

Abstract
Platelet activation has long been postulated to contribute to the development of atherosclerotic plaques, although the mechanism by which this might occur remains unknown. Thrombin is a potent platelet activator and transfusion of thrombin-activated platelets into mice increases plaque formation, suggesting that thrombin-induced platelet activation might contribute to platelet-dependent atherosclerosis. Platelets from protease-activated receptor 4-deficient (Par4-/-) mice fail to respond to thrombin. To determine whether thrombin-activated platelets play a necessary role in a model of atherogenesis, we compared plaque formation and progression in Par4+/+ and Par4-/- mice in the atherosclerosis-prone apolipoprotein E-deficient (ApoE-/-) background. Littermate Par4+/+ and Par4-/- mice, all ApoE-/-, were placed on a Western diet (21% fat, 0.15% cholesterol) for 5 or 10 weeks. The percent of aortic lumenal surface covered by plaques in Par4+/+ and Par4-/- mice was not different at either time point (2.2 ± 0.3% vs. 2.5 ± 0.2% and 5.1 ± 0.4% vs. 5.6 ± 0.4% after 5 and 10 weeks, respectively). Further, no differences were detected in the cross-sectional area of plaques measured at the aortic root (1.53 ± 0.17 vs. 1.66 ± 0.16 × 105 μm2 and 12.56 ± 1.23 vs. 13.03 ± 0.55 × 105 μm2 after 5 and10 weeks, respectively). These findings indicate that thrombin-mediated platelet activation is not required for the early development of atherosclerotic plaques in the ApoE-/- mouse model and suggest that, if platelet activation is required for plaque formation under these experimental conditions, platelet activators other than thrombin suffice.
Keywords: atherosclerosis, protease-activated receptor, thrombin, platelets
Introduction
The pathogenesis of atherosclerosis involves a complex interaction among lipids, plasma proteins and monocytes, vascular endothelial and smooth muscle cells, lymphocytes and platelets (for review see 1). Platelets play a central role in thrombus formation in response to erosion or rupture of atherosclerotic plaques, and such thrombi participate in the evolution of complex plaques and cause the major complications of atherosclerosis, myocardial infarction and stroke. Whether platelets make a significant contribution to the genesis of the early atherosclerotic plaque has been less certain. Whilst early work posited a role for platelet-released chemokines and growth factors in recruiting monocyte/macrophages to the vessel wall and promoting smooth muscle cell proliferation to cause plaque formation,2,3 later work emphasized a key role for adhesion molecules, chemokines and growth factors expressed by activated endothelial cells and monocytes/macrophages themselves in driving these processes.4 Yet strong evidence has emerged over recent years suggesting that activated platelets do indeed contribute to atherosclerotic plaque progression in well-defined animal models.5-8 Despite this, the mechanism(s) by which platelets become activated in the setting of atherogenesis remains unknown. Clues to possible platelet activation mechanisms come from studies demonstrating that tissue factor, the trigger for thrombin generation, is expressed by circulating monocytes in response to inflammatory stimuli9,10 and by monocyte-derived macrophages in atheroma.11,12 Further, transfusion of thrombin-activated platelets enhances plaque development in atherosclerosis-prone mice.13 Thrombin is a potent activator of platelets and stimulation of endothelial cells by chemokines released by thrombin-activated platelets and the consequent expression of leukocyte-binding adhesion molecules by endothelial cells provides a potential mechanism by which platelets might promote monocyte recruitment and atheroma formation.13 Platelet tethering of leukocytes to endothelial cells is well known to enhance leukocyte interactions with the vessel wall,14 and activated platelets and platelet-monocyte aggregates circulate in increased numbers in blood of atherosclerotic patients.15-17 Together, these observations provide a possible mechanism by which thrombin-activated platelets might contribute to the formation of atherosclerotic plaques.
Thrombin triggers cellular responses via protease-activated receptors (PARs), and platelets from protease-activated receptor 4-deficient (Par4-/-) mice are unresponsive to thrombin.18 Thus, such mice provide an opportunity to assess the contribution of thrombin-induced platelet activation in models of disease. To directly assess the importance of thrombin-induced platelet activation in atherogenesis, we examined the effect of Par4-deficiency on atherosclerotic plaque development in ApoE-/- mice. Our results demonstrate that thrombin-mediated platelet activation is not required for the early development of atherosclerotic plaques in the ApoE-/- mouse model and suggest that, if platelet activation contributes to atherogenesis under these experimental conditions, platelet activators other than thrombin are involved.
Materials and Methods
Mice
Par4-/- mice (C57Bl/6 genetic background, N6; 18) were crossed twice to ApoE-/- mice (C57Bl/6, N10, JAX) to generate ApoE-/-:Par4+/- mice (>99% C57Bl/6 genetic background). These mice were then intercrossed to generate littermate ApoE-/-:Par4+/+ and ApoE-/-:Par4-/- mice which were used in the study. The genotypes were determined by PCR analysis using tail biopsy DNA, as previously described 18. Mice of all genotypes were housed together in a specific pathogen-free facility, weaned at age 3 weeks, fed standard rodent chow (6.5% fat and 0.028% cholesterol) until five weeks of age, then switched to a high-fat chow (21% fat and 0.15% cholesterol; Harlan-Teklad) for an additional five or ten weeks.
Serum cholesterol
Serum cholesterol levels were determined in mice fasted overnight. Blood was drawn via retro-orbital venous puncture and allowed to clot. Serum was collected after centrifugation at 600 × g for 5 min and cholesterol levels were measured using a standard enzyme-based colorimetric cholesterol kit (Boehringer Mannheim Biochemicals, Indianapolis, IN), as per the manufacturer's instructions.
Platelet function analyses
Platelets were isolated from whole blood collected from the inferior vena cava of anesthetized mice and anticoagulated with a combination of low molecular weight heparin (40 U/mL) and acid-citrate-dextrose (13 mm sodium citrate, 1 mm citric acid, 20 mm dextrose, and 10 mm theophylline). Anticoagulated blood was mixed with 300 μL of platelet wash buffer (4.3 mm K2HPO4, 4.3 mm Na2HPO4, 24.3 mm NaH2PO4, 113 mm NaCl, 5.5 mm glucose, 0.5% bovine serum albumin, 10 mm theophylline, and 0.02 U/mL apyrase) and centrifuged at 250 × g for 3 min. This was performed three times, and each time the platelet rich plasma was removed. The pooled platelet rich plasma was centrifuged at 2,000 × g for 1 min and resuspended in Tyrode's buffer (10 mm HEPES, 12 mm NaHCO3, 137 mm NaCl, 2.7 mm KCl, 1mm CaCl2, and 5 mm glucose) for functional analyses. For aggregation studies, isolated platelets were resuspended at 2 × 108/mL, stirred at 600 rpm at 37 °C in an automated platelet analyzer (AggRAM, Helena Laboratories), and responses to α-thrombin (bovine; 1 or 10 U/mL) or ADP (1 or 10 μm) recorded as percent change in optical density of the platelet suspension. For P-selectin expression studies, isolated platelets were resuspended at 5 × 107/mL, stimulated with α-thrombin (1 or 10 U/mL) for 2 to 20 min, and fixed with 2% paraformaldehyde for 30 min. Fixed platelets were incubated with a FITC-labeled anti–P-selectin antibody (BD, 1:100) for 20 min, washed once in PBS, and mean fluorescence determined by flow cytometry on a FACSCalibur (Becton Dickinson).
En face analysis
Mice were anesthetized and perfused through the left ventricle with phosphate buffered saline (PBS) followed by 10% neutral buffered formalin (Sigma Chemical Co., St. Louis, MO). For quantitative en face analysis of the entire aortic lumenal surface, aortae (from the aortic sinus to the iliac bifurcation) were isolated and carefully cleared of surrounding adventitia and fatty tissue, then pinned onto wax blocks 19. Sudan IV staining was employed to visualize fatty deposits: the aortae were rinsed in 70% ethanol for 30 s, stained with 0.5% (wt/vol) Sudan IV in 35% ethanol:50% acetone for 20 min at 55°C, destained in 80% ethanol for 5 min, and then stored in PBS. Quantitation of Sudan IV stained surface area was determined from digitized images of stained aortae using Adobe Photoshop with an Image Processing Tool Kit plug-in (Reindeer Graphics, Asheville, NC). Percent area covered by lesion was defined as Sudan IV-positive area (red) divided by total aortic area.
Aortic sinus analysis
Hearts from the above mice were cut approximately in half on a plane below the two atria and perpendicular to the aortic sinus. The segment containing the aortic sinus was incubated in sucrose (30% wt/vol) at least overnight and then embedded in OCT and frozen. 10 μm thick cryostat sections (cut perpendicular to the aorta) were prepared. Sections were discarded until reaching the junction of the heart muscle and aorta where all three valve cusps become visible, then consecutive 10 μm sections were collected for 400 μm toward the aortic arch and exiting the valve region (4 sections/slide; 10 slides/heart). Slides number 1, 4, 7, and 10 were stained with oil red-O and counterstained with hemotoxylin. The area of the lesion (red) was quantified using Image-Pro Plus (Media Cybernetics, Bethesda, MD). Values reported represent the mean lesion area from the four slides (16 sections) for each animal. Slide number 5 was stained immunohistochemically with MOMA-2 (Serotec, Kidlington, UK) to identify macrophages. Briefly, frozen sections were dried, fixed with acetone for 5 min, blocked using 5% goat serum (Vector Laboratories, Burlingame, CA) in 3% BSA for 2 h, stained with MOMA-2 at 1:25 for 1 h followed by a HRP-conjugated goat anti-rat IgG (1:1000, Vector) for 1 h, and visualized using Vectastain ABC-AP Red reagent (Vector). Sections were counterstained with hematoxylin.
Statistics
Mean lesion areas were compared using a two-sided unpaired Student's t-test, with P < 0.05 defined as significant.
Experimental Results
ApoE-/-:Par4-/- mice
Mice with combined deficiencies in ApoE and Par4 were born at the expected Mendelian rates from ApoE-/-:Par4+/- intercrosses. ApoE-/-:Par4-/- mice were indistinguishable from ApoE-/-:Par4+/+ littermate controls in appearance, body weight, and degree of hypercholesterolemia (Table 1). Platelets from Par4-/- mice fail to respond to thrombin18 and we confirmed that platelets isolated from ApoE-/-:Par4-/- mice remained unresponsive to thrombin-induced aggregation and P-selectin expression at thrombin concentrations of up to 10 U/mL, yet exhibited aggregation responses to ADP (10 μm) that were indistinguishable from littermate ApoE-/-:Par4+/+ controls (Fig. 1).
Table 1. Body weight and serum cholesterol levels in Par4+/+ and Par4-/- mice.
| Age (weeks) | 10 | 15 | ||||||
| Gender | M | F | M | F | ||||
| Par4 Genotype | +/+ | -/- | +/+ | -/- | +/+ | -/- | +/+ | -/- |
| Body Weight (g) | 24.7±0.4 | 27.3±0.5 | 21.4±0.2 | 23.8±0.3 | 28.0±1.8 | 32.1±1.1 | 23.8±0.4 | 23.5±0.6 |
| Serum Cholesterol (mg/dL) | 1012±64 | 1007±157 | 1114±95 | 1083±75 | 1034±60 | 1184±81 | 1073±60 | 1153±92 |
| N | 9 | 9 | 15 | 10 | 6 | 4 | 8 | 8 |
Fig. 1. Platelet functional analyses.

Platelets were isolated from littermate ApoE-/- mice that were either Par4+/+ or Par4-/-. Shown are platelet aggregations in response to A: thrombin (10 U/mL; N = 4) and B: ADP (10 μm; N = 6), and C: platelet exposure of P-selectin in response to thrombin (10 U/mL; N = 3). All data are mean ± sem.
En face analysis
Littermate Par4+/+ and Par4-/- mice of either sex in an ApoE-/- background were fed a high-fat diet (21% fat, 0.15% cholesterol) for five weeks and the percent of aorta covered by lipid-rich atheroma was assessed by Sudan IV staining. Par4 deficiency did not effect lesion area. 2.2 ± 0.2 (N=24) percent of the aortic surface was covered by atheroma in Par4+/+ mice versus 2.5 ± 0.2 (N=19) percent in Par4-/- mice (Fig. 2). No significant difference in lesion area was observed between sexes at this early timepoint, as previously reported. In male mice, 1.9 ± 0.2 (N=9) and 2.5 ± 0.3 (N=9) percent was occupied by atheroma in Par4+/+ and Par4-/- mice, respectively. In female mice, the area covered by atheroma was 2.4 ± 0.4 (N=15) and 2.5 ± 0.4 (N=10) percent in Par4+/+ and Par4-/- mice, respectively. Both Par4+/+ and Par4-/- mice developed lesions throughout the aorta from ascending to iliac bifurcation, and in both cases these ranged in appearance from simple fatty streaks to complex fibrous plaques (Fig. 2). Analysis of different aortic segments according to the methods of Tilley et al 23 failed to reveal any regional differences in lesion sizes between Par4+/+ and Par4-/- mice.
Fig. 2. Coverage of the lumenal surface of the aorta with atheroma.
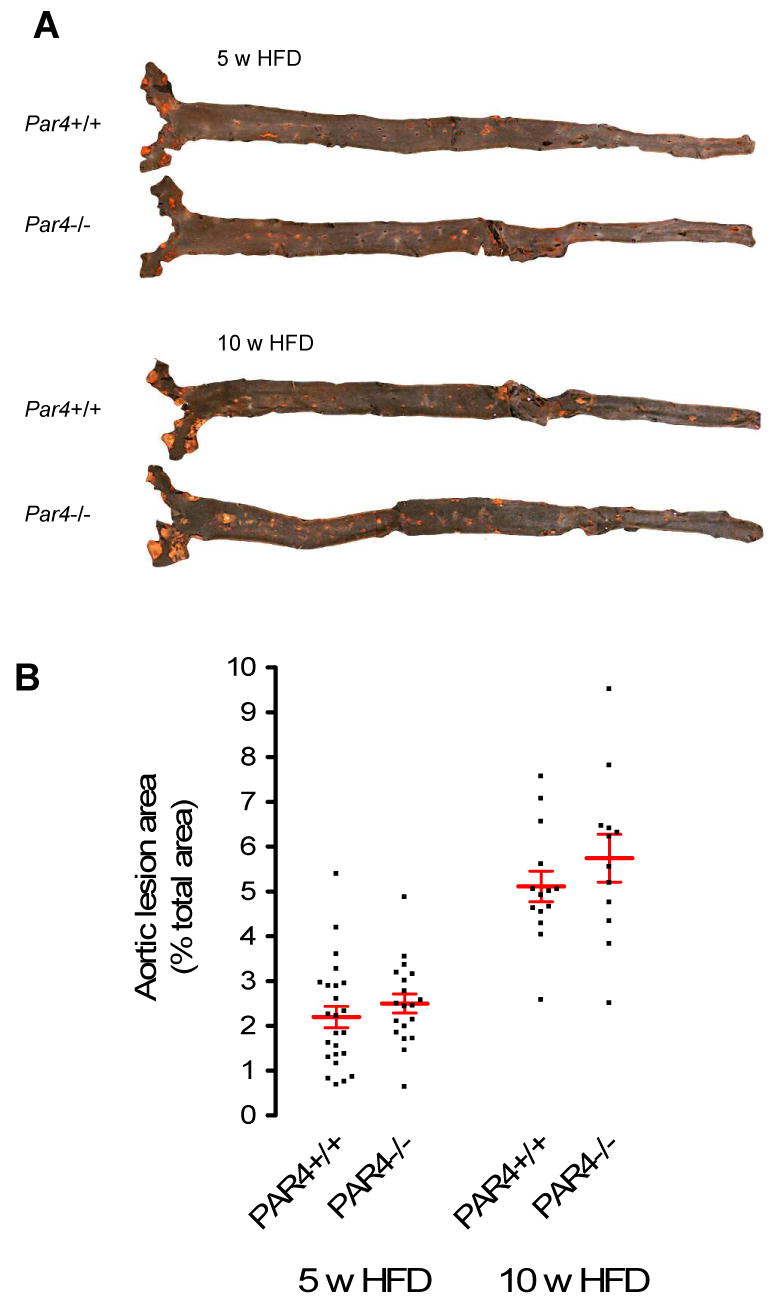
Littermate ApoE-/- mice that were Par4+/+ or Par4-/- were maintained for 5 or 10 weeks on high fat diet before aortae were analyzed. A: Representative en face images of Sudan IV stained aortae. Red represents Sudan IV-positive lipid deposits. B: Percent aortic lumenal surface occupied by Sudan IV-positive lesions. Individual data points, each representing an individual mouse, are shown. HFD = High fat diet (21% fat, 0.15% cholesterol). Bars are mean ± sem. N = 19-24.
In order to examine the role of thrombin-induced platelet activation on the progression of atherosclerosis, we examined littermate Par4+/+ and Par4-/- mice fed a high-fat diet for ten weeks instead of five. The area of the aortic surface occupied by atheroma increased to 5.1 ± 0.4 (N=14) and 5.7 ± 0.6 (N=12) percent in Par4+/+ and Par4-/- mice, respectively (Fig. 2). Again, there was no significant difference by Par4 genotype and no difference in lesion area was found by sex or aortic region.
Aortic sinus analysis
The hearts of twelve mice from each group at each time point were randomly selected for analysis of the relatively large atherosclerotic lesions that occur at the aortic sinus in this model. Sample size was chosen to permit a power of 80% in detecting a 20% difference in means at a confidence level of P = 0.05 (assuming a standard deviation of approximately 30% of the mean, as frequently observed in this model 24). No difference in atheroma size by Par4 genotype was detected at either time point: mean lesion cross sectional area after five weeks of high fat diet was 1.53 ± 0.17 × 105 μm2 vs. 1.66 ± 0.16 × 105 μm2 for Par4+/+ and Par4-/- mice, respectively, and 12.56 ± 1.23 × 105 μm2 vs. 13.03 ± 0.55 × 105 μm2 after 10 weeks on diet (Fig. 3). Furthermore, no consistent differences in the degree of macrophage content in aortic root lesions were detected between the groups at either time point when assessed using immunohistochemical staining for macrophages (Fig. 4).
Fig. 3. Size of aortic sinus atheroma.
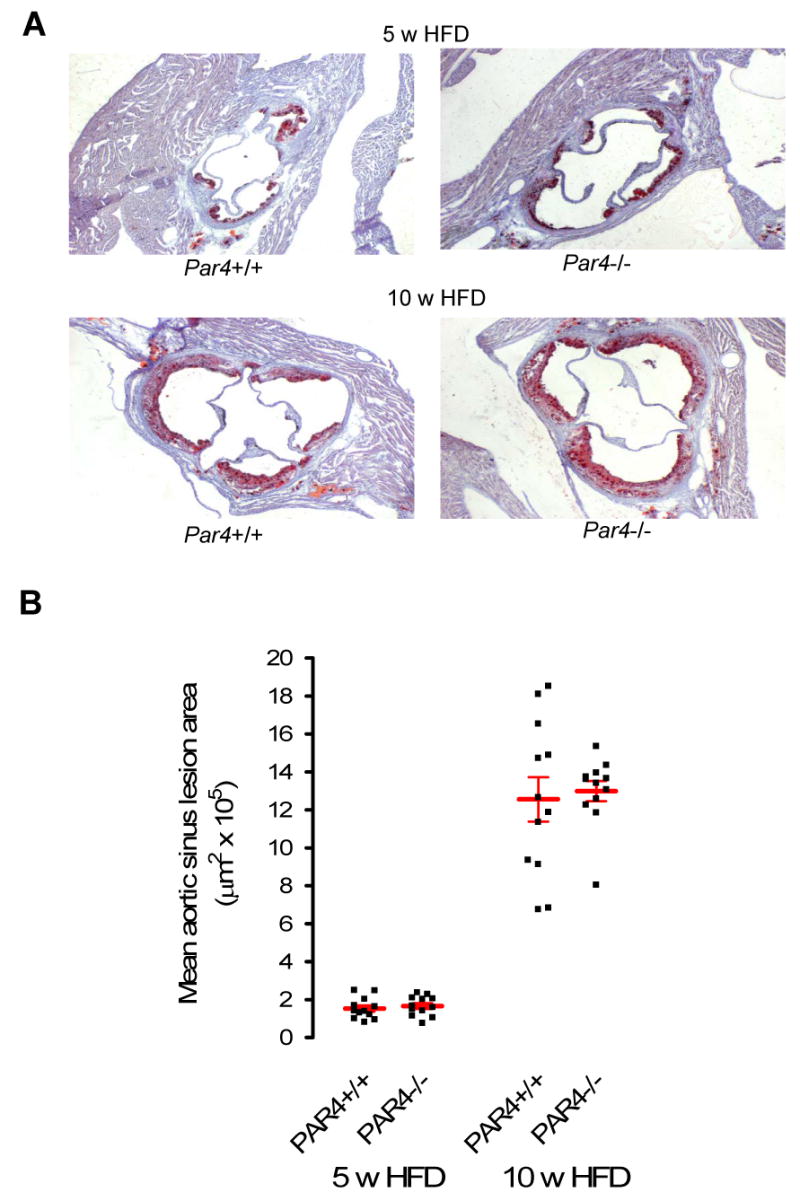
Aortic sinus lesions were analyzed in littermate ApoE-/- mice that were Par4+/+ or Par4-/- and treated as in Fig.2. A: Representative images of oil red-O staining of aortic sinuses, counterstained with hemotoxylin. Red represents positive staining by oil red-O and therefore lipid deposits. B: Cross sectional area of sinus lesions. Individual data points, each representing an individual mouse, are shown. HFD = High fat diet (21% fat, 0.15% cholesterol). Bars are mean ± sem. N = 12 per group.
Fig. 4. Macrophage content within aortic sinus atheroma.
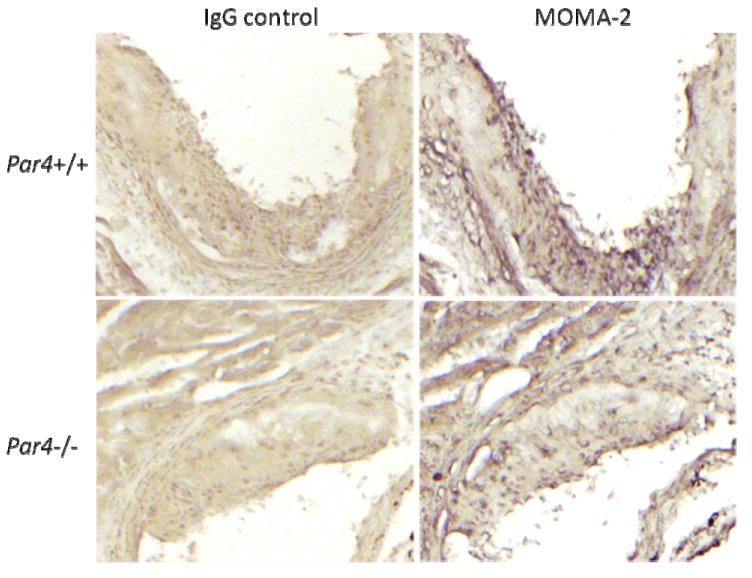
Aortic sinus lesions were analyzed in littermate ApoE-/- mice that were Par4+/+ or Par4-/- and treated as in Fig.2. Shown is MOMA-2 (macrophage) and isotype (IgG) staining of aortic sinuses (red). Images are representative of N = 12 per group.
Discussion
Several lines of evidence have pointed to an association between platelet activation and atherogenesis. Activated platelets are detected in increased numbers in the circulation of patients with atherosclerosis, coronary artery disease, and hypercholesterolemia. Under the relatively high shear rates that occur in arteries, tethering of monocytes to the vessel wall by activated platelets might help support monocyte-endothelial cell interaction and monocytic infiltration of the vessel wall, a key initiating event in atherosclerosis. A role for the leukocyte-binding molecule, platelet P-selectin, in promoting atherosclerosis has been described in the ApoE-/- mouse model.6 P-selectin is mobilized to the platelet surface upon platelet activation. Thus, platelet activation should be required for this pro-atherogenic activity. Similarly, GPIb/vWF interaction facilitates platelet-vessel wall interactions under high shear conditions, and genetic25 or pharmacologic5 disruption of this system limits atherosclerotic lesion formation and adhesion of leukocytes in mouse models. Stimulation of endothelial cells by RANTES released by activated platelets13 with consequent expression of leukocyte-binding adhesion molecules by endothelial cells provides yet another potential mechanism by which platelets might promote monocyte recruitment and atheroma formation. The observation that ApoE-/- mice lacking αIIb (a platelet-specific subunit of the receptor that binds fibrinogen upon platelet activation) show decreased atherosclerosis provides additional evidence for a role for activated platelets in this process.7
Despite these findings, the mechanism by which platelets promote atherogenesis in the models described above remains unknown. Two recent studies suggest platelets activated by thrombin can promote atherogenesis. First, repeated transfusion with thrombin-activated platelets increased plaque formation of ApoE-/- mice, a function ascribed to tethering of monocytes to the vessel wall by activated platelets.13 Monocytes can express tissue factor (the trigger for thrombin generation), particularly in response to inflammatory stimuli.9,10 Thus, a role for thrombin in promoting monocyte-platelet interactions was plausible. Second, fibrinogen-deficiency was associated with an increased number of activated platelets in the circulation and increased plaque formation in LDL-/-:APOBEC-/- mice.26 In the latter study, platelet activation and accelerated atherosclerosis was hypothesized to result from the absence of fibrinogen competition for thrombin, leading to increased thrombin interaction with platelet PARs. Although suggestive of a role for thrombin-induced platelet activation in atherogenesis, such gain-of-function experiments do not address the question of whether activation of platelets by thrombin plays any necessary role in atherosclerotic plaque formation. To directly address this question, we utilized Par4-/- mice. Platelets from these mice show no calcium mobilization, secretion, or aggregation in response to even micromolar concentrations of thrombin.17
Our studies showed no effect of Par4 deficiency on the size, extent, or location of atheroma development in ApoE-/- mice. Thus, platelet activation by thrombin does not play a necessary role in atherogenesis in the ApoE-/- model under the conditions we utilized. Several of the studies outlined above suggest that platelets do play a role in the ApoE-/- model, and we did attempt to directly assess the necessary role for platelets by examining the effect of Nfe2 deficiency, which causes a lack of circulating platelets.27,28 Unfortunately, Nfe2-/-:ApoE-/- mice were not viable, at least not in the C57Bl/66 background used (data not shown).
The atheroma in the ApoE-/- model used for our studies develop over a relatively short time and are comprised of macrophages and lipid. Thus, our studies strongly suggest that activation of platelets by thrombin is unimportant for monocyte/macrophage recruitment and accumulation in the vessel wall in this most common mouse model of atherogenesis. Given the multiple studies discussed above that suggest some role for platelets in this same model, our findings suggest that platelet activators other than thrombin are sufficient for promoting atherogenesis in the experimental conditions employed. The relative importance of various leukocyte recruitment and endothelial and platelet activation mechanisms may vary from model to model depending upon lipoprotein levels and other factors, and mouse models do not recapitulate the repeated plaque rupture and repair with incorporation of thrombus seen in human atherosclerosis.29 Our studies do not address the role of platelet activation by thrombin in such settings, and such a role is certainly plausible.
Acknowledgments
This work was supported in part by NHMRC (Australia) grant numbers 491143 and 166903 (J.R.H.) and NIH grant numbers HL59202, HL65185, and HL44907 (S.R.C.). J.R.H. is a Career Development Fellow of the National Heart Foundation of Australia. The authors thank Drs Judy De Haan, Ara Aslanian and Prof. Israel Charo for advice on atherosclerosis models and techniques, Rommel Advincula and Cherry Concengo for mouse husbandry and histology, and Leigh Herrick for valuable contributions during the course of this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huo Y, Ley KF. Role of platelets in the development of atherosclerosis. Trends Cardiovasc Med. 2004;14:18–22. doi: 10.1016/j.tcm.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 2.Harker LA, Ross R, Glomset J. Role of the platelet in atherogenesis. Ann N Y Acad Sci. 1976;275:321–329. doi: 10.1111/j.1749-6632.1976.tb43364.x. [DOI] [PubMed] [Google Scholar]
- 3.Ross R, Glomset J, Harker L. Response to injury and atherogenesis. Am J Pathol. 1977;86:675–684. [PMC free article] [PubMed] [Google Scholar]
- 4.Peters W, Charo IF. Involvement of chemokine receptor 2 and its ligand, monocyte chemoattractant protein-1, in the development of atherosclerosis: lessons from knockout mice. Curr Opin Lipidol. 2001;12:175–180. doi: 10.1097/00041433-200104000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Massberg S, Brand K, Gruner S, et al. A critical role of platlet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196:887–896. doi: 10.1084/jem.20012044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003;101:2661–2666. doi: 10.1182/blood-2002-07-2209. [DOI] [PubMed] [Google Scholar]
- 7.Massberg S, Schurzinger K, Lorenz M, et al. Platelet adhesion via glycoprotein IIb integrin is critical for atheroprogression and focal cerebral ischemia: an in vivo study in mice lacking glycoprotein IIb. Circulation. 2005;112:1180–1188. doi: 10.1161/CIRCULATIONAHA.105.539221. [DOI] [PubMed] [Google Scholar]
- 8.Sachais BS, Turrentine T, Dawicki McKenna JM, Rux AH, Rader D, Kowalska MA. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE-/- mice. Thromb Haemost. 2007;98:1108–1113. [PubMed] [Google Scholar]
- 9.Brand K, Banka CL, Mackman N, Terkeltaub RA, Fan ST, Curtiss LK. Oxidized LDL enhances lipopolysaccharide-induced tissue factor expression in human adherent monocytes. Arterioscler Thromb. 1994;14:790–797. doi: 10.1161/01.atv.14.5.790. [DOI] [PubMed] [Google Scholar]
- 10.Sanguigni V, Ferro D, Pignatelli P, et al. CD40 ligand enhances monocyte tissue factor expression and thrombin generation via oxidative stress in patients with hypercholesterolemia. J Am Coll Cardiol. 2005;45:35–42. doi: 10.1016/j.jacc.2004.09.047. [DOI] [PubMed] [Google Scholar]
- 11.Landers SC, Gupta M, Lewis JC. Ultrastructural localization of tissue factor on monocyte-derived macrophages and macrophage foam cells associated with atherosclerotic lesions. Virchows Arch. 1994;425:49–54. doi: 10.1007/BF00193948. [DOI] [PubMed] [Google Scholar]
- 12.Wada H, Kaneko T, Wakita Y, et al. Effect of lipoproteins on tissue factor activity and PAI-II antigen in human monocytes and macrophages. Int J Cardiol. 1994;47:S21–25. doi: 10.1016/0167-5273(94)90322-0. [DOI] [PubMed] [Google Scholar]
- 13.Huo Y, Schober A, Forlow SB, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 14.Diacovo TG, Puri KD, Warnock RA, Springer TA, von Andrian UH. Platelet-mediated lymphocyte delivery to high endothelial venules. Science. 1996;273:252–255. doi: 10.1126/science.273.5272.252. [DOI] [PubMed] [Google Scholar]
- 15.Shoji T, Koyama H, Fukumoto S, et al. Platelet-monocyte aggregates are independently associated with occurrence of carotid plaques in type 2 diabetic patients. J Atheroscler Thromb. 2005;12:344–352. doi: 10.5551/jat.12.344. [DOI] [PubMed] [Google Scholar]
- 16.Shoji T, Koyama H, Fukumoto S, et al. Platelet activation is associated with hypoadiponectinemia and carotid atherosclerosis. Atherosclerosis. 2006;188:190–195. doi: 10.1016/j.atherosclerosis.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 17.Furman MI, Benoit SE, Barnard MR, et al. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol. 1998;31:352–358. doi: 10.1016/s0735-1097(97)00510-x. [DOI] [PubMed] [Google Scholar]
- 18.Sambrano GR, Weiss EJ, Zheng YW, Huang W, Coughlin SR. Role of thrombin signaling in platelets in haemostasis and thrombosis. Nature. 2001;413:74–78. doi: 10.1038/35092573. [DOI] [PubMed] [Google Scholar]
- 19.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 20.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 21.Reddick RL, Zhang SH, Maeda N. Atherosclerosis in mice lacking apo E: Evaluation of lesional development and progression. Arterioscler Thromb. 1994;14:141–147. doi: 10.1161/01.atv.14.1.141. [DOI] [PubMed] [Google Scholar]
- 22.Zhang SH, Reddick RL, Burkey B, Maeda N. Diet-induced atherosclerosis in mice heterozygous and homozygous for apolipoprotein E gene disruption. J Clin Invest. 1994;94:937–945. doi: 10.1172/JCI117460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tilley RE, Pedersen B, Pawlinski R, et al. Atherosclerosis in mice is not affected by a reduction in tissue factor expression. Arterioscler Thromb Vasc Biol. 2006;26:555–562. doi: 10.1161/01.ATV.0000202028.62414.3c. [DOI] [PubMed] [Google Scholar]
- 24.Terasawa Y, Ladha Z, Leonard SW, et al. Increased atherosclerosis in hyperlipidemic mice deficient in alpha -tocopherol transfer protein and vitamin E. Proc Natl Acad Sci U S A. 2000;97:13830–13834. doi: 10.1073/pnas.240462697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Methia N, Andre P, Denis CV, Economopoulos M, Wagner DD. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood. 2001;98:1424–1428. doi: 10.1182/blood.v98.5.1424. [DOI] [PubMed] [Google Scholar]
- 26.Iwaki T, Sandoval-Cooper MJ, Brechmann M, Ploplis VA, Castellino FJ. A fibrinogen deficiency accelerates the initiation of LDL cholesterol-driven atherosclerosis via thrombin generation and platelet activation in genetically predisposed mice. Blood. 2006;107:3883–3891. doi: 10.1182/blood-2005-09-3780. [DOI] [PubMed] [Google Scholar]
- 27.Shivdasani RA, Rosenblatt MF, Zucker-Franklin D, et al. Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell. 1995;81:695–704. doi: 10.1016/0092-8674(95)90531-6. [DOI] [PubMed] [Google Scholar]
- 28.Levin J, Peng JP, Baker GR, et al. Pathophysiology of thromboctyopenia and anemia in mice lacking transcription factor NF-E2. Blood. 1999;94:3037–3047. [PubMed] [Google Scholar]
- 29.Davies MJ. Stability and instability: two faces of coronary atherosclerosis. The Paul Dudley White Lecture 1995. Circulation. 1996;94:2013–2020. doi: 10.1161/01.cir.94.8.2013. [DOI] [PubMed] [Google Scholar]