

Abstract
It is recognized that an ideal anti-cocaine treatment is to accelerate cocaine metabolism by producing biologically inactive metabolites via a route similar to the primary cocaine-metabolizing pathway, i.e., butyrylcholinesterase (BChE)-catalyzed hydrolysis of cocaine. BChE mutants with a higher catalytic activity against (-)-cocaine are highly desired for use as an exogenous enzyme in humans. To develop a rational design for high-activity mutants, we carried out free-energy perturbation (FEP) simulations on various mutations of the transition-state structures in addition to the corresponding free-enzyme structures by using an extended FEP procedure. The FEP simulations on the mutations of both the free-enzyme and transition-state structures allowed us to calculate the mutation-caused shift of the free-energy change from the free enzyme (BChE) to the transition state, and thus to theoretically predict the mutation-caused shift of the catalytic efficiency (kcat/KM). The computational predictions are supported by the kinetic data obtained from the wet experiments, demonstrating that the FEP-based computational design approach is promising for rational design of high-activity mutants of an enzyme. One of the BChE mutants designed and discovered in this study has an ∼1800-fold improved catalytic efficiency against (-)-cocaine compared to wild-type BChE. The high-activity mutant may be therapeutically valuable.
Introduction
Cocaine is well known as the most reinforcing drug of abuse (1–3). Currently, there is no anti-cocaine medication available. The disastrous medical and social consequences of cocaine addiction have made the development of an anti-cocaine medication a high priority (4,5). Unfortunately, the classical pharmacodynamic approach, i.e., designing small molecules to target the related transporters or receptors in the central nervous system (CNS), has failed to yield a really useful antagonist because of the difficulties inherent in blocking a blocker like cocaine (1–4,6). An alternative approach is to interfere with the delivery of cocaine to its receptors or accelerate its metabolism in the body (4). An ideal anti-cocaine medication would be to accelerate cocaine metabolism, producing biologically inactive metabolites via a route similar to the primary cocaine-metabolizing pathway, i.e., cocaine hydrolysis catalyzed by plasma enzyme butyrylcholinesterase (BChE) (4,7–11). However, wild-type BChE has a low catalytic efficiency against naturally occurring (-)-cocaine (12–16). Here we report a new computational design and discovery of a high-activity mutant of human BChE with an ∼1800-fold improved catalytic efficiency against (-)-cocaine, which could be used as an exogenous enzyme in humans to prevent (-)-cocaine from reaching the CNS. The results not only provide an encouraging new candidate for anti-cocaine medication, they also demonstrate that the computational protocol used in this study is a promising technique for rational enzyme redesign and drug discovery.
The primary pathway for cocaine metabolism in primates is hydrolysis at the benzoyl ester or methyl ester group (4,11). Benzoyl ester hydrolysis generates ecgonine methyl ester (EME), whereas methyl ester hydrolysis yields benzoylecgonine (BE). The major cocaine-metabolizing enzymes in humans are butyrylcholinesterase (BChE), which catalyzes benzoyl ester hydrolysis, and two liver carboxylesterases (denoted hCE-1 and hCE-2), which catalyze hydrolysis at the methyl ester and the benzoyl ester, respectively. Among the three, BChE is the principal cocaine hydrolase in human serum. Hydrolysis accounts for ∼95% of cocaine metabolism in humans. The remaining 5% is deactivated through oxidation by the liver microsomal cytochrome P450 system, producing norcocaine (4,17). EME appears to be the least pharmacologically active of the cocaine metabolites and may even cause vasodilation (4), whereas both BE and norcocaine appear to cause vasoconstriction and lower the seizure threshold, similarly to cocaine itself. Norcocaine is hepatotoxic and a local anesthetic (4). Thus, hydrolysis of cocaine at the benzoyl ester by BChE is the most suitable metabolic pathway for amplification.
Generally speaking, for rational design of an enzyme mutant with a higher catalytic activity for a given substrate, one must design a mutation that can accelerate the rate-determining step of the entire catalytic reaction process while at the same time the other steps are not slowed down by the mutation. Reported computational modeling and experimental data indicate that the formation of the prereactive BChE-(-)-cocaine complex (ES) is the rate-determining step of (-)-cocaine hydrolysis (Fig. 1) catalyzed by wild-type BChE (12,13,18–20), whereas the rate-determining step of the unnatural, biologically inactive (+)-cocaine hydrolysis catalyzed by the same enzyme is a chemical reaction process consisting of four individual reaction steps (20,22). Based on this mechanistic understanding, previous studies on the rational design of BChE mutants focused on how to improve the ES formation process, and several BChE mutants (12,13,21) were found to have an ∼9- to 34-fold improved catalytic efficiency (kcat/KM) against (-)-cocaine. Recent findings from computational modeling also suggest that the formation of the prereactive BChE-(-)-cocaine complex (ES) is hindered mainly by some amino acid residues in wild-type BChE, but this hindrance can be removed by certain mutations on those residues (13). Combined computational and experimental data (12,19,22–25) reveal that the rate-determining step of (-)-cocaine hydrolysis catalyzed by the A328W/Y332A and A328W/Y332G mutants is the first step in the chemical reaction process. Therefore, starting from the A328W/Y332A or A328W/Y332G mutant, we sought to further improve the catalytic efficiency of BChE against (-)-cocaine by decreasing the energy barrier for the first reaction step without significantly affecting the ES formation and other chemical reaction steps (22,24,26,27). Rational design of BChE mutants starting from the A328W/Y332A or A328W/Y332G mutant has led to the discovery of several BChE mutants that have a considerably improved catalytic efficiency against (-)-cocaine (18,22,24,26,27). As confirmed by an independent group of researchers (28), our previously reported high-activity mutant, A199S/S287G/A328W/Y332G BChE (24), can indeed selectively block cocaine toxicity and reinstatement of drug seeking in rats.
Figure 1.
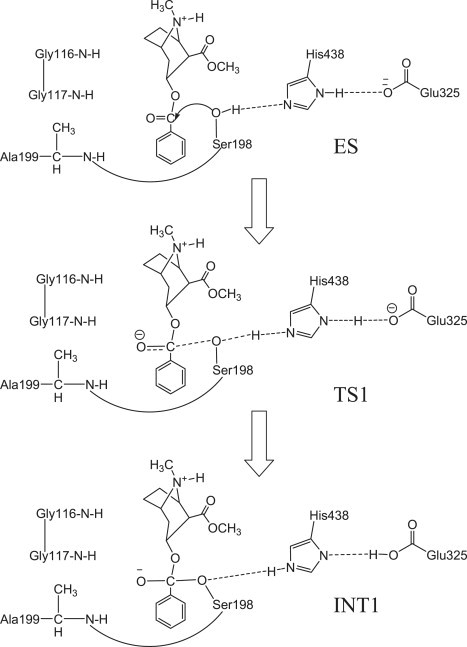
Schematic representation of the first reaction step of the chemical reaction process for (-)-cocaine hydrolysis catalyzed by BChE.
In the study presented here, the computational design of new BChE mutants started from the A328W mutant instead of the A328W/Y332A mutant. This is because the A328W mutant has a higher catalytic efficiency than the A328W/Y332A mutant against (-)-cocaine. The A328W mutant (12) has a ∼17.7-fold improved catalytic efficiency compared to the wild-type, whereas the A328W/Y332A mutant has a ∼9.4-fold improved catalytic efficiency compared to the wild-type. Our design protocol is based on examination of the overall structure of the protein combined with an extension of the well-known free-energy perturbation (FEP) approach (29,30) to predict the catalytic activity change caused by a mutation on an enzyme. FEP can be performed to evaluate the free-energy change caused by a small structural perturbation. In principle, it can only be used to simulate the change of a stable structure associated with a local minimum on the potential energy surface (31–35). By using our recently developed computational strategy that is appropriate for transition-state simulation (i.e., with the lengths of transition bonds frozen) (22,24,26,27), we were also able to simulate the mutations of the rate-determining transition-state structure (in addition to the free-enzyme structure) by using the FEP procedure. In contrast to previous FEP calculations concerning the effects of protein mutation on catalytic efficiency of enzymes (36–38), FEP simulations on mutations of both the free-enzyme and transition-state structures allow us to calculate the mutation-caused shift of the free-energy change from the free enzyme (BChE) to the rate-determining transition state (i.e., TS1; see Fig. 1) (26), and thus to theoretically evaluate/estimate the mutation-caused shift of the catalytic efficiency (kcat/KM). The computational predictions are consistent with the kinetic data obtained from the wet experiments, demonstrating that the FEP-based computational design approach is a promising technique for the rational design of high-activity mutants of an enzyme.
Materials and Methods
Computational studies
Before conducting FEP simulations on each mutation, we first performed molecular-dynamics (MD) simulations on the unperturbed free-enzyme and TS1 structures. The initial structures of both the free-enzyme and transition-state TS1 used in MD simulations were prepared based on our previous MD simulations (24,25,27) on the structures of wild-type BChE and its mutants, which were derived from the x-ray crystal structure (39) deposited in the Protein Data Bank (40) with PDB code 1P0P. Each MD simulation was carried out for at least 1 ns or longer until a stable MD trajectory was obtained. The FEP simulations were carried out starting from the MD-simulated structures of the unperturbed systems.
The schematic structure of the transition state TS1 is shown in Fig. 1. Details of the determination of the TS1 structure were described previously (26,27). Briefly, the key features of the TS1 structure involve the partially formed and partially broken covalent bonds, i.e., transition bonds denoted in this article formed within the catalytic triad (including Ser198, Glu325, and His438) and the transition bond between the hydroxyl oxygen of Ser198 side chain and a carbonyl carbon of the cocaine. The transition bonds were restrained by defining the bond length parameters of new atom types of specific atoms in the TS1 structure based on the geometry obtained from the ab initio reaction coordinate calculations on the cocaine hydrolysis catalyzed by wild-type BChE (20). The partial charges of the cocaine atoms in the TS1 structures were calculated by using the RESP protocol implemented in the antechamber module of the AMBER 7 package (41) after electrostatic potential (ESP) calculations at the ab initio HF/6-31G∗ level using the GAUSSIAN 03 program (42). The geometries used in the ESP calculations were obtained from the previous ab initio reaction coordinate calculations (20). The charges of the residue atoms of the TS1 structure were from the standard AMBER force field used in the AMBER 7 package. The details of the parameter development, the force-field parameters of the unusual atom types in the TS1 structure, and the charges of the cocaine atoms are provided in the Supporting Material (Fig. S1).
All of the MD simulations were performed with the use of the Sander module of Amber7 package (41), using the same procedures as described in our previous computational studies (24,27). For free-enzyme structures, the +1 charge of the enzyme was neutralized by adding one chloride counterion. For the TS1 structure, the +2 charge of the system was neutralized by adding two chloride counterions. Both the free-enzyme and TS1 structures were solvated in a rectangular box of TIP3P water molecule (43) with a solute-wall distance of 10 Å. The solvated systems were carefully equilibrated and fully energy-minimized. We gradually heated these systems from T = 10 K to T = 298.15 K in 40 ps before running the MD simulation at T = 298.15 K for 1 ns or longer, making sure that we obtained a stable MD trajectory for each of the simulated structures. The time step used for the MD simulations was 2 fs. A periodic boundary condition was used in the NPT ensemble at T = 298.15 K with Berendsen temperature coupling and P = 1 atm with isotropic molecular-based scaling (44). The SHAKE algorithm (45) was used to fix all covalent bonds containing hydrogen atoms. The nonbonded pair list was updated every 25 steps. The particle mesh Ewald (PME) method (46) was used to treat long-range electrostatic interactions, and 10 Å was used as the nonbonded cutoff.
The outcomes of the FEP simulations on the mutations of both the free-energy and TS1 structures can be used to predict the mutation-caused changes in the catalytic efficiency for BChE-catalyzed hydrolysis of (-)-cocaine. Depicted in Fig. 2 are the free-energy changes associated with two reaction systems: one is the (-)-cocaine hydrolysis catalyzed by a BChE mutant (or the wild-type), denoted by enzyme 1 or E(1); the other is the (-)-cocaine hydrolysis catalyzed by another BChE mutant, denoted by enzyme 2 or E(2). As shown in Fig. 2, the catalytic efficiency, i.e., kcat(i)/KM(i), for the (-)-cocaine hydrolysis catalyzed by enzyme E(i) is determined by the Gibbs free-energy change ΔG(i) of the reaction system from E(i) plus substrate S, i.e., (-)-cocaine, to the corresponding rate-determining transition state TS1(i). ΔG(i) is the sum of the enzyme-substrate binding free energy ΔGES(i) and the activation free energy ΔGav(i):
| (1) |
When a mutation on an amino acid residue can change the enzyme from E(1) to E(2), we want to know the corresponding free-energy change from ΔG(1) to ΔG(2) for the computational design of the high-activity mutants of the enzyme. There are two possible paths to determine the free-energy change ΔΔG(1→2) ≡ ΔG(2) − ΔG(1). One path is to directly calculate ΔGES(i) and ΔGav(i) associated with E(1) and E(2), which is very computationally demanding in terms of the level of theory. For an alternative path, the relatively less-demanding FEP simulations allow us to estimate ΔΔG(1→2) by determining the free-energy changes ΔGE and ΔGTS1 from E(1) to E(2):
| (2) |
where ΔGE and ΔGTS1 are the free-energy changes from E(1) to E(2) for the free enzyme and TS1, respectively. ΔGE and ΔGTS1 were estimated by performing the FEP simulations in the study presented here. By using the calculated ΔΔG(1→2), the ratio of the catalytic efficiency associated with E(2) to that associated with E(1) can be evaluated via:
| (3) |
Figure 2.
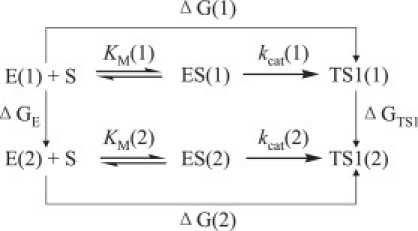
The relationship between different free-energy changes for the reactions catalyzed by two enzymes. E(i) is the free enzyme, ES(i) represents the enzyme-substrate complex, and TS1(i) refers to the transition state. ΔG(i) is the sum of the enzyme-substrate binding free energy ΔGES(i) and the activation free energy ΔGav(i) (i = 1, 2).
Equation 3 can also be used to derive the experimental ΔΔG(1→2) value from the experimental ratio of the catalytic efficiency associated with E(2) to that associated with E(1).
We tested the above FEP-based computational approach for various mutations, including those associated with the following enzyme changes:
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
The mutation A199S associated with the BChE mutant change from A328W to A328W/A199S increases the size of the side chain, whereas the other mutations all decrease the size of the side chain. For technical reasons, it is always more convenient to perform an FEP simulation on the change from a larger side chain to a smaller side chain. So, for A328W → A328W/A199S, we actually carried out the FEP simulation from A328W/A199S to A328W and used the following energy relationship:
| (9) |
For the diminishing atoms during the perturbation simulation from a larger side chain to a smaller side chain, the dummy atoms were added to the perturbed residue to keep the number of atoms constant. Thus, some normal atoms in the starting residue were gradually mutated to the dummy atoms in the perturbed residue. For the dummy atoms, a new atom type, DH, was given to the diminished hydrogen, and atom type DC was given to the diminished carbon atom in the mutations F227A and E441D. The charges and the nonbond parameters of the dummy atoms were set to zero so that they would not have electrostatic or van der Waals interactions with other atoms. At the same time, all of the bond and angle parameters involving the dummy atoms were the same as their counterparts in the initial structure to keep the structural skeletons unchanged. The residue structures before and after perturbations, and the force-field parameters of the dummy atoms are provided in the Supporting Material. The default choice (INTPRT = 0) was used to make sure that the bonded interactions of the dummy atoms would be excluded from the final calculation on the total energy of the perturbed system. The FEP simulations (with a time step of 1 fs) were carried out by using the “fixed width window growth” method implemented in the Gibbs module of AMBER7 (41).
To enlarge the phase space searched by the FEP calculation, for each perturbation, 10 different conformations were extracted from the stable MD trajectory with an interval of 100 ps (one snapshot per 100 ps). The equally distributed 10 snapshots of the simulated structure within the stable MD trajectory were used as the initial structures of the perturbation simulations. The finally calculated ΔGTS1 or ΔGE value is the average of the ΔGTS1 or ΔGE values associated with the initial structures. Each initial structure was first energy-minimized for 1000 cycles followed by 40 ps MD simulation for the heating and equilibration to obtain a better starting structure for the FEP calculation. For FEP calculations on all mutations except two, we used 51 windows (Δλ = 0.02) with 1250 steps of equilibration and 1250 steps for data collection, with both forward and backward directions in each FEP calculation. The exceptions were the enzyme changes A328W/A199S→A328W/A199S/F227A and A328W/A199S/F227A→A328W/A199S/F227A/E441D. For the mutation F227A or E441D in these two enzyme changes, 101 windows (Δλ = 0.01) were used to obtain a more stable FEP performance. Thus, for the FEP simulation on each mutation, we performed the MD simulations for a total of 2500 × 51 × 10 = 1,275,000 (or 2500 × 101 × 10 = 2,525,000) steps or 1.275 (or 2.525) ns.
Experimental studies
Cloned pfu DNA polymerase and Dpn I endonuclease were obtained from Stratagene (La Jolla, CA). [3H](-)-cocaine (50 Ci/mmol) was purchased from PerkinElmer Life Sciences (Boston, MA). The expression plasmid pRc/CMV was a gift from Dr. O. Lockridge, University of Nebraska Medical Center (Omaha, NE). All oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA). QIAprep spin plasmid miniprep, Qiagen plasmid purification, and QIAquick PCR purification kits were obtained from Qiagen (Santa Clarita, CA). Human embryonic kidney 293T/17 cells were obtained from ATCC (Manassas, VA). Dulbecco's modified Eagle's medium (DMEM) was purchased from Fisher Scientific (Fairlawn, NJ). 3, 3′, 5, 5′-Tetramethylbenzidine (TMB) was obtained from Sigma (St. Louis, MO). Anti-butyrylcholinesterase (mouse monoclonal antibody, product No. HAH002-01) was purchased from AntibodyShop (Gentofte, Denmark), and goat anti-mouse IgG HRP conjugate was obtained from Zymed (San Francisco, CA).
Site-directed mutagenesis of human BChE cDNA was performed by means of the QuikChange method (47). Mutations were generated from wild-type human BChE in a pRc/CMV expression plasmid (48). Using plasmid DNA as the template and primers with specific basepair alterations, mutations were made by polymerase chain reaction (PCR) with Pfu DNA polymerase, for replication fidelity. The PCR product was treated with Dpn I endonuclease to digest the parental DNA template. Modified plasmid DNA was transformed into Escherichia coli, amplified, and purified. The DNA sequences of the mutants were confirmed by DNA sequencing. BChE mutants were expressed in human embryonic kidney cell line 293T/17. Cells were grown to 80% to 90% confluence in six-well dishes and then transfected by Lipofectamine 2000 complexes of 4 μg plasmid DNA per each well. Cells were incubated at 37°C in a CO2 incubator for 24 h; they were then moved to a 60-mm culture vessel and cultured for 4 more days. The culture medium (10% fetal bovine serum in DMEM) was harvested for a BChE activity assay. To measure (-)-cocaine and benzoic acid, the product of (-)-cocaine hydrolysis catalyzed by BChE, we used sensitive radiometric assays based on toluene extraction of [3H]-(-)-cocaine labeled on its benzene ring (49). In brief, to initiate the enzymatic reaction, 100 nCi of [3H]-(-)-cocaine was mixed with 100 μL of culture medium. The enzymatic reactions proceeded at room temperature (25°C) with varying concentrations of (-)-cocaine. The reactions were stopped by adding 300 μL of 0.02 M HCl, which neutralized the liberated benzoic acid while ensuring a positive charge on the residual (-)-cocaine. [3H]benzoic acid was extracted by 1 mL of toluene and measured by scintillation counting. Finally, the measured (-)-cocaine concentration-dependent radiometric data were analyzed by using the standard Michaelis-Menten kinetics so that the catalytic parameters (kcat and KM) were determined along with the use of an enzyme-linked immunosorbent assay (ELISA) (50) as described below.
The ELISA buffers used in this study were the same as those described in the literature (50). The coating buffer was 0.1 M sodium carbonate/bicarbonate buffer, pH 9.5. The diluent buffer (EIA buffer) was potassium phosphate monobasic/potassium phosphate dibasic buffer, pH 7.5, containing 0.9% sodium chloride and 0.1% bovine serum albumin. The washing buffer (PBS-T) was 0.01 M potassium phosphate monobasic/potassium phosphate dibasic buffer, pH 7.5, containing 0.05% (v/v) Tween-20. All of the assays were performed in triplicate. Each well of an ELISA microtiter plate was filled with 100 μL of a mixture buffer consisting of 20 μL culture medium and 80 μL coating buffer. The plate was covered and incubated overnight at 4°C to allow the antigen to bind to the plate. The solutions were then removed and the wells were washed four times with PBS-T. The washed wells were filled with 200 μL diluent buffer and shaken for 1.5 h at room temperature (25°C). After the wells were washed four times with PBS-T, they were filled with 100 μL antibody (1:8,000) and incubated for 1.5 h, followed by four more washings. Then the wells were filled with 100 μL goat anti-mouse IgG HRP conjugate complex diluted to a final 1:3000 dilution, incubated at room temperature for 1.5 h, and washed four more times. The enzyme reactions were started by addition of 100 μL substrate (TMB) solution (50). The reactions were stopped after 15 min by the addition of 100 μL of 2 M sulfuric acid, and the absorbance was read at 460 nm using a Bio-Rad ELISA plate reader.
Results and Discussion
To predict the ΔΔG(1→2) values and the catalytic efficiency changes for the mutations A199S, F227A, E441D, S287A, and A287G associated with enzyme changes (4)–(8) or the change from the A328W mutant to the A328W/A199S/F227A/E441D/S287G mutant of BChE, we performed MD simulations on both the free enzyme and TS1 structures of mutants A328W/A199S, A328W/A199S/F227A, A328W/A199S/F227A/E441D, and A328W/A199S/F227A/E441D/S287A. Stable MD trajectories with the simulation time of 1 ns or longer were obtained for these structures. Depicted in Fig. 3 are the MD-simulated TS1 structures of A328W and A328W/A199S/F227A/E441D/S287G mutants of BChE. Indicated in the figure are the locations of mutated residues 199, 227, 287, and 441 relative to cocaine, along with other key residues forming the catalytic triad (residues 198, 325, and 438) and oxyanion hole (residues 116, 117, and 328) in the active site. The yellow dashed lines represent the hydrogen bonds between the oxyanion hole and the carbonyl oxygen of benzoyl ester of cocaine, whereas the white lines refer to the transition bonds. More figures about the time-dependence of some key internuclear distances and the root mean-square deviation (RMSD) of the simulated positions of the protein backbone atoms from those in the initial structure in the MD-simulated TS1 structure are provided in the Supporting Material. The equally distributed 10 snapshots of the MD-simulated free-enzyme or TS1 structure extracted from the stable trajectory with a time interval of 100 ps were used as the starting structures to perform the FEP simulations.
Figure 3.
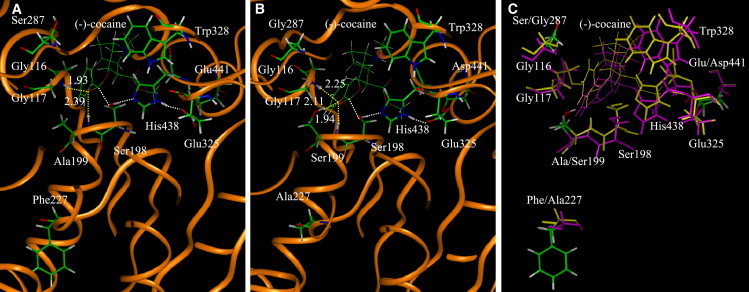
(A) The MD-simulated TS1 structure associated with the A328W mutant. The dashed lines in yellow indicate the hydrogen bonds between (-)-cocaine and the oxyanion hole of the mutant. The simulated average distances are given for the hydrogen bonds. The dashed lines in white refer to the transition bonds. (B) The MD-simulated TS1 structure associated with the A328W/A199S/F227A/E441D/S287G mutant. (C) Superimposition of the two TS1 structures associated with the A328W mutant (yellow) and A328W/A199S/F227A/E441D/S287G mutant (pink).
Important energetic results of the FEP simulations for enzyme changes (4)–(8) are summarized in Table 1. More detailed energetic results obtained from the FEP simulations are provided in the Supporting Material, including the incremental changes of the ΔGTS1 and ΔGE values associated with each mutation as a function of λ parameter (window number), showing the smooth changes of the calculated ΔGTS1 and ΔGE values. In Table 1, the values of the root mean-square fluctuation (RMSF) among the individual energy changes associated with different initial structures are given in parentheses. As shown in Table 1, the absolute values of ΔGTS1 and ΔGE range from 5.0 to 11.1 kcal/mol, whereas the RMSF values range from 0.3 to 0.8 kcal/mol. The RMSF values of our FEP calculations are comparable to previously reported fluctuation values associated with different independent FEP runs on the same system in other computational studies (36,51,52). It should be pointed out that the RMSF values do not necessarily reflect the computational errors of the FEP calculations. In general, the greater the number of starting structures used in FEP simulations, the more reliable are the average ΔGTS1 and ΔGE values obtained. Our previous computational studies (27) demonstrated that the average energetic results calculated by using five different starting structures are close to the corresponding average results calculated by using 10 different starting structures, suggesting that the use of five or 10 different starting structures for FEP calculations on BChE-cocaine systems is adequate.
Table 1.
FEP-calculated free-energy changes (in kcal/mol) in comparison with experimental kinetic data for (-)-cocaine hydrolysis catalyzed by BChE mutants
| Mutation | Calc∗ |
Expt.† |
||
|---|---|---|---|---|
| ΔGE | ΔGTS1 | ΔΔG(1→2) | ΔΔG(1→2) | |
| A328W→A328W/A199S | −9.0 (0.6) | −11.1(0.8) | −2.1 | −1.04 |
| A328W/A199S→A328W/A199S/F227A | 5.9(0.5) | 5.0(0.4) | −0.9 | −0.74 |
| A328W/A199S/F227A→A328W/A199S/F227A/E441D | 7.6(0.3) | 7.5(0.6) | −0.1 | N.D.§ |
| A328W/A199S/F227A/E441D→A328W/A199S/F227A/E441D/S287A | 8.2(0.4) | 7.9(0.5) | −0.3 | N.D.§ |
| A328W/A199S/F227A/E441D/S287A→A328W/A199S/F227A/E441D/ S287G | −5.6(0.4) | −6.5(0.6) | −0.9 | N.D.§ |
| A328W/A199S/F227A →A328W/A199S/F227A/E441D/ S287G‡ | 10.2 | 8.9 | −1.3 | −0.96 |
| Experimental kinetic parameters and total free-energy change from A328W | |||||
|---|---|---|---|---|---|
| Mutant | kcat (min−1) | KM (μM) | kcat/KM (min−1M−1) | Relative kcat/KM∗∗ | ΔΔG†† |
| Wild-type BChE¶ | 4.1 | 4.5 | 9.11 × 105 | 1 | N.A. |
| A328W¶ | 50 | 3.1 | 1.61 × 107 | 17.7 | 0 |
| A199S/A328W‖ | 173 | 1.86 | 9.28 × 107 | 102 | −1.04 |
| A199S/A328W/F227A‖ | 472 | 1.46 | 3.23 × 108 | 355 | −1.78 |
| A328W/A199S/F227A/E441D/S287G‖ | 1326 | 0.81 | 1.64 × 109 | 1800 | −2.74 |
N.A., Not applicable.
ΔGE and ΔGTS1 are the mutation-caused free-energy changes for the free-enzyme and transition states, respectively. The number in parentheses after the ΔGE or ΔGTS1 value refers to the RMSF of the ΔGE or ΔGTS1 values calculated starting from the 10 initial structures.
The experimental ΔΔG(1→2) values were derived from the experimental kcat/KM values.
The theoretical results were derived from the FEP results obtained for the enzyme changes A328W/A199S/F227A → A328W/A199S/F227A/E441D → A328W/A199S/F227A/E441D/S287A → A328W/A199S/F227A/E441D/ S287G.
The experimental data were not determined.
Experimental data from Sun et al. (12).
The kinetic parameters determined in this study.
The relative kcat/KM refers to the ratio of the catalytic efficiency of the BChE mutant to that of the wild-type against (-)-cocaine.
The cumulative ΔΔG derived from experimental kinetic data from the A328W mutant to the mutant under consideration.
As shown in Table 1, the ΔΔG(1→2) values associated with enzyme changes (4)–(8) were all predicted to have a minus sign, which means that the mutations associated with enzyme changes (4)–(8) can all increase the catalytic efficiency of the enzyme against (-)-cocaine. The energetic data summarized in Table 1 can be understood by examining the simulated structures. Residue 199 belongs to the oxyanion hole in the active site that provides a hydrogen-bond-donor environment for the enzyme to form hydrogen bonds with the carbonyl oxygen of benzoyl ester of cocaine during the enzymatic hydrolysis. The ΔΔG(A328W→A328W/A199S) value of −2.1 kcal/mol is mainly attributed to the formation of the additional hydrogen bond between the hydroxyl group of the S199 side chain and the carbonyl oxygen of cocaine in the mutated TS1 structure, as shown in Fig. 3, A and B. Residue F227 is located on the end of the turn between two loops that form the boundary of the active site pocket of the enzyme. The ΔΔG(A328W/A199S→A328W/A199S/F227A) value of −0.9 kcal/mol is due to the indirect effects of the F227A mutation on the active site cavity. After the F227A mutation (from a larger residue to a smaller one), the active site cavity becomes slightly larger so that it can better accommodate (-)-cocaine, which is larger in size than the substrate butyrylcholine. Residue E441 is two residues away from the boundary of the active site pocket. When E441 was mutated to a smaller residue, i.e., from Glu to Asp, the residue slightly shifted further away from the active site pocket such that the overall size of the active site pocket slightly increased during the simulation. Consistent with the effects on the steric interactions, the mutation E441D corresponding to the change A328W/A199S/F227A→A328W/A199S/F227A/E441D slightly decreased the free-energy change by 0.1 kcal/mol. For the ΔΔG(A328W/A199S/F227A/E441D→A328W/A199S/F227A/E441D/S287A) and ΔΔG(A328W/A199S/F227A/E441D/S287A→A328W/A199S/F227A/E441D/S287G values (−0.3 and −0.9 kcal/mol) with a minus sign, residue 287 is close to the benzene ring of (-)-cocaine, and the S287G mutation makes more room and a hydrophobic environment to better accommodate the benzene ring of (-)-cocaine. The slightly enlarged active site cavity of the TS1 structure associated with mutant A328W/A199S/F227A/E441D/S287G relative to mutant A328W can be seen in a figure provided in the Supporting Material.
The computational results predict that the A328W/A199S/F227A/E441D/S287G mutant is expected to have the highest catalytic efficiency within all of the mutants examined in this study. The computational predictions based on the FEP simulations are supported by our experimental kinetic parameters for the mutants. The A328W BChE is known to have a ∼17.7-fold improved catalytic efficiency against (-)-cocaine (12). In the study presented here, we produced the A328W/A199S/F227A/E441D/S287G mutant of human BChE, for the first time to our knowledge, by performing site-directed mutagenesis, protein expression, and activity assays. For comparison, we also produced and tested the A328W/A199S and A328W/A199S/F227A mutants, along with wild-type BChE, by using the same experimental protocol.
The experimental kinetic parameters are summarized in Table 1 for comparison with the calculated free-energy changes. As one can see in Table 1, kcat = 173 min−1 and KM = 1.86 μM for A328W/A199S BChE, kcat = 472 min−1 and KM = 1.46 μM for A328W/A199S/F227A BChE, and kcat = 1326 min−1 and KM = 0.81 μM for A328W/A199S/F227A/E441D/S287G BChE against (-)-cocaine. Thus, compared to wild-type BChE against (-)-cocaine (kcat = 4.1 min−1 and KM = 4.5 μM) (12), A328W/A199S BChE has a ∼102-fold improved catalytic efficiency (kcat/KM), A199S/A328W/F227A BChE has a ∼355-fold improved catalytic efficiency, and A328W/A199S/F227A/E441D/S287G BChE has a ∼1800-fold improved catalytic efficiency. The experimental kinetic data are all qualitatively consistent with the computational predictions based on the FEP simulations.
Conclusions
In summary, this integrated computational-experimental study resulted in a new high-activity mutant of human BChE with an ∼1800-fold improved catalytic efficiency against (-)-cocaine. Using the designed A328W/A199S/F227A/E441D/S287G BChE as an exogenous enzyme in human, when the concentration of this mutant is kept the same as that of wild-type BChE in plasma, the half-life of (-)-cocaine in plasma should be reduced from ∼45−90 min (in the presence of wild-type BChE) to only ∼1.5–3.0 s (in the presence of A328W/A199S/F227A/E441D/S287G BChE). The computational and experimental data also demonstrate that the computational design protocol based on the FEP simulations of transition states is promising for rational design of high-activity mutants of an enzyme. The general computational design protocol may be used to study the effects of a mutation on the activation free energy for any enzymatic reaction, although it is necessary to further test the reliability of the protocol for other enzyme systems. Experimental studies on the mechanisms of enzymatic reactions usually include site-directed mutagenesis on some key amino acid residues to determine the effects of the mutations on the catalytic activity. FEP-based transition-state simulations may provide a convenient computational approach to reliably evaluate such effects by directly comparing them with experimental data.
Acknowledgments
This work was supported by a research grant from the National Institutes of Health (R01 DA013930 to C.-G.Z.). The entire work was performed at the University of Kentucky. W. Yang worked in C.-G. Zhan's laboratory at the University of Kentucky as an exchange graduate student from Central China Normal University. The authors also acknowledge the Center for Computational Sciences, University of Kentucky, for providing supercomputing time on the Superdome (an HP shared-memory supercomputer, with four nodes for 256 processors) and the IBM X-series Cluster (with 340 nodes and 1360 processors).
Footnotes
Wenchao Yang and Yongmei Pan contributed equally to this work.
Supporting Material
References
- 1.Mendelson J.H., Mello N.K. Management of cocaine abuse and dependence. N. Engl. J. Med. 1996;334:965–972. doi: 10.1056/NEJM199604113341507. [DOI] [PubMed] [Google Scholar]
- 2.Singh S. Chemistry, design, and structure-activity relationship of cocaine antagonists. Chem. Rev. 2000;100:925–1024. doi: 10.1021/cr9700538. [DOI] [PubMed] [Google Scholar]
- 3.Paula S., Tabet M.R., Farr C.D., Norman A.B., Ball W.J., Jr. Three-dimensional quantitative structure-activity relationship modeling of cocaine binding by a novel human monoclonal antibody. J. Med. Chem. 2004;47:133–142. doi: 10.1021/jm030351z. [DOI] [PubMed] [Google Scholar]
- 4.Gorelick D.A. Enhancing cocaine metabolism with butyrylcholinesterase as a treatment strategy. Drug Alcohol Depend. 1997;48:159–165. doi: 10.1016/s0376-8716(97)00119-1. [DOI] [PubMed] [Google Scholar]
- 5.Redish A.D. Addiction as a computational process gone awry. Science. 2004;306:1944–1947. doi: 10.1126/science.1102384. [DOI] [PubMed] [Google Scholar]
- 6.Sparenborg S., Vocci F., Zukin S. Peripheral cocaine-blocking agents: new medications for cocaine dependence. Drug Alcohol Depend. 1997;48:149–151. doi: 10.1016/s0376-8716(97)00072-0. [DOI] [PubMed] [Google Scholar]
- 7.Meijler M.M., Kaufmann G.F., Qi L.W., Mee J.M., Coyle A.R. Fluorescent cocaine probes: a tool for the selection and engineering of therapeutic antibodies. J. Am. Chem. Soc. 2005;127:2477–2484. doi: 10.1021/ja043935e. [DOI] [PubMed] [Google Scholar]
- 8.Carrera M.R.A., Kaufmann G.F., Mee J.M., Meijler M.M., Koob G.F. Treating cocaine addiction with viruses. Proc. Natl. Acad. Sci. USA. 2004;101:10416–10421. doi: 10.1073/pnas.0403795101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landry D.W., Zhao K., Yang G.X., Glickman M., Georgiadis T.M. Antibody-catalyzed degradation of cocaine. Science. 1993;259:1899–1901. doi: 10.1126/science.8456315. [DOI] [PubMed] [Google Scholar]
- 10.Zhan C.-G., Deng S.-X., Skiba J.G., Hayes B.A., Tschampel S.M. First-principle studies of intermolecular and intramolecular catalysis of protonated cocaine. J. Comput. Chem. 2005;26:980–986. doi: 10.1002/jcc.20241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamendulis L.M., Brzezinski M.R., Pindel E.V., Bosron W.F., Dean R.A. Metabolism of cocaine and heroin is catalyzed by the same human liver carboxylesterases. J. Pharmacol. Exp. Ther. 1996;279:713–717. [PubMed] [Google Scholar]
- 12.Sun H., Pang Y.P., Lockridge O., Brimijoin S. Re-engineering butyrylcholinesterase as a cocaine hydrolase. Mol. Pharmacol. 2002;62:220–224. doi: 10.1124/mol.62.2.220. [DOI] [PubMed] [Google Scholar]
- 13.Hamza A., Cho H., Tai H.-H., Zhan C.-G. Molecular dynamics simulation of cocaine binding with human butyrylcholinesterase and its mutants. J. Phys. Chem. B. 2005;109:4776–4782. doi: 10.1021/jp0447136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gateley S.J. Activities of the enantiomers of cocaine and some related compounds as substrates and inhibitors of plasma butyrylcholinesterase. Biochem. Pharmacol. 1991;41:1249–1254. doi: 10.1016/0006-2952(91)90665-r. [DOI] [PubMed] [Google Scholar]
- 15.Darvesh S., Hopkins D.A., Geula C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003;4:131–138. doi: 10.1038/nrn1035. [DOI] [PubMed] [Google Scholar]
- 16.Giacobini E., editor. Butyrylcholinesterase: Its Function and Inhibitors. Dunitz Martin Ltd; London, UK: 2003. [Google Scholar]
- 17.Poet T.S., McQueen C.A., Halpert J.R. Participation of cytochromes P4502B and P4503A in cocaine toxicity in rat hepatocytes. Drug Metab. Dispos. 1996;24:74–80. [PubMed] [Google Scholar]
- 18.Zheng F., Yang W., Ko M.-C., Liu J., Cho H. Most efficient cocaine hydrolase designed by virtual screening of transition states. J. Am. Chem. Soc. 2008;130:12148–12155. doi: 10.1021/ja803646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun H., Yazal J.E., Lockridge O., Schopfer L.M., Brimijoin S. Predicted Michaelis-Menten complexes of cocaine-butyrylcholinesterase. Engineering effective butyrylcholinesterase mutants for cocaine detoxication. J. Biol. Chem. 2001;276:9330–9336. doi: 10.1074/jbc.M006676200. [DOI] [PubMed] [Google Scholar]
- 20.Zhan C.-G., Zheng F., Landry D.W. Fundamental reaction mechanism for cocaine hydrolysis in human butyrylcholinesterase. J. Am. Chem. Soc. 2003;125:2462–2474. doi: 10.1021/ja020850+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao Y., Atanasova E., Sui N., Pancook J.D., Watkins J.D. Gene transfer of cocaine hydrolase suppresses cardiovascular responses to cocaine in rats. Mol. Pharmacol. 2005;67:204–211. doi: 10.1124/mol.104.006924. [DOI] [PubMed] [Google Scholar]
- 22.Zhan C.-G., Gao D. Catalytic mechanism and energy barriers for butyrylcholinesterase-catalyzed hydrolysis of cocaine. Biophys. J. 2005;89:3863–3872. doi: 10.1529/biophysj.105.070276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao D., Zhan C.-G. Modeling effects of oxyanion hole on the ester hydrolysis catalyzed by human cholinesterases. J. Phys. Chem. B. 2005;109:23070–23076. doi: 10.1021/jp053736x. [DOI] [PubMed] [Google Scholar]
- 24.Pan Y., Gao D., Yang W., Cho H., Yang G.-F. Computational redesign of human butyrylcholinesterase for anticocaine medication. Proc. Natl. Acad. Sci. USA. 2005;102:16656–16661. doi: 10.1073/pnas.0507332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao D., Zhan C.-G. Modeling evolution of hydrogen bonding and stabilization of transition states in the process of cocaine hydrolysis catalyzed by human butyrylcholinesterase. Proteins. 2006;62:99–110. doi: 10.1002/prot.20713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao D., Cho H., Yang W., Pan Y., Yang G.-F. Computational design of a human butyrylcholinesterase mutant for accelerating cocaine hydrolysis based on the transition-state simulation. Angew. Chem. Int. Ed. 2006;45:653–657. doi: 10.1002/anie.200503025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan Y., Gao D., Yang W., Cho H., Zhan C.-G. Free energy perturbation (FEP) simulation on the transition states of cocaine hydrolysis catalyzed by human butyrylcholinesterase and its mutants. J. Am. Chem. Soc. 2007;129:13537–13543. doi: 10.1021/ja073724k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brimijoin S., Gao Y., Anker J.J., Gliddon L.A., LaFleur D. A cocaine hydrolase engineered from human. butyrylcholinesterase selectively blocks cocaine toxicity and reinstatement of drug seeking in rats. Neuropsychopharmacology. 2008;33:2715–2725. doi: 10.1038/sj.npp.1301666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kollman P. Free energy calculations: applcations to chemical and biochemical phenomena. Chem. Rev. 1993;93:2395–2417. [Google Scholar]
- 30.Beveridge D.L., Dicapua F.M. Free-energy via molecular simulation—applications to chemical and biomolecular systems. Annu. Rev. Biophys. Biophys. Chem. 1989;18:431–492. doi: 10.1146/annurev.bb.18.060189.002243. [DOI] [PubMed] [Google Scholar]
- 31.Jarmula A., Cieplak P., Les A., Rode W. Relative free energies of binding to thymidylate synthase of 2-and/or 4-thio and/or 5-fluoro analogues of dUMP. J. Comput. Aided Mol. Des. 2003;17:699–710. doi: 10.1023/b:jcam.0000017377.07094.2e. [DOI] [PubMed] [Google Scholar]
- 32.Udier-Blagovic M., Tirado-Rives J., Jorgensen W.L. Structural and energetic analyses of the effects of the K103N mutation of HIV-1 reverse transcriptase on efavirenz analogues. J. Med. Chem. 2004;47:2389–2392. doi: 10.1021/jm0303507. [DOI] [PubMed] [Google Scholar]
- 33.Zhang W., Hou T.J., Qiao X.B., Huai S., Xu X.J. Binding affinity of hydroxamate inhibitors of matrix metalloproteinase-2. J. Mol. Model. 2004;10:112–120. doi: 10.1007/s00894-004-0181-1. [DOI] [PubMed] [Google Scholar]
- 34.Danciulescu C.B.N., Wortmann F.J. Structural stability of wild type and mutated α-keratin fragments: molecular dynamics and free energy calculations. Biomacromolecules. 2004;5:2165–2175. doi: 10.1021/bm049788u. [DOI] [PubMed] [Google Scholar]
- 35.Funahashi J., Sugita Y., Kitao A., Yutani K. How can free energy component analysis explain the difference in protein stability caused by amino acid substitutions? Effect of three hydrophobic mutations at the 56th residue on the stability of human lysozyme. Protein Eng. 2003;16:665–671. doi: 10.1093/protein/gzg083. [DOI] [PubMed] [Google Scholar]
- 36.Rao S.N., Singh U.C., Bash P.A., Kollman P.A. Free energy perturbation calculations on binding and catalysis after mutating Asn 155 in subtilisin. Nature. 1987;328:551–554. doi: 10.1038/328551a0. [DOI] [PubMed] [Google Scholar]
- 37.Hwang J.K., Warshel A. Semiquantitative calculations of catalytic free energies in genetically modified enzymes. Biochemistry. 1987;26:2669–2673. doi: 10.1021/bi00384a003. [DOI] [PubMed] [Google Scholar]
- 38.Xiang Y., Oelschlaeger P., Florian J., Goodman M.F., Warshel A. Simulating the effect of DNA polymerase mutations on transition-state energetics and fidelity: evaluating amino acid group contribution and allosteric coupling for ionized residues in human pol β. Biochemistry. 2006;45:7036–7048. doi: 10.1021/bi060147o. [DOI] [PubMed] [Google Scholar]
- 39.Nicolet Y., Lockridge O., Masson P., Fontecilla-Camps J.C., Nachon F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003;278:41141–41147. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- 40.Bernstein F.C., Koetzle T.F., Williams G.J.B., Meyer E.F., Brice M.D. Protein Data Bank—computer-based archival file for macromolecular structures. J. Mol. Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 41.Case D.A., Pearlman D.A., Caldwell J.W., Cheatham T.E.I., Wang J. University of California; San Francisco, CA: 2002. AMBER7. [Google Scholar]
- 42.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A. Gaussian, Inc; Pittsburgh, PA: 2003. GAUSSIAN 03, revision A.1. [Google Scholar]
- 43.Jorgensen W.L., Chandrasekhar J., Madura J.D. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 44.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 45.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constrains: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 46.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 47.Braman J., Papworth C., Greener A. Site-directed mutagenesis using double-stranded plasmid DNA templates. Methods Mol. Biol. 1996;57:31–44. doi: 10.1385/0-89603-332-5:31. [DOI] [PubMed] [Google Scholar]
- 48.Masson P., Xie W., Froment M.-T., Levitsky V., Fortier P.-L. Interaction between the peripheral site residues of human butyrylcholinesterase, D70 and Y332, in binding and hydrolysis of substrates. Biochim. Biophys. Acta. 1999;1433:281–293. doi: 10.1016/s0167-4838(99)00115-6. [DOI] [PubMed] [Google Scholar]
- 49.Sun H., Shen M.L., Pang Y.P., Lockridge O., Brimijoin S. Cocaine metabolism accelerated by a re-engineered human butyrylcholinesterase. J. Pharmacol. Exp. Ther. 2002;302:710–716. doi: 10.1124/jpet.302.2.710. [DOI] [PubMed] [Google Scholar]
- 50.Brock A., Mortensen V., Loft A.G.R., Nørgaard-Pedersen B. Enzyme immunoassay of human cholinesterase (EC 3.1.1.8). Comparison of immunoreactive substance concentration with catalytic activity concentration in randomly selected serum samples from healthy individuals. J. Clin. Chem. Clin. Biochem. 1990;28:221–224. doi: 10.1515/cclm.1990.28.4.221. [DOI] [PubMed] [Google Scholar]
- 51.Florian J., Goodman M.F., Warshel A. Free-energy perturbation calculations of DNA destabilization by base substitutions: the effect of neutral guanine•thymine, adenine•cytosine and adenine•difluorotoluene mismatches. J. Phys. Chem. B. 2000;114:10092–10099. [Google Scholar]
- 52.Bren U., Martinek V., Florian J. Free energy simulations of uncatalyzed DNA replication fidelity: structure and stability of T•G and dTTP•G terminal DNA mismatches flanked by a single dangling nucleotide. J. Phys. Chem. B. 2006;110:10557–10566. doi: 10.1021/jp060292b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.