

Abstract
Proteins can be influenced strongly by the electrolyte in which they are dissolved, and we wish to model, understand, and ultimately control such ionic effects. Relatively detailed Monte Carlo (MC) ion simulations are needed to capture biologically important properties of ion channels, but a simpler treatment of ions, the linearized Poisson-Boltzmann (LPB) theory, is often used to model processes such as binding and folding, even in settings where the LPB theory is expected to be inaccurate. This study uses MC simulations to assess the reliability of the LPB theory for such a system, the constrained, anionic active site of HIV protease. We study the distributions of ions in and around the active site, as well as the energetics of displacing ions when a protease inhibitor is inserted into the active site. The LPB theory substantially underestimates the density of counterions in the active site when divalent cations are present. It also underestimates the energy cost of displacing these counterions, but the error is not consequential because the energy cost is less than kBT, according to the MC calculations. Thus, the LPB approach will often be suitable for studying energetics, but the more detailed MC approach is critical when ionic distributions and fluxes are at issue.
Introduction
Many protein functions involve the binding of molecules to specialized sites on proteins that include one or more ionized groups. The energetics and dynamics of such binding reactions are modulated by the nature of the electrolyte in which they occur; ionic strength and the magnitudes of ionic charges are particularly important determinants of electrolyte effects. The relationship between the ionic environment and the thermodynamics of molecular recognition is thus of profound biological importance, as shown by the tight regulation of the ionic compositions of different compartments in cells and tissues. This relationship is also of practical interest. For example, a drug that works by binding a protein must act in the appropriate physiological compartment, with its specific ionic concentrations; and an enzyme used in an industrial process may need to be engineered to function in a highly nonphysiological environment. Moreover, changes in ionic concentrations can trigger many physiological actions (1–4). Changes in the concentrations of Ca2+ ions, in particular, are used widely by nature to carry specific biological information. For example, Ca2+ is the activator of contraction in skeletal and cardiac muscle.
The analysis of ionic solutions began nearly a century ago with Poisson-Boltzmann (PB) theory, which was introduced by Gouy and Chapman to describe charged planar interfaces, and then used by Debye and Hückel in its linearized form to account for the nonideality of solutions of spherical ions. The derivation and limitations of PB theory have been discussed in textbooks of physical chemistry (5), electrochemistry (6–11), and biophysical chemistry (12–14) for a long time. The generalization of PB to nonequilibrium, which leads to the drift-diffusion equations, was introduced in plasma physics (15–19) and has formed the foundation of computational electronics (20–25). The nonequilibrium version has been extended to deal with ion channels as well under the name Poisson-Nernst-Planck (26–33). Concurrently, the development of fast computers and numerical methods have led to broad application of the linearized Poisson-Boltzmann (LPB) theory to molecules of complex shape, like proteins and nucleic acids (34–37).
LPB theory represents a specific implementation of the primitive model (PM) of electrolytes, in which the solvent is represented by a dielectric continuum and the ions are considered as charged hard spheres. LPB further simplifies the ions by representing them as a continuous distribution of charge whose density responds to the electrostatic field produced by the biomolecule of interest. The effect of the size of the ions on their interaction with the biomolecule is described by the distance of closest approach of the ionic atmosphere to its atoms. (This distance effectively defines a Stern layer in the sense of Gouy-Chapman double-layer theory.) The LPB theory neglects steric interactions and correlations among individual ions; it includes only correlations of the ions with the imposed mean electric field. The neglect of these two effects compensates to a considerable extent for some properties of bulk solution (38), so the PB approach is more successful than one might expect. However, the balance between these two effects depends on conditions: size effects become more important at high concentrations, whereas ion correlations due to electrostatics seem to be more important for low concentrations and higher valence ions. It is also important to recognize that the linearizing approximation of the LPB holds only when the interaction of an ion with the potential field is less than kBT, or ∼25 mV for a 1:1 electrolyte. Based on these considerations, one may anticipate that the LPB theory will be severely stressed in conditions of high electrostatic potential and a confined environment, where both nonlinearity and steric interactions become important. This has, in fact, been the conclusion of many workers in the field of electrochemistry (9–11,39,40).
This study aims to evaluate the validity of LPB theory by comparing it with a more detailed implementation of the PM that includes nonlinearity along with steric interactions and correlations among individual ions. A number of modern statistical mechanical theories of ionic solution might be used for this purpose, including modifications of the Poisson-Boltzmann theory (38), the mean spherical approximation MSA (41), the hypernetted chain HNC approximation (42–44), and second-order integral equations (45). Density functional theories (46–48) have been especially successful lately, with recent developments to account for large spatial variations in concentration (49) and for strong ionic coupling (50). The applicability of these theories is currently limited to systems described by one spatial coordinate, such as one dimensional or spherically symmetrical systems, so they are not readily applied to biomolecular systems described in full atomic detail. (They have, however, been applied with some success, to complex biomolecular systems represented by reduced models (30,49,51–62).) In this study, we use Monte Carlo (MC) simulations of the electrolyte because they provide exact results for a given model, apart from statistical uncertainties, and can be applied straightforwardly to complex, inhomogeneous ionic systems.
MC simulations of the PM have been used to model bulk electrolytes (63) and the double-layer geometry (63). The full PM implemented by such simulations has been shown to reproduce faithfully the structure of the diffuse double layer near a charged surface or macromolecule and to accurately account for many experimental observations. One example is charge inversion, which accounts for attractive interactions between like-charged macroparticles (64,65) in phenomena such as the condensation of DNA molecules (66) and cement cohesion (67), and also for the reversal of the sign of electrophoretic mobility (68). The presence of divalent ions and ionic correlations are crucial in these situations. In addition, simulations of ionic selectivity in Ca2+ (30,51,52,56–60,62) and Na+ (61) channels highlight the importance of accounting for the finite size of ions. The success of the PM in reproducing experimental phenomena is, perhaps, counterintuitive, given that it treats water as a dielectric continuum. Evidently, much of the physics is accounted for by long ranged electrostatic effects manifest in the structure of the diffuse double layer around charged solutes, and ion-ion interactions manifest in the activity coefficients of ions in solution and the selectivity of Ca2+ and Na+ channels. The details of water structure seem to be less important, and averaging over the details of water structure does not change the main conclusions.
HIV protease (HIVP) is a particularly interesting test system for evaluating the LPB theory against a more detailed MC implementation of the PM. For one thing, HIVP is a key drug target for the treatment of HIV infection and AIDS, so learning more about its physical chemistry may ultimately contribute to the development of new therapeutics. In addition, the enzymatic activity of HIVP is sensitive to the concentration of salt (69) and inhibition assays are typically carried out in 1 M NaCl solution. Finally, HIVP poses a particular challenge to the LPB theory: its tunnel-like binding site is confining and furthermore contains two central aspartyl groups whose net charge can range up to −2e.
This study is based on the PM and uses both MC simulations and LPB theory to study the distribution of ions in and around the active site of HIV protease. The representation of the protease is simplified somewhat to facilitate these initial MC calculations. In addition, the active-site charge is allowed to range up to the nonphysical value of −3e to provide a glimpse of how LPB deals with highly charged systems. In addition, calculations with and without a bound ligand bear on the impact of the electrolyte on the thermodynamics of ligand-binding.
Methods
Model of HIV protease and inhibitor KNI-272
Monte Carlo and LPB calculations were carried out for a simplified model of HIV protease with and without a bound inhibitor, KNI-272 (Fig. 1). Atomic coordinates were drawn from PDB (70,71) file 1HPX (72), and each atom was assigned a hard-sphere radius equal to the value of Rmin from the Lennard-Jones term of the CHARMM force field (73) as implemented in the program Quanta (Accelrys, San Diego, CA), except that all hydrogen radii were set to 1.2 Å. The protein comprises 1844 atoms and the ligand 87. All atomic charges of the protein and ligand were set to zero, except that a single point-charge of −1e, −2e, or −3e was placed at the center of the gamma carbon of Asp25 (Fig. 1) to model realistic (−1e, −2e), and unrealistic but informative (−3e), ionization states of the two active site aspartates. This is one of the two catalytic aspartyl groups, and it is located in the wall of the active site tunnel essentially equidistant from the two openings of the active site. The dielectric constant of the molecular interior was set to equal that of the aqueous solvent, 78.46. This value, which overestimates the polarizability of the protein and ligand, was chosen to isolate the most basic consequences of steric interactions among ions and electrostatics beyond the mean field. The conformations of the protein and the ligand were held fixed during all calculations; only the dissolved ions of the electrolyte were allowed to move. Calculations were carried out for all combinations of:
Protein alone or protein-ligand complex.
1:1 or 2:1 electrolyte.
Ionic radius of 1 Å, 1.5 Å or 2 Å; cations and anions are given the same size in our calculations.
Anion concentration of 0.058 M, 0.103 M, 0.148 M, or 0.208 M.
Protein charge of −1e, −2e, or −3e
Figure 1.
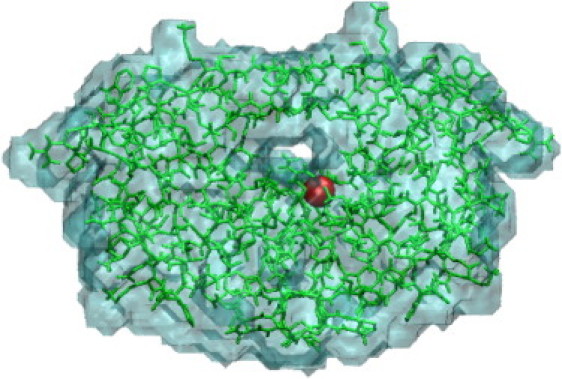
Crystal structure of HIV protease drawn from PDB entry 1HPX. The bound inhibitor, KNI-272 (122), is not shown here so that the tunnel-like binding-site is visible. The surface of the ion-excluding volume for an ion of diameter d= 2 Å, computed with UHBD, is shown in cyan; nonhydrogen atoms are shown in green; and the location of the protein charge in these calculations, Cg of Asp25, is highlighted in red. Graphics generated with the VMD program (121).
for a total of 2 × 2 × 3 × 4 × 3 = 144 calculations by each method.
MC simulations of the electrolyte
MC simulations of the electrolyte were carried out in the canonical (NVT) ensemble; that is, the volume of the simulation cell, the temperature and the numbers of ions were held fixed. The energy function comprised hard-sphere interactions among the ions and between the ions and the protein or protein-ligand complex, along with Coulombic interactions among the mobile ions and between the mobile ions and the protein charge. Coulombic interactions were uniformly reduced by the solvent dielectric constant 78.46 and temperature was set to 300 K. The protein was placed at the center of a cubic simulation cell with the solitary charge at the origin. The dimensions of the simulation cell were set large enough to allow the formation of bulk electrolyte far from the protein at the edge of the box; the length ranged between 148 Å and 226 Å, depending on ionic concentration. The total number of ions was set to make the full system, ions and protein, electroneutral. Edge effects were eliminated by the use of periodic boundary conditions. Long-range corrections are negligible at this value of the dielectric constant and system size.
Sampling was accelerated by using a biased importance sampling method that has been used before in ion channel simulations (55). In this technique, ion exchange between a small subvolume—a sphere containing the protein and centered on the solitary charge—and the large bath is oversampled. When the ion selected for a trial move is located in the sphere, the trial move is into the bath; when the ion to be moved is located in the bath, the trial move is into the sphere. The resulting bias is removed by adjusting the acceptance probability of the MC step according to the ratio of the two subvolumes. To avoid expensive computational loops involving all atoms, we used the linked-cell method (74,75), which reduces the number of ions that must be checked for overlap with the ion subjected to the trial move. The simulations were continued until they yielded smooth radial density profiles of the dissolved ions as a function of the distance from the protein charge.
LPB calculations
The LPB equation was solved for the same protein and protein-ligand systems by the method of finite differences with the program UHBD (76). The dielectric constant was set to 78.46 both in solvent and in the protein and ligand, to match the MC calculations. Mobile ions were excluded from the interior of the protein and ligand by preventing the ionic atmosphere from entering a region defined by the union of their atoms with their hard-sphere radii incremented by the ionic radius. The ionic strength was set to match that of the corresponding MC run.
Finite difference calculations used a 2-stage “focusing” method (77) starting with a 100 Å × 100 Å × 100 Å finite difference grid with 1 Å spacing centered ∼2 Å from the gamma carbon of Asp25. Boundary potentials were computed by applying the Debye-Hückel equation to the source charge, with the approximation of a uniform electrolyte of the appropriate ionic strength. The second stage used a 40 Å × 40 Å × 40 Å grid with 0.4 Å spacing, centered on the same point as the coarse initial grid and with boundary conditions drawn from the coarse grid potentials.
Calculation of ionic and potential distributions
For the MC calculations, radial distribution functions (RDFs) were computed by averaging ionic concentrations in spherical shells of radius r centered on the protein charge to yield the mean concentration as a function of distance, c(r). The RDF graphs in Results are all reported as c(r)/c0, where c0 is the bulk concentration of the ionic species. Thus, for a 2:1 electrolyte with 0.15 M anions, the anion RDFs are divided by 0.15 M and the cation RDFs are divided by 0.075 M. As a consequence, all the RDF graphs approach 1 at long distance from the protein charge. Note that those portions of each spherical shell that lie in the protein or ligand interior have zero ionic concentration because the protein and ligand sterically exclude the ions. The ion-accessible volume of a spherical shell is termed Veff(r), the total volume of a shell is termed Vtot(r), and the ion-accessible fraction of a shell is Veff(r)/Vtot(r). The RDFs reported in Results are averages over Vtot(r), except for the bottom panel of Fig. 2, where the averages are taken over only Veff(r); the two averages are related by the factor Veff(r)/Vtot(r).
Figure 2.

Radial density functions c(r) for 1:1 electrolyte and a weakly charged binding site, Q = −1e. Plots are shown for cations, anions, and net charge. Ionic diameter is set to d= 3 Å, and distances r are measured from the source charge in the protein. All densities are normalized to the bulk concentration c0 = 0.09 M. (Upper panel) Normalized concentrations are obtained by averaging over the entire spherical shell at each distance r, so that the total volume of the shell includes the body of the protein from which ions are excluded. (Lower panel) Same as upper panel except that concentrations are averaged over only the part of the spherical shell accessible to ions. This distribution is obtained from c(r)/c0 by dividing by Veff(r)/Vtot(r), the ratio of the ion-accessible volume of the shell to the total volume of the shell. Inset graphs the ratio Veff(r)/Vtot(r).
For the LPB calculations, the concentrations of cations and anions were computed at each point of the finite difference grid, the grid points were grouped into radial shells around the source charge, and the mean concentration within each shell was computed. Following the LPB theory, the concentration of each ionic species i was computed at grid point (j,k,l) as
| (1) |
where kB is Boltzmann's constant, T the temperature, zi the ionic charge and ϕtot the electrostatic potential generated by the source charge in the protein and the ion atmosphere.
In the MC calculations, the mean concentration in each spherical shell was computed as a simple average over the ensemble of ionic configurations generated by the simulations. The charge profile was computed as , where zi is the valence of ionic species i, e is the charge on a proton and r is the distance from the source charge of the protein. The electrostatic potential ϕ(r) due solely to the ionic atmosphere as a function of the distance from the origin was estimated by applying Poisson's equation to this averaged charge density:
| (2) |
where ɛ is the dielectric constant. We use the boundary condition that the total potential is zero at the edge of the simulation cell r = L/2, where L is the length of the cubic simulation box. The total potential is the sum of the potential of the ionic cloud, ϕ(r), and the potential of the central charge, Q/ɛr. The boundary condition then can be expressed as ϕ(L/2) + 2Q/(ɛL) = 0. The solution is
| (3) |
where
| (4) |
The potential generated by the ions at the protein's source charge at the origin is given by
| (5) |
Work of displacing ions on ligand-binding
The displacement of counterions from the binding site when an inhibitor binds incurs a free energy cost and hence reduces the inhibitor's binding affinity. The work of displacing ions can be obtained as the difference between the work of hypothetically charging the protein source charge in the presence and absence of the bound inhibitor. For LPB theory, the work of charging is simply , where ϕQ(0) is the reaction field potential of the ion atmosphere when the source charge is fully charged. For the nonlinear MC simulations, the work integrals are estimated by applying the trapezoid rule to the electrostatic potentials associated with stepwise increases in the source charge. For example, the work of charging the protein to Q = ±3e is given by:
| (6) |
Results
Spatial distributions of ions in and around the binding site
Optimal agreement between MC and LPB calculations is expected when the protein charge is small, the ions in solution are all monovalent, and their concentration is low. Fig. 2 compares the spatial distributions, i.e., the RDFs, of ions computed by both methods under such conditions: Q = −1e; 1:1 electrolyte at ionic strength 0.09 M, with ion diameter 3 Å. The MC (symbols) and LPB (lines) results agree closely. This consistency is gratifying because the two calculations were carried out entirely independently, and no parameters were adjusted in any way to bring them into agreement.
The upper and lower panels of Fig. 2 plot the same ionic density data in two different ways. In the upper panel, the concentration is averaged over the entire volume of the spherical shell, including the interior of the protein, which sterically excludes ions. This accounts for the dip in concentration at about r = 12 Å. In the lower panel, the concentration is averaged over only the volume of the spherical shell accessible to ions, so the dip in concentration has disappeared. The inset shows Veff(r)/Vtot(r), the ion-accessible fraction of the volume as a function of r. This fraction was calculated from trial ion insertions into the spherical shell and is the ratio of the number of trial when no overlap with the protein occurred and the total number of trials. It is evident that the concentration profiles in the upper panel follow closely the ion-accessible volume fraction. Removing this effect (as in the lower panel) magnifies the differences between the MC and LPB results. Subsequent graphs use the method of the upper panel to display results; nonetheless, it will be seen that differences between MC and LPB become apparent.
The LPB theory is expected to differ from MC when the binding site is highly charged and multivalent ions are present, as these conditions lead to greater nonlinearity and crowding of ions, which the LPB theory neglects. The nonlinearity results primarily from the exponential form of Boltzmann factor with respect to the charge-charge interactions in the system. Crowding of ions tends to occur when their bulk concentrations are high, their diameters are large, and a highly charged binding site attracts a large concentration of counterions into a small space. The following subsections probe the agreement between the two models with calculations spanning the following parameter ranges: bulk anion concentration c0 = 0.058 M, 0.103 M, 0.148 M, and 0.208 M; ionic diameters d = 2 Å, 3 Å, and 4 Å; protein binding charge Q = −1e, −2e, and −3e; and 1:1 and 2:1 electrolytes, like NaCl and CaCl2, respectively.
Larger protein charges and divalent cations
The consequences of increasing the binding site charge and replacing monovalent by divalent cations are examined in Fig. 3, which shows Q = −1e, −2e, and −3e reading top to bottom and 1:1 and 2:1 electrolytes reading left to right, for various ionic concentrations. These data are for ionic diameter 3 Å; other ion sizes are examined subsequently. Here and in subsequent figures, MC and LPB results are shown respectively by lines with and without data symbols. In Fig. 3, different colors represent different concentrations of ions.
Figure 3.
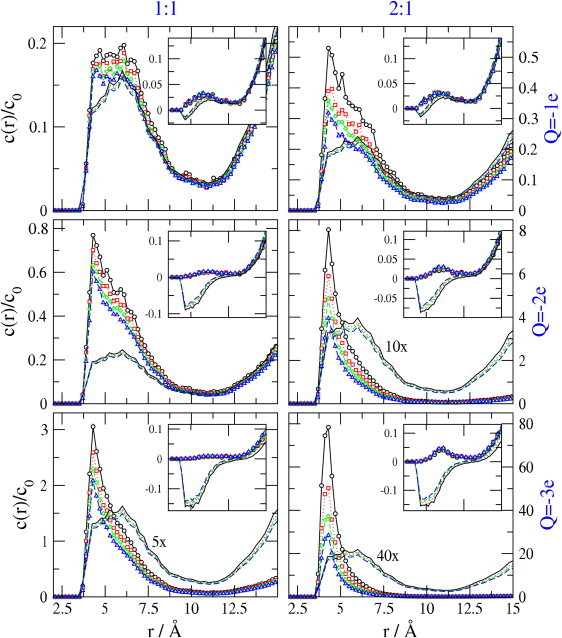
Concentration profiles, normalized to bulk concentrations, for various bulk concentrations, with ionic diameter 3 Å. Binding site charges are Q = −1e, −2e, and −3e reading top to bottom; left and right panels are for 1:1 and 2:1 electrolytes respectively. Ionic concentrations are 0.058 M, 0.103 M, 0.148 M, or 0.208 M for solid black, dotted red, short dashed green, and long dashed blue curves, respectively. MC results are shown as lines decorated with symbols in this and following figures. LPB results are shown by lines without symbols. The main panels show cation profiles, whereas the insets show anion profiles.
The LPB results agree reasonably well with MC in the top-left graph (Q = −1e, 1:1), but the two methods diverge markedly as charge and ion valence increase. For high charge and valence, the MC method yields concentration profiles that are more sharply peaked and more localized near the source charge; i.e., at small values of r. Also, the LPB results scale linearly with Q as required for this linear model, whereas the MC results show marked nonlinearity in Q. In particular, for the 2:1 electrolyte with bulk anion concentration of 0.058 M (black curves, right panels), the peak concentration of cations rises ∼16-fold as Q increases from −1e to −2e, and then another ∼10-fold as Q rises to −3e. (In contrast, the linear model yields only 2-fold and 1.5-fold jumps in ionic concentration for these respective changes in Q.) Interestingly, the nonlinearity of the MC results is somewhat less marked when the bulk concentration of ions is higher. For example, for the 2:1 electrolyte with bulk anion concentration now 0.208 M (blue curves, right panels), the peak concentration increases not by 16- and 10-fold, but by ∼13- and ∼7.5-fold, as Q goes from −1e to −2e and then −3e. This effect of the bulk concentration on the degree of nonlinearity probably stems, at least in part, from the higher degree of steric crowding and weaker electrostatic potentials associated with higher bulk concentrations of ions. However, the complex interactions of terms make it difficult to arrive at a definite explanation of this observation.
The LPB results in Fig. 3 include negative concentrations of anions (see insets). This nonphysical result is traceable to the linearizing approximation of the theory: e−x will never yield negative values, but its linearized approximation, 1 − x, is negative if x exceeds 1. It is worth noting in this regard that the LPB equation for the charge density or potential does not treat cation and anion concentrations separately; instead, the important variable in the LPB equation is charge density, and separate cation and anion densities are computed with Eq. 1 from the electrostatic potential obtained by solving the LPB equation.
Consequences of ion size
Fig. 4 examines the consequences of increased ionic crowding as the sizes of the ions are increased. Lines decorated with symbols are the results of MC calculations. Solid, dashed, or dotted lines without symbols describe results of LPB calculations. The layout of charts is the same as in Fig. 3, with protein charge Q increasing top to bottom and the 1:1 and 2:1 electrolytes in the left and right panels, respectively. However, now the curves in each chart correspond to ionic diameters 2 Å, 3 Å, and 4 Å (black, red, and green, respectively). The bulk concentration of anions is 0.148 M for all results in this figure, so the red graphs recapitulate data shown in Fig. 3.
Figure 4.
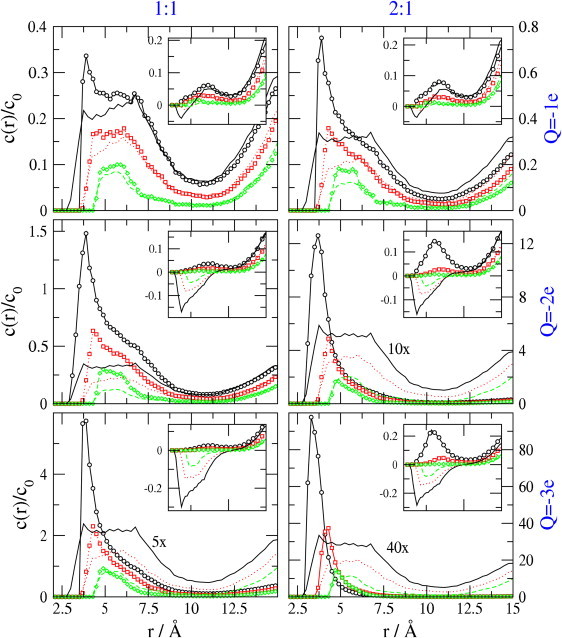
Concentration profiles, normalized to bulk concentrations, for various ionic diameters, with bulk anion concentrations set to 0.148 M. Binding site charges are Q = −1e, −2e, and −3e reading top to bottom; left and right panels are for 1:1 and 2:1 electrolytes respectively. Ionic diameters are 2 Å, 3Å, and 4 Å for solid black, dotted red, and dashed green curves, respectively. The main panels show cation profiles, whereas the insets show anion profiles.
Comparison of the three curves in each chart shows that ion size strongly influences the concentration profiles. For Q = −1e with a 1:1 electrolyte (top left), the LPB and MC methods agree rather well. However, as in Fig. 3, the methods give different results as the electrostatic forces increase; i.e., as Q increases in magnitude to −3e and the cationic charge increases in magnitude to +2e. The discrepancies are least marked when the ions are largest (d = 4 Å, green curves), presumably because then steric interactions among the ions oppose the nonlinear rise in cation density, as Q goes from −1e to −3e, in the MC calculations.
Increasing ionic diameter shifts the peaks in cation concentration to greater distance (r), because then steric interactions keep the ion further away from the protein charge. The peak heights also fall with greater ionic diameter. This probably results from greater steric crowding, along with the fact that the electrostatic potential generated by the protein charge is smaller in magnitude at the distance of closest approach of the larger cations.
Occupancy of the binding site by counterions
Binding in biological systems is usually summarized by a single parameter. The association of mobile ions from the electrolyte with the active site of HIV protease can be summarized by the net occupancy of the binding site by cations or, alternatively, by the net ionic charge in the binding site. These quantities were computed by integrating the cation concentration or the net ionic charge density out to the location where the plot of concentration vs. distance has its minimum, ∼11.2 Å. Fig. 5 plots these occupancies as a function of the protein charge (Q = −1e, −2e, or −3e), for anion concentrations of 0.148 M; note that the ordinates (y axes) have different scales for the 1:1 and 2:1 results. As already seen in the RDF results (above), the LPB results agree reasonably well with MC for a 1:1 electrolyte with a singly charged binding site Q = −1e. Deviations appear when electrolytes and/or the binding site have more than one charge. Also, the LPB results for net charge again agree better with MC than the results for a single ionic species, here the cations.
Figure 5.

Average occupancies of the binding site (r < 11.2 Å) for bulk anion concentration 0.148 M and various ionic diameters. (Upper panels) Mean net charge. (Lower panels) Cation occupancy. Left and right panels refer to 1:1 and 2:1 electrolytes, respectively.
It is interesting to observe that, even under the most extreme conditions examined in this study—an artificially high protein charge of −3e, an ionic strength greater than physiological, and a pure 2:1 electrolyte that corresponds to a far higher concentration of divalent cations than is physiologically normal—the HIV protease binding site is still predicted by the MC calculations to contain less than one cation on average. The occupancy of 0.8 under these conditions corresponds to a net charge of 1.6, barely half of that required to neutralize the −3e charge of the protein. The net charge might be greater if the dielectric constant of the protein interior were modeled as lower than that of the solvent, because this would lead to a stronger source potential in the binding site. Such a trend was previously observed in calculations of ion binding in the selectivity filter of Ca2+ and Na+ channels, where ionic occupancy increases as the dielectric constant inside the protein decreases (56,60,61).
Electrostatic potentials
The influence of the electrolyte on biochemical processes, such as the diffusion and binding of a charged substrate to an enzyme's active site, is determined in large part by the electrical potential. Fig. 6 plots the electrostatic potential generated by the ions within the HIV protease binding site, for cases where Q = −2e and the bulk concentration of anions is 0.148 M for both the 1:1 (left panel) and 2:1 (right panel) electrolytes. This potential does not contain the contribution of the binding charge Q/ɛr. Just as for the RDFs (shown previously), LPB and MC differ significantly when the electrolyte is 2:1, and the discrepancy is enhanced in the case of smaller ionic diameters. On the other hand, the maximal value of the discrepancy is only ∼0.4 kBT/e.
Figure 6.

Radial profiles of the mean electrostatic potential generated by the mobile ions for bulk anion concentration 0.148 M, Q = −2e, and various ionic diameters. Left and right panels show the results for 1:1 and 2:1 electrolytes, respectively. Potential is zero at long range.
The influence of the electrolyte on the energetics of the protein can be characterized by a single number, the electrostatic potential generated by the ions at the source charge Q. Fig. 7 plots these potentials as a function of the source charge for the 1:1 (left panel) and 2:1 (right panel) electrolytes and bulk concentration of anions 0.148 M. As expected, the LPB and MC results diverge for higher values of Q and for the 2:1 electrolyte. However, the absolute magnitude of the differences between the two models remain modest: they are well below kBT/e for Q = −2e, rising to 2 kBT/e only for the nonphysical charge of Q = −3e.
Figure 7.

Mean electrostatic potential at the protein binding site charge due to the mobile ions, as a function of Q, for bulk anion concentration 0.148 M and various ionic diameters. Left and right panels show the results for 1:1 and 2:1 electrolytes, respectively.
Displacement of ions by an HIV protease inhibitor
When an HIV protease inhibitor binds in the active site, it displaces the electrolyte from the solvent-occupied binding site near the protein charge (see Fig. 1), forcing the displaced ions to rearrange into a distribution around the protein that is higher in energy than when they were allowed to accumulate in the binding site. Displacing the ions requires work and thus has implications for the energetics of inhibitor binding. We repeated many of the calculations detailed above for the case where a neutral model of the protease inhibitor KNI-272 occupies the binding site.
Ionic distributions
The consequences of a bound ligand for the ionic RDFs and the electrostatic potential due to the mobile ions are shown in Fig. 8, for the MC simulations. Occupation of the binding site by the inhibitor eliminates the peak of the RDF within the binding site (left panel), and reduces the magnitude of the electrostatic potential in solution due to the electrolyte (right panel). The bound inhibitor fills the binding site and thereby prevents ions from closely approaching the protein's charge and from occupying the spatially constrained binding site. Not surprisingly, then, all results with the inhibitor bound are less sensitive to ionic diameter d and charge Q (data not shown).
Figure 8.

MC simulation results for the effect of inhibitor binding on the concentration (left) and the potential profiles (right) for a 2:1 electrolyte with bulk anion concentration of 0.148 M, d = 3 Å and Q = −1e. Lines and symbols denote the profiles without and with the ligand, respectively.
Energetics
The work done by the inhibitor in displacing the counterions leads to an unfavorable contribution to the binding free energy of the inhibitor, and hence reduces the inhibitor's binding affinity. This work, W, is estimated from the electrostatic potential differences at the source charge, as described in Methods. Fig. 9 plots W as a function of Q for the 1:1 and 2:1 electrolytes with bulk anion concentrations of 0.148 M, for various ionic diameters. As expected, the work of binding the inhibitor and thereby displacing the mobile ions significantly increases when Q is increased, the ionic diameter d is decreased, and the electrolyte goes from 1:1 to 2:1. Thus, the detailed properties of the electrolyte can influence ligand binding by determining the energetic cost of displacing the ions in the binding site. On the other hand, the absolute values of this work are not large. Even under the artificial conditions of Q = −3e, with a pure 2:1 electrolyte at super-physiological concentration, the value of the energy penalty is only 2 kBT. For the more realistic case where Q = −2e, the work is only ∼0.25 kBT, a small quantity in comparison with the other energies normally involved in inhibitor-binding.
Figure 9.

Work of displacing electrolyte on binding of inhibitor, computed with MC and LPB methods, as a function of the protein charge, for bulk anion concentration 0.148 M and various ionic diameters. Top and bottom panels show results for 1:1 and 2:1 electrolytes, respectively.
Discussion
The LPB theory has been applied to proteins since 1924, when Linderstrøm-Lang introduced the smeared-charge model (78), which treats a protein as a sphere with a uniform surface charge to compute protein titration curves. Tanford and Kirkwood (79) subsequently developed a more detailed model of protein titration that treated a protein as a low dielectric sphere with discrete ionization sites. With the development of high-resolution protein crystallography, the electrostatic properties of the protein (80–83) and their energetic, structural, and functional consequences (84–90) became major foci of theoretical study, driving the development of numerical methods for solving the LPB equation (84,91–93) and the full PB equation (94–97) for molecular systems of complex structure. This work ultimately led to implicit solvent models (86,98–101) based on the LPB equation, which have been incorporated into energy models and applied to problems including pH titration, protein folding, and protein-small molecule binding, as reviewed in the Introduction. These applications have focused largely on energetics, and especially the energetic consequences of the protein-solvent dielectric interface.
Accounting for the complex properties of transmembrane ion channels requires more detailed implementations of the PM, due to the large electrostatic potentials and the obvious requirement to account for finite ion size as a basis for ionic selectivity. Such implementations provide a surprisingly powerful framework for understanding these complex systems and predicting their detailed properties. For example, the selectivity of the L-type calcium channel can be understood by the primitive model (30,51,52,56–59,62,102–107), especially via MC simulations of the channel that include dielectric boundaries (i.e., polarization effects) (61). In fact, it is possible to convert nonselective bacterial channels—that have no homology or relation to vertebrate channels—into calcium selective channels using designs constructed by the primitive model (58,59,108,109). Such implementations of the PM also can account for the selectivity of Na+ channels (55,61) and of the ryanodine receptor (110,111) over a wide range of conditions and solutions.
The study presented here was motivated by our recognition that the active sites of many enzymes share some of the key features of ion channels that lead them to require a more detailed form of the PM than LPB theory; in particular, they are often sterically constrained and highly charged. We were thus motivated to ask whether the LPB provides an adequate description of such systems. We also aim to further delineate what types of problems require what types of electrolyte model and, more generally, to highlight potentially valuable links between the field of protein electrostatics and that of the physical chemistry of electrolytes (9,40). The enzyme studied here, HIVP, has a highly constrained binding site with a localized charge of up to −2e, depending on the pH, so we anticipated marked differences between LPB and a more detailed MC implementation of the PM. We examined not only the ionic distributions in and around the active site, but also the energetic cost of displacing ions from the binding site when an inhibitor binds. To our knowledge, this is first study to examine this issue, which relates directly to the applicability of LPB to the calculation of protein-ligand binding energetics.
We find that the ionic distributions from the MC and LPB calculations agree rather well under conditions where the linearizing approximation of LPB is most applicable, and that the two theories differ more and more widely as the strength of the Coulombic interaction between the protein and the ions increases; i.e., for divalent cations and strong source charges Q. Thus, MC and LPB deviate more and more going from the Q = −1e, 1:1 case in the top left corner of Fig. 3 to the Q = −3e, 2:1 case in the lower right. These results point to the linearizing approximation of LPB as a major source of error, because it is precisely these high-charge conditions where the linearizing approximation breaks down. A previous study, which found that a similar MC method and the nonlinear PB theory both yield similar ionic distributions around a protein, suggests that capturing nonlinearity by using the full PB equation should go a long way to generating agreement with the MC calculations in our system (112). On the other hand, the prior study does not analyze a highly charged and constrained binding site, as done in this study, so its results cannot be assumed to apply. As a consequence, it is not yet clear how much of the deviation we find between MC and LPB the two models results specifically from the linearization of the LPB, and how much results from its neglect of steric interactions and correlations among ions. This issue could be addressed by additional studies of HIVP using software that provides a numerical implementation of the full nonlinear PB equation for complex biomolecules (97).
We also find that LPB underestimates the work of displacing ions from the charged binding site of HIV protease when a neutral ligand binds. This observation is broadly consistent with a prior study showing that the potential of mean force for the approach of superoxide anion to a spherical model of the enzyme superoxide dismutase is more strongly attractive when computed with the MC versus the LPB model (113). However, according to both the LPB and MC calculations this energy cost is modest on the scale of the other forces involved in protein-ligand binding. Thus, for the extreme case of Q = −3e and a pure 2:1 electrolyte, MC yields a work of only 1.8 kBT, and the value falls below 1 kBT in all other cases considered here. For comparison, published protein-ligand binding energies typically range between −8 kBT and 30 kBT (114). As a consequence, the energetic differences between LPB and MC are arguably less troublesome than the differences in ionic distributions discussed above. Our results are consistent with a prior study showing that the LPB and full PB equations yield protein-ion interaction free energies that agree with each other to within ∼kBT (94). Our study extends this prior work by testing LPB for a highly charged and sterically constrained binding site of biomedical relevance, and by comparing LPB with MC results, rather than with the less detailed nonlinear PB equation. These results are also broadly consistent with a prior study showing that several different electrolyte models perform equally well in reproducing changes in the affinity of Ca2+ for calbindin and subtilisin due to charge-changing mutations (115).
The present calculations assigned the protein interior a dielectric constant equal to that of the solvent. In general, the value of the dielectric constant arguably depends on what charge rearrangement processes are considered to contribute to the dielectric response (116–120) so the optimal choice of this parameter is case- and model-dependent. Lowering the dielectric constant of the protein in this system, based on the restricted orientational polarizability of dipolar groups (83,117) would modify the energetics of displacing ions from the binding site, but the net effect on the work of displacing counterions is difficult to predict without detailed calculations and can depend on the system. For example, in Na+ channels, the dielectric constant controls the occupancy but not the selectivity between Na+ and K+ ions (61), whereas in Ca2+ channels, the effects of dielectric constant depend on ionic conditions (56). In this HIV protease system, the electrostatic consequences of lowering the protein dielectric constant are likely to be complex, involving a stronger attractive potential generated by the anionic binding site, as well as a greater energy cost for the presence of charge in the binding site due to the lowering of the dielectric constant of this region. Moreover, the many-body and nonlinear nature of the MC simulations makes the ionic consequences of these electrostatic changes difficult to predict.
A central insight of this study is that an electrolyte theory can yield very inaccurate ionic distributions while yielding accurate energetics. This is probably because freely mobile ions are easily redistributed by even weak electrical forces. This observation is directly relevant to the choice of theory for a given biomolecular application. A detailed approach, such as MC simulation, is essential for ion channels, where local ionic concentrations determine biologically relevant conductances and selectivity. Detailed models may prove to be important in other delicately poised molecular systems as well. For example, relatively subtle energetics might determine the conformational state of a protein that is perched on the edge of conformational disorder or transition. On the other hand, the simple and computationally efficient LPB theory appears to be suitable for applications that focus on relatively robust energy changes, such as protein-ligand binding.
Acknowledgments
We thank the Ira and Marylou Fulton Supercomputing Center of Brigham Young University for use of its computer facilities.
This study was made possible by the National Institute of General Medical Sciences of the National Institutes of Health grants GM61300 (M.K.G.) and GM076013 (B.E.), and Hungarian National Research Fund grant OTKA K63322 (D.B., M.V.).
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
References
- 1.Di Cera E. Thrombin: a paradigm for enzymes allosterically activated by monovalent cations. C.R. Biol. 2004;327:1065–1076. doi: 10.1016/j.crvi.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 2.Di Cera E. A structural perspective on enzymes activated by monovalent cations. J. Biol. Chem. 2006;281:1305–1308. doi: 10.1074/jbc.R500023200. [DOI] [PubMed] [Google Scholar]
- 3.Di Cera E., Guinto E.R., Vindigni A., Dang Q.D., Ayala Y.M. The Na binding site of thrombin. J. Biol. Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 4.Suelter C.H. Enzymes activated by monovalent cations. Science. 1970;168:789–795. doi: 10.1126/science.168.3933.789. [DOI] [PubMed] [Google Scholar]
- 5.McQuarrie D.A. Harper and Row; New York: 1976. Statistical Mechanics. [Google Scholar]
- 6.Harned H.S., Owen B.B. Reinhold Publishing; New York: 1958. The Physical Chemistry of Electrolytic Solutions. [Google Scholar]
- 7.Robinson R.A., Stokes R.H. Butterworth Scientific Publications; London: 1959. Electrolyte Solutions. [Google Scholar]
- 8.Friedman H.L. InterScience Publishers; New York: 1962. Ionic Solution Theory. [Google Scholar]
- 9.Barthel J., Krienke H., Kunz W. Springer; New York: 1998. Physical Chemistry of Electrolyte Solutions: Modern Aspects. [Google Scholar]
- 10.Bockris J.O.M., Reddy A.K.N. Plenum; New York: 1988. Modern Electrochemistry. Ionics. [Google Scholar]
- 11.Davis H.T. Wiley-VCH; New York: 1996. Statistical Mechanics of Phases, Interfaces, and Thin Films. [Google Scholar]
- 12.Cohen E.J., Edsall J. Reinhold; New York: 1943. Proteins, Amino Acids, and Peptides. [Google Scholar]
- 13.Edsall J., Wyman J. Academic Press; New York: 1958. Biophysical Chemistry. [Google Scholar]
- 14.Tanford C. Wiley; New York: 1961. Physical Chemistry of Macromolecules. [Google Scholar]
- 15.Mason E., McDaniel E. John Wiley and Sons; New York: 1988. Transport Properties of Ions in Gases. [Google Scholar]
- 16.Kulsrud R.M. Princeton; Princeton, NJ: 2005. Plasma Physics for Astrophysics. [Google Scholar]
- 17.Ichimura S. Addison-Wesley; New York: 1992. Statistical Plasma Physics. Basic Principles. [Google Scholar]
- 18.Ichimura S. Addison-Wesley; New York: 1994. Statistical Plasma Physics. Condensed Plasmas. [Google Scholar]
- 19.Boyd T.J.M., Sanderson J.J. Cambridge University Press; New York: 2003. The Physics of Plasmas. [Google Scholar]
- 20.Selberherr S. Springer-Verlag; New York: 1984. Analysis and Simulation of Semiconductor Devices. [Google Scholar]
- 21.Jerome J.W. Springer-Verlag; New York: 1995. Analysis of Charge Transport. Mathematical Theory and Approximation of Semiconductor Models. [Google Scholar]
- 22.Lundstrom M. Addison-Wesley; New York: 2000. Fundamentals of Carrier Transport. [Google Scholar]
- 23.Assad F., Banoo K., Lundstrom M. The drift-diffusion equation revisited. Solid-State Electron. 1998;42:283–295. [Google Scholar]
- 24.Chazalviel J.-N. Birkhauser; New York: 1999. Coulomb Screening by Mobile Charges. [Google Scholar]
- 25.Lundstrom M.S. Elementary scattering theory of the Si MOSFET. IEEE Electron Device Lett. 1997;18:361–363. [Google Scholar]
- 26.Barcilon V., Chen D.P., Eisenberg R.S. Ion flow through narrow membranes channels: Part II. SIAM J. Appl. Math. 1992;52:1405–1425. [Google Scholar]
- 27.Eisenberg R.S. Atomic biology, electrostatics and ionic channels. In: Elber R., editor. New Developments and Theoretical Studies of Proteins. World Scientific; Philadelphia: 1996. pp. 269–357. [Google Scholar]
- 28.Hollerbach U., Chen D., Nonner W., Eisenberg B. Three-dimensional Poisson-Nernst-Planck theory of open channels. Biophys. J. 1999;76:A205. [Google Scholar]
- 29.Kurnikova M.G., Coalson R.D., Graf P., Nitzan A. A lattice relaxation algorithm for 3D Poisson-Nernst-Planck theory with application to ion transport through the gramicidin A channel. Biophys. J. 1999;76:642–656. doi: 10.1016/S0006-3495(99)77232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nonner W., Catacuzzeno L., Eisenberg B. Binding and selectivity in L-type Ca channels: a mean spherical approximation. Biophys. J. 2000;79:1976–1992. doi: 10.1016/S0006-3495(00)76446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Straaten T.A., Eisenberg R.S., Tang J.M., Ravaioli U., Aluru N. Three dimensional Poisson Nernst Planck simulation of ompF porin. Biophys. J. 2001;80:115a. [Google Scholar]
- 32.Im W., Roux B. Ion permeation and selectivity of OmpF porin: a theoretical study based on molecular dynamics, Brownian dynamics, and continuum electrodiffusion theory. J. Mol. Biol. 2002;322:851–869. doi: 10.1016/s0022-2836(02)00778-7. [DOI] [PubMed] [Google Scholar]
- 33.Eisenberg, B. 2005. Living Transistors: a Physicist's View of Ion Channels. http://arxiv.org/ with PaperID q-bio.BM/0506016.
- 34.Baker N.A. Poisson-Boltzmann methods for biomolecular electrostatics. Methods Enzymol. 2004;383:94–118. doi: 10.1016/S0076-6879(04)83005-2. [DOI] [PubMed] [Google Scholar]
- 35.Fogolari F., Brigo A., Molinari H. The Poisson-Boltzmann equation for biomolecular electrostatics: a tool for structural biology. J. Mol. Recognit. 2002;15:377–392. doi: 10.1002/jmr.577. [DOI] [PubMed] [Google Scholar]
- 36.Simonson T. Macromolecular electrostatics: continuum models and their growing pains. Curr. Opin. Struct. Biol. 2001;11:243–252. doi: 10.1016/s0959-440x(00)00197-4. [DOI] [PubMed] [Google Scholar]
- 37.Honig B., Nichols A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 38.Outhwaite C.W., Bhuiyan L.B. Improved modified Poisson-Boltzmann equation in electric double layer theory. J. Chem. Soc. Faraday Trans. 2. 1983;79:707–718. [Google Scholar]
- 39.Bockris J., Reddy A.M.E. Plenum Press; New York: 1970. Modern Electrochemistry. [Google Scholar]
- 40.Fawcett W.R. Oxford University Press; New York: 2004. Liquids, Solutions, and Interfaces: From Classical Macroscopic Descriptions to Modern Microscopic Details. [Google Scholar]
- 41.Blum L. Theory of electrified interfaces. J. Phys. Chem. 1977;81:136–147. [Google Scholar]
- 42.Henderson D., Blum L., Smith W.R. Application of the hypernetted chain approximation to the electric double layer at a charged planar interface. Chem. Phys. Lett. 1979;63:381–383. [Google Scholar]
- 43.Carnie S.L., Chan D.Y.C.D., Mitchell J., Ninham B.W. The structure of electrolytes at charged surfaces: the primitive model. J. Chem. Phys. 1981;74:1472–1478. [Google Scholar]
- 44.Lozada-Cassou M., Saavedra-Barrera R., Henderson D. The application of the hypernetted chain approximation to the electrical double layer: Comparison with Monte Carlo results for symmetric salts. J. Chem. Phys. 1982;77:5150–5156. [Google Scholar]
- 45.Plischke M., Henderson D. Pair correlation functions and density profiles in the primitive model of the electric double layer. J. Chem. Phys. 1988;88:2712–2718. [Google Scholar]
- 46.Kierlik E., Rosinberg M.L. Density-functional theory for inhomogeneous fluids: adsorption of binary mixtures. Phys. Rev. A. 1991;44:5025–5037. doi: 10.1103/physreva.44.5025. [DOI] [PubMed] [Google Scholar]
- 47.Rosenfeld Y. Free energy model for inhomogeneous fluid mixtures: Yukawa-charged hard spheres, general interactions, and plasmas. J. Chem. Phys. 1993;98:8126–8148. [Google Scholar]
- 48.Rosenfeld Y., Schmidt M., Lowen H., Tarazona P. Fundamental-measure free-energy density functional for hard spheres: dimensional crossover and freezing. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 1997;55:4245–4263. [Google Scholar]
- 49.Gillespie D., Nonner W., Eisenberg R.S. Coupling Poisson-Nernst-Planck and density functional theory to calculate ion flux. J. Phys. Condens. Matter. 2002;14:12129–12145. [Google Scholar]
- 50.Pizio O., Patrykiejew A., Sokolowski S. Phase behavior of ionic fluids in slitlike pores: a density functional approach for the restricted primitive model. J. Chem. Phys. 2004;121:11957–11964. doi: 10.1063/1.1818677. [DOI] [PubMed] [Google Scholar]
- 51.Nonner W., Eisenberg B. Ion permeation and glutamate residues linked by Poisson-Nernst-Planck theory in L-type calcium channels. Biophys. J. 1998;75:1287–1305. doi: 10.1016/S0006-3495(98)74048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nonner W., Gillespie D., Henderson D., Eisenberg B. Ion accumulation in a biological calcium channel: effects of solvent and confining pressure. J. Phys. Chem. B. 2001;105:6427–6436. [Google Scholar]
- 53.Gillespie D., Nonner W., Henderson D., Eisenberg R.S. A physical mechanism for large-ion selectivity of ion channels. Phys. Chem. Chem. Phys. 2002;4:4763–4769. [Google Scholar]
- 54.Gillespie D., Eisenberg R.S. Physical descriptions of experimental selectivity measurements in ion channels. Eur. Biophys. J. 2002;31:454–466. doi: 10.1007/s00249-002-0239-x. [DOI] [PubMed] [Google Scholar]
- 55.Boda D., Busath D., Eisenberg B., Henderson D., Nonner W. Monte Carlo simulations of ion selectivity in a biological Na+ channel: charge-space competition. Phys. Chem. Chem. Phys. 2002;4:5154–5160. [Google Scholar]
- 56.Boda D., Valiskó M., Eisenberg B., Nonner W., Henderson D. Effect of protein dielectric coefficient on the ionic selectivity of a calcium channel. J. Chem. Phys. 2006;125:034901. doi: 10.1063/1.2212423. [DOI] [PubMed] [Google Scholar]
- 57.Boda D., Varga T., Henderson D., Busath D., Nonner W. Monte Carlo simulation study of a system with a dielectric boundary: application to calcium channel selectivity. Mol. Simul. 2004;30:89–96. [Google Scholar]
- 58.Miedema H., Vrouenraets M., Wierenga J., Eisenberg B., Gillespie D. Ca2+ selectivity of a chemically modified OmpF with reduced pore volume. Biophys. J. 2006;91:4392–4440. doi: 10.1529/biophysj.106.087114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miedema H., Meter-Arkema A., Wierenga J., Tang J., Eisenberg B. Permeation properties of an engineered bacterial OmpF porin containing the EEEE-locus of Ca2+ channels. Biophys. J. 2004;87:3137–3147. doi: 10.1529/biophysj.104.041384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boda D., Valiskó M., Eisenberg B., Nonner W., Henderson D. The combined effect of pore radius and protein dielectric coefficient on the selectivity of a calcium channel. Phys. Rev. Lett. 2007;98:168102. doi: 10.1103/PhysRevLett.98.168102. [DOI] [PubMed] [Google Scholar]
- 61.Boda D., Nonner W., Valiskó M., Henderson D., Eisenberg B. Steric selectivity in Na channels arising from protein polarization and mobile side chains. Biophys. J. 2007;93:1960–1980. doi: 10.1529/biophysj.107.105478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boda D., Nonner W., Henderson D., Eisenberg B., Gillespie D. Volume exclusion in calcium selective channels. Biophys. J. 2008;94:3486–3496. doi: 10.1529/biophysj.107.122796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Torrie G.M., Valleau J.P. Electrical double layers. I. Monte Carlo study of a uniformly charged surface. J. Chem. Phys. 1980;73:5807–5816. [Google Scholar]
- 64.Lyubartsev A.P., Tang J.X., Janmey P.A., Nordenskiöld L. Electrostatically induced polyelectrolyte association of rodlike virus particles. Phys. Rev. Lett. 1998;81:5465–5468. [Google Scholar]
- 65.Lobaskin V., Qamhieh K. Effective macroion charge and stability of highly asymmetric electrolytes at various salt conditions. J. Phys. Chem. B. 2003;107:8022–8029. [Google Scholar]
- 66.Pelta J., Durand D., Doucet J., Livolant F. DNA mesophases induced by spermidine: structural properties and biological implications. Biophys. J. 1996;71:48–63. doi: 10.1016/S0006-3495(96)79232-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jönsson B., Nonat A., Labbez C., Cabane B., Wennerström H.L. Controlling the cohesion of cement paste. Langmuir. 2005;21:9211–9221. doi: 10.1021/la051048z. [DOI] [PubMed] [Google Scholar]
- 68.Martín-Molina A., Quesada-Pérez M., Galisteo-González F., Hidalgo-Álvarez R. Probing charge inversion in model colloids: electrolyte mixtures of multi- and monovalent counterions. J. Phys. Condens. Matter. 2003;15:S3475–S3483. [Google Scholar]
- 69.Tropea J.E., Nashed N.T., Louis J.M., Sayer J.M., Jerina D.M. Effect of salt on the kinetic parameters of retroviral and mammalian aspartic acid proteases. Bioorg. Chem. 1992;20:67–76. [Google Scholar]
- 70.Bernstein F.C., Koetzle T.F., Williams T.F., Meyer G.J.B., Jr., Brice M.D. Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 71.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baldwin E.T., Bhat T.N., Gulnik S., Liu B., Topol I.A. Structure of HIV-1 protease with KNI-272, a tight-binding transition-state analog containing allophenylnorstatine. Structure. 1995;3:581–590. doi: 10.1016/s0969-2126(01)00192-7. [DOI] [PubMed] [Google Scholar]
- 73.Momany F.A., Rone R. Validation of the general-purpose Quanta 3.2/Charmm force-field. J. Comput. Chem. 1992;13:888–900. [Google Scholar]
- 74.Allen M.P., Tildesley D.J. Oxford; New York: 1987. Computer Simulation of Liquids. [Google Scholar]
- 75.Boda D., Chan K.Y., Szalai I. Determination of vapor-liquid equilibrium using cavity-biased grand canonical Monte Carlo method. Mol. Phys. 1997;92:1067–1072. [Google Scholar]
- 76.Davis M.E., Madura J.D., Sines J., Luty B.A., Allison S.A. Diffusion-controlled enzymatic reactions. Methods Enzymol. 1991;202:473–497. doi: 10.1016/0076-6879(91)02024-4. [DOI] [PubMed] [Google Scholar]
- 77.Gilson M.K., Sharp K.A., Honig B.H. Calculating the electrostatic potential of molecules in solution: method and error assessment. J. Comput. Chem. 1988;9:327–335. [Google Scholar]
- 78.Linderstrom-Lang K. On the ionization of proteins. Compt. Rend. Trav. Lab. Carlsberg. 1924;15:1–29. [Google Scholar]
- 79.Tanford C., Kirkwood J.G. Theory of protein titration curves. I. General equations for impenetrable spheres. J. Am. Chem. Soc. 1957;79:5333–5339. [Google Scholar]
- 80.Warshel A., Levitt M. Theoretical studies of enzymatic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 81.Honig B., Sharp K., Gilson M. Electrostatic interactions in proteins. Prog. Clin. Biol. Res. 1989;289:65–74. [PubMed] [Google Scholar]
- 82.Nakamura H., Sakamoto T., Wada A. A theoretical study of the dielectric constant of protein. Protein Eng. 1988;2:177–183. doi: 10.1093/protein/2.3.177. [DOI] [PubMed] [Google Scholar]
- 83.Simonson T., Perahia D., Brunger A.T. Microscopic theory of the dielectric properties of proteins. Biophys. J. 1991;59:670–690. doi: 10.1016/S0006-3495(91)82282-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bashford D., Karplus M. pKa's of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- 85.Honig B., Sharp K. Macroscopic models of aqueous solutions: biological and chemical applications. J. Phys. Chem. 1993;97:1101–1109. [Google Scholar]
- 86.Gilson M.K., Honig B. Calculation of the total electrostatic energy of a macromolecular system: solvation energies, binding energies, and conformational analysis. Proteins. 1988;4:7–18. doi: 10.1002/prot.340040104. [DOI] [PubMed] [Google Scholar]
- 87.Hendsch Z.S., Tidor B. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 1994;3:211–226. doi: 10.1002/pro.5560030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rashin A.A., Honig B. On the environment of ionizable groups in globular proteins. J. Mol. Biol. 1984;173:515–521. doi: 10.1016/0022-2836(84)90394-2. [DOI] [PubMed] [Google Scholar]
- 89.Warwicker J., Watson H.C. Calculation of the electric potential in the active site cleft due to alpha-helix dipoles. J. Mol. Biol. 1982;157:671–679. doi: 10.1016/0022-2836(82)90505-8. [DOI] [PubMed] [Google Scholar]
- 90.Klapper I., Hagstrom R., Fine R., Sharp K., Honig B. Focusing of electric fields in the active site of Cu-Zn superoxide dismutase: effects of ionic strength and amino-acid modification. Proteins. 1986;1:47–59. doi: 10.1002/prot.340010109. [DOI] [PubMed] [Google Scholar]
- 91.Nicholls A., Honig B. A rapid finite difference algorithm utilizing successive over-relaxation to solve the Poisson-Boltzmann equation. J. Comput. Chem. 1991;12:435–445. [Google Scholar]
- 92.Davis M.E., Madura J.D., Luty B.A., McCammon A. Electrostatics and diffusion of molecules in solution: simulations with the University of Houston Brownian Dynamics Program. Comput. Phys. Comm. 1991;62:187–197. [Google Scholar]
- 93.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou H.-X. Macromolecular electrostatic energy within the nonlinear Poisson-Boltzmann equation. J. Chem. Phys. 1994;100:3152–3162. [Google Scholar]
- 95.Oberoi H., Alewell N.M. Macromolecular electrostatic energy within the nonlinear Poisson-Boltzmann equation. Biophys. J. 1993;65:48–55. doi: 10.1016/S0006-3495(93)81032-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Holst M., Kozack R.E., Saied F., Subramaniam S. Treatment of electrostatic effects in proteins: multigrid-based Newton iterative method for solution of the full nonlinear Poisson-Boltzmann equation. Proteins. 1994;18:231–245. doi: 10.1002/prot.340180304. [DOI] [PubMed] [Google Scholar]
- 97.Rocchia W., Alexov E., Honig B. Extending the applicability of the nonlinear Poisson-Boltzmann equation: multiple dielectric constants and multivalent ions. J. Phys. Chem. 2001;105:6507–6514. [Google Scholar]
- 98.Sitkoff D., Lockhart D.J., Sharp K.A., Honig B. Calculation of electrostatic effects at the amino terminus of an alpha-helix. Biophys. J. 1994;67:2251–2260. doi: 10.1016/S0006-3495(94)80709-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gilson M.K., McCammon A., Madura J.D. Molecular dynamics simulation with a continuum electrostatic model of the solvent. J. Comput. Chem. 1995;16:1081–1095. [Google Scholar]
- 100.Horvath D., van Belle D., Lippens G., Wodak S.J. Development and parametrization of continuum solvent models. 1. Models based on the boundary element method. J. Chem. Phys. 1996;104:6679–6695. [Google Scholar]
- 101.Sitkoff D., Sharp K., Honig B. Accurate calculation of hydration free-energies using macroscopic solvent models. J. Phys. Chem. 1994;98:1978–1988. [Google Scholar]
- 102.Boda D., Busath D.D., Henderson D., Sokolowski S. Monte Carlo simulations of the mechanism of channel selectivity: the competition between volume exclusion and charge neutrality. J. Phys. Chem. B. 2000;104:8903–8910. [Google Scholar]
- 103.Boda D., Henderson D., Busath D.D. Monte Carlo study of the effect of ion and channel size on the selectivity of a model calcium channel. J. Phys. Chem. B. 2001;105:11574–11577. [Google Scholar]
- 104.Boda D., Henderson D., Patrykiejew A., Sokolowski S. Density functional study of a simple membrane using the solvent primitive model. J. Colloid Interface Sci. 2001;239:432–439. doi: 10.1006/jcis.2001.7560. [DOI] [PubMed] [Google Scholar]
- 105.Boda D., Henderson D., Busath D. Monte Carlo study of the selectivity of calcium channels: improved geometrical mode. Mol. Phys. 2002;100:2361–2368. [Google Scholar]
- 106.Gillespie D., Nonner W., Eisenberg R.S. Density functional theory of charged, hard-sphere fluids. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2003;68:0313503. doi: 10.1103/PhysRevE.68.031503. [DOI] [PubMed] [Google Scholar]
- 107.Boda D., Gillespie D., Nonner W., Henderson D., Eisenberg B. Computing induced charges in inhomogeneous dielectric media: application in a Monte Carlo simulation of complex ionic systems. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2004;69:046702. doi: 10.1103/PhysRevE.69.046702. [DOI] [PubMed] [Google Scholar]
- 108.Miedema H., Vrouenraets M., Wierenga J., Eisenberg B., Schirmer T. Conductance and selectivity fluctuations in D127 mutants of the bacterial porin OmpF. Eur. Biophys. J. 2006;36:13–22. doi: 10.1007/s00249-006-0084-4. [DOI] [PubMed] [Google Scholar]
- 109.Vrouenraets M., Wierenga J., Meijberg W., Miedema H. Chemical modification of the bacterial porin OmpF: gain of selectivity by volume reduction. Biophys. J. 2006;90:1202–1211. doi: 10.1529/biophysj.105.072298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gillespie D. Energetics of divalent selectivity in a calcium channel: the ryanodine receptor case study. Biophys. J. 2008;94:1169–1184. doi: 10.1529/biophysj.107.116798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gillespie D., Xu L., Wang Y., Meissner G. (De)constructing the ryanodine receptor: modeling ion permeation and selectivity of the calcium release channel. J. Phys. Chem. 2005;109:15598–15610. doi: 10.1021/jp052471j. [DOI] [PubMed] [Google Scholar]
- 112.Boström M., Taveres F.W., Bratko D., Ninham B.W. Specific ion effects in solutions of globular proteins: comparison between analytical models and simulation. J. Phys. Chem. B. 2005;109:24489–24494. doi: 10.1021/jp0551869. [DOI] [PubMed] [Google Scholar]
- 113.Bacquet R.J., McCammon J.A., Allison S.A. Ionic strength dependence of enzyme-substrate interactions: Monte Carlo and Poisson-Boltzmann results for superoxide dismutase. J. Phys. Chem. 1988;92:7134–7141. [Google Scholar]
- 114.Liu T., Lin Y., Wen X., Jorissen R.N., Gilson M.K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35:D198–D201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Penfold R., Warwicker J., Jonsson B. Electrostatic models for calcium binding proteins. J. Phys. Chem. 1998;102:8599–8610. [Google Scholar]
- 116.Warshel A., Russell S.T. Calculations of electrostatic interactions in biological systems and in solutions. Q. Rev. Biophys. 1984;17:283–422. doi: 10.1017/s0033583500005333. [DOI] [PubMed] [Google Scholar]
- 117.Gilson M.K., Honig B. The dielectric constant of a folded protein. Biopolymers. 1985;25:2097–2119. doi: 10.1002/bip.360251106. [DOI] [PubMed] [Google Scholar]
- 118.Smith P.E., Brunne R.M., Mark A.E., Van Gunsteren W.F. Dielectric properties of trypsin inhibitor and lysozyme from molecular dynamics simulations. J. Phys. Chem. 1993;97:2009–2014. [Google Scholar]
- 119.Schutz C.N., Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 120.Pitera J.W., Falta M., van Gunsteren W.F. Dielectric properties of proteins from simulation: the effects of solvent, ligands, pH, and temperature. Biophys. J. 2001;80:2546–2555. doi: 10.1016/S0006-3495(01)76226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Humphrey W., Dalke A., Schulten K. VMD—visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 122.Mimoto T., Imai J., Kisanuki S., Enomoto H., Hattori N. Kynostatin (KNI)-227 and (KNI)-272, highly potent anti-HIV agents—conformationally constrained tripeptide inhibitors of HIV protease containing allophenylnorstatine. Chem. Pharm. Bull. Tokyo. 1992;40:2251–2253. doi: 10.1248/cpb.40.2251. [DOI] [PubMed] [Google Scholar]