

Abstract
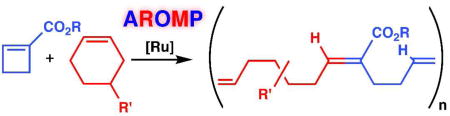
The alternating polymerization of cyclobutene 1-carboxylic esters and cyclohexene derivatives with the precatalyst [(H2IMes)(3-Br-pyr)2Cl2Ru=CHPh] is described. This reaction is synthetically accessible and provides (AB)n heteropolymers with an alternating backbone and alternating functionality. The regiocontrol of heteropolymer formation derives from the inability of the cyclobutene ester and cyclohexene monomers to undergo homopolymerization in combination with the favorable kinetics of cross polymerization.
Copolymers are employed in applications ranging from the biomedical to the electronic.1 Among the most commonly used are block copolymers that require phase separation of the two blocks for their function, e.g., drug delivery,2 and random copolymers in which two functional moieties communicate, e.g., organic light emitting diodes.3 Regularly alternating polymers should allow optimal positioning of functional substituents and be useful in a variety of applications.
Alternating polymers are generally synthesized by radical polymerization with kinetic control of the order of monomer incorporation.4 However, there are isolated examples of their synthesis by ring opening metathesis polymerization (ROMP). Early on, it was reported that the ROMP of racemic 1-methylbicyclo[2.2.1]hept-2-ene with ReCl5 gave polymer in which the two enantiomers alternate.5 More recently, several reports of alternating polymers as the products of ROMP have appeared. With the exception of Grubbs’s ring opening insertion metathesis (ROIMP) approach, in which alternation is controlled by equilibration,6 these rely on the pairing of a bulky but strained monomer with an unhindered and only slightly strained monomer. In all cases except one,7 a significant excess of one of the monomers is required for high levels of alternation.8
We now describe the highly alternating polymerization of cyclobutene 1-carboxylic esters with cyclohexene derivatives with the precatalyst 1 [(H2IMes)(3-Br-pyr)2Cl2Ru=CHPh].9 The success of this reaction derives from the combination of two monomers neither of which forms a homopolymer under the ROMP reaction conditions.
Our laboratory recently developed cyclobutene 1-carboxamides as monomers that undergo ruthenium-catalyzed ring-opening metathesis to yield translationally invariant polymers.10 During the course of extending the ROMP to cyclobutenecarboxylic acid derivatives, we observed that cyclobutene methyl ester 2 underwent ring-opening metathesis without polymerization to afford, with 10 mole % of catalyst, approximately 10% of the α-methylene ester 2a1 (Scheme 1). As in the ring-opening metathesis of 1-substituted cyclobutene amides,10 this reaction is regiospecific. However with ester 2a, the key enoic ruthenium carbene 3 does not react with additional substrate; rather, it survives to react with the quenching agent, providing ester 2a1.
Scheme 1.

Alternating Ring-Opening Polymerization, AROMP.
Cyclohexene is a ring-opening metathesis inactive substrate with ruthenium catalysts.11 However, it undergoes ring opening cross metathesis with acrylates.12 On the basis of this result and the observation noted above, we postulated that ester 2a and cyclohexene, subjected together to an active ruthenium catalyst, would undergo alternating ring-opening metathesis polymerization (AROMP).
In order to test this premise, we examined the fate of a mixture of cyclobutene ester 2a and cyclohexene (4a) in the presence of precatalyst 1. Indeed, polymerization occurred with 74–98% conversion (Table 1). Taken together with the regioselective ROM (but not ROMP) of ester 2a and the lack of reactivity of cyclohexene, this result strongly suggested that AROMP had occurred. 1H-NMR spectroscopic analysis (Figure S1) of each of the polymers revealed the phenyl protons and two sets of vinyl protons. The ratio of the signal for the protons on the disubstituted, non-conjugated olefin (δ = 5.4 ppm) to the signal for the protons on the trisubstituted, conjugated olefin (δ = 6.8 ppm) was approximately 2:1. This result is consistent with polymer structure (2a–4a)n which contains nearly equal numbers of repeating units A and B generated from monomers 2a and 4a respectively. 1H-1H gCOSY spectroscopy of (2a–4a)20 clearly showed internal connectivity between repeating units A and B (Figure S3), further establishing the alternating nature of the polymer backbone.
Table 1.
AROMP Polymers Synthesized.a
| A | B | [Ru] (M) | [A]:[B]:[Ru] | Rxn time (h) | Prod. | % convb |
|---|---|---|---|---|---|---|
| 2a | 4a | 0.05 | 3:6:1 | 6 | (2a-4a)3 | 74c |
| 2a | 4a | 0.01 | 10:20:1 | 3 | (2a-4a)10 | 98 |
| 2a | 4a | 0.01 | 20:40:1 | 3 | (2a-4a)20 | 98 |
| 2a | 4a | 0.01 | 50:100:1 | 3 | (2a-4a)50 | 98 |
| 2a | 4a | 0.01 | 100:200:1 | 3 | (2a-4a)100 | 97 |
| 2a | 4a | 0.005 | 200:400:1 | 6 | (2a-4a)200 | 73 |
| 2a | 4a-D10 | 0.01 | 20:24:1 | 3 | (2a-4a- D10)20 | 97 |
| 2a | 4a-D10 | 0.01 | 20:160:1 | 3 | (2a-4a- D10)20 | 97 |
| 2b | 4a | 0.01 | 20:40:1 | 4 | (2b-4a)20 | 96 |
| 2a | 4b | 0.01 | 20:40:1 | 4 | (2a-4b)20 | 95 |
All ROMP reactions were performed in CD2Cl2 and monitored by 1H-NMR
spectroscopy at rt.
Percent conversion determined by integration of 1H-NMR spectra unless specified otherwise.
Reaction was performed in CH2Cl2 and the isolated yield was determined after flash column chromatography purification.
In order to ascertain with greater accuracy the extent of alternation in the sequence of monomer units, we undertook an isotopic labeling experiment. Cyclohexene-D10, 4a-D10, and cyclobutene 2a were subjected to AROMP, the 1H-NMR spectra of the crude polymers acquired, and the intensities of the olefinic peaks integrated against the phenyl end group (Figure 1). As expected, for a deuterated alternating AB copolymer (2a-4a-D10)20, the relative intensity of the signal at δ = 5.4 ppm was reduced to half of that in the spectrum of polymer (2a-4a)20 (Figure 2A). This halved intensity is consistent with the absence of BB repeat in the polymer (Figure 2B). Moreover, the intensity of the signal at δ = 6.8 ppm in the spectrum of (2a-4a-D10)20 was reduced to 9% of its relative intensity in the spectrum of the undeuterated polymer ((2a-4a)20), showing that only 9% of the dyads are of type AA (Figure 2B). We observed that the fraction of AA dyad was constant regardless of the original A:B feed ratio in the AROMP reaction. Therefore, the polymer chain grows with alternation by a mechanism that does not depend on monomer concentration.
Figure 1.

Alkene region of 1H-NMR spectra (CD2Cl2) of polymers (2a-4a)20 and (2a-4a-D10)20. Proton integrations and assignments are indicated above the peaks.
Figure 2.

Possible Substructures Generated in the Copolymerization of 2a with Cyclohexene-D10.a
aREd carbons are perdeuterated. Blue carbons bear hydrogen.
The concentration-independent population of the AA dyad and the absence of BB dyad suggested that the AA dyads result from intramolecular backbiting of the enoic ruthenium carbene 5 on the disubstituted alkene.13 Partial separation of the components of the polymer by flash chromatography gave a fraction that contained the AA substructure (as indicated by a clean triplet at 6.8 ppm) but not the phenyl end group (Figure S5). Isolation of this material supports a model in which the AA repeat is generated at the backbiting junction during the cyclization step. We found that the Mw’s were shorter than expected and the poly-dispersities (PDIs) were large (> 2) (SI and Figure S6). Polymer (2-4a)200 had a bimodal molecular weight distribution in which the higher molecular weight peak corresponded to the desired polymer with a PDI of 1.2 (Figure S7). These data are consistent with backbiting to form cyclic polymer during chain growth.
In order to explore the breadth of applications of AROMP products, we examined the effects of varying the structures of the monomers. We found, for example, that AROMP of 4a and phenyl ester 2b proceeded with high conversion (>95%) at room temperature in 4 hours to yield (2b-4a)20 (Table 1). This type of product is viewed as a precursor to polymers with a variety of sidechains because of the electrophilicity of phenyl esters. Next, we investigated the effect of substituents on the cyclohexene. Neither 1-methylcyclohexene nor 1-methoxy-cyclohexene generated AROMP polymer with 2a. We inferred that increased substitution at the alkene prevents addition. Indeed, 4-(methoxymethyl)-cyclohexene, 4b underwent AROMP with 2a to generate the corresponding alternating polymer (2a-4b)20, with 95% conversion in 4 hours (Table 1). The regiochemistry of metathesis could not be determined in the polymerization of 4b and is most likely random. 4-Substituted cyclohexenes are attractive monomers for AROMP because they are readily available through Diels-Alder chemistry.
In conclusion, we have demonstrated that synthetically accessible, select monomer pairs undergo AROMP with the reactive precatalyst 1 to form (AB)n heteropolymers with an alternating backbone and alternating functionality. The regiocontrol of heteropolymer formation derives from the inability of the cyclobutene ester and cyclohexene monomers to undergo homopolymerization in combination with the favorable kinetics of cross polymerization.
Supplementary Material
Acknowledgments
We acknowledge NIH grants R01HD38519 S10RR021008 (NSS), and R01GM39287 (KAP), NYSTAR grant (FDP C040076, NSS) and NSF grant CHE0131146 (NMR). We thank Dr. F. Picart for his assistance with NMR spectroscopy.
Footnotes
Supporting Information Available: Experimental procedures and characterization data (PDF). This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Klok HA, Lecommandoux S. Adv Mat. 2001;13:1217–1229. [Google Scholar]; (b) Grayson SM, Frechet JMJ. Chem Rev. 2001;101:3819–3867. doi: 10.1021/cr990116h. [DOI] [PubMed] [Google Scholar]; (c) Cho I. Prog Polym Sci. 2000;25:1043–1087. [Google Scholar]
- 2.(a) Murphy JJ, Furusho H, Paton RM, Nomura K. Chem Eur J. 2007;13:8985–8997. doi: 10.1002/chem.200700291. [DOI] [PubMed] [Google Scholar]; (b) Kumar R, Chen MH, Parmar VS, Samuelson LA, Kumar J, Nicolosi R, Yoganathan S, Watterson AC. J Am Chem Soc. 2004;126:10640–10644. doi: 10.1021/ja039651w. [DOI] [PubMed] [Google Scholar]; (c) Bronich TK, Keifer PA, Shlyakhtenko LS, Kabanov AV. J Am Chem Soc. 2005;127:8236–8237. doi: 10.1021/ja043042m. [DOI] [PubMed] [Google Scholar]
- 3.Furuta PT, Deng L, Garon S, Thompson ME, Frechet JMJ. J Am Chem Soc. 2004;126:15388–15389. doi: 10.1021/ja0446247. [DOI] [PubMed] [Google Scholar]
- 4.(a) Braunecker WA, Matyjaszewski K. Progr Polymer Sci. 2007;32:93–146. [Google Scholar]; (b) Maki Y, Mori H, Endo T. Macromolecules. 2008;41:8397–8404. and references therein. [Google Scholar]
- 5.Hamilton JG, Ivin KJ, Rooney JJ, Waring LC. J Chem Soc, Chem Commun. 1983;4:159–61. [Google Scholar]
- 6.Choi TL, Rutenberg IM, Grubbs RH. Angew Chem Int Ed Engl. 2002;41:3839–3841. doi: 10.1002/1521-3773(20021018)41:20<3839::AID-ANIE3839>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 7.Ilker MF, Coughlin EB. Macromolecules. 2002;35:54–58. [Google Scholar]
- 8.(a) Bornand M, Torker S, Chen P. Organometallics. 2007;26:3585–3596. [Google Scholar]; Bornand M, Chen P. Angew Chem, Int Ed. 2005;44:7909–7911. doi: 10.1002/anie.200502606. [DOI] [PubMed] [Google Scholar]; (b) Vehlow K, Wang D, Buchmeiser MR, Blechert S. Angew Chem Int Ed Engl. 2008;47:2615–2618. doi: 10.1002/anie.200704822. [DOI] [PubMed] [Google Scholar]; (c) Amir-Ebrahimi V, Rooney JJ. J Mol Catal A. 2004;208:115–121. [Google Scholar]; Al Samak B, Carbill AG, Hamilton JG, Rooney JJ, Thompson JM. Chem Comm. 1997:2057–2058. [Google Scholar]
- 9.Love JA, Morgan JP, Trnka TM, Grubbs RH. Angew Chem Int Ed Engl. 2002;41:4035–4037. doi: 10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 10.Lee J, Parker KA, Sampson NS. J Am Chem Soc. 2006;128:4578–4579. doi: 10.1021/ja058801v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivin KJ, Mol JC. Olefin metathesis and metathesis polymerization. Academic Press; London: 1997. [Google Scholar]
- 12.(a) Ulman M, Belderrain TR, Grubbs RH. Tetrahedron Lett. 2000;41:4689–4693. [Google Scholar]; (b) Choi TL, Lee CW, Chatterjee AK, Grubbs RH. J Am Chem Soc. 2001;123:10417–10418. doi: 10.1021/ja016386a. [DOI] [PubMed] [Google Scholar]; (c) Fomine S, Tlenkopatchev MA. Organometallics. 2007;26:4491–4497. [Google Scholar]
- 13.Thorn-Csanyi E, Ruhland K. Macromol Chem Phys. 1999;200:1662–1671. [Google Scholar]; (b) Kamau SD, Hodge P, Hall AJ, Dad S, Ben-Haida A. Polymer. 2007;48:6808–6822. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.