

Abstract
To understand the thermodynamic exclusion of Na+ relative to K+ from the S2 site of the selectivity filter, the distribution PX(ɛ) (X = K+ or Na+) of the binding energy (ɛ) of the ion with the channel is analyzed using the potential distribution theorem. By expressing the excess chemical potential of the ion as a sum of mean-field 〈ɛ〉 and fluctuation μexflux,X contributions, we find that selectivity arises from a higher value of relative to . To understand the role of site-site interactions on μexflux,X, we decompose PX(ɛ) into n-dependent distributions, where n is the number of ion-coordinating ligands within a distance λ from the ion. For λ comparable to typical ion-oxygen bond distances, investigations building on this multistate model reveal an inverse correlation between favorable ion-site and site-site interactions: the ion-coordination states that most influence the thermodynamics of the ion are also those for which the binding site is energetically less strained and vice versa. This correlation motivates understanding entropic effects in ion binding to the site and leads to the finding that μexflux,X is directly proportional to the average site-site interaction energy, a quantity that is sensitive to the chemical type of the ligand coordinating the ion. Increasing the coordination number around Na+ only partially accounts for the observed magnitude of selectivity; acknowledging the chemical type of the ion-coordinating ligand is essential.
Introduction
The KcsA K+ channel is highly permeable to K+ relative to Na+ (1). The reversible work of transferring a Na+ ion from the aqueous phase to a site (S) in the selectivity filter relative to the case for a K+ ion provides a thermodynamic measure of this selectivity. Thus,
| (1) |
has been sought using the protein structure (2) and molecular simulations (3–5). The quantity μex is the excess chemical potential of the ion: (aq) (X = K+, Na+) is the usual hydration free energy, and (S) is the analogous quantity in the site S in the selectivity filter. In Eq. 1, it is understood that a common reference state is used in specifying μex in the aqueous phase and in the channel. Of the five possible ion-binding sites S0–S4 in the selectivity filter (6), the S2 site is the most selective with Δμex ≈ 6 kcal/mol (5). (Sites are labeled according to (5).)
In studying the role of protein flexibility and electrostatic properties of the ion-binding site on selectivity, Noskov et al. (5) made an interesting observation. In computer experiments, suppressing the interaction between the carbonyl ligands comprising the S2 binding site shifted the selectivity towards Na+, a behavior that was also reproduced in a model of the binding site with eight carbonyl ligands confined in a sphere of radius 3.5 Å. This suggests that the correlation among ion-binding ligands impacts selectivity. However, the ligand-ligand correlation is expected to be sensitive to the chemical type of the ligand. Indeed, Noskov et al. (5,7,8) suggested that, for the given binding configuration, the higher dipole moment of the carbonyl ligand relative to water was essential in conferring selectivity toward K+.
Other researchers have investigated the role of ion coordination and the electrostatic properties of the coordinating ligand on selectivity using various models of the ion-binding site (9–11). One study in particular concluded that a model site with eight water molecules confined in a sphere of radius 3.5 Å is as selective for K+ over Na+ as a site consisting of eight carbonyl ligands (11). This observation suggests that enhancing the coordination around Na+ to values similar to the more probable coordination numbers seen for K+(aq) itself confers selectivity.
In this work we explore how the energetics of the binding site influences the thermodynamics of the ion. Our aim is to understand better ion coordination and the role of the type of ion-coordinating ligand on selectivity. Earlier, we studied a model of the selectivity filter (12) which showed that relative to K+ the interaction energy of Na+ with the selectivity filter fluctuates more strongly and is responsible for the observed exclusion of Na+ from the selectivity filter. Here we simulate the entire protein in a realistic membrane environment, and, drawing upon the insightful idea of multiple substates in hydration (13), explore the relation between coordination states and the thermodynamics of the ion. We find that ion-site interactions are inversely correlated with site-site interactions. By obtaining an expression for the excess entropy of ion binding to the S2 site, we show that the contribution to the excess chemical potential of the ion due to ion-site energy fluctuations is directly related to the average site-site interactions, i.e., the average excess internal energy of the eight-carbonyl ligands comprising the S2 site. Further, the site-site interactions are quantitatively different for a model site consisting of eight water molecules in a defined volume in bulk solvent, and, relative to the S2 site, this model site is less selective for K+. Thus increasing the coordination number around Na+ only partially accounts for the observed magnitude of Δμex; acknowledging the chemical type of the coordinating ligand is essential.
Theory
The ion binds to a configuration of the channel with an energy ɛ. If PX(ɛ) is the probability density distribution of the binding (interaction) energies, then (14–16)
| (2) |
Here, β = 1/kBT, where kB is Boltzmann's constant and T is the temperature. Because of the exponential factor, eβɛ, the high energy (high-ɛ) tail of PX(ɛ), and thus the less favorable ion-protein interaction, dominates the thermodynamics.
Expanding Eq. 2 into cumulants (17) with respect to β provides a way to analyze the distribution PX(ɛ). Thus,
| (3) |
The first three cumulants are, for example, C0, X = 0, the mean C1, X = 〈ɛ〉X, and the variance C2, X = σ2X = 〈(ɛ − 〈ɛ〉)2〉X. Thus, in principle, the excess chemical potential can be written as
| (4) |
where, by definition, the fluctuation contribution comprises all the effects beyond the mean-field contribution 〈ɛ〉X. In particular, if PX(ɛ) is a Gaussian distribution, .
Another way to analyze PX(ɛ) is to decompose the distribution based on the coordination states (13,15,16). The various coordination structures around the ion can be visualized as potential energy basins of attraction for the coordination shell ligands, in the spirit of Stillinger-Weber inherent structures (18). In studying ion hydration, Hummer et al. (13) used the perspective of inherent structures to model the non-Gaussian fluctuations of the electrostatic potential of ion-water interaction as a sum of Gaussian fluctuations for each coordination state of the ion. Here we adapt these ideas to gain insights into the fluctuation contribution in terms of the ligand coordination structure around the ion.
The coordination number, n, is the number of oxygen atoms (from either water or the carbonyl ligand) within a distance λ of the ion X in the nth coordination state. The region within λ defines the coordination sphere of the ion. The probability of observing the nth coordination state, p(n), is the fraction of configurations with n oxygens within the coordination sphere. These configurations also specify the conditional binding energy distribution PX(ɛ|n). Thus,
| (5) |
From Eqs. 2 and 5, we have
| (6) |
Equation 6 is a multistate organization of the excess chemical potential (13,15,16). The value p(n) is related to the equilibrium constant for forming a cluster comprising the ion and n ligands (15,16), but here its use as the marginal probability associated with PX(ɛ|n) is implied. The quantity (n) is the excess chemical potential of the ion in the nth coordination state. Operationally, (n) can be calculated just as one would calculate , but with the added proviso that the n-coordinate state of the ion is always maintained. Alternatively, from a given simulation record, one need only extract those configurations obeying the coordination number constraint to construct PX(ɛ|n).
To highlight the joint role of coordination number and the chemical potential in that coordination state, Eq. 6 can be rewritten as
| (7) |
where , and K(n) measures the weight of each term of this sum relative to the maximum. It is apparent that a high probability p(n) does not guarantee the dominance of that state in the thermodynamics of the ion. Conversely, to avoid biasing the results from states with very low population (limited statistical information) but very high (n), we consider only states with p(n) ≥ 2.5 × 10−2. (See also (13).) An alternative approach to an equation similar to Eq. 7 can be found in Deng and Roux (19).
Equation 6 is an identity, and is independent of λ. However, due to practical limitations in computing (n) with sufficient accuracy, the estimate of does vary with λ. But as shown below, this deficiency does not limit the usefulness of Eq. 6 in providing qualitative insights into the factors determining the thermodynamics of the ion.
A multi-Gaussian form of Eq. 6 results if each PX(ɛ|n) is well described by a Gaussian. Here we find that PX(ɛ|n) has a small negative skew, but the high-ɛ tail of the distribution is best described by a Gaussian. We model this behavior by a skew normal distribution and set (20). The quantity is the normal density distribution with a location parameter and a scale parameter σ. (These are the mean and variance if the distribution is strictly Gaussian.) The quantity α, the shape parameter, specifies the skewness; α = 0 reproduces a Gaussian. For distributions with a negative skew (α < 0) in the region where , the distribution approaches a normal distribution since Erf(x) → 0 for x → −∞. When we refer to the multi-Gaussian model below, we imply use of the skew normal for each substate PX(ɛ|n).
The multi-Gaussian form developed here is motivated by the multi-Gaussian model of hydration investigated by Hummer et al. (13). But there is an important distinction. Hummer et al. (13) use the particle insertion form of the potential distribution theorem; that is, the binding energy distribution is obtained with the solute and the solvent thermally uncoupled. Here we use the particle extraction form of the potential distribution theorem, for the particle insertion approach is not suitable as an ion-free (and water-free) S2 site is structurally unstable.
Methods
We use the CHARMM27 force field (21) and the NAMD v2.6 program (22) for simulations. Construction of the simulation cell was facilitated by VMD (23).
Simulation system
We closely follow the procedure in Bernéche and Roux (24) for constructing the simulation system. The protein structure was obtained from the Protein Data Bank (1BL8 (2)). The N-terminal groups of all the four subunits were acylated. Glu-80 was constructed in the protonated state. Residues 74–79 comprise the selectivity filter. Potassium ions were placed in the S0 and S4 binding sites, and the S1 and S3 sites had a water molecule each. Either K+ or Na+ was placed in the S2 site. (Ion occupancy and site labels follow earlier studies (5,7,8).) A well-equilibrated box of water molecules was overlaid on the vestibule and the channel entrance region below the selectivity filter. After removing water molecules within 3 Å of protein heavy atoms, ∼80 waters occupied the vestibule and the channel entrance. Water was modeled with the TIP3P potential (25,26). This system was energy-minimized for 2000 steps and then inserted into a well-equilibrated palmitoyloleoyl phosphatidylcholine lipid bilayer ∼94 × 94 Å2 in size and with ∼15 Å layer of water molecules on either side of the membrane. The position of the protein with respect to the bilayer was as in Bernéche and Roux (24). Lipids within 3 Å of protein heavy atoms were removed. To prevent translation drift in the z direction, harmonic restraints of stiffness constant 5 kcal/mol/Å2 are imposed on the z coordinate of the phosphorus atoms of the lipids. The initial equilibration of 150 ps allowed the membrane to pack around the protein. During this period, stiff harmonic restraints were placed on the pore and the ions to preclude large movement of the ions. Then, over a period of 140 ps, these restraints were progressively removed. The system was then equilibrated for 10.15 ns. After equilibration, a 4-ns production run was analyzed by sampling configurations every 100 fs. The initial Na+-bound structure was obtained from the end-point of the parametric integration procedure described below. For the Na+-bound structure, a 1-ns equilibration was followed by a 4-ns production.
Equilibration and production were conducted at a pressure of 1 bar using a Langevin-piston barostat (27,28) and a temperature of 310 K using a Langevin thermostat. The barostat piston period was 200 fs and the decay time was 100 fs. The decay constant for the thermostat was 1 ps−1. Covalent bonds to hydrogen were held rigid using the SHAKE algorithm (29). The equations of motion were integrated with a 2-fs time step. Lennard-Jones interactions were smoothly switched to zero between 10.215 Å and 10.715 Å. Nonbonded electrostatics were treated using particle-mesh Ewald summation. The grid size for the particle mesh Ewald was always <0.75 Å.
Energy calculations
K+ was transformed to Na+ in 20 steps and the free energy change calculated via coupling parameter integration. Each step comprises an equilibration of 5 ps (10 ps in the case of the aqueous simulation) followed by 20 ps of production. The coupling parameter result was the standard against which the multi-Gaussian model was compared.
The ion-protein interaction energy, ɛ, is a sum of Lennard-Jones interactions and electrostatics evaluated using Ewald summation. We considered ion interactions with:
-
(A1)
all protein and solvent atoms;
-
(A2)
the carbonyl groups of the S2 site and the two adjacent water molecules; and
-
(A3)
only the carbonyl groups.
Approaches A2 and A3 are motivated by quasichemical considerations that seek to explicate the role of the local neighborhood on the thermodynamics of the ion (see Eq. 6) and are expected to be sensitive to the distribution of coordinating ligands around the ion. We anticipate that the response of the material outside the local neighborhood will be similar for K+ and Na+.
All the approaches above led to quantitatively similar results within the coupling parameter approach. The first two yield quantitatively similar results within the multi-Gaussian approach, while the third gave energy distributions that were skewed enough to render inappropriate a multi-Gaussian description. The latter observation is in contrast to our earlier study on a minimal model that used artificial restraints on the system and considered only ion-carbonyl interaction energies (12) and found even a single Gaussian description to be appropriate. But, in principle, for all of the approaches a fluctuation contribution () to the chemical potential can be defined. Thus, on this basis, the results below, unless otherwise noted, are founded on coupling parameter integration and the decomposition of the chemical potential according to Eq. 4. The mean-field contribution 〈ɛ〉X (Eq. 4) is obtained from PX(ɛ) and the fluctuation contribution is obtained from by subtraction of the mean-field contribution. We emphasize that the multi-Gaussian model is used solely to obtain qualitative insights into the interplay of ion-site and site-site interactions, quantitative studies based on these insights always used the coupling parameter approach.
Results and Discussion
Table 1 collects results obtained using approaches A1 and A2 (see Methods; A3 is considered later together with a site comprising water molecules as the ion-coordinating ligands). The difference in hydration free energies in the bulk against which selectivity is measured is Δμex(aq) = −20.7 kcal/mol. Thus, relative to K+, dehydration itself presents a barrier to the entry of Na+ into the filter. Observe that Na+ is better bound in the S2 site, and mean binding energies alone do not predict the observed magnitude of selectivity; the fluctuation contribution to the chemical potential is an essential determinant of selectivity, consistent with the earlier analysis based on a simple model of the binding site and a Gaussian description of the binding energy distributions (12). The estimate of selectivity presented in Table 1 is consistent with earlier estimates of selectivity (5,8).
Table 1.
Mean-field and fluctuation contributions to the selectivity (Eq. 1) obtained from the coupling transformation of K+ to Na+ in the S2 site
| A1 | A2 | ||
|---|---|---|---|
| Δμex | 6.0 | 4.9 | |
| Δ〈ɛ〉 – Δμex(aq) | −0.2 | 1.9 | |
| 6.2 | 3.0 |
To obtain insights into the factors influencing the fluctuation contribution (Table 1), we consider a multi-Gaussian model for various values of the coordination radius λ (Fig. 1). For the chosen values of λ, water oxygen atoms do not contribute substantially to the coordination state of the ion. Thus all the n-coordinate states involve only ion-carbonyl oxygen bonding.
Figure 1.

The excess chemical potential of the ion obtained using the multi-Gaussian model for various values of λ and approach A2 (see Methods) for computing energies. Averaging across λ, kcal/mol and kcal/mol. The error given at the 2σ level is within 3% of the mean value, but because μex is itself large, obtaining agreement within 1 kcal/mol of the coupling parameter estimate is difficult. In particular, Δμex(S2) = −18 ± 3 kcal/mol, and Δμex = −18.0 + 20.7 = 2.7 kcal/mol.
The multi-Gaussian model predicts Δμex = 2.7 kcal/mol (Fig. 1). Although the numerical agreement with coupling parameter integration is poor, it must be noted that the net selectivity arises due to the balancing of terms that are themselves large (Eq. 1). Thus even a low standard error in the estimate of μexX(S2) by the multi-Gaussian model is not sufficient to match the results of coupling parameter integration. The same concerns apply to a single Gaussian model, a point that was less well appreciated in our earlier study (12). Despite these limitations, the multi-Gaussian model provides valuable insights.
Consider λ = 2.7 Å for Na+ and λ = 3.1 Å for K+, values based on typical ion-oxygen coordination distances observed in biophysical and inorganic chemistry situations (30,31). For these coordination radii, the distribution of the probability of observing a n-coordinate state is shown in Fig. 2, and the relative contribution of various coordination states to the excess chemical potential is shown in Fig. 3. The binding energy distribution and its decomposition into the least and most dominant substate distributions are shown in Fig. 4.
Figure 2.

Log p(n) for λ = 2.7 Å for Na+ and λ = 3.1 Å for K+. For both ions, a maximum of eight carbonyl oxygens are observed within the coordination sphere. The most probable state for Na+ is n = 6 and for K+ it is n = 8.
Figure 3.

The kBT log K(n) (Eq. 7) for various n-coordinate states of the ion. For the state that contributes maximally in Eq. 7, K(n) = 1. λ = 2.7 Å for Na+ (○) and λ = 3.1 Å for K+ (□).
Figure 4.

Normalized distribution of ion binding energies using approach A2 (see Methods) and its multistate decomposition. Following Hummer et al. (13) the substate distributions are given by log [p(n)P(ɛ|n)] (Eq. 5). (Left panel, □) Simulation results for K+. (Right panel, ○) Simulation results for Na+. The unbroken lines are the multistate fits. The skew normal fits to the individual states are labeled by the state index n. (Dashed curve) Thermodynamically dominant state. (Dotted curve) Least dominant state. For clarity, the raw substate distributions are not shown.
The n = 8 state for Na+ does not contribute substantially to the partition sum in Eq. 7. Relative to the n = 5 state, the more probable n = 6 state contributes e−2 ≈ 10−1 and the nearly equiprobable n = 7 state contributes ∼e−10 ≈ 5 × 10−5 to the partition sum in Eq. 7 (Figs. 2 and 3). For K+, relative to the n = 7 state, the more probable n = 8 state contributes ∼e−6 ≈ 2 × 10−3 to the partition sum. The substate distributions in Fig. 4 provide a more visual representation of these quantitative observations.
Observe that p(n) alone is not sufficient to infer the contribution of that n-coordinate state to the thermodynamics of the ion. Further, for the chosen λ-values, coordination states with n smaller than the most probable value for that λ-value dominate the thermodynamics of the ion, suggesting that the preference for states with a low value of n must be related to the behavior of the coordinating ligands. We expect that in states with a low value of n the coordinating ligands will experience weaker energetic (less unfavorable) interactions among themselves and vice versa. The joint probability distribution P(ɛ, UCO–CO) (Fig. 5) confirms this expectation: the ion-site binding energy (ɛ) and the excess internal energy of the site (UCO–CO) are inversely correlated.
Figure 5.
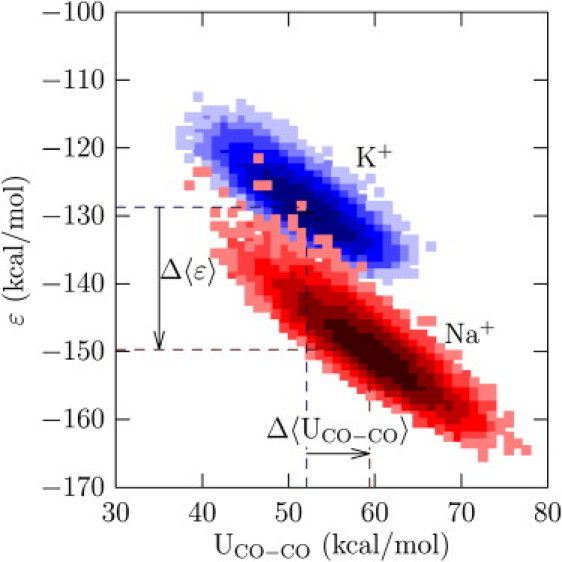
Density plot of the joint probability distribution log P(ɛ, UCO–CO). The value ɛ is the binding energy of the ion with the eight carbonyl ligands comprising the S2 site (see description of A3 in Methods). The quantity UCO–CO is the interaction energy (the excess internal energy) of the eight carbonyl ligands comprising the S2 site. The high-ɛ ion-site interactions (top-left corner of the density plot) occur for less unfavorable site-site interactions. The states are normalized relative to the most frequently observed states and the dashed lines locate the mean values along the given axis. Every factor-of-e change in density (relative to the mean) corresponds to a change in shade. Δ〈ɛ〉 = −21.0 kcal/mol and Δ〈UCO–CO〉 = 7.3 kcal/mol.
The high-ɛ states of the ion are correlated with the states of low ligand-ligand interaction energies in the binding site. However, it is these high-ɛ states that are pertinent to the thermodynamics of the ion (Figs. 3 and 4). Further, the dispersion in energies along the ɛ-axis is correlated with the dispersion in energies along the UCO–CO axis. But the dispersion in ɛ—the variance in a Gaussian model, for example—determines the fluctuation contribution to the excess chemical potential of the ion (Eqs. 3 and 4). This fluctuation contribution is larger with Na+ than with K+ in the site. Thus, from the perspective of the ion-binding site, Na+ energetically strains the binding site much more than K+, the ion for which the site evolved. (See also (8).)
By considering entropic effects in ion binding to the site, we can quantify the correlation in Fig. 5. Following Asthagiri et al. (32) and ignoring pressure-volume effects we have, for the process of adding one ion to the (ion-free) binding site,
| (8) |
since (S2), the excess internal energy of the ion-site complex, comprises the average ion-site interaction, 〈ɛ〉X, and the average site-site interaction, 〈UCO–CO〉X. (We ignore the role of material outside the ion-site complex; see description of A3 in Methods.) Here the excess internal energy is relative to the state when the ligands are infinitely far apart. If the excess energy is measured relative to the energy of the site without the ion, then the first two members of Eq. 8 acquire an additional constant. Also a factor of kBT is required in the first two members of Eq. 8 (see also Eq. 8 in (32)). But in comparing Na+ with K+, these constant factors cancel.
Coupling parameter transformation of K+ to Na+ in the S2 site considering the ion-carbonyl interactions solely (see description of A3 in Methods) shows that the S2 site is selective for K+ (Table 2). Mean energies alone are insufficient to predict selectivity for K+; the fluctuation contribution is decidedly important: the net selectivity of the site for K+ is nearly equal to the fluctuation contribution. Following Eq. 8, this fluctuation contribution is seen to arise largely from the change in the mean site-site energy (Δ〈UCO–CO〉 = 7.3 kcal/mol) that is balanced somewhat by entropic effects (TΔsex(S2) = 2.2 kcal/mol). (The small magnitude of the entropic effects is consistent with the earlier study by (7) on a minimal model.) Thus selectivity arises due to energy fluctuations and these fluctuations are intimately tied to the site-site interactions (Fig. 5 and Table 2).
Table 2.
Thermodynamic quantities for the coupling transformation of K+ to Na+ in the S2 and W8 sites
| S2 | W8 | ||
|---|---|---|---|
| Δμex | 4.8 | 1.0 | |
| Δ〈ɛ〉 – Δμex(aq) | −0.3 | −2.0 | |
| Δ〈UL–L〉 | 7.3 | 4.3 | |
| TΔsex(Site) | 2.2(1.9) | 1.3 | |
| 5.1 | 3.0 |
The quantity Δμex(S2) is obtained by considering only the interaction between the ion and the eight carbonyl ligands in the S2 site (see description of A3 in Methods). The quantity Δμex(W8) is obtained by including the interaction of the ion with all the water molecules in the simulation cell, but with the added constraint that eight (and only eight) water molecules be within 3.5 Å of the ion (the W8 site). For the W8 site, Δ〈UL–L〉 is based only on the eight water molecules in the site. The entropic contribution is obtained from Eq. 8. The value in parenthesis is obtained using coupling parameter integration for the difference in excess entropies. The close agreement between these different estimates provides an additional check on the energy estimates. The quantities Δμex(aq) = −20.7 kcal/mol (by molecular dynamics) and Δμex(aq) = −20.1 kcal/mol (by Monte Carlo) are used as reference values for the S2 and W8 sites, respectively. Site = S2 or W8 and L = CO or W(≡ H2O). All values are in kcal/mol.
To study how these fluctuation contributions change if water is used as a ligand, we consider a spherical volume in bulk water that is restricted to contain eight water molecules. Using Monte Carlo simulations (S. Merchant and D. Asthagiri, unpublished), the free energy of hydrating an ion in the bulk and in this site (denoted by W8) is obtained using Gaussian quadrature estimates for the electrostatic contribution (33) and the histogram overlap approach for the nonelectrostatic contribution (16,32). For the difference in hydration free energies between Na+ and K+, this approach gives a value in good agreement with that obtained using molecular dynamics and coupling parameter integration (Table 2).
Calculations show that the W8 site is substantially less selective for K+ than the S2 site (Table 2). Relative to the difference in bulk hydration free energies, mean binding energy favors Na+, more so than in the S2 site (Table 2 and Fig. 6). Selectivity for K+ emerges primarily due to the unfavorable change in mean site-site energy (Δ〈UW–W〉 = 4.3 kcal/mol), balanced by a small change in excess entropy (TΔsex(W8) = 1.3 kcal/mol). Comparing the S2 and W8 sites, we observe that the change in the average site-site interactions is substantially more when the site is composed of carbonyl ligands than when it is composed of water molecules (Table 2 and Figs. 5 and 6).
Figure 6.
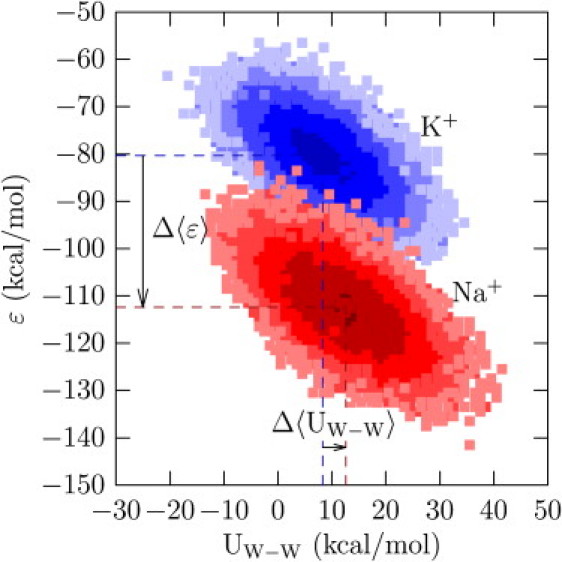
Density plot of the joint probability distribution log P(ɛ, UW–W), where W ≡ H2O. The value ɛ is the binding energy of the ion with the eight water molecules in a sphere of radius 3.5 Å embedded in bulk water. The quantity UW–W is the interaction energy (the excess internal energy) of the ligands comprising the site. Δ〈ɛ〉 = −22.1 kcal/mol and Δ〈UW–W〉 = 4.3 kcal/mol. Rest as in Fig. 5.
Figs. 5 and 6 and the analysis above suggest the importance of ligand type on selectivity. But a strict comparison is confounded by the presence of the protein around S2 (and the bulk around W8). To secure a more direct comparison, we pursue Monte Carlo simulations on a minimal model with eight carbonyl or water ligands restricted to a 3.5 Å sphere with the ion at the center. (For conciseness we quote energies relative to the corresponding value in the minimal model with water.) We find that the carbonyl cluster is more selective for K+ by 3.9 kcal/mol, of which 3.2 kcal/mol results from mean energy changes, 1.4 kcal/mol results from changes in ligand-ligand interactions, and 0.7 kcal/mol results from entropic effects. These results agree with the conclusions based on the S2 and W8 systems. Relative to water as the ligand, and for the given coordination state of n = 8, the site-site unfavorable interaction is enhanced with a carbonyl ligand. Since the ion-site energy fluctuation is tied to these site-site interactions, selectivity is amplified with carbonyl as a ligand.
An earlier study (11) had concluded that a site with eight water molecules confined in a spherical cavity (in bulk water) is selective for K+ by ∼4.0 kcal/mol, substantially higher than the 1 kcal/mol obtained above. (See Fig. 3 B in (11); slightly different coordination radii are used for Na+ and K+, but that distinction appears small in this context. Further, following the analysis in (11), with the parameters used here a value of ∼4.5 kcal/mol, instead of the 4.0 kcal/mol noted above, is obtained.) We suggest a possible reason for this difference. In Bostick and Brooks (11), the logarithm of the ratios of the probabilities of observing the most probable and eight-coordinate states (for the aqueous ions) is equated to the free energy of transferring the ion from bulk water to an eight-coordinate state. But note that both p(n) and μex(n) jointly define the chemical potential of the ion (Eq. 7) and the most probable state need not solely determine the thermodynamics in the bulk. In particular, in the notation developed here, the free energy change in transferring an ion from the bulk to a site with eight water molecules is given by (n = 8) – (aq). This perhaps explains the difference between the earlier study (11) and this work.
The analysis above shows that increasing the coordination has the effect of increasing the chemical potential of the ion, more so for the smaller Na+ ion. The importance of number of ligands was noted before (5,7–11). Equation 8 and the results above explain this from the perspective of the ion-binding site. In the presence of the smaller Na+, unfavorable ligand-ligand interaction energy increases and this shifts the selectivity toward the larger K+. The same analysis transparently reveals the importance of ligand type, consistent with observations made before ((5,7,8) and see also (9)).
Conclusions
Ion-selectivity in the S2 site of the ion channel arises due to the balance of two effects: ion-site interactions and site-site interactions. For the S2 site, mean ion-site interactions are not sufficient to quantify selectivity. Relative to K+, the smaller Na+ causes the site-site interaction energy to increase, and this is a key factor in the selectivity for K+. But, relative to K+, the entropy of Na+ binding to the site is small, because of compensation between binding-energy fluctuations and matrix strain associated with ligand-ligand repulsions. Comparing the S2 site and a model site comprising eight water molecules embedded in bulk water shows that increasing the coordination of the smaller Na+ ion does shift the selectivity toward K+. But this effect alone does not explain the magnitude of selectivity observed in the S2 site. The chemical nature of the ligands comprising the binding site is important in explaining the observed magnitude of selectivity.
Acknowledgments
We thank L. R. Pratt, M. E. Paulaitis, and B. Roux for helpful discussions.
Financial support from National Science Foundation grant No. 0736000 is gratefully acknowledged.
References
- 1.Latorre R., Miller C. Conduction and selectivity in potassium channels. J. Membr. Biol. 1983;71:11–30. doi: 10.1007/BF01870671. [DOI] [PubMed] [Google Scholar]
- 2.Doyle D.A., Cabral J.M., Pfuetzner R.A., Kuo A., Gulbis J.M. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 3.Allen T.W., Bliznyuk A., Rendell A.P., Kuyucak S., Chung S.H. The potassium channel: structure, selectivity and diffusion. J. Chem. Phys. 2000;112:8191–8204. [Google Scholar]
- 4.Luzhkov V.B., Åqvist J. K+/Na+ selectivity of the KcsA potassium channel from microscopic free energy perturbation calculations. Biochim. Biophys. Acta. 2001;1548:194–202. doi: 10.1016/s0167-4838(01)00213-8. [DOI] [PubMed] [Google Scholar]
- 5.Noskov S.Y., Bernéche S., Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–834. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Y., Morais-Cabral J.H., Kaufman A., MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 7.Noskov S.Y., Roux B. Ion selectivity in potassium channels. Biophys. Chem. 2006;124:279–291. doi: 10.1016/j.bpc.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 8.Noskov S.Y., Roux B. Importance of hydration and dynamics on the selectivity of the KcsA and NaK channels. J. Gen. Physiol. 2007;129:135–143. doi: 10.1085/jgp.200609633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varma S., Rempe S.B. Tuning ion coordination architectures to enable selective partitioning. Biophys. J. 2007;93:1093–1099. doi: 10.1529/biophysj.107.107482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas M., Jayatilaka D., Corry B. The predominant role of coordination number in potassium channel selectivity. Biophys. J. 2007;93:2635–2643. doi: 10.1529/biophysj.107.108167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bostick D.L., Brooks C.L., III Selectivity in K+ channels is due to topological control of the permeant ion's coordinated state. Proc. Natl. Acad. Sci. USA. 2007;104:9260–9265. doi: 10.1073/pnas.0700554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asthagiri D., Pratt L.R., Paulaitis M.E. Role of fluctuations in a snug-fit mechanism of KcsA channel selectivity. J. Chem. Phys. 2006;125:024701. doi: 10.1063/1.2205853. [DOI] [PubMed] [Google Scholar]
- 13.Hummer G., Pratt L.R., Garcia A.E. Multistate Gaussian model for electrostatic solvation free energies. J. Am. Chem. Soc. 1997;119:8523–8527. [Google Scholar]
- 14.Widom B. Potential-distribution theory and the statistical mechanics of fluids. J. Phys. Chem. 1982;86:869–872. [Google Scholar]
- 15.Beck T.L., Paulaitis M.E., Pratt L.R. Cambridge University Press; Cambridge, UK: 2006. The Potential Distribution Theorem and Models of Molecular Solutions. [Google Scholar]
- 16.Pratt L.R., Asthagiri D. Potential distribution methods and free energy models of molecular solutions. In: Chipot C., Pohorille A., editors. Free Energy Calculations: Theory and Applications in Chemistry and Biology. Chapt. 9, of Springer Series in Chemical Physics. Vol. 86. Springer; New York: 2007. [Google Scholar]
- 17.Kubo R. Generalized cumulant expansion method. J. Phys. Soc. Jpn. 1962;17:1100–1120. [Google Scholar]
- 18.Stillinger F.H., Weber T.A. Hidden structures in liquids. Phys. Rev. A. 1982;25:978–989. [Google Scholar]
- 19.Deng Y., Roux B. Computation of binding free energy with molecular dynamics and grand canonical Monte Carlo simulations. J. Chem. Phys. 2008;128:115103. doi: 10.1063/1.2842080. [DOI] [PubMed] [Google Scholar]
- 20.Azzalini A. A class of distributions which includes the normal ones. Scand. J. Stat. 1985;12:171–178. [Google Scholar]
- 21.MacKerell A.D., Jr., Bashford D., Bellott M., Dunbrack R.L., Jr., Evanseck J.D. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 22.Kale L., Skeel R., Bhandarkar M., Brunner R., Gursoy A. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999;151:283–312. [Google Scholar]
- 23.Humphrey W., Dalke A., Schulten K. VMD—visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 24.Bernéche S., Roux B. Molecular dynamics of the KcsA K+ channel in a bilayer membrane. Biophys. J. 2000;78:2900–2917. doi: 10.1016/S0006-3495(00)76831-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jorgensen W., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 26.Neria E., Fischer S., Karplus M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996;105:1902–1921. [Google Scholar]
- 27.Martyna G.J., Tobias D.J., Klein M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 28.Feller S.E., Zhang Y., Pastor R.W., Brooks B.R. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 29.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 30.Tieleman D.P., Biggin P.C., Smith G.R., Sansom M.S.P. Simulation approaches to ion channel structure-function relationships. Q. Rev. Biophys. 2001;34:473–561. doi: 10.1017/s0033583501003729. [DOI] [PubMed] [Google Scholar]
- 31.Shrivastava I.H., Tieleman D.P., Biggin P.C., Sansom M.S.P. K+ versus Na+ in a K channel selectivity filter: a simulation study. Biophys. J. 2002;83:633–645. doi: 10.1016/s0006-3495(02)75197-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asthagiri D., Merchant S., Pratt L.R. Role of attractive methane-water interactions in the potential of mean force between methane molecules in water. J. Chem. Phys. 2008;128:244512. doi: 10.1063/1.2944252. [DOI] [PubMed] [Google Scholar]
- 33.Hummer G., Szabo A. Calculation of free-energy differences from computer simulations of initial and final states. J. Chem. Phys. 1996;105:2004–2010. [Google Scholar]