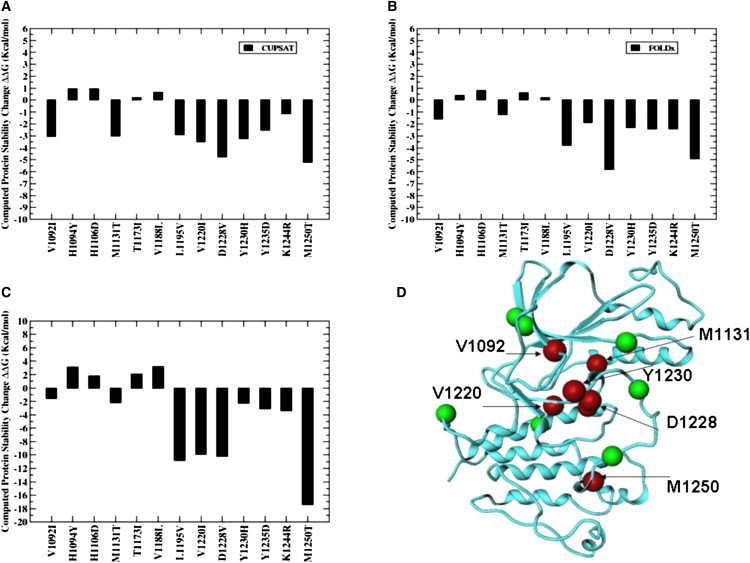
Structural mapping of cancer mutations and modeling protein stability effects in the MET kinase. Protein stability differences between the WT RET and RET mutants using CUPSAT (A), FOLDx (B), and MM-GBSA (C). Mapping of cancer mutations into the structure of the RET kinase (D). Negative values of protein stability changes correspond to destabilizing mutations. Mutational sites are in green CPK Ca models. Mutations with the largest destabilization effect are shown in red.
Indeed, M918T RET and M12509T MET mutations, which have the highest oncogenic activity, may result in the largest destabilization effect on the inactive kinase structure (Figs. 11 and 12). Similarly, mutations of D1228 and M1250, which cause a significant oncogenic transformation (61,97), are predicted to have a significant destabilization effect on the protein structure. In contrast, the transforming potential of mutations at H1094 and H1106 positions is known to be very small (98). In accordance with these experimental data, we have observed a small stabilization effect of H1094Y and H1106D mutations on the autoinhibited MET structure. Interestingly, M918T, Y806C, S891A, and A919V mutations, which display a high transforming potential (62), have the greater destabilization effect on the computed protein stability changes (Fig. 11). Furthermore, P973L and M980T mutations, which have markedly decreased the expression of the RET protein with normal kinase activity and impair the RET kinase activity (99), are shown to have a significant destabilizing effect on the inactive form of RET kinase.
Although a detailed quantitative analysis and validation of the computed protein stability changes would be warranted only when the complete experimental data set becomes available, it is consistent with previous knowledge (100) and may be instructive to note, in the context of this study, that the effect of cancer mutations on normal kinase activity may be associated with their impact on protein stability. Hence, the results suggest that structural effects of activating cancer mutations in RET and MET kinases may manifest in reduced protein stability in the inactive state of the enzyme, thereby triggering a detrimental imbalance in the dynamic equilibrium shifted toward the constitutively active kinase form.
Protein stability and structural effects of cancer mutations: the energy landscape perspective
Our results suggest that structural topology of protein kinase tends to preserve globally similar protein flexibility profiles in the WT and mutational forms, whereas allowing for functionally important local divergences near the mutational site. This result is consistent with the recent studies, which have revealed protein flexibility profiles that can diverge slowly, being conserved both at family and superfamily levels (101,102). From an evolutionary point of view, structural conservation of global protein flexibility and local deviations at functionally critical regions for the mutants may be determined by natural selection of dynamical features that are most important for function, as evolution may partly determine the functional motions for proteins. Thus, in case of protein kinases, natural selection would tend to preserve protein flexibility features that retain the ability of the protein to fluctuate normally between active and inactive states. In contrast, cancer kinase mutations may result in the increased conformational space to be explored in the inactive state, therefore disrupting optimal equilibrium balance between active and inactive enzyme forms. The results of our study can be interpreted from the perspective of the energy landscape theory (103–106), which suggests that less stable and less selective protein states may be a result of a more rugged bottom of the energy funnel, while functionally relevant and specific protein structures are likely to be relatively rigid, with a steep funnel of conformations leading to the native state. In the framework of the energy landscape theory, we can speculate that detrimental activating mutations in protein kinases may act by shifting the accessible conformational space away from regions that facilitate normal functional performance of the kinase and perturb the funneled nature of the free energy landscape. This analysis is consistent with a recently proposed mechanism (107), in which proteins can modulate their function by altering the accessible conformational space, while deleterious mutations may increase the probability of exploring additional conformational regions that are incompatible with the normal functional attributes. While the structural diversity of WT protein kinases has been illuminated in recent years given the rapidly increasing body of crystal structures, structural knowledge of functionally important kinase mutants is limited and presents an important challenge for structural pathology studies of protein kinases (34). It is evident that the accuracy of computational structure predictions for kinase cancer mutants and the atomic details of activation mechanisms can be fully understood only when the respective high resolution crystal structures become available. However, considering current experimental challenges in dissecting the molecular basis of cancer-causing mutations (34), the presented computational analysis provides useful insights into the mechanistic basis of activating kinase mutations, which agree with available experimental data. Computational predictions from this study can inform future experiments exploring the molecular pathology of tumorigenesis and facilitate rationale drug design of personalized cancer therapies.
Conclusions
Computer simulations of the conserved cancer mutations in the RET and MET kinases have allowed us to elucidate at atomic resolution the impact of these mutations in altering protein kinase structure, dynamics, and stability. The functional role of a structurally conserved mutational hotspot in the RET and MET kinases, shared by M918T RET and M1250T MET, has been deciphered by simulating the differential effect of these mutations on active and inactive kinase states. We have found that the mechanistic basis of the activating RET and MET cancer mutations may be driven by an appreciable free energy destabilization of the inactive kinase state in the mutational forms. The computed protein stability differences between the WT and cancer mutants are consistent with circular dichroism spectroscopy and differential scanning calorimetry experiments, and therefore provide a molecular rationale of the observed phenomenon. These results support the molecular mechanism of activation, which causes a detrimental imbalance in the dynamic equilibrium shifted toward the active form of the enzyme. Furthermore, structural mapping of cancer mutations in RET and MET kinases and the computed protein stability changes using three different approaches have revealed a consistent trend and reinforced the proposed mechanism of activation, whereby the mutations that display the higher oncogenic activity tend to have the greatest destabilization effect on the inactive kinase structure. Ultimately, understanding structural and functional effects of conserved cancer mutations can aid in the development of therapeutics to specifically combat mutation-dependent tumorigenesis, while avoiding potential side effects related to normal functions of the targeted kinase.
Acknowledgments
N.J.S. and his laboratory are supported in part by the following research grants: The National Heart Lung and Blood Institute Family Blood Pressure Program (grant No. U01 HL064777-06); The National Institute on Aging Longevity Consortium (grant No. U19 AG023122-01); the National Institute of Mental Health Consortium on the Genetics of Schizophrenia (grant No. 5 R01 HLMH065571-02); The National Institute of Mental Health-funded Genetic Association Information Network Study of Bipolar Disorder (grant No. 1 R01 MH078151-01A1); National Institutes of Health grants: Nos. N01 MH22005, U01 DA024417-01, and P50 MH081755-01; and the Scripps Genomic Medicine and the Scripps Translational Science Institute. A.T. is a Scripps Genomic Medicine Dickinson Scholar.
References
- 1.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Hahn W.C., Weinberg R.A. Modeling the molecular circuitry of cancer. Nat. Rev. Cancer. 2002;2:331–341. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- 3.Davies H., Bignell G.R., Cox C., Stephens P., Edkins S. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 4.Bardelli A., Parsons D.W., Silliman N., Ptak J., Szabo S. Mutational analysis of the tyrosine kinome in colorectal cancers. Science. 2003;300:949. doi: 10.1126/science.1082596. [DOI] [PubMed] [Google Scholar]
- 5.Samuels Y., Wang Z., Bardelli A., Silliman N., Ptak J. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z., Shen D., Parsons D.W., Bardelli A., Sager J. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 7.Futreal P.A., Coin L., Marshall M., Down T., Hubbard T. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas R.K., Baker A.C., Debiasi R.M., Winckler W., Laframboise T. High-throughput oncogene mutation profiling in human cancer. Nat. Genet. 2007;39:347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 9.Hanks S.K., Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 10.Hunter T. Signaling—2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 11.Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 12.Scheeff E.D., Bourne P.E. Structural evolution of the protein kinase-like superfamily. PLoS Comput Biol. 2005;1:e49. doi: 10.1371/journal.pcbi.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kannan N., Taylor S.S., Zhai Y., Venter J.C., Manning G. Structural and functional diversity of the microbial kinome. PLoS Biol. 2007;5:e17. doi: 10.1371/journal.pbio.0050017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knighton D.R., Zheng J.H., Eyck L.F.T., Ashford V.A., Xuong N.H. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 15.Knighton D.R., Zheng J.H., Eyck L.F.T., Xuong N.H., Taylor S.S. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 16.Johnson L.N., Noble M.E., Owen D.J. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 17.Huse M., Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 18.Nolen B., Taylor S., Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 19.Stephens P., Edkins S., Davies H., Greenman C., Cox C. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat. Genet. 2005;37:590–592. doi: 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- 20.Sjoblom T., Jones S., Wood L.D., Parsons D.W., Lin J. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 21.Wood L.D., Parsons D.W., Jones S., Lin J., Sjoblom T. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 22.Greenman C., Stephens P., Smith R., Dalgliesh G.L., Hunter C. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma S.V., Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214–3231. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- 24.Torkamani A., Schork N.J. Accurate prediction of deleterious protein kinase polymorphisms. Bioinformatics. 2007;23:2918–2925. doi: 10.1093/bioinformatics/btm437. [DOI] [PubMed] [Google Scholar]
- 25.Torkamani A., Schork N.J. Prediction of cancer driver mutations in protein kinases. Cancer Res. 2008;68:1675–1682. doi: 10.1158/0008-5472.CAN-07-5283. [DOI] [PubMed] [Google Scholar]
- 26.Torkamani A., Kannan N., Taylor S.S., Schork N.J. Congenital disease SNPs target lineage specific structural elements in protein kinases. Proc. Natl. Acad. Sci. USA. 2008;105:9011–9016. doi: 10.1073/pnas.0802403105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 28.Paez J.G., Janne P.A., Lee J.C., Tracy S., Greulich H. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 29.Pao W., Miller V., Zakowski M., Doherty J., Politi K. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greulich H., Chen T.-H., Feng W., Janne P.A., Alvarez J.V. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X., Gureasko J., Shen K., Cole P.A., Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 32.Yun C.-H., Boggon T.J., Li Y., Woo M.S., Greulich H. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yun C.-H., Mengwasser K.E., Toms A.V., Woo M.S., Greulich H. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar A., Petri E.T., Halmos B., Boggon T.J. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 2008;26:1742–1751. doi: 10.1200/JCO.2007.12.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Modugno M., Casale E., Soncini C., Rosettani P., Colombo R. Crystal structure of the T315I AbI mutant in complex with the aurora kinases inhibitor PHA-739358. Cancer Res. 2007;67:7987–7990. doi: 10.1158/0008-5472.CAN-07-1825. [DOI] [PubMed] [Google Scholar]
- 36.Zhou T., Parillon L., Li F., Wang Y., Keats J. Crystal structure of the T315I mutant of AbI kinase. Chem. Biol. Drug Des. 2007;70:171–181. doi: 10.1111/j.1747-0285.2007.00556.x. [DOI] [PubMed] [Google Scholar]
- 37.Wong C.F. Flexible ligand-flexible protein docking in protein kinase systems. Biochim. Biophys. Acta. 2008;1784:244–251. doi: 10.1016/j.bbapap.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 38.Cavasotto C.N., Abagyan R.A. Protein flexibility in ligand docking and virtual screening to protein kinases. J. Mol. Biol. 2004;337:209–225. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Rockey W.M., Elcock A.H. Rapid computational identification of the targets of protein kinase inhibitors. Curr. Opin. Drug Discov. Devel. 2006;9:326–331. [PubMed] [Google Scholar]
- 40.Young M.A., Gonfloni S., Superti-Furga G., Roux B., Kuriyan J. Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell. 2001;105:115–126. doi: 10.1016/s0092-8674(01)00301-4. [DOI] [PubMed] [Google Scholar]
- 41.Liu B., Bernard B., Wu J.H. Impact of EGFR point mutations on the sensitivity to gefitinib: insights from comparative structural analyses and molecular dynamics simulations. Proteins. 2006;65:331–346. doi: 10.1002/prot.21111. [DOI] [PubMed] [Google Scholar]
- 42.Zou J., Wang Y.-D., Ma F.-X., Xiang M.-L., Shi B. Detailed conformational dynamics of juxtamembrane region and activation loop in c-Kit kinase activation process. Proteins. 2008;72:323–332. doi: 10.1002/prot.21928. [DOI] [PubMed] [Google Scholar]
- 43.Pricl S., Fermeglia M., Ferrone M., Tamborini E. T315I-mutated Bcr-Abl in chronic myeloid leukemia and imatinib: insights from a computational study. Mol. Cancer Ther. 2005;4:1167–1174. doi: 10.1158/1535-7163.MCT-05-0101. [DOI] [PubMed] [Google Scholar]
- 44.Lee T.-S., Potts S.J., Kantarjian H., Cortes J., Giles F. Molecular basis explanation for imatinib resistance of BCR-ABL due to T315I and P-loop mutations from molecular dynamics simulations. Cancer. 2008;112:1744–1753. doi: 10.1002/cncr.23355. [DOI] [PubMed] [Google Scholar]
- 45.Verkhivker G.M. Imprint of evolutionary conservation and protein structure variation on the binding function of protein tyrosine kinases. Bioinformatics. 2006;22:1846–1854. doi: 10.1093/bioinformatics/btl199. [DOI] [PubMed] [Google Scholar]
- 46.Verkhivker G.M. Computational proteomics of biomolecular interactions in the sequence and structure space of the tyrosine kinome: deciphering the molecular basis of the kinase inhibitors selectivity. Proteins. 2007;66:912–929. doi: 10.1002/prot.21287. [DOI] [PubMed] [Google Scholar]
- 47.Verkhivker G.M. In silico profiling of tyrosine kinases binding specificity and drug resistance using Monte Carlo simulations with the ensembles of protein kinase crystal structures. Biopolymers. 2007;85:333–348. doi: 10.1002/bip.20656. [DOI] [PubMed] [Google Scholar]
- 48.Verkhivker G.M. Exploring sequence-structure relationships in the tyrosine kinome space: functional classification of the binding specificity mechanisms for cancer therapeutics. Bioinformatics. 2007;23:1919–1926. doi: 10.1093/bioinformatics/btm277. [DOI] [PubMed] [Google Scholar]
- 49.Stenberg K.A., Riikonen P.T., Vihinen M. KinMutBase, a database of human disease-causing protein kinase mutations. Nucleic Acids Res. 1999;27:362–364. doi: 10.1093/nar/27.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stenberg K.A., Riikonen P.T., Vihinen M. KinMutBase, a database of human disease-causing protein kinase mutations. Nucleic Acids Res. 2000;28:369–371. doi: 10.1093/nar/28.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santoro M., Melillo R.M., Carlomagno F., Fusco A., Vecchio G. Molecular mechanisms of RET activation in human cancer. Ann. N. Y. Acad. Sci. 2002;963:116–121. doi: 10.1111/j.1749-6632.2002.tb04102.x. [DOI] [PubMed] [Google Scholar]
- 52.Santoro M., Melillo R.M., Carlomagno F., Vecchio G., Fusco A. Minireview: RET: normal and abnormal functions. Endocrinology. 2004;145:5448–5451. doi: 10.1210/en.2004-0922. [DOI] [PubMed] [Google Scholar]
- 53.Cranston A.N., Carniti C., Oakhill K., Radzio-Andzelm E., Stone E.A. RET is constitutively activated by novel tandem mutations that alter the active site resulting in multiple endocrine neoplasia type 2B. Cancer Res. 2006;66:10179–10187. doi: 10.1158/0008-5472.CAN-06-0884. [DOI] [PubMed] [Google Scholar]
- 54.Gujral T.S., Singh V.K., Jia Z., Mulligan L.M. Molecular mechanisms of RET receptor-mediated oncogenesis in multiple endocrine neoplasia 2B. Cancer Res. 2006;66:10741–10749. doi: 10.1158/0008-5472.CAN-06-3329. [DOI] [PubMed] [Google Scholar]
- 55.Gujral T.S., Mulligan L.M. Molecular implications of RET mutations for pheochromocytoma risk in multiple endocrine neoplasia 2. Ann. N. Y. Acad. Sci. 2006;1073:234–240. doi: 10.1196/annals.1353.025. [DOI] [PubMed] [Google Scholar]
- 56.Lai A.Z., Gujral T.S., Mulligan L.M. RET signaling in endocrine tumors: delving deeper into molecular mechanisms. Endocr. Pathol. 2007;18:57–67. doi: 10.1007/s12022-007-0009-5. [DOI] [PubMed] [Google Scholar]
- 57.Maritano D., Accornero P., Bonifaci N., Ponzetto C. Two mutations affecting conserved residues in the Met receptor operate via different mechanisms. Oncogene. 2000;19:1354–1361. doi: 10.1038/sj.onc.1203431. [DOI] [PubMed] [Google Scholar]
- 58.Nakaigawa N., Weirich G., Schmidt L., Zbar B. Tumorigenesis mediated by MET mutant M1268T is inhibited by dominant-negative Src. Oncogene. 2000;19:2996–3002. doi: 10.1038/sj.onc.1203628. [DOI] [PubMed] [Google Scholar]
- 59.Morotti A., Mila S., Accornero P., Tagliabue E., Ponzetto C. K252a inhibits the oncogenic properties of Met, the HGF receptor. Oncogene. 2002;21:4885–4893. doi: 10.1038/sj.onc.1205622. [DOI] [PubMed] [Google Scholar]
- 60.Berthou S., Aebersold D.M., Schmidt L.S., Stroka D., Heigl C. The Met kinase inhibitor SU11274 exhibits a selective inhibition pattern toward different receptor mutated variants. Oncogene. 2004;23:5387–5393. doi: 10.1038/sj.onc.1207691. [DOI] [PubMed] [Google Scholar]
- 61.Miller M., Ginalski K., Lesyng B., Nakaigawa N., Schmidt L. Structural basis of oncogenic activation caused by point mutations in the kinase domain of the MET proto-oncogene: modeling studies. Proteins. 2001;44:32–43. doi: 10.1002/prot.1069. [DOI] [PubMed] [Google Scholar]
- 62.Knowles P.P., Murray-Rust J., Kjaer S., Scott R.P., Hanrahan S. Structure and chemical inhibition of the RET tyrosine kinase domain. J. Biol. Chem. 2006;281:33577–33587. doi: 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- 63.Stamos J., Sliwkowski M.X., Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 64.Plaza-Menacho I., Mologni L., Sala E., Gambacorti-Passerini C., Magee A.I. Sorafenib functions to potently suppress RET tyrosine kinase activity by direct enzymatic inhibition and promoting RET lysosomal degradation independent of proteasomal targeting. J. Biol. Chem. 2007;282:29230–29240. doi: 10.1074/jbc.M703461200. [DOI] [PubMed] [Google Scholar]
- 65.Schiering N., Knapp S., Marconi M., Flocco M.M., Cui J. Crystal structure of the tyrosine kinase domain of the hepatocyte growth factor receptor c-Met and its complex with the microbial alkaloid K-252a. Proc. Natl. Acad. Sci. USA. 2003;100:12654–12659. doi: 10.1073/pnas.1734128100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carlomagno F., Vitagliano D., Guida T., Napolitano M., Vecchio G. The kinase inhibitor PP1 blocks tumorigenesis induced by RET oncogenes. Cancer Res. 2002;62:1077–1082. [PubMed] [Google Scholar]
- 67.Carlomagno F., Guida T., Anaganti S., Vecchio G., Fusco A. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene. 2004;23:6056–6063. doi: 10.1038/sj.onc.1207810. [DOI] [PubMed] [Google Scholar]
- 68.Carlomagno F., Vitagliano D., Guida T., Ciardiello F., Tortora G. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284–7290. [PubMed] [Google Scholar]
- 69.Carlomagno F., Vitagliano D., Guida T., Basolo F., Castellone M.D. Efficient inhibition of RET/papillary thyroid carcinoma oncogenic kinases by 4-amino-5-(4-chloro-phenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) J. Clin. Endocrinol. Metab. 2003;88:1897–1902. doi: 10.1210/jc.2002-021278. [DOI] [PubMed] [Google Scholar]
- 70.Marti-Renom M.A., Stuart A.C., Fiser A., Sanchez R., Melo F. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 71.Fiser A., Do R.K., Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Canutescu A.A., Shelenkov A.A., Dunbrack R.L.J. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 75.Eargle J., Wright D., Luthey-Schulten Z. Multiple alignment of protein structures and sequences for VMD. Bioinformatics. 2006;22:504–506. doi: 10.1093/bioinformatics/bti825. [DOI] [PubMed] [Google Scholar]
- 76.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 77.Verkhivker G.M., Bouzida D., Gehlhaar D.K., Rejto P.A., Arthurs S. Deciphering common failures in molecular docking of ligand-protein complexes. J. Comput. Aided Mol. Des. 2000;14:731–751. doi: 10.1023/a:1008158231558. [DOI] [PubMed] [Google Scholar]
- 78.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general AMBER force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 79.Srinivasan J., Miller J., Kollman P.A., Case D.A. Continuum solvent studies of the stability of RNA hairpin loops and helices. J. Biomol. Struct. Dyn. 1998;16:671–682. doi: 10.1080/07391102.1998.10508279. [DOI] [PubMed] [Google Scholar]
- 80.Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 81.Verkhivker G.M., Bouzida D., Gehlhaar D.K., Rejto P.A., Freer S.T. Monte Carlo simulations of the peptide recognition at the consensus binding site of the constant fragment of human immunoglobulin G: the energy landscape analysis of a hot spot at the intermolecular interface. Proteins. 2002;48:539–557. doi: 10.1002/prot.10164. [DOI] [PubMed] [Google Scholar]
- 82.Verkhivker G.M., Bouzida D., Gehlhaar D.K., Rejto P.A., Freer S.T. Computational detection of the binding-site hot spot at the remodeled human growth hormone-receptor interface. Proteins. 2003;53:201–219. doi: 10.1002/prot.10456. [DOI] [PubMed] [Google Scholar]
- 83.Verkhivker G.M. Computational analysis of ligand binding dynamics at the intermolecular hot spots with the aid of simulated tempering and binding free energy calculations. J. Mol. Graph. Model. 2004;22:335–348. doi: 10.1016/j.jmgm.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 84.Gilson M.K., Zhou H.-X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 85.Parthiban V., Gromiha M.M., Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res. 2006;34:W239–W242. doi: 10.1093/nar/gkl190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Parthiban V., Gromiha M.M., Abhinandan M., Schomburg D. Computational modeling of protein mutant stability: analysis and optimization of statistical potentials and structural features reveal insights into prediction model development. BMC Struct. Biol. 2007;7:54. doi: 10.1186/1472-6807-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guerois R., Nielsen J.E., Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 89.Liu X., Vega Q.C., Decker R.A., Pandey A., Worby C.A. Oncogenic RET receptors display different autophosphorylation sites and substrate binding specificities. J. Biol. Chem. 1996;271:5309–5312. doi: 10.1074/jbc.271.10.5309. [DOI] [PubMed] [Google Scholar]
- 90.Yuan Z.-L., Guan Y.-J., Wang L., Wei W., Kane A.B. Central role of the threonine residue within the p+1 loop of receptor tyrosine kinase in STAT3 constitutive phosphorylation in metastatic cancer cells. Mol. Cell. Biol. 2004;24:9390–9400. doi: 10.1128/MCB.24.21.9390-9400.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Durney M.A., Wechselberger R.W., Kalodimos C.G., Kaptein R., Vorgias C.E. An alternate conformation of the hyperthermostable HU protein from Thermotoga maritima has unexpectedly high flexibility. FEBS Lett. 2004;563:49–54. doi: 10.1016/S0014-5793(04)00247-9. [DOI] [PubMed] [Google Scholar]
- 92.LeMaster D.M., Tang J., Paredes D.I., Hernandez G. Enhanced thermal stability achieved without increased conformational rigidity at physiological temperatures: spatial propagation of differential flexibility in rubredoxin hybrids. Proteins. 2005;61:608–616. doi: 10.1002/prot.20594. [DOI] [PubMed] [Google Scholar]
- 93.Bishop S.M., Ross J.B., Kohanski R.A. Autophosphorylation dependent destabilization of the insulin receptor kinase domain: tryptophan-1175 reports changes in the catalytic cleft. Biochemistry. 1999;38:3079–3089. doi: 10.1021/bi982546s. [DOI] [PubMed] [Google Scholar]
- 94.Ablooglu A.J., Kohanski R.A. Activation of the insulin receptor's kinase domain changes the rate-determining step of substrate phosphorylation. Biochemistry. 2001;40:504–513. doi: 10.1021/bi002292m. [DOI] [PubMed] [Google Scholar]
- 95.Wang W., Marimuthu A., Tsai J., Kumar A., Krupka H.I. Structural characterization of autoinhibited c-Met kinase produced by coexpression in bacteria with phosphatase. Proc. Natl. Acad. Sci. USA. 2006;103:3563–3568. doi: 10.1073/pnas.0600048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van der Vaart A., Karplus M. Simulation of conformational transitions by the restricted perturbation-targeted molecular dynamics method. J. Chem. Phys. 2005;122:114903. doi: 10.1063/1.1861885. [DOI] [PubMed] [Google Scholar]
- 97.Jeffers M., Schmidt L., Nakaigawa N., Webb C.P., Weirich G. Activating mutations for the met tyrosine kinase receptor in human cancer. Proc. Natl. Acad. Sci. USA. 1997;94:11445–11450. doi: 10.1073/pnas.94.21.11445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schmidt L., Junker K., Nakaigawa N., Kinjerski T., Weirich G. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. 1999;18:2343–2350. doi: 10.1038/sj.onc.1202547. [DOI] [PubMed] [Google Scholar]
- 99.Iwashita T., Kurokawa K., Qiao S., Murakami H., Asai N. Functional analysis of RET with Hirschsprung mutations affecting its kinase domain. Gastroenterology. 2001;121:24–33. doi: 10.1053/gast.2001.25515. [DOI] [PubMed] [Google Scholar]
- 100.Yue P., Li Z., Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 2005;353:459–473. doi: 10.1016/j.jmb.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 101.Maguid S., Fernandez-Alberti S., Parisi G., Echave J. Evolutionary conservation of protein backbone flexibility. J. Mol. Evol. 2006;63:448–457. doi: 10.1007/s00239-005-0209-x. [DOI] [PubMed] [Google Scholar]
- 102.Maguid S., Fernandez-Alberti S., Ferrelli L., Echave J. Exploring the common dynamics of homologous proteins. Application to the globin family. Biophys. J. 2005;89:3–13. doi: 10.1529/biophysj.104.053041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shoemaker B.A., Portman J.J., Wolynes P.G. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Levy Y., Wolynes P.G., Onuchic J.N. Protein topology determines binding mechanism. Proc. Natl. Acad. Sci. USA. 2004;101:511–516. doi: 10.1073/pnas.2534828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsai C.J., Ma B., Nussinov R. Folding and binding cascades: shifts in energy landscapes. Proc. Natl. Acad. Sci. USA. 1999;96:9970–9972. doi: 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Verkhivker G.M., Bouzida D., Gehlhaar D.K., Rejto P.A., Freer S.T. Complexity and simplicity of ligand-macromolecule interactions: the energy landscape perspective. Curr. Opin. Struct. Biol. 2002;12:197–203. doi: 10.1016/s0959-440x(02)00310-x. [DOI] [PubMed] [Google Scholar]
- 107.Thorpe I.F., Brooks C.L., III Molecular evolution of affinity and flexibility in the immune system. Proc. Natl. Acad. Sci. USA. 2007;104:8821–8826. doi: 10.1073/pnas.0610064104. [DOI] [PMC free article] [PubMed] [Google Scholar]