

Abstract
Motivation: The task of engineering a protein to perform a target biological function is known as protein design. A commonly used paradigm casts this functional design problem as a structural one, assuming a fixed backbone. In probabilistic protein design, positional amino acid probabilities are used to create a random library of sequences to be simultaneously screened for biological activity. Clearly, certain choices of probability distributions will be more successful in yielding functional sequences. However, since the number of sequences is exponential in protein length, computational optimization of the distribution is difficult.
Results: In this paper, we develop a computational framework for probabilistic protein design following the structural paradigm. We formulate the distribution of sequences for a structure using the Boltzmann distribution over their free energies. The corresponding probabilistic graphical model is constructed, and we apply belief propagation (BP) to calculate marginal amino acid probabilities. We test this method on a large structural dataset and demonstrate the superiority of BP over previous methods. Nevertheless, since the results obtained by BP are far from optimal, we thoroughly assess the paradigm using high-quality experimental data. We demonstrate that, for small scale sub-problems, BP attains identical results to those produced by exact inference on the paradigmatic model. However, quantitative analysis shows that the distributions predicted significantly differ from the experimental data. These findings, along with the excellent performance we observed using BP on the smaller problems, suggest potential shortcomings of the paradigm. We conclude with a discussion of how it may be improved in the future.
Contact: fromer@cs.huji.ac.il
1 INTRODUCTION
The engineering objective of ‘creating’ a protein to perform a target biological function is known as protein design. Since the pursuit of a protein with a specific function without additional constraints is perceived as overly difficult, the function-design problem is usually restricted to the search for an amino acid sequence that assumes a target 3D structure (Kuhlman et al., 2003; Park et al., 2004), assuming that this structure will entail a specific function. The potential applications of such design are diverse and numerous (for a recent review, see Rosenberg and Goldblum 2006). Specific objectives include the modest ambition of slight modifications of existing proteins to affect such characteristics as stability or binding affinity (Shifman and Mayo, 2002). A loftier goal is to design protein sequences that will assume novel structures (Kuhlman et al., 2003) or acquire new functionalities. Such functionalities may be therapeutic (e.g. Bewley et al., 2002, where an HIV inhibitor protein was designed, or Lazar et al., 2003) or industrial (e.g. Arnold 2001, which discusses the design of biocatalysts). Furthermore, the success (or failure) of such protein design experiments will aid us in the validation of our understanding of the physics of protein stability and structure.
Most recent work on structural protein design adopts a paradigmatic representation of a protein as a fixed backbone structure. Also, the amino acid side chain conformations are not permitted to move continuously in space; rather, the allowed conformations consist of a library of discrete, energetically favorable empirical observations [termed ‘rotamers’ (Dunbrack and Karplus, 1993)]. The two former assumptions artificially limit the spatial degrees of freedom of the protein structure but are adopted for computational tractability. Additionally, pairwise atomic energy terms are used to confer pseudo-physical energetic values to pairs of atoms (Gordon et al., 1999). These energies are employed in scoring individual sequences, either through per-sequence consideration of the minimal-energy rotamer conformation (as in Shifman and Mayo, 2002) or of the rotamer-based free energy partition function (Kamisetty et al., 2007; Lilien et al., 2005). It should also be noted that there exists a line of work that attempts to overcome the use of a fixed backbone by iterating between cycles of fixed backbone design and backbone improvements, e.g. Kuhlman et al., (2003).
Ultimately, the results of computational methods for this task must be experimentally verified in the wet lab. This typically leads to cycles of computational prediction of top-scoring sequences, synthesis of these sequences and biological characterization, and fine-tuning of the computational methods and inputs based on these empirical results (Kuhlman et al., 2003). However, this strategy only permits the biophysical screening of relatively few sequences (conceivably, hundreds at the most). Computational design of a small set of candidate sequences has been termed directed protein design (Park et al., 2005).
1.1 Probabilistic protein design
On the other hand, recent revolutionary advances in molecular biology have provided scientists with combinatorial sequence-screening techniques, such as phage display (Pal et al., 2006), which can produce 109–1010 randomized protein sequences that are then simultaneously tested for relevant biological function. This randomized synthesis and screening process is schematically illustrated in Fig. 1. This technology has lead to an alternative protein design protocol, designated probabilistic protein design (Park et al., 2005), which incorporates powerful computational tools into large-scale biological screening of candidate protein sequences. Overall, it involves two steps:
Computational prediction of amino acid probabilities at each position for the given design problem (considering all other possible amino acid sequences at other positions).
Use of these predicted probabilities to ‘bias’ the synthesis of random gene sequence libraries, which can be experimentally selected for relevant biological function (Hecht et al., 2004; Kono and Saven 2001; Park et al., 2005).
Fig. 1.

font The principle of combinatorial sequence screening is depicted for a protein of length 4, where amino acid types are represented by green, yellow, blue and red. The wild-type (WT) protein is mutated at its leftmost and rightmost positions, according to position-specific probabilities, yielding random proteins to be screened through a selection assay; dashed boxes indicate mutations from WT. In this scenario, four of the seven random protein sequences were positively selected.
Despite the ability of the aforementioned experimental methods to screen billions of sequences, the computational aspect remains of utmost importance, since many relevant design problems involve tens of positions where all 20 amino acids could potentially be placed, exceeding the biological screening capability by many orders of magnitude. The goal of this two-step technique is that the computationally predicted positional probabilities will significantly enrich the randomized sequences with those that are highly likely to possess the target biological characteristics and thus pass the screening stage, resulting in much higher success rates. Nevertheless, we emphasize that the predicted probabilities are only intended to bias the sequence space towards optimality, but do not guarantee the exclusive synthesis of low-energy sequences; this derives from the use of site-specific amino acid probabilities, independently employed for the various design positions.
Computational methods for calculation of positional amino acid probabilities based on structural information have been developed in the context of both directed protein design (Calhoun et al., 2003; Delarue and Koehl 1997) (where they were intended to build highly probable sequences) and probabilistic protein design (Biswas et al., 2005; Kono and Saven 2001; Park et al., 2005; Voigt et al., 2001; Yang and Saven 2005). These methods include the use of mean field (Delarue and Koehl, 1997; Voigt et al., 2001), Monte Carlo sampling techniques (Yang and Saven, 2005), and Lagrangian optimization based on simplifying approximations (Biswas et al., 2005; Calhoun et al., 2003; Kono and Saven 2001; Park et al., 2005). Overall, these approaches have been met with some success (e.g. Park et al., 2006), depending on the criterion utilized to assess performance.
In this paper, we describe a novel formulation for modeling the probabilistic design problem, following the structural design paradigm and the corresponding assumptions described above. Furthermore, we apply an innovative technique for efficient and accurate calculation of the marginal amino acid probabilities. We show that our novel method obtains quicker and more accurate results than those derived from previously applied approaches, as compared with exact results on the paradigmatic model. On the other hand, quantitative analysis of the results obtained by this model, as compared to evolutionary and experimental results, indicates inherent deficiencies in the modeling of the probabilistic protein space by this widely used paradigm.
2 PROBABILISTIC PROTEIN DESIGN: THEORY AND EFFICIENT MODELING
The input to the protein design problem consists of a spatial structure, N positions to be designed, and the amino acids (and their respective rotamers) permitted at each position. We denote by r=(r1,…,rN) an assignment of rotamers for all positions. For a given energy function, the energy of assignment r, E(r), is the sum of interaction energies between a rotamer ri at position i and the fixed template (Ei (ri)), and pairwise interaction energies between rotamers ri and rj for neighboring positions i,j (Eij(ri,rj)). We define 𝒯(k) as the amino acid type of rotamer k and let 𝒯(r1,…,rN)=(𝒯(r1),…,𝒯 (rN)). Let 𝒮=(𝒮1,…,𝒮N) denote an assignment of amino acids for all positions.
2.1 Sequence probability space
Consider the Helmholtz free energy of a specific amino acid sequence 𝒮 at system temperature T, as defined in Yedidia et al., (2005):
| (1) |
where
| (2) |
is the per-sequence partition function for sequence 𝒮 (Lilien et al., 2005) at system temperature T.
Since free energy is the operative value in measuring the thermodynamic compatibility of various sequences with the structure, and conceptually following Meyerguz et al. (2004) in assuming the applicability of Boltzmann's law over the sequence space, we define the probability of a given sequence 𝒮 at system temperature T as:
| (3) |
It is easy to see that the normalization factor for Pr(𝒮) is
| (4) |
thus yielding:
| (5) |
Boltzmann's law has been shown to describe physical systems in thermal equilibrium at a given temperature level (Huang, 1987; Yedidia et al., 2005) Since we are dealing with an energetic system where the states and energies are sequence assignments and their free energies, respectively, Equations 3–5 are applicable. In essence, this perspective regards the system of all sequences as competing among themselves to be chosen during the stage of experimental (or possibly evolutionary, e.g. Meyerguz et al., 2004) selection.
2.2 Rotamer probability space
We wish to follow Equation 5 but would like to work directly with the rotamers of all sequences simultaneously. Therefore, we consider a probability space over all rotamer assignments for all sequences, where the probability of rotamer assignment r, at system temperature T, is defined as:
| (6) |
where Z(T), the rotamer space partition function, is as defined in Equation 4. We note that Equation 6 follows Boltzmann's law over the rotamer space and, more significantly, substitution into the intuitive definition for sequence probability:
| (7) |
directly yields Equation 5. Now, by definition, the marginal amino acid probabilities at position i are given by:
| (8) |
However, due to Equation 7, this result could equivalently be generated if Pr (𝒮i) were directly defined as:
| (9) |
where Pr(ri)=∑r∖ri Pr(r) is the marginal probability of rotamer ri at position i. Note that, in fact, Equation 9 was used as the definition for amino acid probabilities in previous work (Biswas, et al., 2005; Kono and Saven, 2001; Park et al., 2005; Voigt et al., 2001; Yang and Saven, 2005). In contrast to the theory developed above, however, no explicit definition of the probability space over the sequences or justification for such amino acid probabilities was given.
At this point, we find it pertinent to emphasize that although our model accounts for the per-sequence free energy partition function (Lilien et al., 2005) in defining the probability for a particular sequence (Equation 5), these values are never actually calculated for any individual sequences. Rather, these per-sequence free energies form the theoretical basis of the site-specific amino acid probabilities (Equation 8), but we do not actually resort to the exponential summation of sequence probabilities (see below).
2.3 Protein design using probabilistic graphical models
With a clearly delineated model for positional amino acid probabilities in hand, we now present our novel approach. A probabilistic graphical model (Lauritzen, 1996) is built to represent this probabilistic protein design problem, where each designed position is a variable in the model, and its values correspond to the allowed rotamers (for all permitted amino acids) at that position. The exponentiated rotamer-backbone energies are the local potentials in the model:
and the rotamer-rotamer energies define the pairwise potentials:
Since the size of the rotamer space is exponential in protein length, the calculation of exact probabilities (Equation 8) using this graphical model is computationally infeasible for large proteins. Therefore, an approximate inference method is required. Herein, we apply sum–product loopy belief propagation (Pearl, 1988), to which we simply refer as BP. Thus, belief messages of the form:
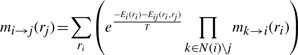 |
are passed from position i to position j, where the contents of mi→j indicate the relative likelihood of each rotameric state at position j. Note that N(i) denotes the set of variables neighboring variable i.
After convergence of BP, the resulting marginal rotamer probabilities are defined as (after normalization):
These rotamer beliefs at each position (which account for all possible sequences at all other designed positions) are then marginalized per amino acid to obtain the per-position amino acid probabilities for the design problem, as described in Equation 9.
We note that the method described here puts to use, and thus assesses, the actual values of the sum–product marginals derived through loopy BP on a graphical model built to describe a protein structure (and all of its possible sequences), and is thus conceptually similar to the method utilized by Moore and Maranas (2003), to predict residue–residue clashes in protein hybrids. On the other hand, most previous related methods seek the minimal-energy assignments (using max-product BP) (Yanover and Weiss, 2003; Yanover et al., 2006) or utilize the sum–product beliefs (Kamisetty et al., 2007) or pruning methods (Lilien et al., 2005) to approximate the free energy for a single protein sequence.
3 METHODS
3.1 Test cases and benchmark probabilities
PDB dataset and evolutionary data
Protein structures were culled from the PDB database (Berman, et al., 2000) in the following manner: experimental X-ray structures containing a single protein chain of length 30−70 amino acids, with a minimal resolution of 2 Å were considered, where a cut-off level of 30% sequence identity was applied to remove homologues. For these cases, the ground truth probabilities were taken to be those derived for the PDB structures from evolutionary data by the HSSP (Homology-derived Secondary Structure of Proteins) database (Dodge et al., 1998). We further required that the HSSP entry for the protein structure contain a minimum of 10 alignments; the final set of 29 structures is listed in Table 1. For each of these structures, all positions were designed using the pairwise atomic energy function of the Rosetta design package (Kuhlman and Baker, 2000) with default parameters and the backbone-dependent rotamer library of (Dunbrack and Karplus 1993). The energy function includes terms for the 12-6 Lennard-Jones potential, implicit interaction with the solvent, electrostatics, a hydrogen-bonding potential, backbone dependent internal free energies of rotamers, and amino acid reference energies (wref).
Table 1.
Dataset of 29 proteins from the PDB database, for which full sequence probabilistic design was computationally performed. Representative structures for each structural class are taken from (Berman et al., 2000).
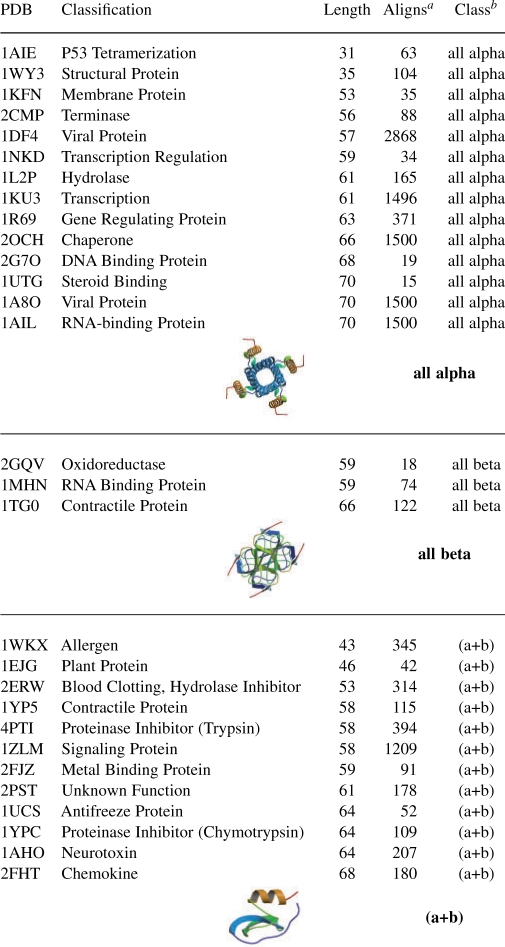 |
aNumber of HSSP alignments from which the evolutionary probabilities were derived.
bStructural class, as defined by the SCOP or CATH databases (as available).
Human growth hormone
As an in-depth case study, we further considered the structure of the human growth hormone (hGH) complexed with the extracellular domain of its receptor (hGHR), PDB code 3HHR. For this case, 35 positions of hGH (Fig. 4), which lie on the interface with hGHR were designed under a variety of scenarios (see Experiments). We attempted to recover the experimentally derived positional probabilities detailed in Pal et al. As above, the Rosetta energy function was employed with default parameters, except where otherwise noted. For this case, we arbitrarily added pseudo-counts of 0.5 to the raw experimental data from Pal et al. in order to compensate for sample bias (unobserved amino acids).
Fig. 4.

Left: The hGH–hGHR complex (PDB code 3HHR). The 35 design residues are colored in cyan (positions included in small-scale testing) and blue. The shell of 106 positions (in hGH and hGHR), allowed to conformationally vary in the large-scale testing, is colored yellow. All other positions are colored red. Structures were drawn using Chimera (Pettersen et al., 2004). Right: The JSD for the designed positions in the small-scale problems, for the methods tested (compared to the exact probabilities): Gibbs sampling after 200 and 2000 samples, MF and BP. Results are depicted in log scale and non-existent columns (e.g. for BP) indicate values lower than 10−10. For position 168, the Gibbs sampler failed to converge to accurate probabilities even after 200 000 samples. Also for this position, depending on the update order, MF often yielded highly inaccurate probabilities (not shown).
Since Rosetta does not fully support the design of the amino acid cysteine, all references to design for all possible amino acids should be interpreted as meaning design of all other 19 natural amino acids; in order to compensate for this, we manually removed all references to cysteine from the HSSP-derived probabilities and the experimental results of Pal et al. For all experiments described herein, we use a temperature value corresponding to human body temperature (37○C), scaled by the Boltzmann constant to yield an energetic term (Huang, 1987).
3.2 Prediction algorithms
Belief propagation (BP)
Sum–product loopy belief propagation (Pearl, 1988) was run until numerical convergence, or a maximum of 100 000 messages passed. BP converged for all proteins in the PDB dataset and for almost all cases for the hGH–hGHR problems; in any event, the sum–product marginals at termination were utilized to predict the positional marginal amino acid probabilities (Equation 9).
Mean field (MF)
Self-consistent mean field theory (Delarue and Koehl, 1997) was applied to the problems, using a randomized update order for the positions. The first-order mean field iterative update formula is:
where αi is a normalization factor. At termination, Equation 9 was applied to obtain amino acid probabilities from the rotamer probabilities (qi(ri)).
Gibbs sampling (Gibbs)
Gibbs sampling was performed such that each atomic step consisted of randomly switching all of the positions in the problem (in random order), based on the local posterior probabilities; the sampler was burned in for 50 steps, with an interval of 20 steps between samples. Equation 9 was utilized here as well.
Exact probabilities
For the smaller hGH–hGHR cases, exact results were obtained by iterating over all possible sequences and explicitly calculating the per-sequence partition function defined in Equation 2 [using the junction tree method (Cowell, 1998)] and applying Equation 5. For all other instances, this was not feasible because the junction tree method is exponential in the largest clique size formed, which is intractably large for even moderate-sized design problems.
3.3 Assessment of predicted distributions
To quantitatively compare probability distributions, we use the symmetric Jensen-Shannon divergence (JSD), which ranges from 0 (identical) to 1 (‘distant’ distributions), so that lower JSD scores reflect higher similarity between distributions. The JSD between distributions P and Q is given by:
where 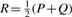 is the average distribution, and
is the average distribution, and
is the Kullback-Leibler divergence between distributions A and B.
4 EXPERIMENTS
We assessed our framework for probabilistic protein design in two main contexts: using wide-ranging evolutionary data and in a case study of experimental data for hGH. For both scenarios, we demonstrate enhanced performance using BP on our modeling of the problem, yet find the ubiquitous structural paradigm to be quite lacking.
4.1 Evolutionary data
Firstly, we applied our novel approach to the comprehensive dataset culled from the PDB (Table 1). It must be noted that we should not expect to fully predict evolutionary profiles exclusively using protein energetics, since there exist additional selective pressures for which we do not account. Nonetheless, due to the sparsity of experimental data (see hGH experiments below) in which the energetic sequence landscape was more directly probed, in this section we resort to utilizing the distribution of amino acids in the HSSP database for a given protein structure as the ‘ground truth’.
All protein positions were designed to all amino acids, and we measured the correspondence of our BP-predicted amino acid probabilities (using the JSD measure) to those derived by the HSSP database (Fig. 2). Furthermore, we used the same paradigmatic models for each structure to compute amino acid probabilities using Gibbs sampling (200 samples) and MF. A naive alternative to predicting positional probabilities for probabilistic design is simply to allow all amino acids with equal probability at all positions (the uniform distribution). We thus also include this distribution in our analysis.
Fig. 2.

The JSD values for 1700 design positions spanning 29 fully designed proteins from the PDB, where the ground truth probabilities are derived from the HSSP database. Left: Comparison of JSD obtained by Gibbs (200 sampling steps), MF, BP and the uniform distribution, where each column denotes the mean JSD for all positions of a single protein in the dataset; proteins are ordered as in Table 1 and structural classes are as marked. Numbers on the right indicate the mean JSD values, over all proteins, for the respective methods. Right: Histogram of JSD values for all 1700 design positions.
Overall, our results (Fig. 2, left) show that for most proteins the mean JSD for the various methods (excluding uniform) are highly correlated; since they all use the same pre-calculated Rosetta energies, this is not completely surprising. Nevertheless, this indicates that for cases where the paradigmatic model corresponds to the evolutionary data, the prediction methods will yield similarly good results (albeit with some performing better than others, see below), and vice versa. In particular, it seems that for the mixed α and β proteins, the paradigmatic model works better to predict the evolutionary data than for other structural classes.
Expectedly, the uniform distribution performs most poorly (highest mean JSD), which attests to the need for computational methods to predict the relevant positional amino acid probabilities for specific proteins. Moreover, BP and Gibbs sampling significantly outperform MF (lower mean JSD and leftward shifted distributions, Fig. 2), despite using the same pre-computed Rosetta energies and requiring approximately the same run-time (Fig. 3). We also found, in most cases, that Gibbs sampling takes slightly longer to achieve results comparable to those of BP.
Fig. 3.

Computational run-times for representative proteins from the dataset in Table 1, where bar colors are as in Fig. 2. m = minutes.
We observed that, for positions where the JSD is larger for the BP probabilities than for the MF ones, it is only slightly higher (mean increase of 0.035 in JSD for 760 positions). On the other hand, there are a very large number of positions where BP has significantly improved the performance of the paradigmatic model in recovering HSSP-derived probabilities, i.e. the JSD is decreased as compared to the state-of-the-art MF method (mean decrease of 0.134 over 884 positions). Note that a comparison of BP with Gibbs sampling did not show significant differences in per-position performance.
Despite the fact that BP and Gibbs sampling outperform the MF method in the prediction of amino acid probabilities, their mean JSD from the HSSP-derived probabilities nonetheless indicate that the paradigmatic model does not strongly correspond to the amino acid probabilities found in nature. We hypothesized that this could be due to other, non-energetic constraints that may exist for natural sequences (e.g. Humphris and Kortemme 2007). To investigate this matter, we proceeded to perform a case study of recently published large-scale experimental probability data, for which it is expected that non-energetic considerations are less significant.
4.2 Experimental design data for hGH
Although some of the techniques described above for probabilistic protein design have been applied with some success (e.g. Park et al., 2006), they have not yet, to the best of our knowledge, been applied to perform large-scale unsupervised combinatorial protein design (as suggested in Park et al., 2005), or even tested on a comprehensive dataset of experimentally-derived probabilities. The dataset presented in Pal et al. is the first such data of its kind, in that it experimentally estimated the site-specific amino acid probabilities for a large number of positions of a given protein structure. Specifically, it calculated the positional amino acid probabilities for 35 positions (Fig. 4) on the interface of hGH with its receptor (hGHR), by grouping them into 6 groups of 5 or 6 positions each and uniformly allowing for all possible sequences within each group. Random sequences were phage displayed by fusion to the M13 coat protein and subsequent biological selection was performed by either binding to hGHR or an antibody that specifically binds the side of the protein opposite this interface. These two selection criteria were used to assay either for hGH functionality or hGH stability, respectively. Herein, we attempt to recover these experimental probabilities by application of our method to the six sub-problems explored in Pal et al.
For all subsequent experiments in this paper, we employ the structure of hGH complexed with the extracellular domain of hGHR. We analyze the probabilities calculated at the hGH positions for the case of binding to hGHR. Qualitatively similar results were obtained for the case of antibody binding (where we removed hGHR from the structure to perform our calculations).
Small-scale problems
Before testing our method by comparison with the large-scale data from Pal et al., we wanted to test the efficacy of our method in recovering the marginal probabilities as compared to an exhaustive enumeration of the exact sequence space, assuming correctness of the paradigmatic model. We created some ‘artificial’ small-scale examples, similar to those assessed below: from each group of six positions in hGH defined in Pal et al., we chose two of the spatially proximal ones to be designed and allowed for a small shell of approximately seven nearby native amino acids in the hGHR protein to change side chain conformations (rotamers). For all other positions, the side chain conformations were held constant. The positions tested (12 in total) are colored cyan in Fig. 4 (left panel).
We compared the results of three approximate inference methods — BP, Gibbs sampling and MF theory — with the exact results (Fig. 4, right). The run-times for MF and BP were comparable (∼1 s), as was the run−time for 200 samples of the Gibbs sampler. We also ran the sampler for 2000 samples, with an approximately proportional increase in run-time.
Figure 4 demonstrates that BP performs best, being the lowest scorer for all positions. MF is next best and does comparably well, since for almost all positions it achieved low JSD just as quickly. The Gibbs sampler fares the worst, since in order to achieve JSD comparable with MF (at best) it had to be run approximately 10 times as long. Overall, we conclude that, for the paradigmatic model, both previous prediction algorithms and BP are relatively successful in yielding accurate positional probabilities.
Large-scale experimental data
To simulate the experiments reported in Pal et al., 2006, we divided the 35 hGH interface positions into the 6 groups defined there. For a given group, its members were permitted to assume all amino acid identities, while other positions in the set of 35 were allowed to change rotameric state for the native amino acid. In addition, we defined a shell of ∼11 Å (7 Å for the longest side chain and 4 Å interaction distance) around each of the group's Cα atoms such that all residues in this shell (in hGH and hGHR) whose side chain could directly interact with a side chain in the designed group were allowed to change rotameric state (Fig. 4). Here, we compare the results of both MF and BP for three different Rosetta parameter sets: default parameters (Default), addition of rotamers for both χ1 and χ2 angles (+Rotamers) and addition of rotamers with nullification of the wref energy term intended to enhance native sequence recovery (+Rotamers, −wref) (Kuhlman and Baker 2000). We do not show the results of Gibbs sampling, since (as observed above for both the evolutionary and small-scale hGH data) it requires more computation time to achieve accurate results comparable to those of BP.
The quantitative comparison of the methods with this experimental data is again performed using the JSD (Fig. 5). Firstly, we emphasize that we apply the JSD measure to the distributions over all amino acids, since certain functional amino acid groupings used in the assessment of results in earlier studies [e.g. Kono and Saven 2001; Biswas et al., 2005] were shown to be somewhat unjustified by the results in Pal et al. The JSD results here indicate large discrepancies between the predicted and observed probability distributions. Nonetheless, using default Rosetta parameters, BP obtains a mean JSD value of 0.435, which is a significant improvement over its performance on the evolutionary data (mean JSD 0.529). Thus, as hypothesized, the paradigmatic model performs better when compared to experimental sequence selection studies. Furthermore, we find that BP significantly outperforms MF for almost all 35 positions. In addition, relatively independent of the algorithm used, there is a trend of decreasing JSD as more rotamers are allowed and when the energetic term intended to recover native amino acid identities (wref) is removed. As in the small-scale problems, the run-times for MF and BP are comparable (an average of 24 and 48 s for the default rotamer library and 695 and 1265 s with extra rotamers added, respectively).
Fig. 5.

Left: The JSD for all 35 designed hGH positions, compared with the experimental data. Each pair of rows portrays the results for MF and BP. The three pairs of rows depict the results using default Rosetta parameters, an enhanced rotamer library, and the enhanced library where the wref term was eliminated. Numbers on the right indicate mean JSD values. Right: The JSD values resulting from the inference performed by BP in the +Rotamers, −wref case (left panel, 5th row) are color mapped onto the Cα atoms of the hGH structure, where the whiter the color, the better the prediction at that position.
In analyzing the results obtained here, it should be noted that the findings of Pal et al. were somewhat unexpected, as indicated by the authors. In some positions, for example, substitutions that are believed to be ‘conservative’ were not permitted. Additionally, they observed an over-abundance of hydrophobic substitutions, even at positions evolutionarily conserved as hydrophilic. In fact, the JSD results using probabilities predicted by evolutionary conservation were significantly worse than those reported here (not shown). The findings in Pal et al. also seem to contradict the results of Kuhlman and Baker (2000), where it was concluded that, for the most part, ‘native protein sequences are close to optimal for their structures’. The unforeseen nature of these results could explain, to some extent, the high degree of inaccuracy of the paradigmatic model (built to describe previously characterized phenomena). Indeed, closer inspection (Fig. 6) reveals that some of the worst predictions by our method are due to hydrophilic (serine) and hydrophobic amino acids (e.g. phenylalanine, valine and leucine), on account of under- or over-prediction of these residue types (large JSD contributions).
Fig. 6.

The various amino acid contributions to the JSD values resulting from the +Rotamers, −wref method on the hGH design. Since the JSD consists of a sum over all amino acids, this decomposition was possible. The contributions over all 35 positions were summed to obtain the results here.
Positional independence within designed groups
Notwithstanding the large discrepancies between the experimental and predicted probabilities, we used our model to test one of the basic assumptions in Pal et al.: the non-cooperativity (independence) of the positions in each of the six groups. In Fig. 7, we regard the positional probabilities computationally predicted by the six groupings as a baseline in comparison with two possible extremes: one group of 35 simultaneous computational design positions (A) and 35 groups of single design positions (I). For each of these 36 new design problems, the shell of amino acid positions allowed to vary conformation was taken to be the same as that in the six groups of design problems, as described above. Pal et al. are clearly precluded from performing the former experiment since the phage display technology can yield at most 1010 sequences (exponentially <2035), and rule out the latter experiment due to its requirement of the synthesis and biochemical analysis of 700 specific sequences. Nonetheless, their stated goal was to simulate the latter situation.
Fig. 7.

The JSD for all positions, for the computational results of the extremal groupings: one group of 35 design positions (A) and 35 groups of single design positions (I), as compared to the computational results obtained by the six groupings from Pal et al. Color map values are limited to 0.2 for presentation purposes. Numbers on the right indicate the mean JSD values for the respective groupings.
Our calculations show that, for most of the positions, the probability distribution obtained through individual design (I) was indeed almost identical to that computationally predicted by the authors’ groupings (low JSD values), especially in the loop regions. On the other hand, the distributions derived from the simultaneous design of all 35 positions (A) agree somewhat less with those calculated by the authors’ groupings (higher JSD values). For more than a third of the positions (found overwhelmingly in the designed helices), the positional JSD value is 0.1 or greater; for four of these positions, the JSD is even greater than 0.3. Nevertheless, the individual probabilities (I) do provide a very reasonable approximation of those obtained from the complete, synchronous design setting (mean JSD 0.1).
We had expected to find larger discrepancies when considering grouping A, since positions have the potential to participate in cooperative interactions with other positions (directly, or through intermediaries in the shell), unavailable when they are individually mutated. Surprisingly, our results imply that, in some cases, the use of probabilities obtained when only considering a single design position at a time (or a group of relatively independent positions, as in the authors’ experimental results) may work together to provide optimal probabilities for simultaneous design of all positions, as required for probabilistic protein design.
5 DISCUSSION
Advances in research biology now permit the analysis of a vast number of protein sequences, on the order of 1010. Nevertheless, the number of possible sequences in a typical design problem surpasses this by many orders of magnitude. Therefore, strategies to enrich the screened library of sequences with potentially promising ones are vital. In this paper, we have proposed a computational technique to predict positional biases to achieve this end. These biases are calculated using approximate inference on graphical models built to represent the paradigmatic protein design problem.
A major phenomenon demonstrated here is that use of the paradigmatic protein model results in probability distributions far from those observed experimentally. A similar conclusion was recently reached (to a much lesser degree) based on a smaller dataset of only six mutated positions (Lassila et al., 2007). However, it must be cautioned that the nature of amino acid preferences experimentally observed for the hGH–hGHR complex in Pal et al. may not be typical of other protein structures. Additional experimental data are required to draw more general conclusions, specifically regarding the accuracy of this model. Nonetheless, since the paradigmatic model fares poorly in predicting both experimental and evolutionary (HSSP) probabilities, it is safe to say that there remains substantial progress to be made in the field.
To put the performance of the paradigmatic model used here into context, we observe that the uniform distribution over all 19 amino acids obtains a mean JSD of 0.163 when compared with the experimental hGH results. Clearly, this would seem to be a much better result than that achieved using the paradigm applied here (Fig. 5), as the best method (BP) yields a mean JSD of 0.358. However, since it is a safe assumption to make that not all positions tolerate all amino acids equally in all proteins (and as also indicated by the poor performance of the uniform distribution on the evolutionary data), more effort needs to be made to refine the inaccuracies and limitations described below.
5.1 Limitations and improvements of the paradigm
The structural design paradigm used here is well-known to embody a number of limitations, including the imprecision of the energy function (especially for protein surfaces and interfaces, Jaramillo et al., 2002) and its decomposition into pairwise terms, the assumption of a fixed backbone Kuhlman et al., 2003, and the discretization of side chain conformation. We review the inadequacies of the paradigm through the prism of the results for the hGH design, though they are equally pertinent to all design problems assessed.
Since addition of rotamers for the hGH design improved the prediction results, it can be suggested that the side chain discretization utilized is an acute limitation of this paradigm. Also, the wref energy term, intended to enhance the correct choice of native amino acids, only seemed to impair performance. However, this is somewhat unsurprising since the results in Pal et al. frequently do not recover the native amino acids with high probability. Nevertheless, the decreased performance realized using the wref term alludes to the fact that refinement of the energy functions used by the paradigmatic model could, to some degree, increase prediction accuracy. The problematic nature of the fixed-backbone assumption was observed for the case of position 61 (in loop 2), where the native amino acid is proline (putting unique constraints on the backbone). The paradigm used here over-predicts the probability of proline, since other amino acids are energetically unfavorable due to the steric constraints of the backbone.
One promising future direction would be to optimize a pairwise energy function for performance of the task of amino acid probability prediction at interfaces (for example, conceptually as in Yanover et al., 2007), since the energy function used here was only optimized for single-position native amino acid recovery in monomers (Kuhlman and Baker 2000). However, this objective can only be fully realized once sufficient experimental probability data have accumulated. Alternatively, incorporation of geometric constraints into this paradigm could be used to produce better predictions, since this has been shown to improve native sequence prediction (Chakrabarti et al., 2005). A complementary goal is to permit deviations of the backbone from the native one (Schueler-Furman et al., 2005), thus allowing for relaxation of some of the computationally predicted steric hindrances that may preclude certain amino acids observed experimentally and in evolutionarily related structures (Saunders and Baker 2005). Finally, since each sequence could potentially fold to any of numerous similar structures, it may be necessary to explicitly consider these ‘competing’ structures when modeling the sequence probabilities of a given structure (Biswas et al., 2005).
In conclusion, although our novel approach for the large-scale prediction of amino acid probabilities for protein design significantly improves the state-of-the-art (both in accuracy and time), there is still considerable room for improvement of the ubiquitous models used. This will require breakthroughs in the field of protein structure energetics, modeling of sequence probabilities, and efficient algorithms for probability prediction.
ACKNOWLEDGEMENTS
The authors would like to thank Michal Linial, Julia Shifman and Ora Schueler-Furman for their discussions and critical reading of the manuscript, and Nati Linial for his analysis of the topic.
Funding: M.F. was supported by the Sudarsky Center for Computational Biology. C.Y. was supported by the Yeshaya Horowitz Association through the Center for Complexity Science.
Conflict of Interest: none declared.
REFERENCES
- Arnold FH. Combinatorial and computational challenges for biocatalyst design. Nature. 2001;409:253–257. doi: 10.1038/35051731. [DOI] [PubMed] [Google Scholar]
- Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewley CA, et al. Design of a novel peptide inhibitor of HIV fusion that disrupts the internal trimeric coiled-coil of gp41. J. Biol. Chem. 2002;277:14238–14245. doi: 10.1074/jbc.M201453200. [DOI] [PubMed] [Google Scholar]
- Biswas P, et al. Statistical theory for protein ensembles with designed energy landscapes. J. Chem. Phys. 2005;123:154908. doi: 10.1063/1.2062047. [DOI] [PubMed] [Google Scholar]
- Calhoun JR, et al. Computational design and characterization of a monomeric helical dinuclear metalloprotein. J. Mol. Biol. 2003;334:1101–1115. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Chakrabarti R, et al. Computational prediction of native protein ligand-binding and enzyme active site sequences. PNAS. 2005;102:10153–10158. doi: 10.1073/pnas.0504023102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell R. Advanced inference in Bayesian networks. In: Jordan M, editor. Learning in Graphical Models. Cambridge, MA, USA: MIT Press; 1998. pp. 27–49. [Google Scholar]
- Delarue M, Koehl P. The inverse protein folding problem: self consistent mean field optimisation of a structure specific mutation matrix. In: Altman RB, et al., editors. Pacific Symposium on Biocomputing. Singapore: World Scientific; [PubMed] [Google Scholar]
- Dodge C, et al. The HSSP database of protein structure-sequence alignments and family profiles. Nucleic Acids Res. 1998;26:313–315. doi: 10.1093/nar/26.1.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack RL, Karplus M. J. Mol. Biol. 1993. Backbone-dependent rotamer library for proteins application to side-chain prediction; pp. 230pp. 543–574. [DOI] [PubMed] [Google Scholar]
- Gordon DB, et al. Energy functions for protein design. Curr. Opin. Struc. Biol. 1999;9:509–513. doi: 10.1016/s0959-440x(99)80072-4. [DOI] [PubMed] [Google Scholar]
- Hecht MH, et al. De novo proteins from designed combinatorial libraries. Protein Sci. 2004;13:1711–1723. doi: 10.1110/ps.04690804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K. Statistical mechanics. New York: John Wiley and Sons, Inc; 1987. [Google Scholar]
- Humphris EL, Kortemme T. Design of multi-specificity in protein interfaces. PLoS Computational Biology. 2007;3:e164. doi: 10.1371/journal.pcbi.0030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo A, et al. Folding free energy function selects native-like protein sequences in the core but not on the surface. PNAS. 2002;99:13554–13559. doi: 10.1073/pnas.212068599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisetty H, et al. Free energy estimates of all-atom protein structures using generalized belief propagation. In: Speed T, Huang H, editors. RECOMB. Heidelberg, Germany: Springer; 2007. pp. 366–380. [DOI] [PubMed] [Google Scholar]
- Kono H, Saven JG. Statistical theory for protein combinatorial libraries. packing interactions, backbone flexibility, and sequence variability of main-chain structure. J. Mol. Biol. 2001;306:607–628. doi: 10.1006/jmbi.2000.4422. [DOI] [PubMed] [Google Scholar]
- Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. PNAS. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- Lassila J, et al. Exhaustive mutagenesis of six secondary active-site residues in Escherichia coli chorismate mutase shows the importance of hydrophobic side chains and a helix n-capping position for stability and catalysis. Biochemistry. 2007;46:6883–6891. doi: 10.1021/bi700215x. [DOI] [PubMed] [Google Scholar]
- Lauritzen S. Graphical Models. Oxford, UK: Oxford University Press; 1996. [Google Scholar]
- Lazar GA, et al. Designing proteins for therapeutic applications. Curr. Opin. Struc. Biol. 2003;13:513–518. doi: 10.1016/s0959-440x(03)00104-0. [DOI] [PubMed] [Google Scholar]
- Lilien RH, et al. A novel ensemble-based scoring and search algorithm for protein redesign and its application to modify the substrate specificity of the gramicidin synthetase a phenylalanine adenylation enzyme. J. Com. Biol. 2005;12:740–761. doi: 10.1089/cmb.2005.12.740. [DOI] [PubMed] [Google Scholar]
- Meyerguz L, et al. The evolutionary capacity of protein structures. In: Bourne PE, Gusfield D, editors. RECOMB. New York, NY, USA: ACM Press; 2004. pp. 290–297. [Google Scholar]
- Moore GL, Maranas CD. Identifying residue-residue clashes in protein hybrids by using a second-order mean-field approach. PNAS. 2003;100:5091–5096. doi: 10.1073/pnas.0831190100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal G, et al. Comprehensive and quantitative mapping of energy landscapes for protein–protein interactions by rapid combinatorial scanning. J. Biol. Chem. 2006;281:22378–22385. doi: 10.1074/jbc.M603826200. [DOI] [PubMed] [Google Scholar]
- Park S, et al. Advances in computational protein design. Curr. Opin. Struc. Biol. 2004;14:487–494. doi: 10.1016/j.sbi.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Park S, et al. Progress in the development and application of computational methods for probabilistic protein design. Comput. Chem. Eng. 2005;29:407–421. [Google Scholar]
- Park S, et al. Limitations of yeast surface display in engineering proteins of high thermostability. Protein Eng. Des. Sel. 2006;19:211–217. doi: 10.1093/protein/gzl003. [DOI] [PubMed] [Google Scholar]
- Pearl J. Probabilistic reasoning in intelligent systems: networks of plausible inference. San Fransisco, CA, USA: Morgan Kaufmann; 1988. [Google Scholar]
- Pettersen EF, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rosenberg M, Goldblum A. Computational protein design: a novel path to future protein drugs. Curr. Pharm. Des. 2006;12:3973–3997. doi: 10.2174/138161206778743655. [DOI] [PubMed] [Google Scholar]
- Saunders CT, Baker D. Recapitulation of protein family divergence using flexible backbone protein design. J. Mol. Biol. 2005;346:631–644. doi: 10.1016/j.jmb.2004.11.062. [DOI] [PubMed] [Google Scholar]
- Schueler-Furman O, et al. Progress in modeling of protein structures and interactions. Science. 2005;310:638–642. doi: 10.1126/science.1112160. [DOI] [PubMed] [Google Scholar]
- Schueler-Furman O. Progress in modeling of protein structures and interactions. Science. 2005;310:638–642. doi: 10.1126/science.1112160. [DOI] [PubMed] [Google Scholar]
- Shifman JM, Mayo SL. Modulating calmodulin specificity through computational protein design. J. Mol. Biol. 2002;323:417–423. doi: 10.1016/s0022-2836(02)00881-1. [DOI] [PubMed] [Google Scholar]
- Voigt CA, et al. Computational method to reduce the search space for directed protein evolution. PNAS. 2001;98:3778–3783. doi: 10.1073/pnas.051614498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Saven JG. Computational methods for protein design and protein sequence variability: biased monte carlo and replica exchange. Chem. Phys. Lett. 2005;401:205–210. [Google Scholar]
- Yanover C, Weiss Y. Approximate inference and protein-folding. In: Becker S, et al., editors. Advances in Neural Information Processing Systems15. MIT Press: Cambridge, MA; 2003. pp. 1457–1464. [Google Scholar]
- Yanover C, et al. Linear programming relaxations and belief propagation – an empirical study. J. Mach. Learn. Res. 2006;7:1887–1907. [Google Scholar]
- Yanover C, et al. In RECOMB. Suzanna Becker and Sebastian Thrun and Klaus Obermayer: 2007. Minimizing and learning energy functions for side-chain prediction; pp. 381–395. [DOI] [PubMed] [Google Scholar]
- Yedidia JS, et al. Constructing free-energy approximations and generalized belief propagation algorithms. IEEE Trans. Inf. Theory. 2005;51:2282–2312. [Google Scholar]