

Abstract
To maintain normal metal metabolism, bacteria use metal-sensing metalloregulators to control transcription of metal resistance genes. Depending on their metal-binding states, the MerR-family metalloregulators change their interactions with DNA to suppress or activate transcription. To understand their functions fundamentally, we study how CueR, a Cu1+-responsive MerR-family metalloregulator, interacts with DNA, using an engineered DNA Holliday junction (HJ) as a protein-DNA interaction reporter in single-molecule fluorescence resonance energy transfer measurements. By analyzing the single-molecule structural dynamics of the engineered HJ in the presence of various concentrations of both apo- and holo-CueR, we show how CueR interacts with the two conformers of the engineered HJ, forming variable protein-DNA complexes at different protein concentrations and changing the HJ structures. We also show how apo- and holo-CueR differ in their interactions with DNA, and discuss their similarities and differences with other MerR-family metalloregulators. The surprising finding that holo-CueR binds more strongly to DNA than to apo-CueR suggests functional differences among MerR-family metalloregulators, in particular in their mechanisms of switching off gene transcription after activation. The study also corroborates the general applicability of engineered HJs as single-molecule reporters for protein-DNA interactions, which are fundamental processes in gene replication, transcription, recombination, and regulation.
Introduction
Metal ions are essential in biology and play key roles in the structure and function of a large number of proteins (1). Despite their importance, they can also be cytotoxic, especially at high concentrations (2,3). Intracellular metal ion concentrations and their bioavailability must therefore be tightly regulated to maintain normal cell metabolism. Bacteria, being susceptible to either limiting or toxic levels of metal ions in their living environment, have developed highly sensitive and selective metal homeostasis mechanisms (3–15). A key step in bacteria's response to varying levels of metal ions in their environment is through metal-sensing regulatory proteins (4–16). These proteins, also known as metalloregulators, respond to specific metal ions within the cell and regulate gene expression for metal-specific homeostasis (3–6).
A large class of bacterial metalloregulators belongs to the MerR-family; they respond to metal ions such as Hg2+, Pb2+, and Cu1+ with high selectivity and sensitivity (4–6,16–22). All MerR-family regulators are homodimers with two DNA-binding domains. They regulate gene transcription via a unique DNA distortion mechanism (5,17,18,23,24), in which both the apo- and the holo-regulator bind tightly to a dyad-symmetric sequence in the promoter region, with one DNA-binding domain binding to each half of the dyad sequence. In the apo-regulator bound form, DNA is slightly bent and the transcription is suppressed. Upon metal binding, the holo-regulator further unwinds DNA slightly, and transcription is activated. As the regulator-DNA interactions dictate the transcription process, we are interested in defining the associated protein-DNA interactions quantitatively as a fundamental step to understand their detailed structure-dynamics-function relationships.
Single-molecule fluorescence resonance energy transfer (smFRET) measurements are powerful in studying protein-DNA interactions and associated structural changes of proteins and DNA (25–27). Owing to both the FRET mechanism and the fluorescent probes suitable for single-molecule detection, smFRET relies largely on detecting nanometer-scale distance changes (25,26). The structural changes associated with MerR-family regulator-DNA interactions are mainly on the angstrom scale, however (23,24). To detect small structural changes, we recently developed engineered DNA Holliday junctions (HJs) as generalizable single-molecule reporters in smFRET measurements for protein-DNA interaction studies (28).
Our method builds on the intrinsic structural dynamics of DNA HJs and the ease of following the dynamics by smFRET. In the presence of Na+ and Mg2+, each HJ molecule folds into two X-shaped stacked conformers that interconvert dynamically at room temperature (conf-I and conf-II, Fig. 1) (28–33). With a FRET donor-acceptor pair labeled at the ends of two HJ arms, the two conformers have distinct FRET signals, one having high FRET efficiency (EFRET) and the other low EFRET, and their interconversion dynamics are reflected by their two-state FRET fluctuation behaviors (28,29,31). To use a HJ as a single-molecule protein-DNA interaction reporter, we encode in its arms the dyad-symmetric sequence recognized by a metalloregulator (Fig. 1). Because the encoded sequence has distinct spatial orientations in the two conformations, the metalloregulator binds to the two conformers differentially and causes changes in their structures and dynamics, which are readily measurable by smFRET and thus report the associated protein-DNA interactions. As the effects of protein actions on DNA are converted to and amplified by the changes in the structures and dynamics of the engineered HJ, small protein-induced structural changes can be studied (28).
Figure 1.
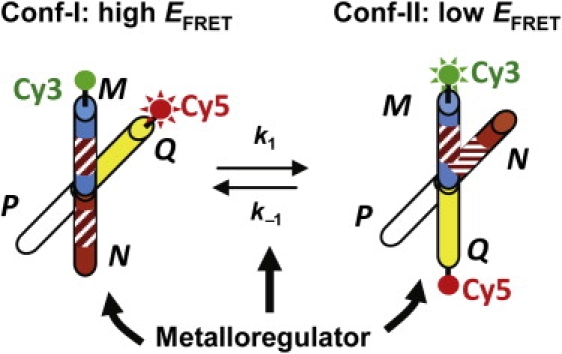
Structural dynamics of engineered Holliday junction (HJ) between its two conformers, conf-I and conf-II. Cy3 and Cy5 are labeled at the ends of arms M and Q to differentiate conf-I (high EFRET) from conf-II (low EFRET). The stripes on arms M and N indicate the encoded dyad-symmetric sequence recognized by a metalloregulator. Protein binding will perturb both the structures and the dynamic equilibrium of the HJ, which are readily followed by the FRET signal.
Using this approach, we have previously shown that a specifically engineered HJ can report how the Pb2+-responsive MerR-family metalloregulator PbrR691 interacts with DNA (28). To test the general applicability of our method and to gain further insight into the functions of MerR-family regulators, here we extend this engineered HJ approach to examine the actions on DNA of a crystallographically defined Cu1+-responsive MerR-family metalloregulator, CueR, which regulates gene expression for copper resistance in Escherichia coli (34–38).
Materials and Methods
Expression and purification of CueR
E. coli CueR protein was expressed and purified as previously described (34). Briefly, CueR was cloned in an expression vector pET30a, transformed and expressed in E. coli BL21(DE3). The cells were grown until the OD600 was 0.6 before isopropyl-beta-D-thiogalactopyranoside (IPTG, 1 mM) was added. After an additional 4 h growth at 37°C, cells were harvested by centrifugation and then disrupted by French press in lysis buffer (10 mM Tris, 300 mM NaCl, 10 mM beta-mercaptoethanol (BME), and 10% glycerol at pH 7.3). The cell debris was removed by centrifugation and the protein in the supernatant was purified first by precipitating with 45% (NH4)2SO4 and then by gel filtration in a Sephadex G-25 column. The collected fractions were further purified through a Heparin affinity column (16/10 Heparin FF, GE Healthcare, Waukesha, WI) and a gel filtration column (HILOAD 26/60 Superdex 200 PR, GE Healthcare). Purified CueR was checked by SDS-PAGE, confirmed by ESI-MS, and quantified via bicinchoninic acid (BCA) assay (Pierce, Thermo Fisher Scientific, Rockford, IL). Purified protein was stored at −80°C in 50 mM pH 8.0 Tris Buffer, with 250 mM NaCl, 5 mM BME, and 20% glycerol. The as-purified CueR is in its apo-form from BCA copper quantitation assay (39).
HJ preparation and purification
CueR-specific HJC2 was designed and purified as described previously (28). The four DNA strands (Fig. 2 A) were purchased from Integrated DNA Technologies (Coralville, IA), and dissolved in 10 mM Tris buffer pH 7.3 with 100 mM NaCl. HJC2 was assembled by annealing strands a, b, and c first at 50°C; after slow cooling to 37°C, strand d was added. The solution was then incubated for 30 min at 37°C before cooling down to room temperature. The annealed HJC2 was purified by electrophoresis in 20% polyacrylamide gel.
Figure 2.
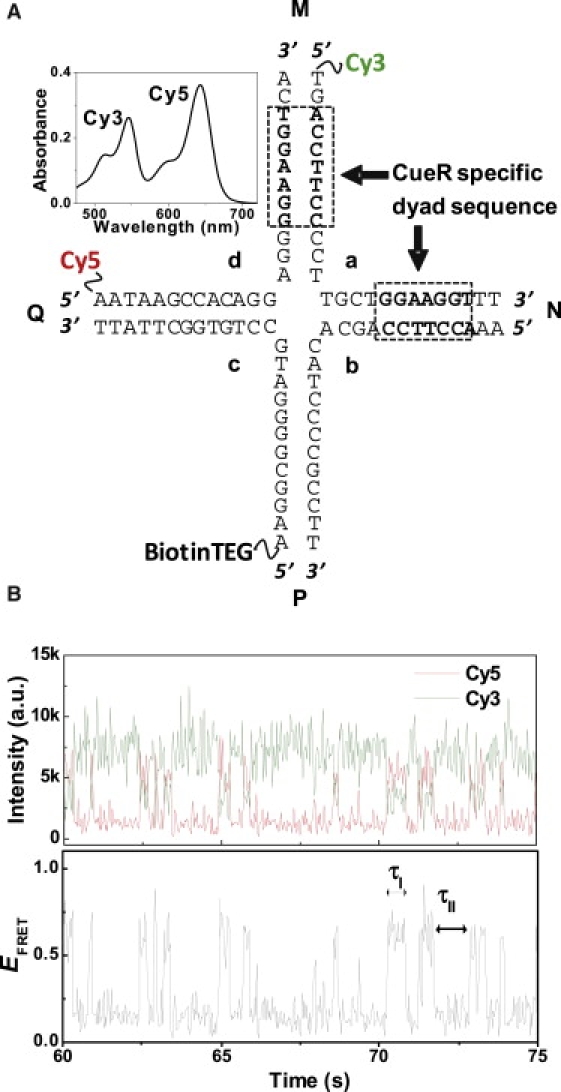
CueR-specific Holliday junction, HJC2. (A) Design of HJC2 with the dyad-symmetric sequence in arms M and N, Cy3 and Cy5 labels on strands a and d, and BiotinTEG on strand c. (Inset) Absorption spectrum of the Cy3- and Cy5-labeled HJC2. (B) Exemplary single-molecule fluorescence (top) and EFRET (bottom) trajectories of HJC2, showing two-state fluctuations. EFRET is approximated as ICy5/(ICy3 + ICy5) (I: fluorescence intensity). The values τI and τII are the waiting times on the EFRET states of conf-I and conf-II, respectively. Note that a.u. = arbitrary units.
Single-molecule fluorescence experiments and data analysis
A prism-type total internal reflection microscope based on an Olympus IX71 inverted microscope (Olympus, Melville, NY) was used for single-molecule fluorescence measurements. The Cy3 probe on HJC2 was directly excited by a continuous-wave circularly polarized 532-nm laser (GCL-025-L-0.5%, CrystaLaser, Reno, NV) of ∼6 mW focused onto an area of ∼150 × 75 μm2 on the sample. The fluorescence of both Cy3 and Cy5 was collected by a 60× NA 1.2 water-immersion objective (UPLSAPO60XW, Olympus), with an extra 1.6× magnification, and split by a dichroic mirror (635DCXR) into two channels using a Dual-View system (Optical Insights, Santa Fe, NM). HQ550LP filter was used to reject the excitation laser light and each channel of fluorescence was further filtered (HQ580-60m or HQ660LP) and projected onto half of the imaging area of a camera (Ixon EMCCD, DV887DCS-BV, Andor, Belfast, N. Ireland), controlled by the Andor IQ software. The time resolution for all the single-molecule experiments is 30 ms. A custom IDL program was then used to extract individual fluorescence trajectories of Cy3 and Cy5 for each HJC2 from the fluorescence movie recorded by the camera.
Single-molecule experiments were carried out using a flow cell, formed by double-sided tapes sandwiched between a quartz slide (Technical Glass, Opus, Snoqualmie, WA) and a borosilicate coverslip (Gold Seal coverslip, Thomas Scientific, Swedesboro, NJ). All samples were in 10 mM Tris buffer, pH 7.3 with 10 mM NaCl and 2 mM MgCl2. To minimize nonspecific protein adsorption on glass surfaces, quartz slides were first amine-functionalized (Vectabond, Vector Laboratories, Burlingame, CA) and then coated with polyethylene glycol (PEG) polymers (100 mg/mL m-PEG-SPA-5000, SunBio, Santa Clara, CA; and 1 mg/mL biotin-PEG-NHS-3400, JenKem Technology, Allen, TX) (26). One-percent of the PEG polymers contain a biotin terminal group to form biotin-streptavidin (Molecular Probes, Eugene, OR) linkages for immobilizing biotinylated HJC2 molecules. Oxygen scavenging system (0.1 mg/mL glucose oxidase (Sigma Chemical, St. Louis, MO), 0.025 mg/mL catalase (Roche, Hoffman-LaRoche, Basel, Switzerland), 4% glucose (Sigma Aldrich, St. Louis, MO)), and 1 mM Trolox (Sigma) were added into the sample solution just before each experiment to prolong the lifetime and suppress the blinking of the fluorescence probes (40).
EFRET trajectories were obtained from Cy3 and Cy5 intensity trajectories as EFRET = ICy5/(ICy3 + ICy5), a good approximation for FRET efficiency. Photobleaching and blinking of either dye were first removed before performing threshold analysis on each trajectory based on the distribution of the EFRET values to obtain individual waiting times (29). The average waiting time was then calculated from all trajectories obtained at a given protein concentration with the standard error of the mean as the error bar.
Fluorescence anisotropy
Fluorescence of Cy3-labeled double-strand DNA was measured using a Cary Eclipse fluorescence spectrophotometer (Varian, Palo Alto, CA). The CueR titration was in 10 mM Tris buffer with 10 mM NaCl and 2 mM MgCl2 at pH 7.3, and PbrR691 titration in 10 mM Tris buffer with 100 mM NaNO3 at pH 7.2. The sequence of the double-stand DNA for PbrR691 titration is 5′-TGACTCTATATCTACTAGAGGTT-3′, where the PbrR691-specific dyad-symmetric sequence is underlined. The fluorescence was excited at 532 nm. Anisotropy (r) was calculated as , where I‖ and I⊥ are the fluorescence intensity parallel and perpendicular to the excitation polarization, respectively, and G is the correction factor for the instrument's different responses to light of parallel and vertical polarizations. The fluorescence anisotropy titration curves were fitted with (41)
| (1) |
where rD and rPD are the anisotropy values for free and protein-bound DNA, respectively, [D]T is the total DNA concentration, [P]T is the total protein concentration, and KD is the dissociation constant of the protein-DNA complex.
Results and Analysis
CueR-specific engineered HJ
Fig. 2 A shows the design of the engineered Holliday junction, HJC2, targeting the metalloregulator CueR and using four oligo-DNA strands. The sequence of strand a is taken from the wild-type promoter that CueR binds, and it contains the CueR-specific dyad-symmetric sequence, which spans the arms M and N. The ends of arms M and Q are labeled with the FRET pair, Cy3 (donor) and Cy5 (acceptor), to distinguish between the two stacked conformers of HJC2: conf-I has a higher EFRET and conf-II has a lower EFRET (see Fig. 1). A biotin is attached at the end of arm P for surface immobilization. The assembly of HJC2 is confirmed by gel electrophoresis in reference to a characterized HJ (28), and by its absorption spectrum, in which the absorption bands of Cy3 and Cy5 indicate their 1:1 labeling ratio (Fig. 2 A, inset).
The intrinsic structural dynamics of a single HJC2 molecule is clear from its anticorrelated two-state fluorescence intensity fluctuations in both the Cy3-donor and the Cy5-acceptor channel (Fig. 2 B, upper). The corresponding EFRET trajectory shows a two-state fluctuation between a high EFRET (∼0.59) and a low EFRET (∼0.17) state, corresponding to the structural interconversions between conf-I and conf-II (Fig. 2 B, bottom). Past studies have shown that the two stochastic waiting times in the EFRET trajectory, τI and τII, follow exponential distributions and the exponential decay constants are the interconversion rate constants (28,29). Therefore, 〈τI〉−1, where 〈..〉 denotes averaging and which represents the time-averaged single-molecule rate of conf-I → conf-II transition, equals k1, the rate constant for conf-I → conf-II transition. In addition, 〈τII〉−1, which represents the time-averaged single-molecule rate of conf-II → conf-I transition, equals k−1, the rate constant for conf-II → conf-I transition (Supporting Material, Section A). For HJC2, the rate constants determined are 〈τI〉−1 = k1 = 5.1 ± 0.1 s−1 and 〈τII〉−1 = k−1 = 0.85 ± 0.02 s−1.
Apo-CueR-HJC2 interaction dynamics
In the presence of CueR without Cu1+ bound, i.e., apo-CueR, significant perturbations are observed in the EFRET trajectory of individual HJC2 molecules (Fig. 3 A), indicating that apo-CueR binding alters HJC2 structural dynamics. The EFRET trajectory shows a shift toward the high EFRET state, i.e., conf-I. This shift in structural equilibrium is clearer in the EFRET histogram (Fig. 3, B and C), where the intensity of the peak corresponding to conf-I increases relative to that of conf-II. This equilibrium shift reports the preferential binding of apo-CueR to conf-I over conf-II.
Figure 3.
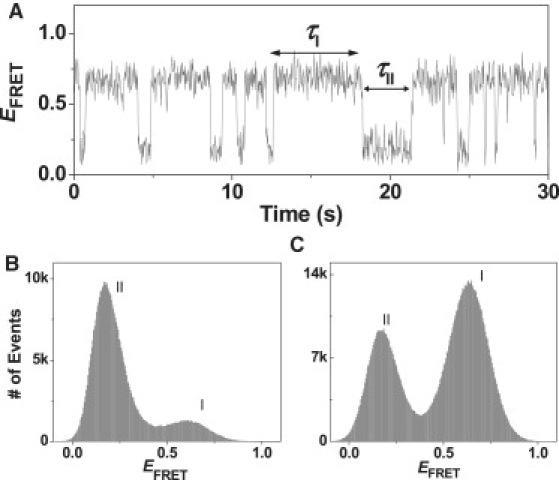
(A) Exemplary HJC2 EFRET trajectory in the presence of 1.0 μM apo-CueR. (B and C) Histograms of HJC2 EFRET trajectories in the absence (B) and presence (C) of 1.0 μM apo-CueR. Bin size: 0.005. Approximately 250 molecules were analyzed for each histogram. Histograms in the presence of other apo-CueR concentrations are given in Fig. S2.
The structural equilibrium shift of HJC2 caused by apo-CueR binding is accompanied by changes in the interconversion kinetics. The time-averaged single-molecule rate of conf-I → conf-II transition, 〈τI〉−1, depends on the apo-CueR concentration, [apo-CueR], decreasing asymptotically to zero with increasing [apo-CueR] (Fig. 4 A). This dependence indicates that apo-CueR binding slows down conf-I → conf-II structural transition, lengthening the lifetime of conf-I. In contrast, the time-averaged single-molecule rate of conf-II → conf-I transition, 〈τII〉−1, increases initially with increasing [apo-CueR], but decays at higher [apo-CueR] after reaching a maximum (Fig. 4 B). This biphasic behavior of 〈τII〉−1 indicates that the initial apo-CueR binding facilitates conf-II → conf-I transition whereas a higher-order apo-CueR interaction slows it down.
Figure 4.
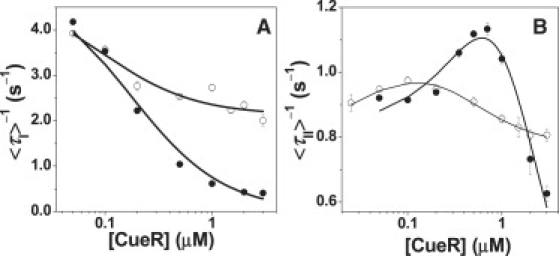
Apo-CueR (•) and holo-CueR (○) concentration dependence of 〈τI〉−1 (A) and 〈τII〉−1 (B) Each data point is an average of the waiting times from ∼250 trajectories. The solid lines are the fits with Eq. 2 (apo) and Eq. 4 (holo) for panel A and Eq. 3 (apo) and Eq. 5 (holo) for panel B. Results from the fit are summarized in Table 1.
The [apo-CueR] dependence of 〈τI〉−1 can be described by a simple kinetic mechanism in which apo-CueR (P) binds to conf-I to form a complex (P-I) that does not lead to structural transition to conf-II (Fig. 5 A, red box). Based on this kinetic scheme and following a single-molecule kinetic analysis (Supporting Material, Section B), we get
| (2) |
Figure 5.
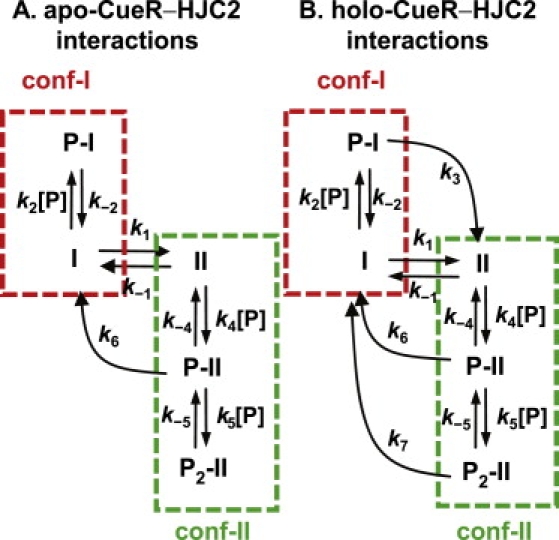
Kinetic schemes for HJC2 interactions with apo-CueR (A) and holo-CueR (B). I, conf-I; II, conf-II; P, apo-CueR or holo-CueR; and k-values, rate constants.
Here KP-I (= k−2/k2) is the dissociation constant for the apo-CueR-conf-I complex, and k2 and k−2 are the protein binding and unbinding rate constants to conf-I, respectively. Equation 2 predicts that with increasing protein concentration, 〈τI〉−1 decreases asymptotically to zero, consistent with the experimental results (Fig. 4 A). Using k1 determined from the free HJC2 and fitting the data in Fig. 4 A give KP-I = 0.17 ± 0.02 μM.
To account for the biphasic [apo-CueR] dependence of 〈τII〉−1, we considered a two-step kinetic mechanism. Apo-CueR initially binds to conf-II to form a complex (P-II) that can lead to structural transition to conf-I; this P-II complex can then bind a second protein molecule to form a tertiary complex (P2-II) that does not lead to structural transition to conf-I (Fig. 5 A, green box). Based on this two-step interaction scheme between apo-CueR and conf-II and following a single-molecule kinetic analysis, we get
| (3) |
Here = (k−4 + k6)/k4, = k−5/k5, and k-values are the rate constants defined in Fig. 5 A. Equation 3 predicts that at low protein concentrations, 〈τII〉−1 increases with increasing protein concentration because the formation of complex P-II facilitates the structural transition to conf-I, and at higher protein concentrations, 〈τII〉−1 decreases because the formation of complex P2-II slows down the transition to conf-I. Using k−1 determined from the free HJC2 and fitting the data in Fig. 4 B give: k6 = 4 ± 3 s−1, the rate constant for P-II → I transition; = 3 ± 3 μM, the apparent protein dissociation constant of complex P-II; and = 0.5 ± 0.5 μM, the first dissociation constant of complex P2-II. The determined k6 is higher than k−1, indicating that the initial binding of apo-CueR to conf-II facilitates its structural transition to conf-I. Table 1 summarizes all the fitting results.
Table 1.
Kinetic parameters for CueR-HJC2 interaction dynamics
| Free HJC2 | ||
|---|---|---|
| k1 | 5.1 ± 0.1 s−1 | |
| k−1 | 0.85 ± 0.02 s−1 | |
| Apo-CueR | Holo-CueR | |
| k3 | — | 2.3 ± 0.2 s−1 |
| k6 | 4 ± 3 s−1 | 1.2 ± 0.3 s−1 |
| k7 | — | 0.77 ± 0.02 s−1 |
| (k−2 + k3)/k2 | 0.17 ± 0.02 μM (KP-I)∗ | 0.05 ± 0.02 μM () |
| (k−4 + k6)/k4 | 3 ± 3 μM () | 0.1 ± 0.1 μM ()† |
| (k−5 + k7)/k5 | 0.5 ± 0.5 μM ()‡ | 0.2 ± 0.2 μM () |
k3 = 0 for apo-CueR.
k−4 is set to zero for holo-CueR (Supporting Material, Section C).
k7 = 0 for apo-CueR.
Holo-CueR-HJC2 interaction dynamics
Holo-CueR, i.e., Cu1+-bound CueR, causes perturbations on HJC2 structural dynamics similar to apo-CueR. With increasing [holo-CueR], 〈τI〉−1, the time-averaged conf-I → conf-II transition rate, decreases gradually (Fig. 4 A), and 〈τII〉−1, the time-averaged conf-II → conf-I transition rate, increases initially and then decays at higher [holo-CueR] (Fig. 4 B). However, significant differences also exist: neither 〈τI〉−1 nor 〈τII〉1 decays to zero at high [holo-CueR], in contrast to those of apo-CueR−HJC2 interactions. The nonzero values of 〈τI〉−1 and 〈τII〉−1 at high [holo-CueR] indicate that the relevant holo-CueR−HJC2 complexes can still allow transitions from one conformer of HJC2 to the other.
To account for these differences observed for holo-CueR-HJC2 interactions, we added two kinetic transitions on top of the kinetic mechanism of apo-CueR-HJC2 interactions (Fig. 5 B). One transition connects P-I to II (i.e., k3), and the other connects P2-II to I (i.e., k7). Based on this kinetic mechanism, the corresponding equations connecting 〈τI〉−1 and 〈τII〉−1 with the kinetic parameters and the protein concentration are (Supporting Material, Section C)
| (4) |
| (5) |
where = (k−2 + k3)/k2, = k6/k4, and = (k−5 + k7)/k5. The individual kinetic parameters are defined in Fig. 5 B. In deriving Eq. 5, we assumed k−4 = 0 to obtain a clean analytical expression; this assumption does not affect our analyses of 〈τI〉−1 (Eq. 4), from which and k3 can be obtained and interpreted quantitatively.
Equation 4 predicts that with increasing [holo-CueR], 〈τI〉−1 decreases and eventually approaches k3, the rate constant for the P-I → II transition, consistent with experimental observations (Fig. 4 A) and resulting from that high [holo-CueR] drives the conversion of I → P-I. Using k1 determined from the free HJC2 and fitting the data in Fig. 4 A give k3 = 2.3 ± 0.2 s−1 and = 0.05 ± 0.02 μM, which is the apparent dissociation constant of P-I. The determined k3 is smaller than k1, consistent with the expectation that holo-CueR binding stabilizes conf-I and slow down its structural transition to conf-II.
Equation 5 predicts the observed [holo-CueR] dependence of 〈τII〉−1, with an initial rise followed by a decay. At high [holo-CueR], 〈τII〉−1 approaches k7, the rate constant of the P2-II → I transition, as high [holo-CueR] drives the formation of P2-II. Using k−1 determined from the free HJC2 and fitting the data in Fig. 4 B give k7 = 0.77 ± 0.02 s−1, = 0.1 ± 0.1 μM, which is the apparent dissociation constant of complex P-II, and = 0.2 ± 0.2 μM, which is the apparent first protein dissociation constant of P2-II. for holo-CueR here is smaller than that (3 ± 3 μM) for apo-CueR; this is consistent with that the maximum of 〈τII〉−1 occurs at lower [holo-CueR], as compared with the maximum in apo-CueR-HJC2 interactions (Fig. 4 B). The kinetic parameters are summarized in Table 1.
To confirm that the interactions between CueR and HJC2 are specific, i.e., due to the encoded specific dyad-symmetric sequence in HJC2, we studied the structural dynamics of HJC2 in the presence of another DNA-binding protein, PbrR691, which is also a MerR-family metalloregulator; no noticeable perturbation was observed (Fig. S1). Additionally, we studied another HJ that does not contain CueR-targeting sequence; expectedly, in the presence of CueR, no noticeable perturbation on this HJ's structural dynamics was observed (28).
CueR-imposed HJC2 structural changes
Our single-molecule kinetic analyses on the structure dynamics of HJC2 indicate that both apo- and holo-CueR can bind to the two conformers of HJC2 to form complexes P-I, P-II, and P2-II (Fig. 5). The changes in the EFRET values of conf-I and conf-II in these complexes relative to those of free HJC2 can inform the structural changes of HJC2 imposed by the CueR. For interactions with conf-I, at high [apo-CueR] (e.g., 2 μM) where I is converted to P-I, Econf-I increases from ∼0.59 to ∼0.64 (Fig. 6, A and B). This increase of Econf-I indicates that in the P-I complex, apo-CueR causes a shortening of the distance between the ends of arms M and Q where Cy3 and Cy5 are located (see Fig. 1, left). This protein-induced structural change of conf-I also confirms the binding of apo-CueR. Similarly, holo-CueR binding also increases Econf-I (Fig. 6 C), indicating a similarly shortened distance between the ends of arms M and Q in conf-I.
Figure 6.
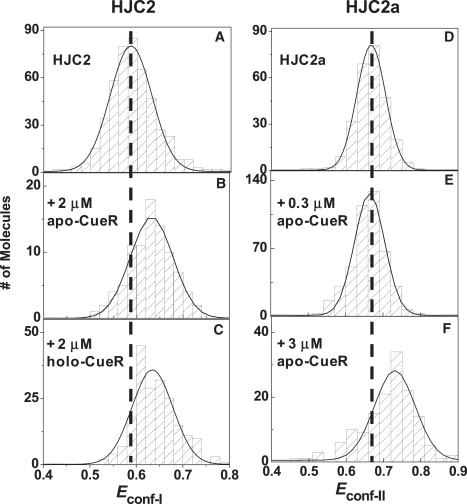
Histograms of Econf-I of HJC2 (A–C) and Econf-II of HJC2a (D–F) in the presence of various apo-CueR and holo-CueR concentrations. Solid lines are Gaussian fits centered at 0.59 ± 0.01 (A), 0.64 ± 0.01 (B), 0.63 ± 0.01 (C), 0.67 ± 0.01 (D), 0.66 ± 0.01 (E), and 0.73 ± 0.01 (F). For each molecule, its Econf-I or Econf-II was obtained by fitting the histogram of its EFRET trajectory with two Gaussian functions. Histograms at another protein concentration are given in Fig. S3.
To determine structural changes of conf-II upon CueR binding, we studied an alternatively labeled HJC2, referred to as HJC2a, which has the Cy5 placed at the end of arm N instead of arm Q (see Fig. 1). This alternative labeling makes conf-II of HJC2a the high EFRET state, rendering its EFRET value (Econf-II) more sensitive to structural changes imposed by protein binding. At low [apo-CueR] (e.g., 0.3 μM), where the complex P-II dominates the population of all forms of conf-II, no significant decrease in Econf-II is observed compared to that of free HJC2a (Fig. 6, D and E). At high [apo-CueR] (e.g., 3 μM), where P2-II dominates, Econf-II increases significantly (Fig. 6 F). This clear increase indicates that in the P2-II complex, where two apo-CueR molecules are bound, the arms M and N are brought closer to each other. These protein-induced structural changes of conf-II also confirm the binding of apo-CueR. For holo-CueR and conf-II interactions, in contrast, no significant changes of Econf-II were observed.
Ensemble CueR-DNA affinity determination
For apo-CueR interaction with conf-I of HJC2, the dissociation constant KP-I (= k−2/k2) directly reflects the binding affinity of apo-CueR to conf-I (Eq. 2). For holo-CueR, only the apparent dissociation constant (= (k−2 + k3)/k2) is obtainable from analyzing the waiting times (Eq. 4), and it gives an upper limit for the dissociation constant of holo-CueR-conf-I interactions, as (k−2 + k3)/k2 > k−2/k2. The binding affinity to conf-I is important, as conf-I has its arms M and N, which contain the dyad-symmetric sequence, coaxially stacked as in a B-form DNA (32,33). Experimentally, KP-I (= 0.17 ± 0.02 μM) for apo-CueR is larger than (= 0.05 ± 0.02 μM) for holo-CueR (Table 1), indicating that apo-CueR binds weaker to conf-I than holo-CueR does.
On the other hand, previous results from the gel-shift assay suggested a slightly stronger binding affinity for apo-CueR than for holo-CueR to a double-strand DNA containing the promoter sequence, with the dissociation constants KD(apo-CueR) = 17 ± 2 nM and KD(holo-CueR) = 25 ± 7 nM (36), although the error bars of these values preclude a reliable comparison. To test whether there is indeed a discrepancy that could come from the DNA used (i.e., using engineered HJ versus using a double-strand DNA), we determined the binding affinity of both apo- and holo-CueR to a double-strand DNA using ensemble fluorescence anisotropy titration, a more accurate quantitation method than the gel-shift assay. We used a Cy3-labeled 25-basepair double-strand DNA with the same sequence as that spanning the M, N arms of HJC2. The results in Fig. 7 A confirm that apo-CueR does bind weaker to DNA than holo-CueR, with KD(apo-CueR) = 6 ± 2 nM and KD(holo-CueR) = 1.9 ± 0.8 nM, consistent with the results using the engineered HJC2 as a single-molecule reporter. The affinities of apo- and holo-CueR to conf-I of HJC2 are weaker than to the double-strand DNA, possibly due to the presence of the other helix or the perturbation of the junction structure of HJC2 on the protein-DNA interactions.
Figure 7.
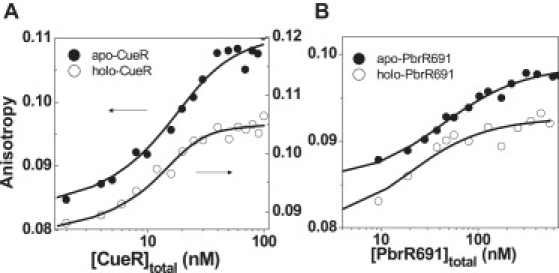
Fluorescence anisotropy titrations of CueR (A) and PbrR691 (B) binding to Cy3-labeled double-strand DNA. Solid lines are the fits with Eq. 1, with dissociation constants of KD(apo-CueR) = 6 ± 2 nM, KD(holo-CueR) = 1.9 ± 0.8 nM, KD(apo-PbrR691) = 43 ± 9 nM, and KD(holo-PbrR691) = 23 ± 9 nM. The x axes are total protein concentrations. Plots with x axes being the free protein concentration are given in Fig. S5.
Discussion
Nature of CueR-HJC2 interactions and relation to protein function
The single-molecule studies of CueR-HJC2 interactions indicate that both apo- and holo-CueR preferentially bind and stabilize conf-I of HJC2. This preferential interaction is directly reflected by HJC2's structural equilibrium shift toward conf-I (Fig. 3, B and C), as well as the decrease of 〈τI〉−1 with increasing protein concentration due to the formation of the P-I complex (Fig. 4 A and Fig. 5). The preferential interaction with conf-I is also reflected by the protein concentration dependence of 〈τII〉−1, which shows that the P-II complex can induce structural transition from conf-II to conf-I with a larger rate constant (k6) than that (k−1) of the intrinsic II → I transition (Table 1 and Fig. 5). All these observations are consistent with CueR's normal function as a double-strand DNA-binding protein, as conf-I mimics the natural substrate of CueR and has its arms M and N, which encodes the dyad-symmetric sequence, coaxially stacked to form a B-form DNA-like structure (Fig. 1, left) (32,33).
Both apo- and holo-CueR binding to conf-I bring the arms M and Q closer, reflected by the increase of Econf-I of HJC2 (Fig. 6, B and C). This structural change can be associated with the bending of the M-N helix of conf-I, similarly observed in our previous study of PbrR691 (28) and typical among MerR-family metalloregulators (5,23,24). For conf-II, no significant structural change is observed upon initial binding of one apo-CueR or holo-CueR molecule.
A distinct feature of CueR-HJC2 interactions is the initial-rise-followed-by-decay of 〈τII〉−1 with increasing protein concentrations (Fig. 4 B). This biphasic protein concentration dependence of 〈τII〉−1 indicates that at high protein concentrations, both apo- and holo-CueR, interact with conf-II to form the P2-II complex, in which two protein molecules are bound (Fig. 5). This tertiary complex was not observed in the study of PbrR691 (28), suggesting that differences exist among MerR-family metalloregulators in their interactions with DNA. The formation of this tertiary complex could be related to the highly bent orientation of the two halves of the dyad-symmetric sequence in conf-II, which largely deviates from the structure of a B-form DNA (Fig. 1, right). Consequently, only half of the dyad sequence could be bound to one of the two DNA-binding domains of CueR, leaving the other half to bind another CueR molecule. The double binding of apo-CueR then leads to the M and N arms being pushed closer in conf-II, reflected by the increase in Econf-II of HJC2a (Fig. 6 F). However, double binding of holo-CueR does not result in observable changes in Econf-II, indicating different interactions with DNA between apo- and holo-CueR.
To support our model that CueR can bind to half of the dyad-symmetric sequence, we constructed a Cy3-labeled double-strand DNA that encodes only half of the CueR-specific dyad-symmetric sequence. We then used fluorescence anisotropy titration to probe CueR binding. The results (Fig. S4) show that apo-CueR can indeed bind to this DNA with a KD of ∼0.7 μM, which is in the concentration range where the P2-II complex forms (Fig. 4 B).
Further differences exist between apo-CueR and holo-CueR. Unlike apo-CueR, holo-CueR binding can still allow P-I → II and P2-II → I transitions (k3 and k7, Fig. 5 B). As conf-I and conf-II are largely different in their spatial arrangements of the dyad-symmetric sequence (Fig. 1), being more accommodating in allowing HJC2 structural interconversion suggests that holo-CueR has more flexible conformation than apo-CueR. This conformational flexibility of holo-CueR could play important roles in its interaction with the RNA polymerase (RNAp) for transcription, as past studies on MerR, the prototype MerR-family metalloregulator, showed that the holo-MerR-DNA-RNAp tertiary complex undergoes structural rearrangements in transcription initiation (5,17,18,42).
Implications for transcriptional suppression after activation
Our single-molecule studies and the ensemble fluorescence anisotropy titration both indicate a stronger binding of holo-CueR to DNA than apo-CueR. The stronger DNA binding of the holo-protein is also observed for PbrR691 from fluorescence anisotropy titrations (Fig. 7 B), with the dissociation constants of KD(apo-PbrR691) = 43 ± 9 nM and KD(holo-PbrR691) = 23 ± 9 nM. Furthermore, BmrR, another MerR-family regulator that responds to organic effectors, also has a higher affinity to its substrate in its holo-form compared to its apo-form (43).
This stronger DNA binding by the holo-protein is surprising, however, as past studies on MerR have shown that the holo-protein binds weaker to DNA than the apo-protein (KD(apo-MerR) = 0.14 ± 0.04 nM, KD(holo-MerR) = 0.42 ± 0.07 nM) (17). As the direct dissociation of the metal ion from the metalloregulator is believed to be difficult due to strong metal coordination, it was thought that the weaker binding of holo-protein would facilitate its replacement from the DNA by the apo-protein, thus switching off the transcription after transcriptional activation once the cell is relieved of the metal stress. Therefore, the opposite relative DNA binding affinity of apo-protein versus holo-protein for CueR (as well as PbrR691 and BmrR) suggests possible differences in the mechanism by which MerR-family regulators switch off transcription.
Moreover, unlike MerR, which might involve another protein MerD to help the dissociation of the holo-MerR-DNA complex (16), no evidence has so far been found for a coregulator role of a MerD homolog in the regulatory mechanism of CueR (16). For CueR to switch off transcription after activation, one simple scenario is a direct dissociation of holo-CueR from DNA followed by binding of apo-CueR, which would be the dominant form of CueR inside the cell after activation of Cu-resistance genes. For this scenario to be viable, the dissociation kinetics of holo-CueR from DNA has to be in a relevant timescale to gene regulation. From our single-molecule kinetic analyses, the rate constants for CueR unbinding (k−2) and binding (k2) to conf-I of HJC2 cannot be obtained. Nevertheless, as the CueR binding and unbinding are contained in the observed structural dynamics of HJC2, which we measure experimentally, CueR binding and unbinding should occur at a comparable timescale to HJC2's structural dynamics, i.e., hundreds of milliseconds to seconds, a relevant timescale for gene expression regulation.
Summary
Using the engineered HJC2 as a single-molecule protein-DNA interaction reporter, we have studied how CueR, a Cu1+-responsive MerR-family metalloregulator, interacts with DNA. Both apo- and holo-CueR preferentially bind conf-I of HJC2, in which the protein-recognition sequence is arranged similarly as in a B-form DNA. This preferential binding stabilizes conf-I and slows down its structural conversion to conf-II. Both also bend the M-N helix of conf-I, reflecting their actions on DNA for transcriptional regulation. In their interactions with conf-II of HJC2, apo- and holo-CueR exhibit a biphasic behavior—at low protein concentrations, a 1:1 protein-conf-II complex is present, whereas at high protein concentrations a 2:1 protein-conf-II tertiary complex dominates. Many differences also exist between apo- and holo-CueR in their interactions with HJC2. Whereas apo-CueR causes clear structural changes of both conf-I and conf-II of HJC2, holo-CueR only causes measurable structural changes of conf-I. Holo-CueR is also more accommodating in allowing the structural interconversions of HJC2, suggesting its more flexible conformation, which could be important for its cooperation with RNAp in initiating transcription. Moreover, holo-CueR binds stronger to DNA than apo-CueR, a surprising finding and contrary to the behaviors of the prototype metalloregulator MerR. This contrast suggests functional differences among MerR-family regulators, in particular possible different mechanisms in switching off transcription after activation. The study also corroborates the general applicability of engineered HJs as single-molecule reporters for protein-DNA interactions, which are fundamental in gene replication, transcription, recombination, and regulation.
Supporting Material
Derivations of equations and additional results are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01033-9.
Supporting Material
Acknowledgments
We thank Prof. Chuan He and Dr. Peng R. Chen for providing the expression plasmids of CueR and PbrR691.
This research is supported by the National Institutes of Health (grant No. GM082939), National Science Foundation (grant No. CHE0645392), the Camille and Henry Dreyfus Foundation, and Cornell University.
References
- 1.Holm R.H., Kennepohl P., Solomon E.I. Structural and functional aspects of metal sites in biology. Chem. Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 2.Finney L.A., O'Halloran T.V. Transition metal speciation in the cell: insights from the chemistry of metal ion receptors. Science. 2003;300:931–936. doi: 10.1126/science.1085049. [DOI] [PubMed] [Google Scholar]
- 3.O'Halloran T.V. Transition metals in control of gene expression. Science. 1993;261:715–725. doi: 10.1126/science.8342038. [DOI] [PubMed] [Google Scholar]
- 4.Barkey T., Miler S.M., Summers A.O. Bacterial mercury resistance from atoms to ecosystems. FEMS Microbiol. Rev. 2003;27:355–384. doi: 10.1016/S0168-6445(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 5.Brown N.L., Stoyanov J.V., Kidd S.P., Hobman J.L. The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 2003;27:145–163. doi: 10.1016/S0168-6445(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 6.Busenlehner L., Pennella M.A., Giedroc D.P. The SmtB/ArsR family of metalloregulatory transcriptional repressors: structural insights into prokaryotic metal resistance. FEMS Microbiol. Rev. 2003;27:131–143. doi: 10.1016/S0168-6445(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 7.Andrews S.C., Robinson A.K., Rodriguez-Quinones F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 8.Cavet J.S., Borrelly G.P.M., Robinson N.J. Zn, Cu and Co in cyanobacteria: selective control of metal availability. FEMS Microbiol. Rev. 2003;27:165–181. doi: 10.1016/S0168-6445(03)00050-0. [DOI] [PubMed] [Google Scholar]
- 9.Kehres D.G., Maguire M.E. Emerging themes in manganese transport, biochemistry and pathogenesis in bacteria. FEMS Microbiol. Rev. 2003;27:263–290. doi: 10.1016/S0168-6445(03)00052-4. [DOI] [PubMed] [Google Scholar]
- 10.Lloyd J.R. Microbial reduction of metals and radionuclides. FEMS Microbiol. Rev. 2003;27:411–425. doi: 10.1016/S0168-6445(03)00044-5. [DOI] [PubMed] [Google Scholar]
- 11.Mergeay M., Monchy S., Vallaeys T., Auquier V., Benotmane A. Ralstonia metallidurans, a bacterium specifically adapted to toxic metals: towards a catalogue of metal-responsive genes. FEMS Microbiol. Rev. 2003;27:385–410. doi: 10.1016/S0168-6445(03)00045-7. [DOI] [PubMed] [Google Scholar]
- 12.Mulrooney S.B., Hausinger R.P. Nickel uptake and utilization by microorganism. FEMS Microbiol. Rev. 2003;27:239–261. doi: 10.1016/S0168-6445(03)00042-1. [DOI] [PubMed] [Google Scholar]
- 13.Nies D.H. Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol. Rev. 2003;27:313–339. doi: 10.1016/S0168-6445(03)00048-2. [DOI] [PubMed] [Google Scholar]
- 14.Rensing C., Grass G. Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol. Rev. 2003;27:197–213. doi: 10.1016/S0168-6445(03)00049-4. [DOI] [PubMed] [Google Scholar]
- 15.Solioz M., Stoyanov J.V. Copper homeostasis in Enterococcus hirae. FEMS Microbiol. Rev. 2003;27:183–195. doi: 10.1016/S0168-6445(03)00053-6. [DOI] [PubMed] [Google Scholar]
- 16.Hobman J.L., Wilkie J., Brown N.L. A design for life: prokaryotic metal-binding MerR family regulators. Biometals. 2005;18:429–436. doi: 10.1007/s10534-005-3717-7. [DOI] [PubMed] [Google Scholar]
- 17.O'Halloran T.V., Frantz B., Shin M.K., Ralston D.M., Wright J.G. The MerR heavy metal receptor mediates positive activation in a topologically novel transcription complex. Cell. 1989;56:119–129. doi: 10.1016/0092-8674(89)90990-2. [DOI] [PubMed] [Google Scholar]
- 18.Frantz B., O'Halloran T.V. DNA distortion accompanies transcriptional activation by the metal-responsive gene-regulatory protein MerR. Biochemistry. 1990;29:4747–4751. doi: 10.1021/bi00472a001. [DOI] [PubMed] [Google Scholar]
- 19.Giedroc D.P., Arunkumar A.I. Metal sensor proteins: Nature's metalloregulated allosteric switch. Dalton Trans. 2007;29:3107–3120. doi: 10.1039/b706769k. [DOI] [PubMed] [Google Scholar]
- 20.Chen P.R., He C. Selective recognition of metal ions by metalloregulatory proteins. Curr. Opin. Chem. Biol. 2008;12:214–221. doi: 10.1016/j.cbpa.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 21.Permina E.A., Kazakov A.E., Kalinina O.V., Gelfand M.S. Comparative genomics of regulation of heavy metal resistance in eubacteria. BMC Microbiol. 2006;6:49. doi: 10.1186/1471-2180-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen P., He C. A general strategy to convert the MerR family proteins into highly sensitive and selective fluorescent biosensors for metal ions. J. Am. Chem. Soc. 2004;126:728–729. doi: 10.1021/ja0383975. [DOI] [PubMed] [Google Scholar]
- 23.Zheleznova E.E., Brennan R.G. Crystal structure of the transcription activator BmrR bound to DNA and a drug. Nature. 2001;409:378–382. doi: 10.1038/35053138. [DOI] [PubMed] [Google Scholar]
- 24.Newberry K.J., Brennan R.G. The structural mechanism for transcription activation by MerR family member multidrug transporter activation, N-terminus. J. Biol. Chem. 2004;279:20356–20362. doi: 10.1074/jbc.M400960200. [DOI] [PubMed] [Google Scholar]
- 25.Michalet X., Weiss S., Jaeger M. Single-molecule fluorescence studies of protein folding and conformational dynamics. Chem. Rev. 2006;106:1785–1813. doi: 10.1021/cr0404343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ha T. Single-molecule fluorescence resonance energy transfer. Methods. 2001;25:78–86. doi: 10.1006/meth.2001.1217. [DOI] [PubMed] [Google Scholar]
- 27.Zhuang X. Single-molecule RNA science. Annu. Rev. Biophys. Biomol. Struct. 2005;34:399–414. doi: 10.1146/annurev.biophys.34.040204.144641. [DOI] [PubMed] [Google Scholar]
- 28.Sarkar S.K., Andoy N.M., Benitez J.J., Chen P.R., Kong J.S. Engineered Holliday junctions as single-molecule reporters for protein-DNA interactions with application to a MerR-family regulator. J. Am. Chem. Soc. 2007;129:12461–12467. doi: 10.1021/ja072485y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKinney S.A., Declais A.C., Lilley D.M.J., Ha T. Structural dynamics of individual Holliday junctions. Nat. Struct. Biol. 2003;10:93–97. doi: 10.1038/nsb883. [DOI] [PubMed] [Google Scholar]
- 30.Lilley D.M.J. Structures of helical junctions in nucleic acids. Q. Rev. Biophys. 2000;33:109–159. doi: 10.1017/s0033583500003590. [DOI] [PubMed] [Google Scholar]
- 31.Karymov M.A., Chinnaraj M., Bogdanov A., Srinivasan A.R., Zheng G. Structure, dynamics, and branch migration of a DNA Holliday junction: a single-molecule fluorescence and modeling study. Biophys. J. 2008;95:4372–4383. doi: 10.1529/biophysj.108.135103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ortiz-Lombardía M., González A., Eritja R., Aymamí J., Azorín F. Crystal structure of a DNA Holliday junction. Nat. Struct. Biol. 1999;6:913–917. doi: 10.1038/13277. [DOI] [PubMed] [Google Scholar]
- 33.Eichman B.F., Vargason J.M., Mooers B.H.M., Ho P.S. The Holliday junction in an inverted repeat DNA sequence: sequence effects on the structure of four-way junctions. Proc. Natl. Acad. Sci. USA. 2000;97:3971–3976. doi: 10.1073/pnas.97.8.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Outten F.W., Outten C.E., Hale J., O'Halloran T.V. Transcriptional activation of an Escherichia coli copper efflux regulation by the chromosomal MerR homologue, CueR. J. Biol. Chem. 2000;275:31024–31029. doi: 10.1074/jbc.M006508200. [DOI] [PubMed] [Google Scholar]
- 35.Petersen C., Moller L.B. Control of copper homeostasis in Escherichia coli by a P-type ATPase CopA, and a MerR-like transcriptional activator, CopR. Gene. 2000;261:289–298. doi: 10.1016/s0378-1119(00)00509-6. [DOI] [PubMed] [Google Scholar]
- 36.Stoyanov J.V., Hobman J.L., Brown N.L. CueR (YBBI) of Escherichia coli is a MerR family regulator controlling expression of the copper exporter CopA. Mol. Microbiol. 2001;39:502–511. doi: 10.1046/j.1365-2958.2001.02264.x. [DOI] [PubMed] [Google Scholar]
- 37.Changela A., Chen K., Xue Y., Holschen J., Outten C.E. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science. 2003;301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto K., Ishihama A. Transcriptional response of Escherichia coli to external copper. Mol. Microbiol. 2004;56:215–227. doi: 10.1111/j.1365-2958.2005.04532.x. [DOI] [PubMed] [Google Scholar]
- 39.Brenner A.J., Harris E.D. A quantitative test for copper using bicinchoninic acid. Anal. Biochem. 1995;226:80–84. doi: 10.1006/abio.1995.1194. [DOI] [PubMed] [Google Scholar]
- 40.Rasnik I., McKinney S.A., Ha T. Nonblinking and long-lasting single molecule fluorescence imaging. Nat. Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 41.Heyduk T., Lee J.C. Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and Lac promoter interaction. Proc. Natl. Acad. Sci. USA. 1990;87:1744–1748. doi: 10.1073/pnas.87.5.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heltzel A., Lee I.W., Totis P.A., Summers A.O. Activator-dependent preinduction binding of σ-70 RNA polymerase at the metal-regulated MerR promoter. Biochemistry. 1990;29:9572–9584. doi: 10.1021/bi00493a011. [DOI] [PubMed] [Google Scholar]
- 43.Ahmed M., Borsch C., Taylor S.S., Vazquez-Laslop N., Neyfakh A.A. A protein that activates expression of a multidrug efflux transporter upon binding the transporter substrates. J. Biol. Chem. 1994;269:28506–28513. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.