

Abstract
The folding of DNA on the nucleosome core particle governs many fundamental issues in eukaryotic molecular biology. In this study, an updated set of sequence-dependent empirical “energy” functions, derived from the structures of other protein-bound DNA molecules, is used to investigate the extent to which the architecture of nucleosomal DNA is dictated by its underlying sequence. The potentials are used to estimate the cost of deforming a collection of sequences known to bind or resist uptake in nucleosomes along various left-handed superhelical pathways and to deduce the features of sequence contributing to a particular structural form. The deformation scores reflect the choice of template, the deviations of structural parameters at each step of the nucleosome-bound DNA from their intrinsic values, and the sequence-dependent “deformability” of a given dimer. The correspondence between the computed scores and binding propensities points to a subtle interplay between DNA sequence and nucleosomal folding, e.g., sequences with periodically spaced pyrimidine-purine steps deform at low cost along a kinked template whereas sequences that resist deformation prefer a smoother spatial pathway. Successful prediction of the known settings of some of the best-resolved nucleosome-positioning sequences, however, requires a template with “kink-and-slide” steps like those found in high-resolution nucleosome structures.
Introduction
One of the most remarkable aspects of DNA packaging is the positioning of nucleosomes on specific base sequences without the direct involvement of the bases. Contacts between the histone proteins and DNA are almost exclusively ionic interactions involving cationic amino acid side groups on the proteins and the negatively charged sugar-phosphate backbone of DNA (1–3). The localized build up of positive charge at the protein-DNA interface creates patches of neutralization on one side of the double helix (4), allowing the DNA to collapse toward the site of neutralization and fold around the multiprotein assembly without specific interactions with the bases (5,6).
The preferential binding of specific “positioning sequences” to the histone core presumably involves an indirect response in DNA related to the intrinsic structure and “deformability” of the constituent basepair (bp) steps. For example, naturally curved DNA sequences with tracts of three to four A·T basepairs, repeated in phase with the ∼10 bp helical period, are strong nucleosome-positioning sequences (7), as are intrinsically deformable sequences with periodic occurrences of pyrimidine-purine (YR) steps (8,9). The former sequences may require less structural distortion than a random, naturally straight piece of DNA to wrap around the nucleosome surface (10), and the latter sequences contain dimeric sites known to be easily deformed by protein interaction (11) and crystal contacts (12).
Many of the DNA sequences found to bias the positioning of nucleosomes exhibit a common 71-bp palindromic “consensus pattern” with short, periodically spaced nucleotide segments showing 50% or greater sequence identity (13). The latter sites, however, show almost no overlap with the palindromic sequences that have been successfully crystallized in the nucleosome core-particle structure (1,2,14–21). Notably missing from the crystalline DNA is the strong ∼10 bp periodicity of TA dinucleotide steps and the out-of-phase alternation of A·T and G·C-rich regions seen in many of the strong nucleosome-binding sequences selected by solution methods (9,22,23). Instead, there is a regular recurrence of CA and CAG fragments in the crystallized DNA and the interruption of AT-rich segments by only a few (1–3) successive G·C basepairs. In addition, there are no CG dimer steps in the DNA on the surfaces of most crystallized nucleosomes despite the regular occurrence of such basepairs in certain well-characterized binding sequences (9). Other features characteristic of known nucleosome-binding sequences, such as the phased repetition of TATA tetrads (22), are absent in both the aforementioned solution consensus pattern and the crystallized sequences. Thus, nucleosome-positioning sequences may fit into distinct classes that take advantage of different sets of nucleotide-deformation properties. By contrast, other nucleotide signals such as TGGA repeats (24), the (G/C)3NN motif (where N refers to any of the four common basepairs) (25), and telomeric repeats (26,27) contain information that seemingly resists nucleosome formation.
Interest in understanding the nucleosome-binding propensities of genomic DNA has prompted the development of novel approaches on different fronts to analyze and depict the binding patterns of arbitrary sequences. Some predictive tools, such as the RECON web server (28) and recent bioinformatics schemes designed to predict nucleosome occupancy in vivo (29), are based on the frequencies of occurrence of different dinucleotides in specific positions along experimentally detected nucleosomal sequences, whereas other schemes are based on selected aspects of DNA structure (7) and/or deformability (30). Notable in this regard are the calculations of nucleosome stability by De Santis et al. based on a simple, sequence-dependent elastic model of DNA (31–33). The intrinsic bending of basepair steps in this treatment is derived from potential-energy calculations and the differential deformabilities of dimers are related to the relative melting temperatures of known sequences. Neither the known sequence-dependent differences in intrinsic twisting (34,35) and stretching (36) nor the known coupling of conformational variables (11) are considered in the model. Predictions of nucleosome stability, tested on a broad range of DNA sequences, nevertheless, account remarkably well for experimental observations (31,32).
The distortions of DNA revealed in the set of solved, high-resolution nucleosome crystal structures (over 30 examples to date in the Nucleic Acid Database (37)) are much more complex than the structural models considered in most studies of nucleosome positioning. Underlying the characteristic left-handed superhelical wrapping of the double helix around the histone assembly is an alternating pattern of directional bending that concomitantly narrows or opens the minor and major grooves of the structure (3,38–40). The bending is accompanied by sequence-dependent variation in both the twisting and the displacement (shearing) of adjacent basepairs as well as in the relative positioning of phosphate groups with respect to the basepairs (3,19,38–40). Significantly, many of the structural changes in nucleosomal DNA conform to conformational patterns found in other protein-DNA complexes (e.g., preferential minor-groove narrowing of A-tracts and major-groove narrowing of GC-rich stretches, localization of sharp bends at CA·TG dimer steps) despite the diverse modes of intermolecular association. Knowledge-based elastic functions, which incorporate the sequence-dependent information in other protein-DNA structures (11), provide useful insights into the conformational mechanics of nucleosome positioning (40). The occurrence of a given sequence in a particular three-dimensional spatial arrangement is scored in terms of the deviation of each basepair step from its preferred equilibrium structure, taking into account not only the precise structural distortions but also the known sequence-dependent anisotropy of deformation and the observed correlations of conformational variables. As we have reported recently (40), this approach accounts remarkably well for the rotational settings of some of the best-characterized nucleosome-positioning sequences.
As a next step in investigating the mechanics of nucleosome positioning, we examine some of the factors that underlie our estimates of the capability of an arbitrary DNA sequence to bind to the histone octamer. We update the data set of protein-DNA structures used to construct our knowledge-based potentials and consider different sets of potential functions corresponding to different categories of duplex deformation, e.g., functions based on configurational states characteristic of room-temperature fluctuations of the B-DNA double helix versus functions that include dimers deformed to alternate double-helical arrangements. We examine the effects of various structural templates on the binding/deformation scores, including i), the 147-bp double-helical fold observed in the best-resolved nucleosome core-particle structure (2); ii), an ideal, smoothly deformed DNA superhelix; and iii), the subtly different crystalline scaffolds found in the presence of different DNA sequences. By contrast, our recent analyses of nucleosome positioning use the set of elastic potentials derived more than a decade ago (11) from a substantially smaller data set and assume that the core of histone proteins imposes exactly the same conformational constraints on DNA regardless of basepair sequence. The present study also considers the effect of template length on the DNA deformation scores, focusing primarily on the central 60 basepair steps (61 bp) in contact with the (H3·H4)2 tetramer and believed to be critical to nucleosome positioning (13,41). Our published predictions of nucleosome positioning in well-characterized sequences reflect the deformation “energies” of DNA over all 146 basepair steps in the best-resolved crystal structure. Here we study the DNA deformation properties of sequences from the mouse genome, some of which enhance (9) and others that resist (24) nucleosome formation, and synthetic constructs (22), which bind the histone core even more tightly than naturally occurring positioning sequences. Because the precise positioning of these sequences is not yet known, we also test the capability of the potentials and templates to mimic the known settings of representative experimentally well-characterized positioning sequences on the nucleosome.
Methods
DNA deformability
The deformability of DNA is based on the range of conformational states observed in a nonredundant set of 135 protein-DNA crystal complexes taken from the Nucleic Acid Database (37) at a resolution cut-off of 2.5 Å or better. The data set (see Table S1 in Supporting Material) excludes duplicate structures, which have been solved independently under slightly different crystallographic conditions, or solved with modifications of a few basepairs or with a mutant protein in place of the wild-type protein. Structures of complexes obtained from different cell types or species (e.g., human versus archaea TATA-box binding protein) or solved in different space groups (e.g., independent complexes of DNA with the Trp repressor protein) are included in the data set. The structure of the best-resolved (1.94 Å) nucleosome core particle (2), however, is not included in the set of reference structures because our aim is the prediction of nucleosome binding using the intrinsic, sequence-dependent conformational properties of DNA found in a wide collection of protein-bound double-helical structures other than the nucleosome. Both the number of protein-DNA complexes and the types of structures representing different kinds of deformations of DNA are increased and the quality of structures is improved compared to the set of protein-DNA structures used in earlier knowledge-based estimates (11) of DNA sequence-dependent deformability. The composite data also provide useful benchmarks for molecular simulations of DNA sequence-dependent structure (42) and deformability (43). Particularly notable in this regard is the generally excellent agreement found in recent molecular-dynamics simulations of the 136 unique DNA tetramers (44) with structural properties of protein-bound DNA sequences found by our group and published only now. Interestingly, the predicted deformability of B DNA exceeds that extracted from protein-free structures (11,45), the disagreement presumably reflecting of restrictions on basepair motions imposed by the three lattices in which high resolution structures have been determined.
The conformation of DNA is described in terms of the relative positions and orientations of neighboring basepairs. The preferred arrangements and likely fluctuations of the basepair steps are derived from the average properties of 2862 dimeric units extracted from the aforementioned high-resolution crystal complexes using the 3DNA software package (46,47). Terminal dimer units, which may adopt alternate conformations or be affected by crystal packing, and steps with single-stranded nicks and mismatches are not considered. “Melted” residues, where the displacements of complementary basepairs deviate significantly from average values, and chemically modified nucleotides are also omitted.
Six rigid-body parameters are used to specify the relative position of each successive pair. The parameters include the two components of bending (θ1,θ2) called Tilt and Roll, the Twist (θ3), the two components of shearing (θ4,θ5) called Shift and Slide, and the out-of-plane displacement Rise (θ6) (48) (see images of representative parameters in Fig. 2). To separate intrinsic deformations from severe protein-induced conformational distortions and to obtain quasi-Gaussian distributions of basepair step parameters, outlying states with extreme bending, twisting, and/or stretching are excluded in a stepwise fashion until there are no parameters that deviate from their average values by more than three standard deviations (3σ) before culling.
Figure 2.
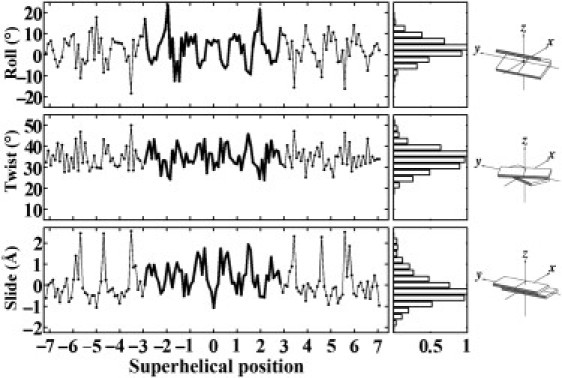
Variation of the three primary basepair step parameters (Roll, Twist, Slide), computed with 3DNA (46,47), as a function of superhelical position of DNA on the 147-bp nucleosome core-particle structure (NDB_ID pd0287) (2). The step parameters of the 60 dimer steps (61 bp) bound to the (H3·H4)2 tetramer are highlighted by heavy lines at the center of the plotted data. Superhelical positions correspond to the number of double-helical turns a dimeric step is displaced from the structural dyad on the central basepair (denoted by 0). Histograms on the right edge of the figure are derived from the distribution of step parameters in 135 other well-resolved protein-DNA complexes (see Methods and Supporting Material) and scaled with respect to a value of unity for the most populated parametric ranges. The fixed angular scales emphasize the preferential deformation of nucleosomal DNA via bending rather than twisting, compared to the similar ranges of Roll and Twist in other protein-DNA complexes. Block images of step parameters obtained with 3DNA (46,47) and illustrated with RasMol (87).
Knowledge-based scoring functions are derived, as described elsewhere (11), from the statistical properties of the protein-DNA complexes. The “energy” E of a basepair step is expressed as a sum of elastic contributions over the six basepair step parameters:
| (1) |
Here E0 is the minimum energy of the step and the fij are elastic constants impeding dimeric deformations of the DNA. The Δθi = θi − 〈θi〉 define the deviation of the ith step parameter θi (i.e., Tilt, Roll, Twist, Shift, Slide, or Rise) from the equilibrium (average) value 〈θi〉 of the given dimer. The fij are extracted from the pairwise covariance of relevant variables for the dimer in the reference data set. The dimeric energy E is thus a (unitless) statistical score measuring the cost of deforming a particular DNA basepair step relative to the observed dispersion of step parameters of the same type of dimer in many structural contexts. The total energy U of a given sequence is a sum of the values of E over all basepair steps N in a given DNA chain (i.e., ).
Elastic parameters for a generic dimer step are based on equal weighting of the mean values of the step parameters of the 16 common dimers, i.e., AA and TT, AG and CT, etc., have identical averages except for different signs of Tilt and Shift (48). The number of generic dimer entries thus exceeds the sum of examples of the 10 unique dimers. The covariance Δθij of the generic step parameters is computed from the weighted mean-square and mean values of all 16 step parameters, i.e., Δθij = (〈θiθj〉 − 〈θi〉〈θj〉)1/2, where the averages are based on equal weighting of the average parameters for all step types.
The range of conformational states adopted by a given basepair type is further characterized by the volume of conformation space V within a given energy contour, here set to E = 1/2. The covariance of observed step parameters is expressed in matrix form, (i.e., a 6 × 6 grid with Δθij entries corresponding to all pairwise combinations of the six step parameters), and V is obtained from the product of the eigenvalues of the array.
Conformational classification
We use analysis routines in the 3DNA software package (46,47) to identify dimer steps of different conformational types in the protein-DNA data set. The binding of certain proteins to DNA is known, for example, to induce a B → A transformation of the double helix at individual basepair steps (49). Moreover, the DNA dimer steps in such complexes may fall into different conformational categories, i.e., some steps may be A-like and others B-like (49). The very different rigid-body parameters of the DNA dimer steps in different helical forms suggest that different input data sets may have varying effectiveness in predicting the positioning of DNA on nucleosomes. The structural composition of the reference protein-DNA dimers is thus taken into consideration.
We use the value of zp, the mean (out-of-plane) z-coordinates of the backbone phosphorus atoms with respect to the dimeric coordinate frame (50), to distinguish A-like from B-like basepair steps. The classification scheme is based on criteria previously established (49): B DNA (zp ≤ 0.5 Å); and A DNA (zp ≥ 1.5 Å). Dimer steps with intermediate zp values are defined as AB conformational intermediates along the B → A transition pathway(s).
The TA form of the double helix, seen in the DNA bound to the TATA-box binding protein (TBP) (51), is distinguished from B-DNA dimers by zp(h), the projection, on the local helical axis, of the vector that links the phosphorus atoms on the two strands of a given basepair step (46). The dimer steps are classified here according to established guidelines (46): B DNA (zp(h) < 4.0 Å); and TA DNA (zp(h) > 4.0 Å).
A series of knowledge-based scoring functions, representative of different categories of DNA structural deformation, is obtained from the average values and covariance of step parameters in the various subsets of data and subsequently applied in the threading of different sequences on a nucleosome scaffold.
Nucleosomal sequences
The knowledge-based models of DNA deformability are tested against collections of sequences known to bind to or to resist uptake in nucleosomes. The cost of DNA deformation is evaluated for all possible positionings of each sequence on the nucleosomal template (see below). The group of binding sequences includes: i), 88 DNA segments (109–151 bp in length) from the mouse genome found in competitive reconstitution experiments to form the most stable nucleosomes (9); and ii), 41 sequences (220–234 bp in length) physically selected from a large pool of random, chemically synthesized DNA molecules (22). The set of nucleosome-resistant sequences includes 40 sequences (77–126 bp in length) from a large pool of DNA fragments from the mouse genome that are not incorporated in nucleosomes (24). The complete list of binding and nonbinding sequences is given in Table S2.
The positioning of the nucleosome on DNA is known in two of the sequences considered: i), the so-called TG-pentamer, a synthetic, 20-bp (TCGGTGTTAGAGCCTGTAAC) repeating sequence designed to have a very high affinity for histone octamers (8); and ii), the well-characterized, 164-bp pGUB nucleosome-positioning sequence (52,53).
Superhelical models
A smooth superhelical template is constructed from basepair step parameters using standard mathematical expressions (54). The DNA bends via smooth sinusoidal variation in Tilt and Roll (θ1,θ2) and deforms out of the plane through the uniform decrease of Twist (θ3) (55). By fixing the net dinucleotide bending angle at 4.46°, equating the per residue Twist to 35.575°, and ignoring the shearing of basepairs, i.e., (θ4,θ5,θ6) = 0, 0, 3.4 Å), we obtain a smooth, left-handed superhelical template of 43.3 Å radius and 32.2 Å pitch with 60 basepair steps spanning 0.75 superhelical turn. The latter parameters are obtained, with the expressions of Miyazawa (56), from the distances and angles between corresponding points along the “chain backbone”. In other words, the superhelical template build with 3DNA (46,47) from the selected set of step parameters is converted by a linear transformation from the coordinate frame embedded in the structure into the global frame of the molecule as a whole. This direct approach avoids the approximate dependence of overall superhelical structure on local conformational parameters incorporated in some models (57). As noted previously (46), the basepair pathway of nucleosomal DNA can be reconstructed exactly from the derived step parameters, e.g., 0.05 Å root mean-square deviations between the reconstructed form with planar bases versus the high-resolution structure with distorted bases. The superhelical axis of the smoothly deformed structure is then superimposed on the DNA pathway of the best-resolved nucleosome structure (2) and rotated such that the origin of the central basepair overlaps that of the basepair on the crystallographic dyad. The x-ray model is oriented in the principal-axis frame defined by the central 81 basepaired nucleotides such that the smallest variance of base and backbone coordinates lies in the direction of the superhelical axis. The latter axis coincides closely with the line, which minimizes the sum , where 〈d〉 is the average of dn, the distance from the n-th basepair center to the line, for n = 1 to 147 (40).
Threading
The deformation energy of a given DNA sequence in a specific setting on the nucleosome, i.e., the cost of threading DNA on a rigid scaffold, is computed as the sum of dimer deformation energies, using the mean sequence-dependent step parameters (obtained from the above analysis of non-nucleosomal protein-DNA structures) as the rest state 〈θi〉, of a given dimer, the step parameters of DNA on the nucleosome as the deformed state θi, and the dispersion of data in the non-nucleosomal protein-bound duplexes as the source of sequence-dependent force constants fij in Eq. 1. This structure-based approach differs from studies of nucleosome positioning, which are carried out at a literal level, e.g., extraction of nucleotide patterns in aligned sequences (22,23,29), or based on a single structural feature of DNA, e.g., sequence-dependent bending (31) or deformability (58). We use both six- and three-parameter scoring functions, based respectively on the observed distributions of all six basepair step parameters or the three primary rigid-body variables (Roll, Twist, Slide). As noted above, the smooth model adopts only the latter states.
The ability of each step in the DNA to conform to the structure of the nucleosome is scored for all possible settings of the selected sequence on the nucleosome. In view of the variable range of chain lengths in the aforementioned library of DNA sequences, we take the central 61 basepairs (i.e., 60 bp steps) of the nucleosome as the structural template in most cases and consider longer structural templates in selected cases. Possible gaps in the bound DNA (i.e., small bubbles of unbound duplex that may loop away from the surface of the nucleosome (59)) are ignored. A continuous stretch of sequence is overlaid on a continuous stretch of structure, and consecutive alignments are obtained by moving by a window of one basepair step along the sequence. Thus, each alignment corresponds to overlaying a dinucleotide in the DNA sequence over a reference point θi (i = 1…6) at basepair step n on the nucleosome core-particle structure, with the next dimer in the sequence fitted to the subsequent point at dinucleotide step n + 1 on the structure. The positioning score U is defined as the sum of the dimer energies of all basepair steps constrained to the nucleosomal template. The scores are based, in most cases, on the 61-bp template running between basepairs −30 to +30 (superhelical positions −3 to +3) of the best-resolved nucleosome core-particle structure (2). Individual sequences are assigned an average positioning score 〈U〉, computed over all possible nucleosomal alignments and an optimal and worst score, U0 and U†, corresponding to the most and least favorable settings of the sequence on the structural template. The minimum energy E0 at each dimer step is set to zero throughout, and a series of scoring functions is considered. DNA template parameters are extracted from various crystal structures using 3DNA (46,47) or taken from the idealized model described above. These two structural extremes are thought to bound the types of pathways that DNA might assume as nucleotide or histone sequence is altered.
The DNAThreader system implements the nucleosome-threading algorithm. The system consists of a program written in C, which carries out the threading calculations, and supporting scripts in a Linux environment. The program supports both six- and three-parameter scoring functions and can, if desired, ignore specific dimer steps based on parametric criteria. The program is available on request.
Results and Discussion
Knowledge-based force fields
Conformational families
The scatter plot and representative structural examples (60–63) in Fig. 1 show the wide variety of conformational states in the protein-bound DNA structures used to generate sequence-dependent dimeric elastic functions. As shown elsewhere (49), the coordinates of backbone phosphorus atoms with respect to the coordinate frame of neighboring basepairs, zp(h) and zp, divide the arrangements of basepair steps into distinct conformational families. The value of zp measures the out-of-plane displacement of the phosphorus atoms with respect to the local dimeric coordinate frame and that of zp(h) the displacement of the cross-strand P···P vector along the helical axis of the basepair step (46). The sample includes 60 dimer steps (20 RR, 25 RY, 15 YR, where R designates purine and Y pyrimidine) with A-like (zp > 1.5 Å) characteristics, 254 dimer steps (98 RR, 71 RY, 85 YR) in the AB transition region (0.5 Å ≤ zp ≤ 1.5 Å) and 1524 steps (540 RR, 511 RY, 473 YR) with B-like character (zp ≤ 0.5 Å).
Figure 1.
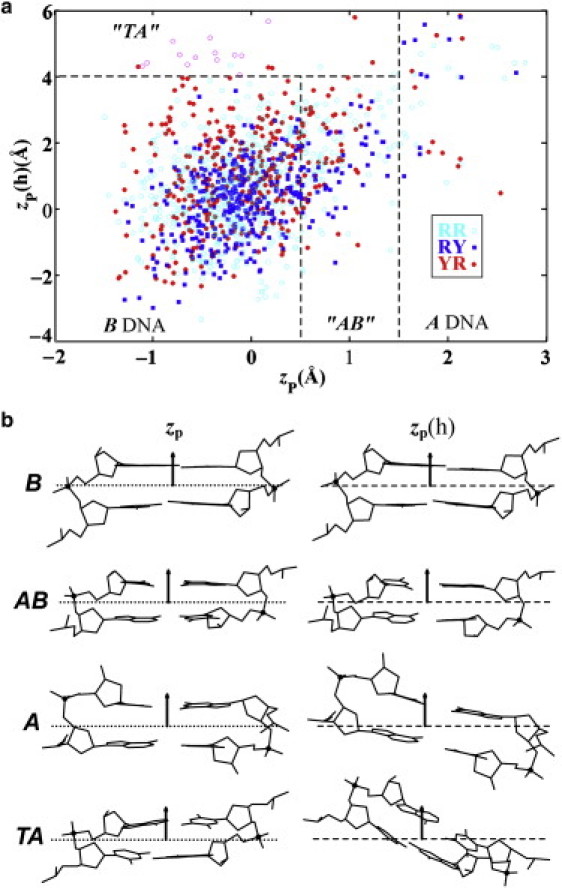
(a) Scatter plot of the coordinates of backbone phosphorus atoms, zp(h) versus zp, used to divide the arrangements of basepair steps in high-resolution protein-DNA complexes into conformational families: B DNA (zp < 0.5 Å, zp(h) < 4.0 Å); AB DNA (0.5 ≤ zp < 1.5 Å, zp(h) < 4.0); TA DNA (zp ≤ 1.5 Å, zp(h) > 4.0 Å); A DNA (zp ≥ 1.5 Å). Whereas zp gives the out-of-plane displacement of the phosphorus atoms with respect to the local dimeric coordinate frame, zp(h) measures the displacement of the cross-strand P···P vector along the helical axis of the basepair step (46); see images in (b). Occurrences of individual dimers are color-coded by sequence type: purine-purine RR, open cyan circles; purine-pyrimidine RY, solid blue squares; pyrimidine-purine YR, solid red circles. Highly deformed basepair steps from complexes with TATA-box binding proteins, which are omitted by the culling of step parameters but included in some calculations, are denoted by open magenta circles. (b) Illustrative examples of the relative positions of phosphorus atoms (black balls) in different DNA conformational states, i.e., zp value with respect to the middle dimeric frame and zp(h) value with respect to the middle helical plane of basepair steps: B-DNA state of CG·CG from the Cre recombinase-DNA structure (NDB_ID pd0003) (61); AB-DNA state of AC·GT from the ternary complex of DNA with the large fragment of Thermus aquaticus DNA polymerase I (NDB_ID pd0032) (62); A-DNA state of AC·GT from the complex of DNA with the D34G mutant of PvuII endonuclease (NDB_ID pd0006) (60); TA-DNA state of CA·TG from the ternary complex of DNA bound to the homeodomain repressor protein MATα2 and the MADS-box transcription factor MCM1 (NDB_ID pdr036) (63). Left: middle frames (dotted lines) of dimer steps determined by the coordinate axes on consecutive basepairs. Right: planes (dashed lines) normal to the local helical axes of the same basepair steps. Arrows denote positive directions of the z-axes in the two frames.
So-called TA-DNA dimers (51), resembling the highly bent and untwisted basepair steps found in the DNA complexed to TBP, are automatically culled in the selection of intrinsic step parameters. The Roll, Twist, and Slide of such steps typically fall outside the 3σ limit used to obtain quasi-Gaussian distributions of parameters (see Methods). These highly distorted basepair steps have, nevertheless, been combined with subsets of the protein-DNA dimers for the purpose of generating knowledge-based DNA potentials with enhanced deformability.
Dimeric properties
General features of the knowledge-based potentials are reported in Table 1. The rest states of the basepair step parameters—Tilt, Roll, Twist, Shift, Slide, Rise, i.e., 〈θi〉, (i = 1…6)—and the deformability V of a generic dimer step are compared with the values reported a decade ago (11) (see Fig. 2 for images of key parameters and Table S3 for a comparison of the rest states and deformabilties of individual dimer steps). The data are labeled in terms of the conformations of dimers that make up the different data sets: i), A + B + AB, the quasi-Gaussian distributions of parameters obtained in the culling of original data (2862 steps for all 16 dimers); ii), B + AB, the preceding culled data minus 92 A-like dimer steps; iii), B, 2374 steps with phosphate-group positioning typical of B DNA, iv); B + AB + TA, the aforementioned mixture of AB conformational intermediates and B-like dimers with 22 added steps of TA-like DNA; and v), B + TA, a composite of B-like and TA-DNA dimer steps. (The smaller number of points plotted in Fig. 1 compared to these numbers reflects the double counting of complementary steps (e.g., AA and TT) required for the determination of generic parameters.)
Table 1.
Rest states and local conformational deformabilities of generic DNA basepair steps in knowledge-based functions based on different types of observed protein-bound DNA conformations
| Olson et al. (11) | A + B + AB | B + AB | B | B + AB + TA | B + TA | |
|---|---|---|---|---|---|---|
| N | 1840 | 2862 | 2770 | 2374 | 2798 | 2402 |
| Tilt 〈θ1〉 | 0.0(3.6) | 0.0(3.1) | 0.0(3.1) | 0.0(3.1) | 0.0(3.1) | 0.0(3.1) |
| Roll 〈θ2〉 | 2.7(5.2) | 2.9(4.9) | 2.9(4.9) | 3.0(4.7) | 3.0(5.0) | 3.1(4.8) |
| Twist 〈θ3〉 | 34.2(5.5) | 33.8(4.9) | 34.0(4.9) | 33.8(4.8) | 33.8(5.0) | 33.6(4.9) |
| Shift 〈θ4〉 | 0.00(0.64) | 0.00(0.61) | 0.00(0.61) | 0.00(0.62) | 0.00(0.61) | 0.00(0.62) |
| Slide 〈θ5〉 | −0.09(0.69) | −0.21(0.67) | −0.17(0.64) | −0.07(0.63) | −0.16(0.65) | −0.07(0.64) |
| Rise 〈θ6〉 | 3.36(0.25) | 3.34(0.23) | 3.33(0.22) | 3.31(0.21) | 3.33(0.22) | 3.31(0.21) |
| V (Å−°)3 | 9.2 | 5.2 | 4.8 | 4.2 | 5.1 | 4.5 |
Average parameters and standard deviations (subscripted values in parentheses) for a generic DNA dimer derived from basepair steps of specified conformational type in high-resolution structures of protein-DNA complexes (Fig. 1). Generic dimers based, as in Olson et al. (11), on equal weighting of average parameters of the 16 common dimers. Dimer types include canonical A and B helical forms, intermediate AB states, and extreme TA arrangements, classified in terms of the relative positioning of the bases and phosphates (46). Number of basepair steps of a given type denoted by N. Deformabilities V correspond to volumes of conformation space within a common energy contour, given for each data set by the product of the eigenvalues of the covariance matrix of average and mean-square step parameters. Tilt, Roll, Twist expressed in degrees and Shift, Slide, Rise in ångstrom units. See text for further details.
As evident from Table 1, the rest states of the new potentials are slightly altered, with more A-like character, compared to the mean step parameters deduced originally with substantially less and poorer resolution data (1860 examples vs. 2862 steps currently from the nonredundant set of better resolved structures described above), i.e., average Roll 〈θ2〉 is now 0.2–-0.4° more positive, average Twist 〈θ3〉 0.2–0.6° lower, average Slide 〈θ5〉 as much as 0.12 Å more negative, and average Rise 〈θ6〉 0.02–0.05 Å smaller. The differences in rest states, however, are not as striking as the changes in flexibility/entropy measured by V. Both the standard deviation of individual step parameters and the dimeric entropies are smaller than the values estimated from the data available in 1998 and collected, of necessity, without regard to resolution or structural duplication. The ranking of dimeric deformabilities also differs from that reported originally, e.g., CG, formerly the most deformable pyrimidine-purine step, now seems to be stiffer than TA and CA·TG dimers and AA·TT dimers are now the stiffest of all basepair steps (see Table S3). Three-parameter deformability profiles, in which the variation of secondary (Tilt, Shift, Rise) conformational variables is ignored, follow the same trends as the six-parameter data shown here, i.e., the B-DNA data set is most restricted in terms of V and the data sets containing the originally culled data or added TA steps in place of A-like steps (A + B + AB and B + AB + TA, respectively) are the most deformable.
Nucleosomal template
The conformational features of the nucleosomal template adopted for DNA threading follow trends seen in other well-resolved protein-DNA structures (Fig. 2). The three primary basepair step parameters (Roll, Twist, Slide) of nucleosomal DNA, here plotted as a function of superhelical position, i.e., the number of complete turns of double helix away from the pseudo-symmetrically positioned central basepair (1), span ranges comparable to those found in the complexes used to generate knowledge-based potentials (histograms on the right edge of the figure). The distribution of Slide, however, tends to be more positive in the nucleosome than in other protein-DNA complexes, not only at the highly skewed steps at either end of the nucleosome but also over the central 60 steps (61 bp) in contact with the (H3·H4)2 tetramer (highlighted by heavy lines). As noted elsewhere (11,40) and evident from the set of scatter plots in Fig. S1, almost all known examples of extreme positive Slide (>1 Å) in other protein-DNA structures entail CA·TG and TA dimer steps.
The extremes of Roll, Twist, and Slide seen in nucleosomal DNA are reminiscent of the values of the step parameters found in the A and C helical forms of DNA (38,46). Whereas A-DNA is untwisted (∼11 bp per helical turn or 32.7° helical twist) with basepairs globally inclined (via positive Roll) and displaced (via negative Slide) with respect to the helical axis (64), C DNA is overtwisted (∼9 bp per turn or ∼40° helical twist) with basepairs inclined and displaced in the opposite sense (via negative Roll and positive Slide) (65,66). Basepair steps with C-like characteristics in nucleosomal DNA occur at positions (half-integral values of superhelical position in Fig. 2) where the minor-groove edges of basepairs face the histone proteins, and dimers with partial A-like characteristics at the steps (integral values of superhelical position) where the major-groove edges are directed toward the protein core (38).
Dimeric deformations
The cost of threading each of the 16 common basepair steps on the DNA dimers wrapped on the surface of the nucleosome core particle is reported in Fig. 3. The scores are based on the structure of DNA in the currently best-resolved nucleosome structure (NDB_ID pd0287) (2), and the potentials are derived from the conformational variables of basepair steps in other high-resolution protein-DNA complexes, here the A + B + AB data set in Table 1. That is, the nucleosome structure serves as the template of step parameters θi at the selected basepair steps and the protein-bound dimers in other protein-DNA structures define the sequence-dependent rest states 〈θi〉 and force constants fij used in the evaluation of the deformation energy (Eq. 1). Sites on the nucleosome lattice are labeled, as above, in terms of superhelical position. The energy score is color-coded such that the color varies from blue to white to red as the computed score increases in value. Data are presented for the central 60 basepair steps (superhelical positions 0 ± 3) using both six- and three-parameter scoring functions (Fig. 3, a and b).
Figure 3.
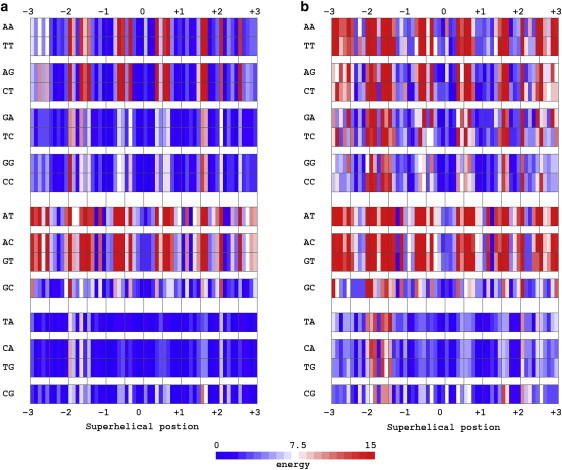
Color-coded image of the cost of threading each of the 16 basepair steps on the positions adopted by the central 61 basepairs (60 bp steps) of the DNA on the 147-bp nucleosome core-particle structure (NDB_ID pd0287) (2). Scores are based on dimer steps from all conformational categories, i.e., the A + B + AB data set in Table 1. Sites on the nucleosome lattice are labeled in terms of superhelical position. Dimeric energies E are color-coded such that the color varies from blue to white and red as the computed scores increase from low to high values. Data are reported for (a) six- and (b) three-parameter scoring functions. See legend to Fig. 2 and text.
As is clear from these images, the cost of DNA deformation is lowest for pyrimidine-purine steps regardless of nucleotide composition and location on the template. The highest (red) deformational barriers occur at C-like basepair steps where Roll and Slide exhibit the large, concerted conformational changes known to accompany the narrowing of the minor-groove and recently shown to control the curvature and pitch of nucleosomal DNA (40). The cost of these deformations is especially large for certain purine-pyrimidine and purine-purine steps in which adenine occurs at the 5′-end of the sequence strand (i.e., AT, AC, AA, and AG dimers). The scores are generally lower for dimers with guanine in the corresponding position (i.e., GC, GA, and GG steps). Some of the latter sequences are more easily deformed, albeit slightly, than the pyrimidine-purine dimers at a few nucleosomal sites (e.g., GC at superhelical position 1.4 in Fig. 3 a). The DNA deformation scores of pyrimidine-purine steps are always among the very lowest (42 of 60 basepair steps on the assumed nucleosomal template) and those of purine-pyrimidines among the very highest (46 of 60 steps). As noted elsewhere (40), the cost of nucleosomal DNA distortions is also substantially lower for pyrimidine-purines at the six highly skewed steps at either end of the core-particle structure (superhelical positions ±5.7, ±4.7, ±3.5).
The slightly different scores of complementary sequences (e.g., rows labeled AA versus TT; Fig. 3 a) reflect the opposing signs of Tilt and Shift assigned to the bases of complementary strands (50) and the effects of these definitions on the rest states and force constants of the potentials. The deviations of Tilt or Shift from the rest state and the resultant energy contributions thus differ for the two settings of a dimer at a specific nucleosomal step, i.e., whether AA or TT occurs in the sequence strand. The differences between complementary sequences, however, disappear when the functions are based on the three primary, strand-independent parameters (Fig. 3 b). The sequence-dependent distinctions in the ease of DNA folding are particularly sharp in the latter set of images. The relatively minor contributions of Tilt, Shift, and Rise to the DNA deformation scores are clear from the similar spread of colors (i.e., similar range of scores) in the two images. The distortions in Tilt and/or Shift at a few dimer steps, however, contribute to some conformational barriers, e.g., Tilt (θ1 ≈ 10°) and Shift (θ4 = −1.25 Å) at superhelical position 2.5 give rise to the higher score (red versus blue bars) in the six- versus three-parameter surfaces of purine-purine steps.
These deformational patterns persist with different knowledge-based functions. As shown in Table 2, not only is the mean cost 〈E〉 of deforming a generic dimer over the central 60 basepair steps of the nucleosome core particle relatively insensitive to the choice of scoring function, but also the scores E0 and E† and the respective superhelical locations SH0 and SH† of the least and most costly deformations. As expected from the lower conformational entropies V of the input data sets (Table 1), the average cost of deforming a generic basepair step is higher with the elastic functions based on the new set of well-resolved protein-DNA structures than with the potentials developed in 1998. The scores are slightly increased when selected subsets of structural data are used in the construction of scoring functions. The highest-scoring site found with these functions is displaced from that found with the older elastic potential. (See Table S4 for the corresponding scores and preferred/disfavored threading sites for each of the 16 dimer steps).
Table 2.
Comparative cost of nucleosomal deformation of generic DNA basepair steps with knowledge-based functions based on different subsets of observed protein-bound DNA conformations
| Score | Olson et al. (11) | A+B+AB | B+AB | B | B+AB+TA | B+TA |
|---|---|---|---|---|---|---|
| 〈E〉 | 7.8 | 9.1 | 9.3 | 9.2 | 9.2 | 9.1 |
| E0 | 0.9 | 0.9 | 0.9 | 1.0 | 0.9 | 0.9 |
| E | 24.3 | 25.2 | 26.3 | 27.6 | 26.5 | 27.7 |
| SH0 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 |
| SH† | −2.0 | −1.5 | −1.5 | −1.5 | −1.5 | −1.5 |
Knowledge-based scores (average cost 〈E〉 over the central 60 basepair steps of the best-resolved nucleosome core particle structure (2) and the least and most costly values, E0 and E) derived from the basepair step parameters of DNA dimers of different conformational types in high-resolution protein-DNA structures. Locations SH0 and SH† of the least and most costly steps are expressed in terms of superhelical position, i.e., number of helical turns with respect to the structural dyad. See text and legend to Table 1.
Threading scores
The total threading scores U in Fig. 4 correlate with selected properties of known nucleosome-binding sequences. For example, the scores of high-affinity nucleosome-binding fragments (Fig. 4, right end) are lower on average than those of known binding sequences from the mouse genome (Fig. 4, left half), whether the mean value 〈U〉 for all possible settings of a given sequence on the structural template (filled-in points connected by solid lines; Fig. 4) or the optimum score U0 for the best setting of each sequence (values listed in Table S5) is considered. (Because the knowledge-based functions are not true potential energies with values scaled to kT, we report the mean values and standard deviations (shaded corridors around the plotted data) for each sequence rather than a Boltzmann-weighted averages.) The scores of sequences known to be refractory to nucleosome formation (points labeled anti-selection), however, are lower than those of most of the naturally occurring binding sequences. Furthermore, although the scoring function distinguishes the 14 anti-selection sequences with TGGA fragments from the 26 poor binding (Bad) sequences with no obvious literal features (24), it assigns a lower score to the TGGA sequences, despite their lesser affinity for the histone-octamer core. Small discrepancies of the same sort, reported on a different energy scale, occur in the nucleosome formation energies predicted by Anselmi et al. (32) and Scipioni et al. (33). On the other hand, the difference U0 − 〈U〉 tends to be lower for binding sequences than for nonbinding sequences (−93 ± 16 vs. −76 ± 16 on average) and even more negative for the strongly associated high-affinity fragments (−102 ± 18), suggesting that the preferences for association may reflect particularly favorable settings of the sequences on the nucleosome and/or the absolute deformation score. The values of U† − 〈U〉 characterizing the least likely settings of the high-affinity fragments (113 ± 21) also exceed those of the nucleosome-binding and nonbinding sequences (104 ± 20 and 81 ± 34) (see Table S5).
Figure 4.
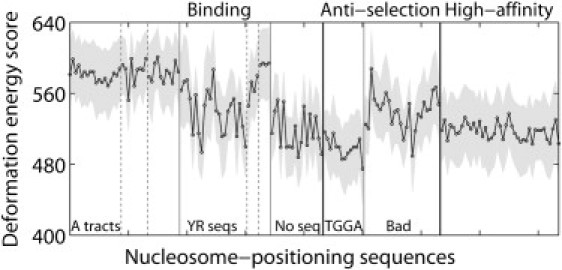
Cost of threading 88 known binding sequences from the mouse genome (9), 40 sequences refractory to nucleosome formation (anti-selection) (24), and 41 high-affinity nucleosome-binding fragments (22) on the central 60 basepair steps of the DNA in the 147-bp nucleosome core-particle structure (NDB_ID pd0287) (2). Sequences are arranged in the order reported in Table S2 and grouped according to literature descriptors (9,22,24). Thin vertical lines highlight sequences of similar types. Total threading scores—mean values 〈U〉 (points connected by dashed lines) and standard deviations σU (values equal to half the width of the gray corridors containing the points) for all possible settings of each sequence on the structural template—based on six-parameter elastic functions for dimer steps from all conformational categories, i.e., the A + B + AB data set in Table 1. See Table S5 for the scores of individual fragments.
As anticipated from the relative cost of dimeric deformations on the nucleosome template (Fig. 3), the scoring function assigns higher values to nucleosome-binding sequences that contain A-tracts and lower values to sequences with regularly phased pyrimidine-purine steps. One of the three sets of A-tracts, however, resembles the anti-selection sequences in terms of the small magnitude of U0 − 〈U〉. The stiff AT dimers in the repeating TATA tetrads contribute to the high threading scores of these four (YR) fragments, which, nevertheless, form the most stable nucleosomes among the mouse sequences (9).
Smooth nucleosomal template
The discrepancies between the DNA threading scores in Fig. 4 and the nucleosome-binding properties of a number of well-characterized sequences (9,24) suggest possible limitations in the knowledge-based potentials and/or errors in the assumption that the core of histone proteins imposes exactly the same conformational constraints on DNA regardless of basepair sequence. As noted above, although there are numerous examples in other high-resolution protein-bound structures of the extreme distortions of DNA seen in the nucleosome core-particle structure (i.e., sharp bending into the minor groove in concert with the shearing and overtwisting of basepairs), the only basepair steps found to exhibit such deformations are CA·TG and TA (11,40). The perturbations of the double helix observed in high-resolution structures of DNA containing A-tracts are much less pronounced than those at the aforementioned (pyrimidine-purine) steps, i.e., the largest distortions in both Roll and Slide are about an order of magnitude smaller in AA·TT steps (38). Furthermore, there is reason to expect that other nucleosome-binding DNA sequences might assume a different structure from the palindromic fragment (67) engineered from the α-satellite DNA in the human X-chromosome (68) that is incorporated in most currently solved nucleosome core-particle structures (1,2,14–19). For example, the substitution of highly kinked pyrimidine-purine steps in contact with the catabolite activator protein (CAP) by purine-purine steps smoothes the duplex locally while preserving the superhelical wrapping of DNA on the surface of the protein (69). The kinking of pyrimidine-purine steps in the CAP-DNA complex, however, is of a different type from the sharp bending of DNA in the nucleosome, i.e., the most severely distorted dimers in the CAP-DNA complex bend into the major groove with concomitant undertwisting and shearing of the opposite sense (via negative Slide).
We constructed an ideal, smoothly deformed scaffold (Fig. 5 a) that roughly mimics the global folding of nucleosomal DNA for further investigation of the threading properties of known histone-octamer binding sequences. Although the global configuration of the hypothetical template is similar to the overall folding of the crystalline nucleosome, the assumed dinucleotide structure is appreciably different from the high-resolution model, e.g., fixed 3.4 Å per residue spacing versus observed center-to-center distances of 3.53 ± 0.50 Å, displacements directed exclusively along basepair normals without consideration of the periodic changes in Slide that contribute to the pitch of nucleosomal DNA (40), uniform bending of ∼4.5° per basepair step versus observed net bending angles of 7.9 ± 4.2°, constant dimeric twisting of ∼35.6° versus experimental values of 34.8 ± 5.3°. The deviation in global structure is especially pronounced near superhelical locations ±3, corresponding to the shortened radius of the real superhelical trajectory at the interfaces of the H2A·H2B dimers and the (H3·H4)2 tetramer (Fig. 5 b).
Figure 5.
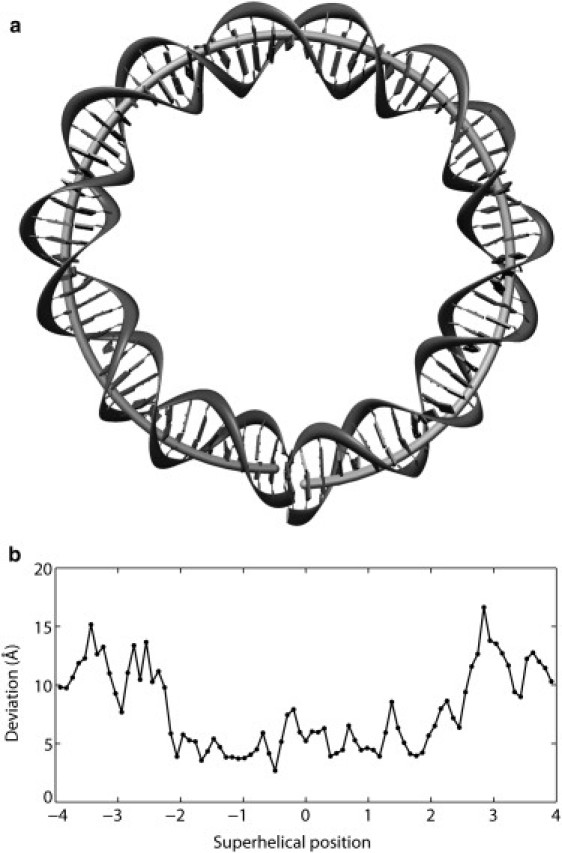
(a) Basepair centers of an ideal, smoothly deformed, left-handed superhelical scaffold of 43.3 Å radius and 32.2 Å pitch (lightly shaded tube) superimposed on the central 80 bp of the DNA in the currently best-resolved nucleosome core-particle structure (gray block/ribbon representation of the observed pathway of bases/phosphates) (2). The idealized DNA bends smoothly via sinusoidal-like variation in Tilt and Roll and deforms out of the plane through the uniform decrease of Twist, differing from the periodic changes in Roll and Slide that contribute to the curvature and pitch of nucleosomal DNA (40). (b) Root mean-square deviation of the base and backbone atoms at each of the central 80 basepair steps of the smooth superhelical model compared to the crystallographically observed positions (2).
The relatively low scores associated with threading known nucleosome-binding sequences on the ideal scaffold (Fig. 6) stem from the limited deformations of DNA imposed by the superhelical pathway. The step parameters of the idealized template, particularly Roll and Slide, are not as far from the equilibrium rest states as those of the crystal structure, e.g., values of Slide (θ5) are null and Roll (θ2) lies between ±4.46° in the smooth superhelix but span broad ranges, −15.6° ≤ θ2 ≤ 21.6° and −1.06 Å ≤ θ5 ≤ 2.32 Å, over the central 60 basepair steps of the core-particle structure. Roughly two-thirds of the difference in threading scores in Fig. 4 versus Fig. 6 reflect the assumed values of Roll and Slide.
Figure 6.
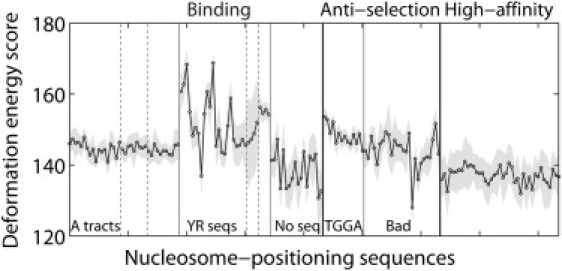
Threading scores of known nucleosome-binding sequences (9,22,24) on the ideal, smooth, 61-bp superhelical template shown in Fig. 5a with global features resembling those of the central 80 basepair steps of the nucleosome core-particle structure but distinctly different modes of local dimeric deformation (see legends to Figs. 4 and 5 and text). Lower scores, compared to Fig. 4, reflect the smaller deformations of step parameters from their equilibrium rest states on the idealized template versus the crystal structure.
Interestingly, the smooth superhelical template preserves the low scores of the highest affinity nucleosome-binding sequences (22) while concomitantly increasing the cost of threading sequences that are known to be refractory to nucleosome formation (24) and bringing the scores of nucleosome-binding sequences that contain A-tracts (9) more in line with the values of sequences with regularly phased pyrimidine-purine steps. The scores of TGGA antiselection sequences are thus appropriately higher than those of poor binding (Bad) sequences with no obvious literal features. The smooth template, however, suppresses the large differences in U0 − 〈U〉 that favor particular settings of the synthetic high-affinity sequences and fragments from the mouse genome on the crystalline template. In addition, the idealized basepair pathway, although of lower deformation energy, does not necessarily preserve the known, potentially stabilizing contacts of the histone proteins with the sugar-phosphate backbone (1,2) that may contribute to the observed structural pathway.
Alternate crystalline templates
To date there are two crystallographic examples of nucleosomes that incorporate sequences substantially different from the human α-satellite DNA found in the best-resolved structures (2): i), a 2.6-Å structure with a different 146-bp human α-satellite repeating sequence (NDB_ID pd0286) (2); and ii), a 3.2-Å structure with a 147-bp sequence containing a 16-bp poly(dA·dT) element (NDB_ID pd0755) (70). Although the basepair content in the former structure is nearly identical to that in the highest resolution (2.0 Å) nucleosome structure containing a DNA of the same chain length (NDB_ID pd0285) (2), i.e., 88 A·T and 58 G·C pairs in pd0285 versus 86 A·T and 60 G·C pairs in pd0286, the sequence differs at nearly half the sites (69 of 146 bp). The shuffling of basepairs, in turn, perturbs the local DNA structure. Although the DNA exhibits a similar pattern of conformational deformation in the two structures, the most pronounced distortions occur at different sequential locations. In other words, whereas the mean step parameters are very similar, e.g., average Slide 〈θ5〉 = 0.17 Å (pd0285) vs. 0.20 Å (pd0286), the root mean-square differences in step parameters over all steps are large, e.g., = 0.87 Å. By contrast, the pattern of DNA deformation is relatively smooth along the 147-bp DNA containing a poly(dA·dT) element, with fewer highly kinked and sheared basepair steps compared to the DNA in the best-resolved nucleosome structure, e.g., average Slide 〈θ5〉 = 0.19 Å (pd0287) vs. 0.07 Å (pd0755) and = 0.85 Å. The poor resolution of the nucleosome containing the A-tract, however, precludes detailed analysis of its DNA folding pattern.
The subtle differences in local basepair structure associated with the binding of different sequences on the nucleosome translate into appreciably different DNA deformation scores (Table 3). Regardless of the assumed length of the nucleosomal template, the score is lower if the crystallized sequence, rather than the shuffled sequence, is threaded on its natural structural template, i.e., I-I versus II-I and II-II versus I-II sequence-template combinations in Table 3. The differences are naturally more pronounced for longer templates, where conformationally stiff dimers must take up the highly skewed C-like states adopted by CA·TG and TA steps in the two crystal structures. The optimum deformation scores coincide with the observed positioning of DNA in these and in other known nucleosome structures. The scores are also consistently lower for better-resolved structures, e.g., 424 vs. 607 for the central 60 basepair steps of the 146-bp DNA in the 2.0-Å (pd0285) vs. 2.6-Å (pd0286) structures and 354 vs. 798 for the corresponding steps of the 147-bp DNA in the 1.94-Å (pd0287) vs. 3.2-Å (pd0755) structures.
Table 3.
Optimum deformation scores of human α-satellite DNA sequences threaded on different nucleosomal templates
| DNA sequence∗ | Structural template† | Template length (basepair steps) |
|||
|---|---|---|---|---|---|
| 60 | 80 | 100 | 120 | ||
| I | I | 424 | 551 | 678 | 819 |
| II | I | 552 | 812 | 989 | 1224 |
| I | II | 658 | 978 | 1206 | 1513 |
| II | II | 607 | 825 | 1033 | 1209 |
I: human α-satellite sequence incorporated in the best-resolved (2.0 Å) nucleosome core-particle structure with 146-bp DNA (NDB_ID pd0285) (2); II: different human α-satellite sequence of comparable AT-content incorporated in the 2.6 Å structure with 146-bp DNA (NDB_ID pd0286) (2).
I: nucleosomal DNA template constructed from structural fragments of variable length centered on the dyad of the 2.0 Å nucleosome structure; II: template containing corresponding fragments from the 2.6 Å structure.
Sequence settings
TG-pentamer
The extent to which the threading scores account for the measured positioning of selected sequences on the nucleosome is reported in Figs. 7 and 8. Fig. 7 a presents the scores associated with all 20 settings of the so-called TG-pentamer, a well-known nucleosome binding sequence (8), on the central 60 basepair steps of the crystalline template (pd0287). The sequence setting is described in terms of the nucleotide that is placed on the structural dyad. The nucleosomal template accommodates the regularly repeating sequence in a number of relatively low-cost settings (circles connected by solid lines). Two of these possible settings, separated by 10 bp and centered respectively on the TCG and AGC trimers in the repeating sequence, are preferentially favored if the cost of sharp bending into the minor groove (via large negative Roll) with concomitant shear (via the increase of Slide) is artificially lowered for CA·TG and TA steps, i.e., E = 0 if θ2 <0° and θ5> 0 Å (circles connected by dashed lines in Fig. 7 a). Such an approximation takes into account the many known C-like states of extreme Roll and Slide adopted by these steps in other protein-DNA complexes but not incorporated in the knowledge-based potentials. The computed scores, however, are not as low as the total deformation score of the DNA sequence that is crystallized on the nucleosome (horizontal line in Fig. 7 a computed without special treatment of CA·TG and TA energies).
Figure 7.

(a) Predicted deformation score versus positioning site of the TG-pentamer (8) on the central 61-bp template (60 basepair steps) of the crystalline nucleosome (2). Scores based on two different potentials: i), the six-parameter elastic function derived from all dimer steps that are accepted from protein-DNA complexes (points connected by straight lines); and ii), the same potential but with no penalty imposed on C-like CA·TG and TA dimers deformed via large negative Roll and positive Slide (points connected by dashed lines). The straight solid line is the deformation score of the human α-satellite DNA sequence in the setting found in the nucleosome core-particle structure (2). See text and legend to Fig. 4. (b) Observed hydroxyl-radical cutting pattern in solution (scatter points) (8) and computed minor-groove width (open diamonds connected by thin solid lines) (46,47) of the currently best-resolved nucleosome structure (2) when the TG-pentamer (8) is centered on the low-scoring TCG trimer site in a. Sites of maximum cutting denoted by filled-in circles and sites of minimum cutting by open circles.
Figure 8.

Predicted DNA deformation score versus positioning site of the pGUB nucleosome-positioning sequence on three-dimensional nucleosomal templates of different length and type. Scores reported for all possible settings of the sequence on templates of 60, 80, 100, 120 basepair steps centered on the dyad of (a) the best-resolved nucleosome core-particle structure (2), and (b) the ideal superhelical scaffold depicted in Fig. 5. Threading energies based on the six-parameter (A + B + AB) elastic function derived from dimer steps of all conformational types in other protein-DNA complexes and compared with the mean deformation scores of all settings of the DNA sequence that is crystallized on the nucleosome template (finely dotted lines).
Fig. 7 b shows the coincidence of the hydroxyl-radical cutting patterns of the TG-pentamer (8) with the minor-groove width of the nucleosomal template when the sequence is centered on one of the two lowest-scoring sites (here the TCG trimer). The points of maximum cutting coincide with the local maxima in minor-groove width, and the points of lowest cutting with the local minima. Only two settings preserve this pattern, the primary and secondary minima in Fig. 7 a. The occurrences of CA·TG or TA dimers at sites of sharp local bending into the minor groove (negative Roll) and large shearing (positive Slide) seemingly contribute to the observed nucleosomal positioning of the TG-pentamer.
pGUB
The 183-bp pGUB fragment is one of the strongest nucleosome-positioning sequences characterized to date (52,53). The observed, single-nucleotide resolution mapping of nucleosomes on this sequence is based on micrococcal nuclease cutting patterns (52) and chemical modification of nucleotides in close spatial proximity to reactive agents placed at specific sites on nearby histone proteins, e.g., site-specific photochemical cross-linking and subsequent cleavage, by heat and alkali treatment, of DNA photo adducts formed with reactive groups on cysteines introduced at specific amino acid sites in histones H2A (R45C), H2B (S53C), and H4 (S47C) (53). Fig. 8 a reports the DNA deformation scores found upon threading the pGUB sequence on crystalline nucleosomal templates of increasing length (the central 60, 80, 100, and 120 basepair steps of the best-resolved nucleosome structure). The shortest template spans the DNA in contact with the (H3·H4)2 tetramer, and each successive template incorporates two of the six highly skewed basepair steps in contact with the H2A·H2B dimers, i.e., the so-called “kink-and-slide” steps (40) with large negative Roll and positive Slide in Fig. 2. Deformation scores are reported for all possible arrangements of the sequence on the structural template, with the positioning site denoted by the number of the basepair on the dyad.
As expected, the threading scores of pGUB are higher in value and the number of possible settings is reduced on longer structural scaffolds. The scores on the 121-bp (120 basepair step) template, however, exceed those reported recently (40) for the threading of pGUB on the full 147-bp crystallographic template, the lower values reflecting the softer force field (11) used in the other calculations. As shown here, the positioning scores also change character with the increase of template length. As with the TG-pentamer, the shortest template accommodates many low-scoring settings, two of which lie within 1 bp of the experimentally characterized nucleosome positions (at basepairs 84 and 104). As the template is lengthened, these settings becomes more important than other sites, with appreciably lower scores as the template includes more of the six kink-and-slide steps. The two settings, however, are less favorable than a setting centered at basepair 117, a positioning site too close to the end of the pGUB sequence to allow formation of a complete 147-bp nucleosome. Although the latter setting better accommodates the kink-and-slide distortions of DNA induced by H2A·H2B, it deforms less easily than the two competing settings on the 61-bp (H3·H4)2-contacted template.
The deformation scores of pGUB are not as low as the optimum scores of the α-satellite DNA sequence that is crystallized on the nucleosome (with threading scores ∼70% those of the pGUB sequence). The latter values are obtained when the crystallized sequence is in perfect register with the nucleosomal template, i.e., the central basepair lies on the structural dyad. As noted previously (40), intrinsically flexible TG or CA dimers take up most of the extreme distortions of DNA in the crystal structure. The computationally predicted settings of pGUB contain few such dimers at these sites, the limited examples contributing to the higher deformation scores. The scores of the preferred pGUB positioning sites, however, are ∼20% lower than the mean deformation scores of the α-satellite DNA threaded over all possible settings on the corresponding crystalline templates (finely dotted lines in Fig. 8).
Threading of pGUB on the smooth superhelical template fails to account for the two known nucleosome positions. These settings, which are predicted with 1–2-bp accuracy, do not stand out from many other low-scoring sites that place the sequence in an equivalent rotational setting, i.e., with the same basepairs translated with respect to the dyad but bent in exactly the same way on the smooth template. Thus, DNA threading on the ideal structure yields a broad sinusoidal profile, with a 9–11 bp period and no deep energy valleys to lock the nucleosome in place (Fig. 8 b). This shortcoming in the smooth template confirms the importance of the kink-and-slide steps found in the crystal structure in distinguishing the preferred translational settings of the pGUB sequence on the nucleosome.
Conclusion
How eukaryotic genomes bias the wrapping of DNA into nucleosomes is of paramount importance to both the packaging and the biological processing of the genetic message. Given the virtual absence of direct contacts of protein with the DNA bases in known high-resolution nucleosome structures, the positioning of nucleosomes on specific sequences reflects the capability of the double-helical molecule to adopt the tightly wrapped, superhelical fold dictated by its association with the histone proteins. This work lends support to this idea in showing i), that DNA exhibits the same intrinsic deformational patterns on the nucleosome as in other high-resolution protein-DNA complexes; and ii), that the cost of deformation, based on knowledge-based elastic potentials that take account of the known sequence-dependent dimeric distortions of double-helical structure, approximates the nucleosome-binding properties of a number of experimentally characterized DNA sequences. The cost of deformation reflects both the deviation of structural parameters at each basepair step of nucleosome-bound DNA from the intrinsic (average) values found in other structural contexts and the sequence-dependent deformability determined by the spread of the non-nucleosomal reference states. The variables in these calculations include the dimer steps used to construct the scoring functions and the three-dimensional structural templates on which the sequences are threaded. Comparison of the deformation scores with experiment points to a subtle interplay of sequence and structure in nucleosomal DNA and the potential importance of long-range interactions in accounting for available experimental data. Different sequences appear to take slightly different paths around the nucleosome, with dimer steps that resist significant deformation seemingly smoothing the fold of the bound duplex.
Scoring functions
The updated set of DNA elastic functions confirms many of the sequence-dependent features extracted in our original set of knowledge-based potentials (11), e.g., pyrimidine-purine (YR) dimers stand out as the most easily deformed basepair steps. With a larger, better-resolved, and more carefully selected set of protein-DNA complexes we can rank the ease of YR deformation, i.e., TA > CA·TG > CG, and identify the dimers apparently most resistant to conformational distortion, namely AX steps where X = A, T, C, G (Fig. 3 a and Table S3). The intrinsic stiffness of the latter steps, particularly the AA dimers, confirms long-held notions (71) of the molecular features that impede the reconstitution of poly dA·poly dT-containing nucleosomes (72–75) and increase the persistence length of DNA (76). In contrast to the hinge-like behavior of YR dimers, the AX steps resist the shearing motions (via Slide and Shift) that accompany large-scale transitions of B-DNA double-helical structure (49) and/or contribute to the tight packing of DNA against protein (2,3). As we have pointed out recently (40), the role of A-tracts in nucleosome positioning may be to bring the DNA sequence in register with the histone octamer, allowing the duplex to kink and dislocate at specific, deformable steps.
These sequence-dependent conformational trends persist in different sets of knowledge-based potentials with different types of structural features, e.g., functions based on configurational states characteristic of room-temperature fluctuations of the B-DNA double helix versus functions that include dimers deformed to other (A, AB, TA) conformational states (Fig. 1, Tables 1 and 2), and in simplified (three-parameter) treatments that ignore the variation of secondary (Tilt, Shift, Rise) parameters (Fig. 3 b). The cost of elastic deformation naturally reflects the selected reference states, i.e., force constants and deformation scores are higher if only B-DNA conformers are considered or if the contributions of all six basepair step parameters are counted.
The dimeric model implicitly incorporates the effects of the sugar-phosphate backbone, including the intervening phosphate groups and immediate chemical environment, but ignores the influence on conformational freedom of flanking nucleotides, e.g., tetrameric sequence content, and longer-range interactions, such as patches of phosphate neutralization on one face of the double helix (4). There are now enough high-resolution protein-DNA structures to provide reliable estimates of the mean dimeric step parameters in all tetrameric contexts (77) but not yet enough data to estimate the deformability of such fragments. Understanding the bending of DNA and the changes in groove widths associated with the neutralization of phosphates requires representative examples of even longer (pentameric and hexameric) structural fragments, e.g., the major- and minor-groove widths are typically defined by the distances between phosphorus atoms in different strands separated by 3–4 basepairs. The incorporation of such information might improve the correspondence between computation and observation. On the other hand, a dimeric representation of DNA is extremely simple to understand and implement in polymeric models (78–80). The effects of local sequence-dependent deformations on global structure and properties are shown immediately from such treatments.
The knowledge-based dimeric potentials, nevertheless, capture other key sequence-dependent features of DNA. For example, the lesser bending of AA steps compared to other dimers in combination with the high positive Roll of CA steps found in protein-bound DNA complexes (Table S3) accounts for both the magnitude and direction of curvature found in phased A-tracts such as the (A6C5A6C4)n repeat (38). The computed end-to-end distributions of flexible chains subject to the knowledge-based potentials (45) also match the cyclization propensities of various short DNAs (81,82). Furthermore, mixed-sequence homopolymers guided by the potentials have persistence lengths more closely resembling those known to characterize polymeric DNA than chains that are subject to the deformations associated with the more restrictive B-like data set (11,45). Thus, the wide variety of configurational states induced by the binding of many proteins appear, from this perspective, to be necessary to account for the solution properties of B DNA.
Nucleosomal templates
The rough correspondence of the nucleosome-binding properties and deformation scores of a number of DNA sequences suggests that these molecules wrap on the surface of the nucleosome much like the DNA in the best-resolved core-particle structure (2) (Fig. 2). That is, high-affinity, synthetic nucleosome-binding fragments score lower on the crystallographically observed scaffold than less tightly bound sequences from the mouse genome (Fig. 4). The high-affinity sequences contain periodically positioned TA dimers that deform at relatively low cost on the kinked structural scaffolds found in high-resolution structures. The large differences between the mean and optimized scores of known binding sequences compared to those of the nonbinding sequences further indicate that the preferences for association may reflect particularly favorable settings of the binding sequences on the nucleosome scaffold. Moreover, two well-resolved nucleosome-positioning sequences, the TG-pentamer (8) and the pGUB (52,53) fragment, show favorable threading scores at the experimentally mapped positions, particularly if the cost of sharp bending into the minor groove (via large negative Roll) and concomitant shear (via the increase of Slide) is lowered for CA·TG and TA steps (Figs. 7 and 8). Correct prediction of the observed positioning of nucleosomes on pGUB requires a template with kink-and-slide steps like those found along the DNA pathway in high-resolution nucleosome structures.
Several lines of evidence, nevertheless, suggest that certain DNA sequences may perturb nucleosomal structure. First of all, the substitution of the TG·CA step found at the sharply kinked recognition site of the catabolic activator protein by a GG·CC or AG·TC dimer smoothes the bending of the bound duplex (69). As noted above and seen in Table S3, the AG·TC step strongly resists deformation of B-DNA structure in other structural contexts. Second, we see that the introduction of an ideal, smooth superhelical scaffold lowers the threading scores of A-tract sequences and disfavors sequences that are refractory to nucleosome binding, reversing some incorrect predictions made with the kinked crystalline template (Fig. 6). The smooth template minimizes the distortions of double-helical structure and takes advantage of the tendency of A-tracts to preserve B-DNA geometry. The idealized pathway (Fig. 5) ignores the contacts of DNA to protein, which persist in all known nucleosome structures (including those with modified histones) and which may contribute to the relatively jagged superhelical pathway deduced in the low-resolution structure of a nucleosome core particle containing a poly dA·poly dT sequence element (70). On the other hand, a smooth pathway does not distinguish the sites of well-positioned nucleosomes, e.g., pGUB (Fig. 8 b). Finally, the energetically costly DNA kinks found in at least two well-resolved nucleosome structures move to neighboring nucleosomal positions to accommodate the shuffling of deformable basepair steps. The threading score of a crystalline sequence on its natural scaffold is thus lower than that of a shuffled sequence on the same template (Table 3). Moreover, the deformational preferences occur with templates as short as 60 basepair steps, corresponding to the DNA in contact with the (H3·H4)2 tetramer that is thought to be critical to nucleosomal positioning (13,41). The DNA pathways in currently solved nucleosomes are sensitive to perturbations of protein structure, in that extreme kink-and-slide steps appear and disappear in complexes assembled from the same DNA and chemically modified or mutant histones, but are insensitive to protein, in that the extreme states occur at the same nucleotide positions (Fei Xu, Wilma K. Olson, unpublished data).
Different types of DNA folding may occur as nucleosomes make use of the unique sequences in different genomes. For example, the dominant repetition of GG·CC dimers in phase with the double-helical repeat in human nucleosomes (83) is suggestive of DNA wrapping that takes advantage of the A-philic character of these basepairs (84), incorporating a different balance of A- versus C-like deformations along the folding pathway. Cisplatin, the anti-cancer agent that covalently locks sequential basepair steps in the A form, may use such a mechanism in fixing the rotational setting of DNA on the nucleosome (85). Indeed, our analysis of the recently reported structure of the nucleosome core particle treated with the platinum complex (86) shows A-like duplex unwinding and kink-and-slide steps (with positive Roll and negative Slide) not found in the absence of ligand (Fig. S2).
Acknowledgments
The authors thank Drs. Michael Tolstorukov and Victor Zhurkin for valuable discussions, Dr. A.R. Srinivasan for assistance with molecular modeling, Drs. Sanford Leuba, Mikael Kubista, and Jonathan Widom for sharing sequence information. S.B. acknowledges support of a predoctoral traineeship from the U.S. Public Health Service (Molecular Biophysics Training grant GM08319).
This work was taken in part from the dissertations of Sreekala Balasubramanian and Fei Xu written in partial fulfillment of the requirements for the degree of Doctor of Philosophy, Rutgers University, 2003 and 2007.
This work was supported by the U.S. Public Health Service (grant GM20861).
Supporting Material
References
- 1.Luger K., Mäder A.W., Richmond R.K., Sargent D.F., Richmond T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Davey C.A., Sargent D.F., Luger K., Mäder A.W., Richmond T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 3.Richmond T.J., Davey C.A. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- 4.Strauss J.K., Maher L.J., III Bending by asymmetric phosphate neutralization. Science. 1994;266:1829–1834. doi: 10.1126/science.7997878. [DOI] [PubMed] [Google Scholar]
- 5.Mirzabekov A.D., Rich A. Asymmetric lateral distribution of unshielded phosphate groups in nucleosomal DNA and its role in DNA bending. Proc. Natl. Acad. Sci. USA. 1979;76:1118–1121. doi: 10.1073/pnas.76.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manning G.S., Ebralidse K.K., Mirzabekov A.D., Rich A. An estimate of the extent of folding of nucleosomal DNA by laterally asymmetric neutralization of phosphate groups. J. Biomol. Struct. Dyn. 1989;6:877–879. doi: 10.1080/07391102.1989.10506519. [DOI] [PubMed] [Google Scholar]
- 7.Kiyama R., Trifonov E.N. What positions nucleosomes?–a model. FEBS Lett. 2002;523:7–11. doi: 10.1016/s0014-5793(02)02937-x. [DOI] [PubMed] [Google Scholar]
- 8.Shrader T.E., Crothers D.M. Artificial nucleosome positioning sequences. Proc. Natl. Acad. Sci. USA. 1989;86:7418–7422. doi: 10.1073/pnas.86.19.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Widlund H.R., Cao H., Simonsson S., Magnusson E., Simonsson T. Identification and characterization of genomic nucleosome-positioning sequences. J. Mol. Biol. 1997;267:807–817. doi: 10.1006/jmbi.1997.0916. [DOI] [PubMed] [Google Scholar]
- 10.Widom J. Role of DNA sequence in nucleosome stability and dynamics. Q. Rev. Biophys. 2001;34:269–324. doi: 10.1017/s0033583501003699. [DOI] [PubMed] [Google Scholar]
- 11.Olson W.K., Gorin A.A., Lu X.-J., Hock L.M., Zhurkin V.B. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. USA. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subirana J.A., Faria T. Influence of sequence on the conformation of the B-DNA helix. Biophys. J. 1997;73:333–338. doi: 10.1016/S0006-3495(97)78073-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thåström A., Bingham L.M., Widom J. Nucleosomal locations of dominant DNA sequence motifs for histone-DNA interactions and nucleosome positioning. J. Mol. Biol. 2004;338:695–709. doi: 10.1016/j.jmb.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 14.Harp J.M., Hanson B.L., Timm D.E., Bunick G.J. Asymmetries in the nucleosome core particle at 2.5 Å resolution. Acta Crystallogr D. Biol. Crystallogr. 2000;56:1513–1534. doi: 10.1107/s0907444900011847. [DOI] [PubMed] [Google Scholar]
- 15.Suto R.K., Clarkson M.J., Tremethick D.J., Luger K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat. Struct. Biol. 2000;7:1121–1124. doi: 10.1038/81971. [DOI] [PubMed] [Google Scholar]
- 16.White C.L., Suto R.K., Luger K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001;20:5207–5218. doi: 10.1093/emboj/20.18.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suto R.K., Edayathumangalam R.S., White C.L., Melander C., Gottesfeld J.M. Crystal structures of nucleosome core particles in complex with minor groove DNA-binding ligands. J. Mol. Biol. 2003;326:371–380. doi: 10.1016/s0022-2836(02)01407-9. [DOI] [PubMed] [Google Scholar]
- 18.Muthurajan U.M., Bao Y., Forsberg L.J., Edayathumangalam R.S., Dyer P.N. Crystal structures of histone Sin mutant nucleosomes reveal altered protein-DNA interactions. EMBO J. 2004;23:260–271. doi: 10.1038/sj.emboj.7600046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsunaka Y., Kajimura N., Tate S., Morikawa K. Alteration of the nucleosomal DNA path in the crystal structure of a human nucleosome core particle. Nucleic Acids Res. 2005;33:3424–3434. doi: 10.1093/nar/gki663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clapier C.R., Chakravarthy S., Petosa C., Fernandez-Tornero C., Luger K. Structure of the Drosophila nucleosome core particle highlights evolutionary constraints on the H2A–H2B histone dimer. Proteins. 2007;71:1–7. doi: 10.1002/prot.21720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakravarthy, S., and K. Luger. 2008. Comparative Analysis of Nucleosome Structures from Different Species. To be published.
- 22.Lowary P.T., Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- 23.Thåström A., Lowary P.T., Widlund H.R., Cao H., Kubista M. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- 24.Cao H., Widlund H.R., Simonsson T., Kubista M. TGGA repeats impair nucleosome formation. J. Mol. Biol. 1998;281:253–260. doi: 10.1006/jmbi.1998.1925. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y.-H., Griffith J.D. The [(G/C)3NN]n motif: a common DNA repeat that excludes nucleosomes. Proc. Natl. Acad. Sci. USA. 1996;93:8863–8867. doi: 10.1073/pnas.93.17.8863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rossetti L., Cacchione S., Fua M., Savino M. Nucleosome assembly on telomeric sequences. Biochemistry. 1998;37:6727–6737. doi: 10.1021/bi9726180. [DOI] [PubMed] [Google Scholar]
- 27.Mechelli R., Anselmi C., Cacchione S., De Santis P., Savino M. Organization of telomeric nucleosomes: atomic force microscopy imaging and theoretical modeling. FEBS Lett. 2004;566:131–135. doi: 10.1016/j.febslet.2004.04.032. [DOI] [PubMed] [Google Scholar]
- 28.Levitsky V.G. RECON: a program for prediction of nucleosome formation potential. Nucleic Acids Res. 2004;32:W346–W349. doi: 10.1093/nar/gkh482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segal E., Fondufe-Mittendorf Y., Chen L., Thåström A., Field Y. A genomic code for nucleosome positioning. Nature. 2006;442:750–752. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sivolob A.V., Khrapunov S.N. Translational positioning of nucleosomes on DNA: the role of sequence-dependent isotropic DNA bending stiffness. J. Mol. Biol. 1995;247:918–931. doi: 10.1006/jmbi.1994.0190. [DOI] [PubMed] [Google Scholar]
- 31.Anselmi C., Bocchinfuso G., De Santis P., Savino M., Scipioni A. Dual role of DNA intrinsic curvature and flexibility in determining nucleosome stability. J. Mol. Biol. 1999;286:1293–1301. doi: 10.1006/jmbi.1998.2575. [DOI] [PubMed] [Google Scholar]
- 32.Anselmi C., Bocchinfuso G., DeSantis P., Savino M., Scipioni A. A theoretical model for the prediction of sequence-dependent nucleosome thermodynamic stability. Biophys. J. 2000;79:601–613. doi: 10.1016/S0006-3495(00)76319-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scipioni A., Pisano S., Anselmi C., Savino M., De Santis P. Dual role of sequence-dependent DNA curvature in nucleosome stability: the critical test of highly bent Crithidia fasciculata DNA tract. Biophys. Chem. 2004;107:7–17. doi: 10.1016/S0301-4622(03)00214-X. [DOI] [PubMed] [Google Scholar]
- 34.Peck L.J., Wang J.C. Sequence dependence of the helical repeat of DNA in solution. Nature. 1981;292:375–378. doi: 10.1038/292375a0. [DOI] [PubMed] [Google Scholar]
- 35.Rhodes D., Klug A. Sequence dependent helical periodicity of DNA twisting. Nature. 1981;292:378–380. doi: 10.1038/292378a0. [DOI] [PubMed] [Google Scholar]
- 36.Rief M., Clausen-Schaumann H., Gaub H.E. Sequence-dependent mechanics of single DNA molecules. Nat. Struct. Biol. 1999;6:346–349. doi: 10.1038/7582. [DOI] [PubMed] [Google Scholar]
- 37.Berman H.M., Olson W.K., Beveridge D.L., Westbrook J., Gelbin A. The Nucleic Acid Database: a comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 1992;63:751–759. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhurkin V.B., Tolstorukov M.Y., Xu F., Colasanti A.V., Olson W.K. Sequence-dependent variability of B-DNA: an update on bending and curvature. In: Ohyama T., editor. DNA Conformation and Transcription. Landes Bioscience/Eurekah; Georgetown, Texas: 2005. pp. 18–34. [Google Scholar]
- 39.Dlakic M., Ussery D.W., Brunak S. DNA bendability and nucleosome positioning in transcriptional regulation. In: Ohyama T T., editor. DNA Conformation and Transcription. Landes Bioscience/Eurekah; Georgetown, Texas: 2005. pp. 187–202. [Google Scholar]
- 40.Tolstorukov M.Y., Colasanti A.V., McCandlish D., Olson W.K., Zhurkin V.B. A novel roll-and-slide mechanism of DNA folding in chromatin. Implications for nucleosome positioning. J. Mol. Biol. 2007;371:725–738. doi: 10.1016/j.jmb.2007.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Widlund H.R., Vitolo J.M., Thiriet C., Hayes J.J. DNA sequence-dependent contributions of core histone tails to nucleosome stability: differential effects of acetylation and proteolytic tail removal. Biochemistry. 2000;39:3835–3841. doi: 10.1021/bi991957l. [DOI] [PubMed] [Google Scholar]
- 42.Dixit S.B., Beveridge D.L., Case D.A., Cheatham T.E., III, Giudice E. Molecular dynamics simulations of the 136 unique tetranucleotide sequences of DNA oligonucleotides. II: Sequence context effects on the dynamical structures of the 10 unique dinucleotide steps. Biophys. J. 2005;89:3721–3740. doi: 10.1529/biophysj.105.067397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lankaš F., Šponer J., Hobza P., Langowsk J. Sequence-dependent elastic properties of DNA. J. Mol. Biol. 2000;299:695–709. doi: 10.1006/jmbi.2000.3781. [DOI] [PubMed] [Google Scholar]
- 44.Fujii S., Kono H., Takenaka S., Go N., Sarai A. Sequence-dependent DNA deformability studied using molecular dynamics simulations. Nucleic Acids Res. 2007;35:6063–6074. doi: 10.1093/nar/gkm627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olson W.K., Colasanti A.V., Czapla L., Zheng G. Insights into the sequence-dependent macromolecular properties of DNA from basepair level modeling. In: Voth G.A., editor. Coarse-Graining of Condensed Phase and Biomolecular Systems. Taylor and Francis Group; LLC, Boca Raton, Florida: 2008. pp. 205–223. [Google Scholar]
- 46.Lu X.-J., Olson W.K. 3DNA: a software package for the analysis, rebuilding, and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu X.-J., Olson W.K. 3DNA: a versatile, integrated software system for the analysis, rebuilding, and visualization of three-dimensional nucleic-acid structures. Nat. Protocols. 2008;3:1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dickerson R.E., Bansal M., Calladine C.R., Diekmann S., Hunter W.N. Definitions and nomenclature of nucleic acid structure parameters. J. Mol. Biol. 1989;208:787–791. doi: 10.1016/0022-2836(89)90324-0. [DOI] [PubMed] [Google Scholar]
- 49.Lu X.-J., Shakked Z., Olson W.K. A-form conformational motifs in ligand-bound DNA structures. J. Mol. Biol. 2000;300:819–840. doi: 10.1006/jmbi.2000.3690. [DOI] [PubMed] [Google Scholar]
- 50.El Hassan M.A., Calladine C.R. Conformational characteristics of DNA: empirical classifications and a hypothesis for the conformational behavior of dinucleotide steps. Philos. Trans. R. Soc. Lond. A. 1997;355:43–100. [Google Scholar]
- 51.Guzikevich-Guerstein G., Shakked Z. A novel form of the DNA double helix imposed on the TATA-box by the TATA-binding protein. Nat. Struct. Biol. 1996;3:32–37. doi: 10.1038/nsb0196-32. [DOI] [PubMed] [Google Scholar]
- 52.An W., Leuba S.H., van Holde K., Zlatanova J. Linker histone protects linker DNA on only one side of the core particle and in a sequence-dependent manner. Proc. Natl. Acad. Sci. USA. 1998;95:3396–3401. doi: 10.1073/pnas.95.7.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kassabov S.R., Henry N.M., Zofall M., Tsukiyama T., Bartholomew B. High-resolution mapping of changes in histone-DNA contacts of nucleosomes remodeled by ISW2. Mol. Cell. Biol. 2002;22:7524–7534. doi: 10.1128/MCB.22.21.7524-7534.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsumoto A., Tobias I., Olson W.K. Normal mode analysis of circular DNA at the basepair level. I. Comparison of computed motions with the predicted behavior of an ideal elastic rod. J. Chem. Theory Comput. 2005;1:117–129. doi: 10.1021/ct049950r. [DOI] [PubMed] [Google Scholar]
- 55.Matsumoto A., Tobias I., Olson W.K. Normal mode analysis of circular DNA at the basepair level. II. Large-scale configurational transformation of a naturally curved molecule. J. Chem. Theory Comput. 2005;1:130–142. doi: 10.1021/ct049949s. [DOI] [PubMed] [Google Scholar]
- 56.Miyazawa T. Molecular vibrations and structure of high polymers. II. Helical parameters of infinite polymer chains as functions of bond lengths, bond angles, and internal rotation angles. J. Polym. Sci. [B] 1961;55:215–231. [Google Scholar]
- 57.Bishop T.C. Geometry of the nucleosomal DNA superhelix. Biophys. J. 2008;95:1007–1017. doi: 10.1529/biophysj.107.122853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brukner I., Sanchez R., Suck D., Pongor S. Trinucleotide models for DNA bending propensity: comparison of models based on DNaseI digestion and nucleosome packaging data. J. Biomol. Struct. Dyn. 1995;13:309–317. doi: 10.1080/07391102.1995.10508842. [DOI] [PubMed] [Google Scholar]
- 59.Kulic I.M., Schiessel H. Nucleosome repositioning via loop formation. Biophys. J. 2003;84:3197–3211. doi: 10.1016/S0006-3495(03)70044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horton J.R., Nastri H.G., Riggs P.D., Cheng X. Asp34 of PvuII endonuclease is directly involved in DNA minor groove recognition and indirectly involved in catalysis. J. Mol. Biol. 1998;284:1491–1504. doi: 10.1006/jmbi.1998.2269. [DOI] [PubMed] [Google Scholar]
- 61.Guo F., Gopaul D.N., Van Duyne G.D. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389:40–46. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- 62.Li Y., Korolev S., Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 1998;17:7514–7525. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tan S., Richmond T.J. Crystal structure of the yeast MATα2/MCM1/DNA ternary complex. Nature. 1998;391:660–666. doi: 10.1038/35563. [DOI] [PubMed] [Google Scholar]
- 64.Chandrasekaran R., Arnott S. The structures of DNA and RNA helices in oriented fibers. In: Saenger W., editor. Landolt-Börnstein Numerical Data and Functional Relationships in Science and Technology, Group VII/1b, Nucleic Acids. Springer-Verlag; Berlin: 1989. pp. 31–170. [Google Scholar]
- 65.Marvin D.A., Spencer M., Wilkins M.H.F., Hamilton L.D. A new configuration of deoxyribonucleic acid. Nature. 1958;182:387–388. doi: 10.1038/182387b0. [DOI] [PubMed] [Google Scholar]
- 66.van Dam L., Levitt M.H. BII nucleotides in the B and C forms of natural-sequence polymeric DNA: a new model for the C form of DNA. with 40° helical twist. J. Mol. Biol. 2000;304:541–561. doi: 10.1006/jmbi.2000.4194. [DOI] [PubMed] [Google Scholar]
- 67.Harp J.M., Uberbacher E.C., Roberson A.E., Palmer E.L., Gewiess A. X-ray diffraction analysis of crystals containing twofold symmetric nucleosome core particles. Acta Crystallogr. D. Biol. Crystallogr. 1996;52:283–288. doi: 10.1107/S0907444995009139. [DOI] [PubMed] [Google Scholar]
- 68.Yang T.P., Hansen S.K., Oishi K.K., Ryder O.A., Hamkalo B.A. Characterization of a cloned repetitive DNA sequence concentrated on the human X chromosome. Proc. Natl. Acad. Sci. USA. 1982;79:6593–6597. doi: 10.1073/pnas.79.21.6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Napoli A.A., Lawson C.L., Ebright R.H., Berman H.M. Indirect readout of DNA sequence at the primary-kink site in the CAP-DNA complex: recognition of pyrimidine-purine and purine-purine steps. J. Mol. Biol. 2006;357:173–183. doi: 10.1016/j.jmb.2005.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bao Y., White C.L., Luger K. Nucleosome core particles containing a poly(dA·dT) sequence element exhibit a locally distorted DNA structure. J. Mol. Biol. 2006;361:617–624. doi: 10.1016/j.jmb.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 71.Nelson H.C.M., Finch J.T., Luisi B.F., Klug A. The structure of an oligo(dA)-oligo(dT) tract and its biological implications. Nature. 1987;330:221–226. doi: 10.1038/330221a0. [DOI] [PubMed] [Google Scholar]
- 72.Rhodes D. Nucleosome cores reconstituted from poly (dA-dT) and the octamer of histones - failed reconstitution of nucleosomes with AT. Nucleic Acids Res. 1979;6:1805–1816. doi: 10.1093/nar/6.5.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kunkel G.R., Martinson H.G. Nucleosomes will not form on double-stranded RNA or over poly dA poly dT tracts in recombinant DNA. Nucleic Acids Res. 1981;9:6869–6888. doi: 10.1093/nar/9.24.6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Prunell A. Nucleosome reconstitution of plasmid-inserted poly dA·poly dT. EMBO J. 1981;1:173–179. doi: 10.1002/j.1460-2075.1982.tb01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Edwards C.A., Firtel R.A. Resistance of homopolymers to wrap around nucleosomes. J. Mol. Biol. 1984;180:73–90. doi: 10.1016/0022-2836(84)90431-5. [DOI] [PubMed] [Google Scholar]
- 76.Théveny B., Coulaud D., LeBret M., Revét B. Local variations of curvature and flexibility along DNA molecules analyzed from electron micrographs. In: Olson W.K., Sarma M.H., Sarma R.H., Sundaralingam M., editors. DNA Bending and Curvature. Adenine Press; Guilderland, New York: 1988. pp. 39–55. [Google Scholar]
- 77.Olson W.K., Colasanti A.V., Li Y., Ge W., Zheng G. DNA simulation benchmarks as revealed by X-ray structures. In: Sponer J., Lankas F., editors. Computational Studies of RNA and DNA. Springer, Dordrecht; The Netherlands: 2006. pp. 235–257. [Google Scholar]
- 78.Matsumoto A., Olson W.K. Sequence-dependent motions of DNA: a normal mode analysis at the basepair level. Biophys. J. 2002;83:22–41. doi: 10.1016/S0006-3495(02)75147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coleman B.D., Olson W.K., Swigon D. Theory of sequence-dependent DNA elasticity. J. Chem. Phys. 2003;118:7127–7140. [Google Scholar]
- 80.Czapla L., Swigon D., Olson W.K. Sequence-dependent effects in the cyclization of short DNA. J. Chem. Theory Comput. 2006;2:685–695. doi: 10.1021/ct060025+. [DOI] [PubMed] [Google Scholar]
- 81.Cloutier T.E., Widom J. Spontaneous sharp bending of double-stranded DNA. Mol. Cell. 2004;14:355–362. doi: 10.1016/s1097-2765(04)00210-2. [DOI] [PubMed] [Google Scholar]
- 82.Cloutier T.E., Widom J. DNA twisting flexibility and the formation of sharply looped protein-DNA complexes. Proc. Natl. Acad. Sci. USA. 2005;102:3645–3650. doi: 10.1073/pnas.0409059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kogan S.B., Kato M., Kiyama R., Trifonov E.N. Sequence structure of human nucleosome DNA. J. Biomol. Struct. Dyn. 2006;24:43–48. doi: 10.1080/07391102.2006.10507097. [DOI] [PubMed] [Google Scholar]
- 84.Ivanov V.I., Minchenkova L.E. The A-form of DNA: in search of biological role (a review) Mol. Biol. 1995;28:780–788. [PubMed] [Google Scholar]
- 85.Danford A.J., Wang D., Wang Q., Tullius T.D., Lippard S.J. Platinum anticancer drug damage enforces a particular rotational setting of DNA in nucleosomes. Proc. Natl. Acad. Sci. USA. 2005;102:12311–12316. doi: 10.1073/pnas.0506025102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu B., Dröge P., Davey C.A. Site selectivity of platinum anticancer therapeutics. Nat. Chem. Biol. 2008;4:110–112. doi: 10.1038/nchembio.2007.58. [DOI] [PubMed] [Google Scholar]
- 87.Sayle R.A., Milnerwhite E.J. RasMol: biomolecular graphics for all. Trends Biochem. Sci. 1995;20:374–376. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.