

Abstract
Amyloid fibril formation is a distinctive hallmark of a number of degenerative diseases. In this process, protein monomers self-assemble to form insoluble structures that are generally referred to as amyloid fibrils. We have induced in vitro amyloid fibril formation of a PDZ domain by combining mechanical agitation and high ionic strength under conditions otherwise close to physiological (pH 7.0, 37°C, no added denaturants). The resulting aggregates enhance the fluorescence of the thioflavin T dye via a sigmoidal kinetic profile. Both infrared spectroscopy and circular dichroism spectroscopy detect the formation of a largely intermolecular β-sheet structure. Atomic force microscopy shows straight, rod-like fibrils that are similar in appearance and height to mature amyloid-like fibrils. Under these conditions, before aggregation, the protein domain adopts an essentially native-like structure and an even higher conformational stability (ΔGU-FH2O). These results show a new method for converting initially folded proteins into amyloid-like aggregates. The methodological approach used here does not require denaturing conditions; rather, it couples agitation with a high ionic strength. Such an approach offers new opportunities to investigate protein aggregation under conditions in which a globular protein is initially folded, and to elucidate the physical forces that promote amyloid fibril formation.
Introduction
Peptides and proteins have a generic tendency to convert from their soluble states into well organized aggregates characterized by a fibrillar morphology and an extended cross-β structure (1,2). The formation of such fibrillar aggregates (generally termed “amyloid fibrils” when they deposit in vivo in the extracellular space) is associated with a number of human diseases, including neurodegenerative conditions, localized amyloidoses, and systemic amyloidoses (3,4). It is becoming apparent, however, that the formation of amyloid-like fibrils is not necessarily deleterious and associated with pathology. Indeed, evidence is mounting that a number of organisms exploit formation of these fibrillar species to carry out specific biological functions (4,5). In addition, amyloid-like fibrils formed in vitro can have a wide range of novel properties that are useful in research, medicine, and industry (6,7).
More than 40 proteins that have no link to protein deposition diseases have been converted into amyloid-like fibrils in vitro under appropriate experimental conditions (1,2). Our ability to reproduce the process of amyloid fibril formation in vitro with nondisease proteins has increased our potential to investigate the fundamentals of this process. It has also created an awareness that amyloid fibrils—and more generally self-assembled polypeptide chains—can in principle be used to produce an impressively high number of new biological nanomaterials, because proteins are very diverse in terms of sequence (6,7). The in vitro formation of amyloid-like fibrils by normally folded proteins generally requires harsh conditions, such as pH extremes (8,9), high temperature values (10,11), high pressure values (12,13), or the presence of organic cosolvents (14,15). Such harsh denaturing conditions are highly favorable for promoting aggregation because they unfold, at least partially, folded proteins and generate ensembles of highly fluctuating conformations that have a high propensity to aggregate (4).
In a recent very interesting report (16), amyloid formation by initially folded hen lysozyme and human β2-microglobulin was obtained by a two-step procedure that consisted of 1), agitating the protein samples at high ionic strength; and 2), heating. The first step allowed the two proteins to form native-like aggregates, whereas the second denaturing step enabled the conversion of the native-like assemblies into amyloid-like fibrils. In a more recent report (17), the same authors showed that amyloid fibril formation by β2-microglobulin can also be achieved in the absence of a heating step by simply incubating the protein at neutral pH and 37°C, initially under agitation at high ionic strength and then for 25–45 days under the same conditions in the absence of agitation. Here we report the formation of amyloid-like fibrils by a novel globular protein, the second PDZ domain of murine Protein Tyrosine Phosphatase-Bas-Like (PDZ2), under conditions that are conceptually distinct from the denaturing procedures previously utilized. The conversion of PDZ2 into amyloid-like fibrils was achieved by agitation at high ionic strength, at neutral pH, 37°C, and atmosphere pressure, and in the absence of organic cosolvents. Unlike generally used procedures, neither heating nor any other denaturing condition was used in this case. In addition to showing evidence of fibril formation by a novel protein and presenting a novel methodology to form amyloid from a normally folded protein, this approach has implications for the study of the amyloidogenesis process. It may facilitate investigation of new driving forces of aggregation and allow the process to be studied under conditions in which the protein is initially folded.
PDZ domains are a large family of proteins that mediate protein-protein interactions (18). These globular protein domains generally consist of 80–100 residues that fold into six β-strands and two α-helices (Fig. 1). The folding mechanism of this fold family has been extensively investigated, and analysis of the folding kinetics of different members led to the depiction of a consensus folding mechanism (19). In the case of PDZ2, the presence of an intermediate was evidenced from complex folding kinetics observed under certain experimental conditions (20), and its kinetic role was recently clarified (21). Moreover, extensive ϕ-value analysis, together with constrained molecular-dynamics simulations, contributed to the structural description of the early and late transition states that characterize PDZ folding (22). A critical review of the PDZ domain family recently highlighted an intriguing correlation between folding and binding parameters (23). Hence, this fold family represents an ideal candidate to explore the role of structural plasticity in tuning the physiological function of proteins.
Figure 1.

Three-dimensional structure of the second PDZ domain from murine Protein Tyrosine Phosphatase Bas-like (PDZ2). The structure was depicted using the software PyMOL v. 0.99 (DeLano Scientific LLC, South San Francisco, CA) and refers to the NMR structure, PDB entry 1GM1 (24).
Despite the large quantity of data collected on the folding mechanism of PDZ2 and other PDZ domains, the process of amyloid fibril formation by PDZ domains has not been investigated in any detail. In this work we will show that incubation of PDZ2 using a novel protocol converts this protein domain from its soluble folded state into fibrillar aggregates that are tinctorially, morphologically, and structurally indistinguishable from both the natural fibrils associated with human pathology and the synthetic fibrils formed in vitro under the “traditional” conditions that make use of chemical or physical denaturants.
Materials and Methods
Protein purification
To express PDZ2, a modified pET28a vector was used that contained a construct constituted by a poli-histidine tag (MH6) fused to the 94-residue sequence coding for the domain (24). Escherichia coli BL21(DE3) cells containing the engineered vector were grown under vigorous shaking in LB medium at 37°C up to 0.7 OD600. Protein expression was induced by incubation with 1 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) overnight at 25°C. Cells were harvested by centrifugation at 2800 g and by resuspending the pellet in 50 mM sodium phosphate, pH 7.0. The resulting cell suspension was sonicated (six to seven cycles of 30 s sonication at 25 kHz and 100 W alternated with 30 s in ice) and centrifuged at 31,000 g. The supernatant was passed through a column packed with His-Select Affinity Gel (Sigma-Aldrich, St. Louis, MO) at room temperature. Bound protein was abundantly washed with 20 mM sodium phosphate, pH 7.0, and then eluted with 700 mM imidazole. The solution containing the eluted protein was subjected to cycles of alternating ultrafiltration (cutoff 10,000) and dilution steps, using 20 mM sodium phosphate, pH 7.0, to eliminate imidazole. By contrast, the protein domain used to acquire Fourier transform-infrared (FTIR) spectra in native conditions was exchanged against 20 mM ammonium carbonate by the same procedure and lyophilized with an Alpha 2-4 LSC freeze dryer (Christ, Osterode, Germany). Protein purity was checked by means of sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Protein concentration was determined by UV absorption using ɛ280 = 5500 M−1 cm−1.
Protein aggregation
A fresh solution containing PDZ2 at a concentration of 0.4 mg mL−1, 20 mM sodium phosphate, 1 M NaCl, pH 7.0, was prepared and poured into a Hellma “reduced-volume” quartz cell (product number 119.004F; Hellma, Müllheim, Germany). The outer dimensions of the cell were 49.5 mm (height), 12.5 mm (width), and 12.5 mm (depth); the pathlength was 10 × 4 mm and the total volume was 1.5 mL. The volume of the added sample was 1.5 mL, a volume equal to the cuvette capacity. The resulting cell containing the sample was preequilibrated at 37°C for 10 min in a J-810 spectropolarimeter (Jasco, Tokyo, Japan) with a thermostated cell holder attached to a Thermo C25P water bath (Haake, Karlsruhe, Germany). The protein sample was agitated for 24 h at 750 rpm using the Electronic Stirrer (model 300; Rank Brothers Ltd., Cambridge, UK) and the analytical magnetic stir bar provided with the cell (length and diameter approximately 6 and 3 mm, respectively). No cleavage of the PDZ2 polypeptide chain occurred on this timescale, as checked with electro-spray ionization mass spectrometry. Agitation at 450 rpm, using the same apparatus and protein sample, is not effective at inducing PDZ2 fibril formation.
Thioflavin-T fluorescence
PDZ2 was incubated under the following conditions: 1), at a PDZ2 concentration of 0.4 mg mL−1 in 20 mM sodium phosphate buffer, 1 M NaCl, pH 7.0, 37°C, under stirring at 750 rpm; 2), under the same conditions in the absence of stirring; 3), under the same conditions in the absence of NaCl; and 4), under the same conditions in the absence of both stirring and NaCl. From each sample 62 μL aliquots were withdrawn at regular times and added to 438 μL of a solution containing 25 μM Thioflavin-T (ThT), 25 mM sodium phosphate buffer, pH 6.0. The resulting sample was excited at 440 nm and the fluorescence emission at 485 nm was measured at 25°C using a 2 × 10-mm-pathlength quartz cuvette (Hellma, Müllheim, Germany) and an LS 55 spectrofluorimeter (Perkin-Elmer, Wellesley, MA) equipped with a thermostated cell holder attached to an F8 water bath (Haake). The ratio between ThT emission in the presence (F) and absence (F0) of PDZ2 was reported. ThT fluorescence emission spectra between 450 and 550 nm were obtained after 0 and 24 h incubation under condition 1 using the same procedure and instrumentation described above.
Far-UV circular dichroism
PDZ2 was incubated at a concentration of 0.4 mg mL−1 in 20 mM sodium phosphate buffer, 1 M NaCl, pH 7.0, 37°C, under stirring at 750 rpm. Far-UV circular dichroism (CD) spectra were recorded at 37°C before agitation and after 24 h agitation, using a 1-mm-pathlength quartz cuvette (Hellma, Müllheim, Germany) and a J-810 spectropolarimeter (Jasco, Tokyo, Japan) equipped with a thermostated cell holder attached to a Thermo C25P water bath (Haake). To analyze the aggregation kinetics, 130 μL aliquots were withdrawn at regular times and the CD signal was measured at 222 nm.
FTIR spectroscopy
In a first experiment, PDZ2 was incubated at a concentration of 0.4 mg mL−1 in 20 mM sodium phosphate buffer, 1 M NaCl, pH 7.0, 37°C, under stirring at 750 rpm, for 24 h. The protein sample underwent cycles of alternating centrifugation at 16,000 g for 10 min and resuspension of the pellet in D2O (first time) and 20 mM deuterated sodium phosphate buffer, 1 M NaCl, pH 7.0 (second and third times). The fourth time, the pellet was resuspended in 20 mM deuterated sodium phosphate buffer, 1 M NaCl, pH 7.0, to achieve a final volume of 20 μL and a final protein concentration of ∼15 mg mL−1. In a second experiment, PDZ2 dissolved in a solution containing 20 mM ammonium carbonate was lyophilized and resuspended in D2O to achieve a final volume of 20 μL and a final protein concentration of ∼35 mg mL−1. In both cases the obtained 20 μL sample was deposited on a CaF2 window in a semipermanent liquid cell. The pathlengths used for the first and second samples were 6 and 12 μm, respectively. The FTIR spectra between 400 and 4000 cm−1 were recorded at room temperature using an FT/IR 4100 spectrophotometer (Jasco). The background spectra were subtracted from the sample spectra. For both samples 64 scans were accumulated at a spectral resolution of 4 cm−1, and the Amide I region (1600–1700 cm−1) was isolated and baseline corrected. The system was purged with N2 for 15 min before spectra were recorded, and then purged constantly during recording to remove water vapor.
Tapping-mode atomic force microscopy
PDZ2 was incubated at a concentration of 0.4 mg mL−1 in 20 mM sodium phosphate buffer, 1 M NaCl, pH 7.0, 37°C, under stirring at 750 rpm for 24 h. To avoid salt crystallization during atomic force microscopy (AFM) measurements, NaCl was removed by centrifuging the protein sample at 16,000 g for 10 min, resuspending the pellet in 20 mM sodium phosphate, pH 7.0, and diluting 100-fold by adding water. After dilution, 20 μL aliquots of this suspension were immediately deposited on a freshly cleaved mica substrate and dried under a gentle nitrogen flux. Tapping-mode (TM) AFM images were acquired in air using a Dimension 3100 microscope (Digital Instruments, Santa Barbara, CA) equipped with a “G” scanning head (maximum scan size 100 μm) and driven by a Nanoscope IIIa controller. Single-beam uncoated silicon cantilevers (type AC160TS, Olympus, Santa Barbara, CA) were used. The drive frequency was ∼300 kHz and the scan rate was 0.3–1.0 Hz. Fibril sizes were measured from the height in cross section of the topographic AFM images. Because of the drying procedure applied to achieve sample adhesion to the mica substrate, the measured heights are underestimated with respect to fully hydrated conditions and should be corrected by multiplying them by a factor that, as a first approximation, can be assumed to be equal to that estimated previously (1.4) for a different amyloidogenic protein (25). The values reported in the Results section (see Fig. 5) are raw data without correction.
Figure 5.
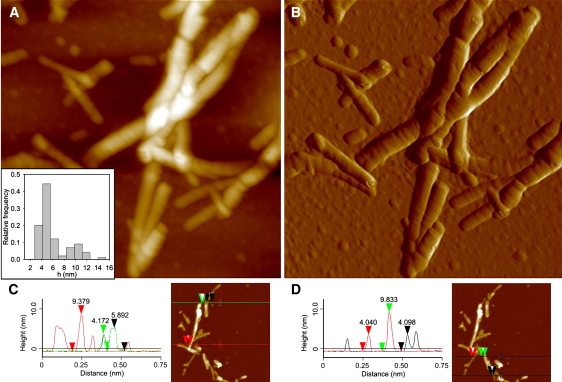
(A and B) TM-AFM image of aggregates formed by PDZ2. Aggregates were produced by incubating the protein domain at a concentration of 0.4 mg mL−1, in 20 mM sodium phosphate, 1 M NaCl, pH 7.0, 37°C, under agitation at 750 rpm for 24 h. (A) Height data (scan size 870 nm, Z range 30 nm); (inset) distribution of fibril height measured from cross sections of TM-AFM topographic images. (B) Amplitude data (scan size 870 nm; not quantitative in height), revealing the fibril ultrastructure in more detail. (C and D) Typical cross-section profiles of PDZ2 fibrils. Red, green, and black arrows facilitate the association between the topographic measurements (left) and the corresponding features in the TM-AFM images (right).
Near-UV CD
PDZ2 was diluted to a final concentration of 0.2 mg mL−1 in 20 mM sodium phosphate buffer, pH 7.0, 25°C, with and without 1 M NaCl. Near-UV CD spectra were acquired at 25°C, shortly after diluting the stocked protein sample into these solutions. An identical sample containing 1 M NaCl was incubated at 37°C under stirring at 750 rpm for 3 h before its near-UV CD spectrum was acquired at 25°C. A 10-mm-pathlength quartz cuvette (Hellma, Müllheim, Germany) and the CD instrument described above (see “Far-UV circular dichroism”) were used. Each spectrum was recorded as the average of 15 scans.
Intrinsic fluorescence
PDZ2 was diluted to a final concentration of 0.4 mg mL−1 in 20 mM sodium phosphate buffer, pH 7.0, 25°C, with and without 1 M NaCl. Fluorescence spectra were acquired at 25°C, shortly after diluting the stocked protein sample into these solutions. An identical sample containing 1 M NaCl was incubated at 37°C under stirring at 750 rpm for 3 h before its fluorescence spectrum was acquired at 25°C. Fluorescence spectra were acquired using the cuvette and instrumental apparatus described above (see “Thioflavin-T fluorescence”). The excitation wavelength was set at 280 nm and the emission range was 300–450 nm. Each spectrum was recorded as the average of three scans.
8-Anilino-1-naphthalenesulfonic acid fluorescence
PDZ2 was diluted to a final concentration of 0.02 mg mL−1 in 55 μM 8-anilino-1-naphthalenesulfonic acid (ANS), 20 mM sodium phosphate buffer, pH 7.0, at 25°C, with and without 1 M NaCl. Spectra were acquired at 25°C, shortly after diluting the stocked protein sample into these solutions. A sample containing 0.2 mg mL−1 PDZ, 20 mM sodium phosphate buffer, 1 M NaCl, no ANS, was incubated for 3 h at 37°C under stirring at 750 rpm. It was then diluted 10-fold in a sample containing ANS. The final conditions were 55 μM ANS, 20 mM sodium phosphate buffer, 1 M NaCl, pH 7.0, 25°C. Fluorescence spectra were acquired using the same cell and equipment described under “Thioflavin-T fluorescence”. The excitation wavelength was set at 380 nm and the emission range was 410–630 nm. Spectra were also acquired for the corresponding solutions in the absence of protein. Each spectrum was recorded as the average of three scans. ANS concentration was determined spectrophotometrically using an ɛ375 value of 8000 mL mmol−1 cm−1.
Equilibrium unfolding
Urea-induced unfolding of PDZ2 at equilibrium was measured by monitoring the decrease in intrinsic fluorescence of the protein at various wavelengths on increasing urea concentration. The final conditions were 0.015 mg mL−1 PDZ2, 20 mM phosphate, pH 7.0, 25°C, and urea concentrations ranging from 0 to 8 M in the presence or absence of 1 M NaCl. In each of the two studied conditions (with and without 1 M NaCl) the plots of intrinsic fluorescence versus urea concentration obtained at 100 different wavelengths were fitted globally to the equation previously described by Santoro and Bolen (26). The resulting ΔGU-FH2O, m, and Cm values were obtained from such a global fitting in both conditions.
Results
Aggregation of PDZ2 in the presence of salt and stirring
To induce aggregation of PDZ2, the protein domain was incubated at a concentration of 0.4 mg mL−1 in 20 mM sodium phosphate buffer, pH 7.0, 37°C, in the presence of 1 M NaCl, under stirring at 750 rpm. At regular time intervals, aliquots of this sample were withdrawn to carry out ThT fluorescence assays. Fig. 2 A shows the ThT spectrum obtained in the presence of the protein domain incubated under these conditions for 24 h. A 21-fold enhancement of ThT fluorescence emission was observed with respect to the blank. In contrast, a sample of PDZ2 that was not preincubated under these conditions did not bind ThT, as demonstrated by the absence of a fluorescence emission increase (Fig. 2 A). The time course of ThT fluorescence reveals a lag-phase of ∼16 h, followed by a rapid increase and a saturation of the signal after ∼26 h (Fig. 2 B). We repeated the experiment by incubating the protein domain under the same conditions in the absence of stirring, under the same conditions in the absence of NaCl, and under the same conditions in the absence of both stirring and agitation. In all three cases the time courses of ThT fluorescence were not characterized by an increase of ThT fluorescence on the investigated timescale, indicating that both agitation and a high ionic strength are necessary factors to promote the observed increase of ThT fluorescence on this timescale (Fig. 2 B).
Figure 2.

(A) ThT fluorescence spectra in the presence of PDZ2 preincubated at a concentration of 0.4 mg mL−1, in 20 mM sodium phosphate, 1 M NaCl, pH 7.0, 37°C, under agitation at 750 rpm for 24 h (solid line). ThT fluorescence spectra in the presence of PDZ2 preincubated for a few minutes in the same conditions but in the absence of agitation (dotted line) and in the presence of the incubation solution without PDZ2 (dashed line) are also reported. (B) Time course of ThT fluorescence emission at 480 nm for PDZ2 preincubated under the same conditions described for panel A (solid circles), under the same conditions in the absence of stirring (solid triangles), under the same conditions in the absence of NaCl (open circles) and under the same conditions in the absence of both stirring and NaCl (open squares). The ratio between the ThT fluorescence emission in the presence (F) and absence (F0) of PDZ2 is reported on the y axis of the figure.
Aggregates of PDZ2 have extensive β-sheet structure
The far-UV CD spectrum of PDZ2, incubated in the absence of agitation in 20 mM sodium phosphate, 1 M NaCl, pH 7.0, 37°C, reveals a negative peak at ∼215–219 nm and shoulders at 209 and 222 nm (Fig. 3 A). This structure is consistent with an α/β protein in which the β-sheet structure is prevalent. In contrast, the spectrum acquired after incubating PDZ2 for 24 h in the same condition under agitation at 750 rpm and in the presence of 1 M NaCl shows only a single broad negative peak around 225 nm (Fig. 3 A). Even though this peak is red-shifted with respect to the canonical β-sheet profile, such a feature is typical of amyloid material organized in large aggregates (16,27,28). The red shift of the peak wavelength and the decrease of signal intensity could be explained by the so-called differential absorption flattening effect arising from heterogeneous samples in which a solid-state material is suspended in solution (29). The time course of the change in mean residue ellipticity at 222 nm reveals a lag phase and a rapid-growth phase substantially superimposable on those detected with ThT fluorescence (Fig. 3 B). Again, the absence of a plateau in the late aggregation phase could be attributed to the rapidly increasing influence of the differential absorption flattening effect. The remarkable parallelism between ThT fluorescence and far-UV CD time courses suggests that very mild structural changes occur during the first 12 h, and that aggregates with ordered β-sheet structure form rapidly later on.
Figure 3.

(A) Far-UV CD spectra of PDZ2 incubated at a concentration of 0.4 mg mL−1, in 20 mM sodium phosphate, 1 M NaCl, pH 7.0, 37°C with (solid line) and without (dotted line) agitation at 750 rpm for 24 h. (B) Change of the mean residue ellipticity at 222 nm as a function of incubation time under agitation in the conditions described for panel A.
To further characterize the conformational state of PDZ2 incubated under the same conditions, we used FTIR spectroscopy. We first analyzed the native protein domain. In this case a protein sample dissolved in 20 mM ammonium carbonate was lyophilized and then resuspended in deuterated water (D2O). The Amide I region of the resulting FTIR spectrum shows a maximum absorption at 1638 cm−1 and a large band typical of an α/β fold with high content in the intramolecular β-sheet, in agreement with the above-mentioned far-UV CD spectrum (Fig. 4 A). We then incubated the protein domain under agitation at 750 rpm for 24 h in 20 mM sodium phosphate, 1M NaCl, 37°C. After incubation, the protein sample was centrifuged and resuspended in 20 mM deuterated sodium phosphate, 1 M NaCl (see Materials and Methods for further details). The Amide I region of the FTIR spectrum recorded for the resulting sample features a maximum absorbance at 1621 cm−1 (Fig. 4 B). This peak is in the range of 1615–1630 cm−1, and is known to arise from intermolecular β-sheet structure typical of amyloid fibrils (30,31).
Figure 4.

FTIR Amide I spectra of PDZ2 obtained in native conditions (A) and after incubation under stirring at high ionic strength (B). (A) PDZ2 domain was lyophilized and dissolved in D2O. (B) PDZ2 was incubated at a concentration of 0.4 mg mL−1 in 20 mM sodium phosphate, 1 M NaCl, pH 7.0, 37°C, under stirring at 750 rpm for 24 h. The resulting solution was centrifuged and the aggregates of the pellet were resuspended in 20 mM deuterated sodium phosphate buffer, 1 M NaCl, pH 7.0 (see Materials and Methods for further details).
Aggregates of PDZ2 have a fibrillar morphology
To assess the nature of the protein species formed after a 24 h incubation under the conditions used here, a morphological analysis was performed by means of TM-AFM. The analysis revealed the presence of aggregates constituted by rather thick rod-like fibrillar structures with a typical length of a few hundreds of nanometers (Fig. 5, A and B). These straight formations show, perpendicularly to their longitudinal axis, a mild streaking on their surface (Fig. 5, A and B); such features are commonly associated with amyloid fibrils with tightly wound protofilaments (32). The statistical analysis of fibril height, measured from the height in cross section of topographic AFM images, yielded a bimodal distribution with a main peak at 4.9 ± 0.1 nm and a smaller one at 10.2 ± 0.4 nm (Fig. 5 A, inset). The highest fibrillar species seem to arise from a partial or complete superimposition of distinct fibrils with a lower height. Typical cross-section profiles are reported in Fig. 5, C and D. However, since the sample was dried to allow its adhesion to the mica substrate before performing the AFM analysis, the reported height measurements underestimate the real fibrillar diameter in solution. To obtain more realistic diameter values, a correction should be applied by multiplying the measured heights by a factor of 1.4 (25). After this correction, the heights of the thinnest fibrils observed are still consistent with amyloid fibrils, typically 7–13 nm. Protofilaments and protofibrils should have lower diameters, typically 2–5 nm.
Hence, the morphological analysis performed here using TM-AFM allows the direct observation of aggregate species with characteristics reminiscent of amyloid-like fibrils. The analysis allows the presence of amorphous, protofilament, and protofibrillar species to be excluded after 24 h of incubation under these conditions. Amorphous aggregates would have a nonfibrillar appearance. Protofibrils would have a lower height than that observed here for the thinnest fibrils, and a spherical, curvilinear, or annular appearance. Protofilaments would appear straight, but again with a much lower height than that of the thinnest species actually observed. It is still possible that all of these species, or some of them, form transiently during the process as intermediate aggregates, but the analysis shows that after 24 h amyloid-like fibrils predominate in the sample.
PDZ2 adopts a native-like structure under conditions promoting aggregation
To explore the conformational state adopted by PDZ2 under the conditions used here to promote amyloid fibril formation, but before the initiation of aggregation, we compared the spectroscopic properties of PDZ2 in 1), 20 mM sodium phosphate buffer, pH 7.0, in the absence of salts and agitation; and 2), 20 mM sodium phosphate buffer, 1 M NaCl, pH 7.0, before and after starting agitation. In the first condition the protein is stable in its native state and does not undergo aggregation. In the second condition PDZ2 undergoes amyloid fibril formation, but the time courses acquired using ThT fluorescence and far-UV CD show that within the first 3 h no fibrils accumulate in the sample (Figs. 2 B and 3 B). The comparison of the spectroscopic properties of PDZ2 under the two conditions therefore allowed us to probe possible conformational changes of PDZ2 after incubation in the presence of salt and agitation, before aggregation was effectively initiated.
We first acquired near-UV CD spectra in the absence of salt and agitation, in the presence of 1 M NaCl before starting agitation and in the presence of 1 M NaCl after 3 h agitation (Fig. 6 A). Unfortunately, the protein has no detectable CD signal in the near-UV region, even at high protein concentrations (Fig. 6 A). We therefore acquired intrinsic fluorescence spectra of the protein in the same conditions, that is, in the absence of salt and agitation, in the presence of 1 M NaCl before agitation, and in the presence of NaCl after 3 h agitation (Fig. 6 B). The spectra are very similar, within experimental error, indicating that the chemical environment around tryptophan and tyrosine residues is not significantly different under aggregating and nonaggregating conditions. We also acquired fluorescence spectra of ANS in the presence of PDZ2 under the different conditions (Fig. 6 C). PDZ2 does not change the intrinsic fluorescence of ANS in either of the conditions tested. The spectra of ANS acquired in the absence or presence of PDZ2, under any of the three tested conditions, are superimposable (Fig. 6 C). This indicates that PDZ2 has no significantly exposed hydrophobic clusters in its native state, and that the situation is maintained in the presence of salts and agitation. Overall, the spectroscopic analysis reported here indicates that monomeric PDZ2 adopts an essentially native-like tertiary structure under conditions promoting aggregation.
Figure 6.

(A) Near-UV CD spectra of 0.2 mg mL−1 PDZ2 in 20 mM sodium phosphate, pH 7.0, 25°C (solid line), 20 mM sodium phosphate, 1 M NaCl, pH 7.0, 25°C, before starting agitation (dashed line), and in 20 mM sodium phosphate, 1 M NaCl, pH 7.0, 25°C, after 3 h agitation at 37°C (dotted line). (B) Intrinsic fluorescence spectra of 0.4 mg mL−1 PDZ2 in 20 mM sodium phosphate, pH 7.0, 25°C (solid line), 20 mM sodium phosphate, 1 M NaCl, pH 7.0, 25°C, before starting aggregation (dashed line), and in 20 mM sodium phosphate, 1 M NaCl, pH 7.0, 25°C, after 3 h agitation at 37°C (dotted line). The inset shows the intrinsic fluorescence at 354 nm versus NaCl concentration before agitation (solid triangles), as well as the corresponding signals after 0 h (open square) and 3 h (solid circle) under agitation in 1 M NaCl at 37°C. (C) Fluorescence spectra of 55 μM ANS in the presence of 0.02 mg mL−1 PDZ2, in 20 mM sodium phosphate, pH 7.0, 25°C (solid line), 20 mM sodium phosphate, 1 M NaCl, pH 7.0, 25°C (dashed line), and in 20 mM sodium phosphate, 1 M NaCl, pH 7.0, after 3 h agitation at 37°C (dotted line). The spectrum of 55 μM ANS in the absence of protein is also shown as a control (line with solid circles). (D) Urea-induced unfolding curves at equilibrium for 0.015 mg mL−1 PDZ2 in 20 mM sodium phosphate, pH 7.0, 25°C in the absence (open circles) and presence (solid circles) of 1 M NaCl. The reported curves were acquired using intrinsic fluorescence at 322 nm and normalized to the fraction of folded protein.
To assess whether the native state of PDZ2 is thermodynamically destabilized or stabilized under conditions promoting aggregation, relative to nonaggregating conditions, we acquired equilibrium urea-induced unfolding curves of PDZ2 in the presence and absence of salt (Fig. 6 D). The urea-induced transitions of PDZ2 appear to be highly cooperative in both conditions, suggesting that urea-induced unfolding occurs via an apparent two-state mechanism. However, it is clear that unfolding occurs at higher concentrations of urea in the presence of salt (Fig. 6 D). The analysis of the two curves with a two-state model (26) yields values of 2.57 ± 0.02 and 3.5 ± 0.1 M for the midpoint of unfolding (Cm) in the absence and presence of salt, respectively. The same analysis yields values of 3.1 ± 0.1 and 3.85 ± 0.3 kcal mol−1 for the free energy change of unfolding (ΔGU-FH2O) in the absence and presence of salt, respectively, further confirming that the native protein is stabilized in the presence of NaCl. By contrast, the cooperativity of the unfolding process is similar in the two conditions, with m values of 1.2 ± 0.03 and 1.1 ± 0.1 kcal M−1 mol−1, respectively.
Discussion
Formation of amyloid-like fibrils by PDZ2 in the presence of nondestabilizing conditions
Incubation of PDZ2 under the conditions explored here allows the conversion of this α/β protein domain from its native, folded state into fibrillar aggregates that appear to be structurally, morphologically, and tinctorially indistinguishable from the amyloid fibrils associated with protein deposition diseases. The morphological inspection carried out using TM-AFM indicates that the aggregates consist of short and straight fibrils, have a characteristic periodic texture, and have a height typical of amyloid-like fibrils (Fig. 5). The aggregates bind ThT, causing a 21-fold enhancement of its intrinsic fluorescence (Fig. 2 A), and are characterized by an extensive β-sheet structure, as indicated by far-UV CD and FTIR spectroscopy (Figs. 3 A and 4 B). The FTIR analysis, in particular, shows that the β-sheet structure is intermolecular in nature and typical of amyloid-like fibrils (Fig. 4 B). The time course of PDZ2 aggregation exhibits the characteristic kinetic profile generally observed for fibril formation, with a lag-phase followed by a rapid growth (Figs. 2 B and 3 B). Hence, using the three criteria commonly used to classify protein aggregates as amyloid fibrils (fibrillar morphology, cross-β structure, and binding to specific diagnostic dyes), the aggregates of PDZ2 formed here have characteristics reminiscent of amyloid-like fibrils.
As shown by a number of studies published in the last decade, harsh denaturing conditions are usually required to induce amyloid fibril formation of initially folded proteins. These include pH extremes (8,9), high temperature (10,11), high pressure (12,13), and cosolvents such as 2,2,2-trifluoroethanol (14,15). Amyloid fibril formation at physiological or nearly physiological conditions generally requires seeding the reaction with fibrillar aggregates that have been preformed under extreme conditions (33–35). The data presented here on PDZ2 indicate that agitation and a high ionic strength in the absence of denaturing agents may be sufficient to promote amyloid fibril formation by a normally folded protein. Neither heating nor a low pH, nor any other chemical or physical denaturant, is required to induce amyloid fibril formation by this initially folded protein. Indeed, under the conditions used here, before aggregation has initiated to any significant extent, PDZ2 retains an essentially native-like structure (Fig. 6, A–C) and the conformational stability of the native fold appears to be even higher relative to a similar buffer devoid of NaCl in which aggregation does not occur (Fig. 6 D).
Amyloidogenic effect of salts and agitation
In a recent elegant study (16), it was shown that stirring and a high ionic strength may be used to trigger native-like aggregation of human β2-microglobulin and hen lysozyme in conditions of physiological pH, temperature, and pressure. The resulting aggregates are native-like and may be converted into amyloid-like fibrils by a further heating step (16). In a more recent report (17), the same authors showed that amyloid fibril formation by β2-microglobulin can also be accomplished in the absence of a heating step by simply incubating the protein at neutral pH and 37°C, initially under agitation at high ionic strength and then for 25–45 days under the same conditions in the absence of agitation. The findings reported previously for β2-microglobulin and those described here for PDZ2 show that amyloid fibril formation in the absence of persistent denaturing agents may be an important and widespread phenomenon.
As shown here for PDZ2 and previously for other systems (36,37), a high ionic strength increases the thermodynamic stability of the folded state of a protein, i.e., it increases the ΔGU-FH2O value. Nor is agitation thought to be an unfolding-inducing agent per se for globular proteins. The aggregation-promoting effects of salts and agitation presumably have a different rationale. High salt concentrations are known to induce aggregation of intrinsically unfolded or pH-unfolded proteins (25,38–40). Salts exert this aggregation triggering effect by several possible mechanisms. They attenuate repulsive electrostatic interactions between protein molecules in solution via a generic Debye-Huckel screening effect (41). Ions can also bind to oppositely charged amino acid residues on the protein and partially neutralize the overall net charge of the protein molecule; this results again in a weakening of the electrostatic repulsion between protein monomers (40). Salts also act indirectly by perturbing the hydration shell of the protein molecule and inducing protein surface desolvation (39,42). Either effect can predominate depending on the conditions and the protein. A combination of both direct protein-salt interactions and changes of water structure has also been proposed (43).
Mechanical forces, such as agitation, shaking, or simple shear flow, have also been reported to accelerate fibril formation from intrinsically disordered proteins (44–46) or globular proteins partially unfolded under nonphysiological conditions (16,47). Only the recent report on β2-microglobulin described amyloid fibril formation of an initially folded protein in the absence of destabilizing agents (17). Regardless of the structural and dynamical nature of the amyloidogenic nuclei, it is reasonable to think that stirring can persistently spread nuclei and growing fibrils over the entire suspension, enhancing the efficiency of seeding. Stirring can also act as a controlled simple shear flow, as reported in a recent study (47). Subsequent to that study, it was proposed that shear flow favors the orientation and deformation of a protein so that aggregates that are able to act as seeds can form more efficiently (47). Agitation can cause transient and local structural fluctuations in a sample of folded protein molecules and create a radial gradient of protein concentration, with areas characterized by high local concentrations. Moreover, similarly to sonication, stirring can cause the fracturing of fibrils to generate new growing ends (44,48). As recently shown for prion propagation, the naturally occurring fragmentation of fibrils could be important to amplify the number of fibrillar ends available for further elongation in a chain-reaction mechanism (49,50). The small length of the PDZ2 fibrils observed here in the late growth phase is consistent with mechanisms of this type and could result from the agitation-induced breakage of growing fibrils or fresh generation of nuclei that act as seeds. In summary, all of the factors mentioned here, including the generation of areas with high protein concentrations, the transient and local deformation of protein molecules, and the fracturing and resuspension of growing fibrils, contribute to the profibrillogenic effect of agitation.
Conclusions
The nature of the chemical and physical agents used here to induce amyloid formation by initially native PDZ2, and the investigation into why amyloid fibril formation by initially folded proteins can occur under these relatively unexplored conditions will allow new aspects of the fundamentals of this process to be determined. Furthermore, the use of physiological values of temperature, pressure, and pH, and the absence of chemical denaturants, such as alcohol cosolvents, will allow the process of aggregation to be investigated under conditions in which the protein is initially native and free to access locally unfolded states by those structural fluctuations that normally occur physiologically. Protocols of this type therefore open new avenues into the exploration of processes of amyloid fibril formation that may shed new light on those that occur physiologically.
Acknowledgments
We thank Matteo Ramazzotti and Elodie Monsellier for helpful and fruitful discussions.
This work was partially supported by grants from the Italian Ministero dell'Istruzione, dell'Università e della Ricerca (projects PRIN 2005027330, PRIN 2006058958, and FIRB RBIN04PWNC), the EMBO Young Investigator Programme (YIP 2005), and the Fondazione ONAOSI. The authors declare the absence of any conflict of interest.
References
- 1.Stefani M., Dobson C.M. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 2.Uversky V.N., Fink A.L. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim. Biophys. Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe D.J. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 4.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 5.Fowler D.M., Koulov A.V., Balch W.E., Kelly J.W. Functional amyloid—from bacteria to humans. Trends Biochem. Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Gazit E. Use of biomolecular templates for the fabrication of metal nanowires. FEBS J. 2007;274:317–322. doi: 10.1111/j.1742-4658.2006.05605.x. [DOI] [PubMed] [Google Scholar]
- 7.Hamley I.W. Peptide fibrillization. Angew. Chem. Int. Ed. Engl. 2007;46:8128–8147. doi: 10.1002/anie.200700861. [DOI] [PubMed] [Google Scholar]
- 8.Guijarro J.I., Sunde M., Jones J.A., Campbell I.D., Dobson C.M. Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. USA. 1998;95:4224–4228. doi: 10.1073/pnas.95.8.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McParland V.J., Kad N.M., Kalverda A.P., Brown A., Kirwin-Jones P. Partially unfolded states of β(2)-microglobulin and amyloid formation in vitro. Biochemistry. 2000;39:8735–8746. doi: 10.1021/bi000276j. [DOI] [PubMed] [Google Scholar]
- 10.Litvinovich S.V., Brew S.A., Aota S., Akiyama S.K., Haudenschild C. Formation of amyloid-like fibrils by self-association of a partially unfolded fibronectin type III module. J. Mol. Biol. 1998;280:245–258. doi: 10.1006/jmbi.1998.1863. [DOI] [PubMed] [Google Scholar]
- 11.Fändrich M., Fletcher M.A., Dobson C.M. Amyloid fibrils from muscle myoglobin. Nature. 2001;410:165–166. doi: 10.1038/35065514. [DOI] [PubMed] [Google Scholar]
- 12.Ferrão-Gonzales A.D., Souto S.O., Silva J.L., Foguel D. The preaggregated state of an amyloidogenic protein: hydrostatic pressure converts native transthyretin into the amyloidogenic state. Proc. Natl. Acad. Sci. USA. 2000;97:6445–6450. doi: 10.1073/pnas.97.12.6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Felice F.G., Vieira M.N., Meirelles M.N., Morozova-Roche L.A., Dobson C.M. Formation of amyloid aggregates from human lysozyme and its disease-associated variants using hydrostatic pressure. FASEB J. 2004;18:1099–1101. doi: 10.1096/fj.03-1072fje. [DOI] [PubMed] [Google Scholar]
- 14.Chiti F., Webster P., Taddei N., Clark A., Stefani M. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmittschmitt J.P., Scholtz J.M. The role of protein stability, solubility, and net charge in amyloid fibril formation. Protein Sci. 2003;12:2374–2378. doi: 10.1110/ps.03152903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasahara K., Yagi H., Naiki H., Goto Y. Heat-induced conversion of β(2)-microglobulin and hen egg-white lysozyme into amyloid fibrils. J. Mol. Biol. 2007;372:981–991. doi: 10.1016/j.jmb.2007.06.088. [DOI] [PubMed] [Google Scholar]
- 17.Sasahara K., Yagi H., Sakai M., Naiki H., Goto Y. Amyloid nucleation triggered by agitation of β2-microglobulin under acidic and neutral pH conditions. Biochemistry. 2008;47:2650–2660. doi: 10.1021/bi701968g. [DOI] [PubMed] [Google Scholar]
- 18.Hung A.Y., Sheng M. PDZ domains: structural modules for protein complex assembly. J. Biol. Chem. 2002;277:5699–5702. doi: 10.1074/jbc.R100065200. [DOI] [PubMed] [Google Scholar]
- 19.Chi C.N., Gianni S., Calosci N., Travaglini-Allocatelli C., Engström K. A conserved folding mechanism for PDZ domains. FEBS Lett. 2007;581:1109–1113. doi: 10.1016/j.febslet.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Gianni S., Calosci N., Aelen J.M., Vuister G.W., Brunori M. Kinetic folding mechanism of PDZ2 from PTP-BL. Protein Eng. Des. Sel. 2005;18:389–395. doi: 10.1093/protein/gzi047. [DOI] [PubMed] [Google Scholar]
- 21.Ivarsson Y., Travaglini-Allocatelli C., Jemth P., Malatesta F., Brunori M. An on-pathway intermediate in the folding of a PDZ domain. J. Biol. Chem. 2007;282:8568–8572. doi: 10.1074/jbc.M611026200. [DOI] [PubMed] [Google Scholar]
- 22.Gianni S., Geierhaas C.D., Calosci N., Jemth P., Vuister G.W. A PDZ domain recapitulates a unifying mechanism for protein folding. Proc. Natl. Acad. Sci. USA. 2007;104:128–133. doi: 10.1073/pnas.0602770104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jemth P., Gianni S. PDZ domains: folding and binding. Biochemistry. 2007;46:8701–8708. doi: 10.1021/bi7008618. [DOI] [PubMed] [Google Scholar]
- 24.Walma T., Spronk C.A., Tessari M., Aelen J., Schepens J. Structure, dynamics and binding characteristics of the second PDZ domain of PTP-BL. J. Mol. Biol. 2002;316:1101–1110. doi: 10.1006/jmbi.2002.5402. [DOI] [PubMed] [Google Scholar]
- 25.Campioni S., Mossuto M.F., Torrassa S., Calloni G., de Laureto P.P. Conformational properties of the aggregation precursor state of HypF-N. J. Mol. Biol. 2008;379:554–567. doi: 10.1016/j.jmb.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Santoro M.M., Bolen D.W. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry. 1998;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]
- 27.Jaikaran E.T., Higham C.E., Serpell L.C., Zurdo J., Gross M. Identification of a novel human islet amyloid polypeptide β-sheet domain and factors influencing fibrillogenesis. J. Mol. Biol. 2001;308:515–525. doi: 10.1006/jmbi.2001.4593. [DOI] [PubMed] [Google Scholar]
- 28.Soldi G., Bemporad F., Torrassa S., Relini A., Ramazzotti M. Amyloid formation of a protein in the absence of initial unfolding and destabilization of the native state. Biophys. J. 2005;89:4234–4244. doi: 10.1529/biophysj.105.067538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castiglioni E., Abbate S., Longhi G., Gangemi R. Wavelength shifts in solid-state circular dichroism spectra: a possible explanation. Chirality. 2007;19:491–496. doi: 10.1002/chir.20404. [DOI] [PubMed] [Google Scholar]
- 30.Zandomeneghi G., Krebs M.R., McCammon M.G., Fändrich M. FTIR reveals structural differences between native β-sheet proteins and amyloid fibrils. Protein Sci. 2004;13:3314–3321. doi: 10.1110/ps.041024904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seshadri S., Khurana R., Fink A.L. Fourier transform infrared spectroscopy in analysis of protein deposits. Methods Enzymol. 1999;309:559–576. doi: 10.1016/s0076-6879(99)09038-2. [DOI] [PubMed] [Google Scholar]
- 32.Harper J.D., Wong S.S., Lieber C.M., Lansbury P.T. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997;4:119–122. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 33.Kihara M., Chatani E., Sakai M., Hasegawa K., Naiki H. Seeding-dependent maturation of β2-microglobulin amyloid fibrils at neutral pH. J. Biol. Chem. 2005;280:12012–12018. doi: 10.1074/jbc.M411949200. [DOI] [PubMed] [Google Scholar]
- 34.Morozova-Roche L.A., Zurdo J., Spencer A., Noppe W., Receveur V. Amyloid fibril formation and seeding by wild-type human lysozyme and its disease-related mutational variants. J. Struct. Biol. 2000;130:339–351. doi: 10.1006/jsbi.2000.4264. [DOI] [PubMed] [Google Scholar]
- 35.Peim A., Hortschansky P., Christopeit T., Schroeckh V., Richter W. Mutagenic exploration of the cross-seeding and fibrillation propensity of Alzheimer's β-amyloid peptide variants. Protein Sci. 2006;15:1801–1805. doi: 10.1110/ps.062116206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cacace M.G., Landau E.M., Ramsden J.J. The Hofmeister series: salt and solvent effects on interfacial phenomena. Q. Rev. Biophys. 1997;30:241–277. doi: 10.1017/s0033583597003363. [DOI] [PubMed] [Google Scholar]
- 37.Taddei N., Chiti F., Paoli P., Fiaschi T., Bucciantini M. Thermodynamics and kinetics of folding of common-type acylphosphatase: comparison to the highly homologous muscle isoenzyme. Biochemistry. 1999;38:2135–2142. doi: 10.1021/bi9822630. [DOI] [PubMed] [Google Scholar]
- 38.Zurdo J., Guijarro J.I., Jiménez J.L., Saibil H.R., Dobson C.M. Dependence on solution conditions of aggregation and amyloid formation by an SH3 domain. J. Mol. Biol. 2001;311:325–340. doi: 10.1006/jmbi.2001.4858. [DOI] [PubMed] [Google Scholar]
- 39.Munishkina L.A., Henriques J., Uversky V.N., Fink A.L. Role of protein-water interactions and electrostatics in α-synuclein fibril formation. Biochemistry. 2004;43:3289–3300. doi: 10.1021/bi034938r. [DOI] [PubMed] [Google Scholar]
- 40.Raman B., Chatani E., Kihara M., Ban T., Sakai M. Critical balance of electrostatic and hydrophobic interactions is required for β2-microglobulin amyloid fibril growth and stability. Biochemistry. 2005;44:1288–1299. doi: 10.1021/bi048029t. [DOI] [PubMed] [Google Scholar]
- 41.Creighton T.E. 2nd ed. W. H. Freeman and Company; New York: 1992. Proteins: Structures and Molecular Properties. 155–156. [Google Scholar]
- 42.Collins K.D., Washabaugh M.W. The Hofmeister effect and the behaviour of water at interfaces. Q. Rev. Biophys. 1985;18:323–422. doi: 10.1017/s0033583500005369. [DOI] [PubMed] [Google Scholar]
- 43.Klement K., Wieligmann K., Meinhardt J., Hortschansky P., Richter W. Effect of different salt ions on the propensity of aggregation and on the structure of Alzheimer's Aβ(1–40) amyloid fibrils. J. Mol. Biol. 2007;373:1321–1333. doi: 10.1016/j.jmb.2007.08.068. [DOI] [PubMed] [Google Scholar]
- 44.Collins S.R., Douglass A., Vale R.D., Weissman J.S. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol. 2004;2:1582–1590. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu J.J., Lindquist S. Oligopeptide-repeat expansions modulate ‘protein-only’ inheritance in yeast. Nature. 1999;400:573–576. doi: 10.1038/23048. [DOI] [PubMed] [Google Scholar]
- 46.Serpell L.C., Berriman J., Jakes R., Goedert M., Crowther R.A. Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc. Natl. Acad. Sci. USA. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hill E.K., Krebs B., Goodall D.G., Howlett G.J., Dunstan D.E. Shear flow induces amyloid fibril formation. Biomacromolecules. 2006;7:10–13. doi: 10.1021/bm0505078. [DOI] [PubMed] [Google Scholar]
- 48.Petkova A.T., Leapman R.D., Guo Z., Yau W.M., Mattson M.P. Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 49.Shorter J., Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 50.Sun Y., Makarava N., Lee C.I., Laksanalamai P., Robb F.T. Conformational stability of PrP amyloid fibrils controls their smallest possible fragment size. J. Mol. Biol. 2008;376:1155–1167. doi: 10.1016/j.jmb.2007.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]