

Abstract
Aggregation and subsequent development of protein deposition diseases originate from conformational changes in corresponding amyloidogenic proteins. The accumulated data support the model where protein fibrillogenesis proceeds via the formation of a relatively unfolded amyloidogenic conformation, which shares many structural properties with the pre-molten globule state, a partially folded intermediate first found during the equilibrium and kinetic (un)folding studies of several globular proteins and later described as one of the structural forms of natively unfolded proteins. The flexibility of this structural form is essential for the conformational rearrangements driving the formation of the core cross-beta structure of the amyloid fibril. Obviously, molecular mechanisms describing amyloidogenesis of ordered and natively unfolded proteins are different. For ordered protein to fibrillate, its unique and rigid structure has to be destabilized and partially unfolded. On the other hand, fibrillogenesis of a natively unfolded protein involves the formation of partially folded conformation; i.e., partial folding rather than unfolding. In this review recent findings are surveyed to illustrate some unique features of the natively unfolded proteins amyloidogenesis.
Keywords: Amyloid fibril, Fibrillation, Amyloidogenesis, Conformational disease, Partially folded conformation, Natively unfolded protein, Intrinsically disordered protein
INTRODUCTION
Proteins are the major components of the living cell, which play crucial role in the maintenance of life, and dysfunction of which may cause development of different pathological conditions. In fact, a broad range of human diseases known as protein conformational or protein misfolding diseases arises from the failure of a specific peptide or protein to adopt its native functional conformational state. The obvious consequences of misfolding are protein aggregation (and/or fibril formation), loss of function, and gain of toxic function. Some proteins have an intrinsic propensity to assume a pathologic conformation, which becomes evident with aging or at persistently high concentrations. Interactions (or impaired interactions) with some endogenous factors (e.g., chaperones, intracellular or extracellular matrixes, other proteins, small molecules) can change conformation of a pathogenic protein and increase its propensity to misfold. Misfolding can originate from point mutation(s) or result from an exposure to internal or external toxins, impaired posttranslational modifications (phosphorylation, advanced glycation, deamidation, racemization, etc.), an increased probability of degradation, impaired trafficking, lost binding partners or oxidative damage. All these factors can act independently or in association with one another.
Misfolding diseases can affect a single organ or be spread through multiple tissues. The largest group of misfolding diseases, including numerous neurodegenerative disorders and the amyloidoses, originates from the conversion of specific proteins from their soluble functional states into stable, highly ordered, filamentous protein aggregates, known as amyloid fibrils, and from the deposition of these aggregated material in the variety of organs and tissues. In each of these pathological states, a specific protein or protein fragment changes from its natural soluble form into insoluble fibrils, which accumulate in a variety of organs and tissues [2–8]. Amyloid-like fibrils display many common properties including a core cross-β-sheet structure in which continuous β-sheets are formed with β-strands running perpendicular to the long axis of the fibrils [9]. Morphologically, they typically consist of 2–6 unbranched protofilaments 2–5 nm in diameter associated laterally or twisted together to form fibrils with 4–13 nm diameter (e.g., see [10–12]). Although the amyloid fibrils from different diseases are structurally and morphologically similar to each other, the amyloidogenic polypeptides causing diseases are extremely diverse and prior to fibrillation may be rich in β-sheet, α-helix, β-helix, or be natively unfolded [8]. For many years it has been generally assumed that the ability to form amyloid fibrils is limited to a relatively small number of proteins, essentially those found in the diseases, and that these proteins posses specific sequence motifs encoding the unique structure of the amyloid core. However, recent studies have established that many diseases unrelated proteins were shown to form fibrils [3, 8, 13, 14]. It is even believed that virtually any protein can be forced to fibrillate if the appropriate conditions are found [3, 8, 13]. The structural diversity of amyloidogenic proteins and close similarity of the resultant fibrils imply that considerable structural rearrangements have to occur in order for fibril formation to happen. In a rigid globular protein, such changes cannot take place due to the constraints of the tertiary structure. Therefore, it has been proposed that fibrillation of a globular protein requires the destabilization of its rigid native structure leading to a partial unfolding and the formation of a partially unfolded conformation [2–8, 15–18]. As natively unfolded (intrinsically unstructured) proteins are devoid of ordered structure, the primary step of their fibrillogenesis requires the stabilization of a partially folded conformation, i.e. partial folding rather than unfolding [8, 13]. Therefore, a general hypothesis of fibrillogenesis states: structural transformation of a polypeptide chain into a partially folded conformation represents an important prerequisite for protein fibrillation [8]. In fact, such partially unfolded conformation enables specific intermolecular interactions, including electrostatic attraction, hydrogen bonding and hydrophobic contacts, which are necessary for oligomerization and fibrillation. These aggregation-prone intermediates would be structurally different for different proteins. Furthermore, intermediate might contain different amount of ordered structure even for the same protein undergoing different aggregation processes. It is believed that the precursor of soluble aggregates is the most structured, whereas amyloid fibrils are formed from the least ordered conformation (cf. [19]). It has been also pointed out that the variations in the amount of the ordered structure in the amyloidogenic precursor might be responsible for the formation of fibrils with distinct morphologies [20].
It is important to remember that there are several investigations favoring the idea that the deposited proteinacous inclusions (such as senile plaques in Alzheimer’s disease or Lewy bodies or Lewy neurites in Parkinson’s disease, etc.) are not toxic, but the formation of some small oligomers, different protofibrillar structures, are responsible for the toxicity [21–25]. However, these issues are outside the primary scope of this review, where the details of the amyloidogenesis of natively unfolded proteins are considered. Table 1 lists natively unfolded or significantly unfolded proteins involved in various amyloid-based clinical disorders, whereas Table 2 represents a set of non-disease-related natively unfolded proteins and peptides. As the major focus of this paper is the amyloidogenesis mechanisms of natively unfolded proteins, subsequent paragraphs are devoted to the brief introduction of these interesting members of the protein kingdom.
Table 1.
Some natively unfolded or significantly disordered amyloidogenic proteins and the corresponding amyloid-based clinical disorders.
| Amyloidogenic protein | Type of structure | Disease |
|---|---|---|
| α-Synuclein | Natively unfolded | Parkinson’s disease (PD) Diffuse Lewy bodies disease (DLBD) Lewy bodies variant of Alzheimer’s disease (LBVAD) Dementia with Lewy bodies (DLB) Multiple system atrophy (MSA) Hallervorden-Spatz disease |
| β-Synuclein | Natively unfolded | Parkinson’s disease (PD) Diffuse Lewy bodies disease (DLBD) |
| γ-Synuclein | Natively unfolded | Parkinson’s disease (PD) Diffuse Lewy bodies disease (DLBD) |
| Islet amyloid polypeptide (IAPP, Amylin) | Natively unfolded | Pancreatic islet amyloidosis in late-onset diabetes (type II diabetes mellitus) |
| Amyloid-β and its fragments | Natively unfolded | Alzheimer disease (AD) Dutch hereditary cerebral hemorrhage with amyloidosis (HCHWA, also known as cerebrovascular amyloidosis) Congophilic angiopathy |
| Tau protein | Natively unfolded | Alzheimer disease (AD), Pick’s disease, Progressive supranuclear palsy (PSP) |
| ABri | Natively unfolded | Familial British dementia |
| ADan | Natively unfolded | Familial Danish Dementia |
| Prion protein and its fragments | N-terminal fragment (23–121) is natively unfolded; C-terminal domain (121–230) is α-helical (predominantly) | Creutzfeld-Jacob disease (CJD) Gerstmann-Straussler-Schneiker syndrome (GSS) Fatal familial insomnia (FFI) Kuru Bovine spongiform encephalopathy (BSE) and scrapie |
| Huntingtin | Exon 1 is unfolded and forms fibrils | Huntington Disease |
| Ataxin-1 | Unknown (Natively unfolded) | Spinocerebellar ataxia (SCA) Neuronal intranuclear inclusion disease (NIID) |
| Androgen receptor protein | Ligand-binding (LBD) and DNA-binding domains (DBD) are α-helical; amino-terminal domain (NTD) is natively unfolded | Spinal and bulbar muscular atrophy (SBMA) |
| DRPLA protein (atrophin-1) | Unknown (probably natively unfolded) | Hereditary dentatorubral-pallidoluysian atrophy (DRPLA) |
| Nuclear poly(A) binding protein | Natively unfolded | Oculopharyngeal Muscular Dystrophy |
| Calcitonin | Natively unfolded | Medullary Carcinoma of the Thyroid (MCT) |
| Gelsolin | Amyloidogenic fragment 173–243 is natively unfolded | Finnish-Type Familial Amyloidosis |
Table 2.
Disease-unrelated natively unfolded proteins known to form amyloid-like fibrils
| Protein | Type of structure |
|---|---|
| Yeast prion Sup35p | α-Helical/unfolded |
| Yeast prion Ure2p | α-Helical/unfolded |
| Prothymosin α | Natively unfolded |
| Apolipoprotein C-II | Natively unfolded |
| Core histones | Natively unfolded |
| Carboxymethylated α-Lactabumin | Unfolded |
| GAGA factor | Fragment 137–519 is natively unfolded |
| αS1-, αS2-, β-, and κ-caseins | Natively unfolded |
| Merozoite surface protein 2 (MSP2) | Natively unfolded |
| Apo cytochrome c552 | Natively unfolded |
| SH3 domain | Unfolded and amyloidogenic at acidic pH |
Recent years show an increasing appreciation of proteins that lack rigid 3-D structure under physiological conditions in vitro, existing instead as dynamic ensembles of interconverting structures. These naturally flexible proteins are currently known as intrinsically disordered [26], natively denatured [27], natively unfolded [28], intrinsically unstructured [29], and natively disordered [30]. These proteins have dynamic structures that interconvert on a number of timescales and were shown to have many similarities to non-native states of “normal” globular proteins, which may exist in at least four different conformations: native (ordered), molten globule, pre-molten globule, and coil-like [13, 31–33]. Using this analogy, it has been established that intrinsically disordered proteins and regions under physiological conditions in vitro might contain collapsed-disorder (i.e., where intrinsic disorder is present in a form of molten globules) and extended-disorder (i.e., regions where intrinsic disorder is present in a form of random coil or pre-molten globule) [13, 26, 30]. The major focus of this review is on the fibrillation of the natively unfolded proteins, which represent a subset of intrinsically disordered proteins with extended disorder.
It has been shown that intrinsically unstructured proteins and regions differ from structured globular proteins and domains with regard to many attributes, including amino acid composition, sequence complexity, hydrophobicity, charge, flexibility, and type and rate of amino acid substitutions over evolutionary time. For example, natively unfolded proteins are significantly depleted in balky hydrophobic (Ile, Leu, and Val) and aromatic amino acid residues (Trp, Tyr, and Phe), which would normally form the hydrophobic core of a folded globular protein, and also possess low content of Cys and Asn residues. These depleted residues, Trp, Tyr, Phe, Ile, Leu, Val, Cys and Asn were proposed to be called order-promoting amino acids. On the other hands, natively unfolded proteins were shown to substantially enriched in polar, disorder-promoting, amino acids: Ala, Arg, Gly, Gln, Ser, Pro, Glu, and Lys [26, 34–36]. Many of the mentioned differences were utilized to develop numerous disorder predictors, including PONDR® (Predictor of Naturally Disordered Regions) [34, 37], charge-hydropathy plots (CH-plots) [38], NORSp [39], GlobPlot [40, 41], FoldIndex© [42], IUPred [43], DisoPred [44–46] to name a few.
Despite lacking any stable secondary and tertiary structure, intrinsically disordered proteins are known to fulfill a great variety of crucial biological functions [13, 26, 29, 30, 38, 47–62]. It has been suggested that the functional diversity provided by disordered regions might complement those of ordered protein regions [59–61]. Intrinsically disordered proteins were shown to have specific functions that can be grouped into four broad classes: (1) molecular recognition; (2) molecular assembly; (3) protein modification; and (4) entropic chain activities.[48] Recently, a novel bioinformatics approach for comprehensive study of functional roles of protein disorder was proposed [59–61]. Applying this novel data mining tool to over 200,000 proteins from Swiss-Prot database and corresponding functional keywords, it has been shown that out of the 711 Swiss-Prot functional keywords that were associated with at least 20 proteins, 262 were found to be strongly positively correlated with long intrinsically disordered regions, whereas 302 were strongly negatively correlated with such regions [59–61]. It is recognized now that despite (or may be due to) their high flexibility, natively unfolded proteins are involved in regulation, signaling and control pathways in which interactions with multiple partners and high-specificity/low-affinity interactions are often requisite [49, 62]. This is further confirmed by the fact that eukaryotic proteomes, with their extensively developed interaction networks, are highly enriched in intrinsically disordered proteins, relative to bacteria and archaea [46, 63, 64]. Another very important feature of the natively unfolded proteins is their unique capability to fold under the variety of conditions. In fact, the folding of these proteins can be brought about by interaction with other proteins, nucleic acids, membranes or small molecules. It also can be driven by changes in the protein environment. The resulting conformations could be either relatively non-compact (i.e., remain substantially disordered) or be tightly folded.
Below, the peculiarities of the amyloidogenesis of natively unfolded proteins are illustrated using results from a detailed analysis of aggregation of human α-synuclein. The generality of major conclusions are further emphasized with a variety of other natively unfolded proteins that are known to form amyloid-like fibrils.
MOLECULAR MECHANISMS OF α-SYNUCLEIN FIBRILLATION
The Center of the Storm: α-Synuclein in Parkinson’s Disease and Other Neurodegenerative Disorders
Synucleinopathies is a group of neurodegenerative disorders characterized by fibrillary aggregates of α-synuclein protein in the cytoplasm of selective populations of neurons and glia [65–68]. Clinically, synucleinopathies are characterized by a chronic and progressive decline in motor, cognitive, behavioral, and autonomic functions, depending on the distribution of the lesions. Because of clinical overlap, differential diagnosis is sometimes very difficult [69]. The neuropathological spectrum of synucleinopathies was intensively discussed [65–76], and the potential mechanisms linking the α-synuclein aggregation with the development of several of these diseases are the major focus of numerous studies. Some of these disorders are briefly discussed below to illustrate a wide range of pathological manifestations in synucleinopathies.
Parkinson’s disease (PD) is a slowly progressive disease that affects neurons of the substantia nigra, a small area of cells in the mid-brain. It is estimated that ~1.5 million Americans are affected by PD. Since only a small percentage of patients are diagnosed before age 50, PD is generally considered as an aging-related disease, and approximately one of every 100 persons over the age of 55 in the US suffers from this disorder [77]. Gradual degeneration of the dopaminergic neurons causes a reduction in the dopamine content and produce classic PD signs: resting tremor on one (or both) side(s) of the body; generalized slowness of movement (bradykinesia); stiffness of limbs (rigidity); and gait or balance problems (postural dysfunction). The precise mechanisms of neuronal death are unknown as of yet. Some surviving nigral dopaminergic neurons contain cytosolic filamentous inclusions known as Lewy bodies (LBs) when found in the neuronal cell body, or Lewy neurites (LNs) when found in axons [78, 79].
Several observations implicate α-synuclein in the pathogenesis of PD. For example, a direct role for α-synuclein in the neurodegenerative processes in PD is demonstrated by genetic evidence and autosomal dominant early-onset PD is associated with three different missense mutations in the α-synuclein gene, corresponding to A30P, E46K, and A53T substitutions in α-synuclein [80–82] or with the hyper-expression of the wild type α-synuclein protein due to gene triplication [83–85]. Antibodies to α-synuclein detect this protein in LBs and LNs, the hallmark lesions of PD, a substantial portion of fibrillar material in these specific inclusions was shown to be comprise of α-synuclein, and insoluble α-synuclein filaments were recovered from purified LBs [86, 87].
Dementia with Lewy bodies (DLB), being the second most frequent neurodegenerative dementing disorder after AD, is a common form of late-onset dementia that exists in a pure form or overlaps with the neuropathological features of AD. This disease is characterized clinically by neuropsychiatric changes often with marked fluctuations in cognition and attention, hallucinations, and parkinsonism [88]. Similar to PD, neurophathological hallmarks of DLB are numerous LBs and LNs in the substantia nigra, which are strongly immunoreactive for α-synuclein [86].
Alzheimer’s disease (AD) is the most common aging-related neurological disorder, which constitutes about two thirds of cases of dementia overall [89, 90] and is characterized by slow, progressive memory loss and dementia due to a gradual neurodegeneration particularly in the cortex and hippocampus [91]. The clinical hallmarks are progressive impairment in memory, judgment, decision making, orientation to physical surroundings, and language [92]. The pathological hallmarks of the AD are neuronal loss, extracellular senile plaques containing the peptide Aβ, and neurofibrillary tangles (NFTs) composed of a hyperphosphorylated form of the microtubular protein tau [93–95]. Detailed analysis of the α-synuclein immunoreactivity in the brains from the patients with sporadic AD revealed the presence of α-synuclein-positive inclusions resembling LBs and LNs in ~50% cases studied [96].
Down’s syndrome is a genetic disorder characterized by an extra chromosome 21 (trisomy 21, i.e., instead of having the normal 2 copies of chromosome 21, the Down’s syndrome patient has 3 copies of this chromosome). The analysis of Down’s syndrome with Alzheimer pathology revealed presence of numerous LBs and LNs in the neurons of the limbic areas, predominantly of the amygdala. Similar lesions were less common in other regions of these brains [97, 98].
Multiple system atrophy (MSA) an adult-onset progressive neurodegenerative disorder of unknown etiology which is characterized clinically by any combination of parkinsonian, autonomic, cerebellar or pyramidal symptoms and signs. The histological hallmarks of MSA are the neuronal loss and the presence of argyrophilic fibrillary inclusions in the oligodendrocytes, referred to as glial cytoplasmic inclusions (GCIs), which are also known as Papp-Lantos bodies [99], the major component of which is fibrillated α-synuclein [100, 101].
Neurodegeneration with brain iron accumulation type 1 (NBIA1) represents a rare progressive neurodegenerative disorder that occurs in both sporadic as well as in familial forms. Clinically, NBIA 1 is characterized by rigidity, dystonia, dyskinesia, and choreoathetosis [102–105], together with dysarthria, dysphagia, ataxia, and dementia [105–107]. The histopathologic hallmarks of NBIA1 include neuronal loss, neuraxonal spheroids, and iron deposition in the globus pallidus and substantia nigra pars compacta, as well as by the presence of the LB-like and GCI-like inclusions and dystrophic neuritis [106].
As it follows from the discussion above, α-synucelin inclusions are present in neurons (both dopaminergic and non-dopaminargic), where they can be deposited in perikarya or in axonal processes of neurons, and in glia. There are at least five morphologically different α-synuclein containing inclusions, Lewy bodies, Lewy neurites (dystrophic neurites), glial cytoplasmic inclusions, neuronal cytoplasmic inclusions and axonal spheroids.
α-Synuclein as a Typical Natively Unfolded Protein
α-Synuclein is a typical intrinsically unstructured, or natively unfolded, or intrinsically disordered protein, possessing little or no ordered structure under the “physiological” conditions (i.e., conditions of neutral pH and low to moderate ionic strength) [28, 108]. It has been already mentioned that the amino acid sequences of natively unfolded proteins are characterized by a number of specific features. For example, the analysis of amino acid sequences based on the normalized net charge and mean hydrophobicity performed for sets of 275 native and 91 natively unfolded proteins has established that a combination of low overall hydrophobicity and high net charge is a specific feature of natively unfolded proteins. Moreover, these proteins have been shown to be specifically localized within a particular region of charge-hydrophobicity phase space, satisfying the following relationship [38, 64]:
where <H> and <R> are the mean hydrophobicity and the mean net charge of the given protein, respectively, whereas <H>b is the “boundary” mean hydrophobicity value, below which a polypeptide chain with a given <R> will be most probably unfolded. The mean hydrophobicity, <H>, is defined as the sum of the normalized hydrophobicities of all residues [calculated according to [109]] divided by the number of residues in the polypeptide.
Interestingly, it has been noted that α-synuclein does not fit the general trend and is located within “native” area of the charge-hydrophobicity phase space [38]. Detailed analysis of this protein amino acid sequence has established that its N- and C- terminal regions are very distinct in overall hydrophobicity and possess charges of opposite sign. The C-terminal fragment (the last 45 residues) of human α-synuclein has parameters typical of natively unstructured proteins, whereas the parameters of the N-terminal 95 residues are typical of native folded globular proteins. It has been suggested that the disordered regions of these molecules prevent the remainder of the protein from normal folding, perhaps through extensive electrostatic attractions [38].
More detailed analysis of the differences in amino acid compositions between ordered and intrinsically disordered proteins constituted a ground for the development of numerous algorithms aiming for the prediction of disordered proteins/regions [reviewed in [110, 111]]. Fig. 1 represents the results of disorder prediction on human α-synuclein sequence using several of these predictors, PONDR® VL3 [34, 37], VSL2 [112], RONN [113] and IUPred [43]. It can be seen that α-synuclein is predicted to be almost completely disordered by all these predictors (as disorder probability scores ≥0.5 correspond to a prediction of disorder), emphasizing that its sequence is typical of the intrinsically disordered proteins.
Fig. 1.
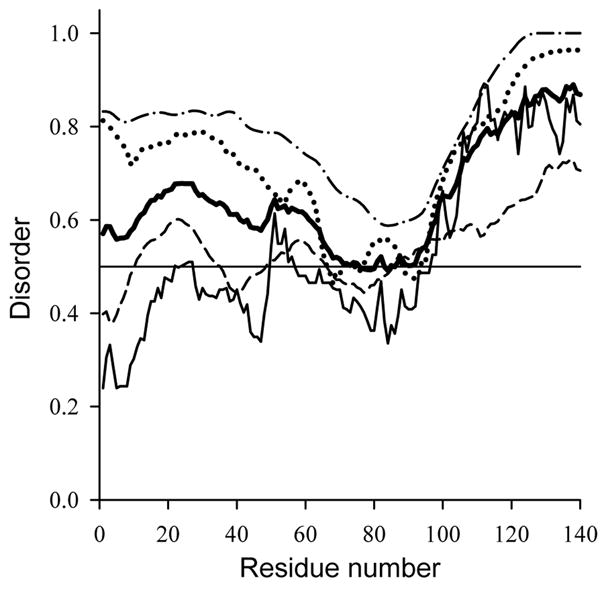
Intrinsic disorder prediction for human α-synuclein using IUPred (solid line); RONN (dashed line); PONDR VSL2 (dotted line) and PONDR VL3 (dash-dotted line). The results averaged over these for predictions are shown as bold line.
In agreement with these predictions, α-synuclein was shown to possess little ordered structure under physiological conditions [28, 108, 114, 115]. For example, at neutral pH it is characterized by far-UV CD and FTIR spectra typical of a substantially unfolded polypeptide chain with a low content of ordered secondary structure [108]. The hydrodynamic properties of α-synuclein are in a good agreement with the results of far-UV CD and FTIR studies. In fact, it has been established that α-synuclein, being essentially expanded, does not have a tightly packed globular structure, but is slightly more compact than expected for a random coil [108, 114, 116]. It has been shown that α-synuclein sedimented more slowly than globular proteins of similar molecular weight, indicating that this protein is not compact [28]. Furthermore, based on the results of pulsed-field gradient NMR (which allows an estimation of the hydrodynamic radii), it has been concluded that α-synuclein is slightly collapsed [117]. Thus, at neutral pH, α-synuclein was shown to be essentially disordered, but slightly more compact than a random coil. In agreement with this conclusion, a high resolution NMR analysis of the protein under these conditions revealed that α-synuclein is largely unfolded in a solution, but exhibits a region between residues 6 and 37 with a preference for helical conformation [115]. Interestingly, the results of recent studies on Raman optical activity spectra were consistent with the conclusion that α-synuclein may contain some helical poly-(L-proline) II-like conformation [118].
Structural Features of the Partially Folded α-Synuclein
A fundamental question is what forces or factors will cause a natively unfolded protein to fold? As natively unfolded proteins are characterized by a unique combination of low overall hydrophobicity and large net charge, it is reasonable to suggest that any alterations in the protein environment leading to an increase in its hydrophobicity and/or decrease in its net charge should be accompanied by at least partial folding of the intrinsically disordered protein. The excess negative charge of α-synuclein at neutral pH (pI = 4.7) would be neutralized at lower pH values, and the overall hydrophobicity of a protein will increase with increasing temperature. Therefore, partial folding of α-synuclein under conditions of high temperature and/or low pH has been predicted [108]. In agreement with this suggestion, Fig. 2 shows that α-synuclein adopts a kind of partially folded conformation at acidic pH or at high temperatures (cf. [108, 114]). At neutral pH the protein possesses a far-UV CD spectrum typical of an unfolded polypeptide chain (Fig. 2A). The spectrum has an intense minimum in the vicinity of 196 nm, with the absence of characteristic bands in the 210–230 nm region. However, as the pH is decreased (or temperature increased) changes were observed in the shape of the spectrum. Fig. 2A shows that the minimum at 196 nm becomes less intense, whereas the negative intensity of the spectrum around 222 nm increases, reflecting pH-induced formation of secondary structure. Fig. 2B compares the FTIR spectra of α-synuclein measured at pH 7.5 and pH 3.0. The FTIR spectrum of α-synuclein at pH 7.5 is typical of a substantially unfolded polypeptide chain, whereas a decrease in pH leads to significant spectral changes, indicative of increased ordered structure. The most evident change is the appearance of a new band in the vicinity of 1626 cm−1, which corresponds to β-sheet. This means that at acidic pH natively unfolded α-synuclein is transformed into a partially folded conformation with a significant amount of β-structure [108, 114]. Furthermore, Fig. 2C shows that a decrease in pH leads to a considerable increase in 1-anilino-8-naphthalene sulfonate (ANS) fluorescence intensity and a large blue shift of the ANS fluorescence maximum (from ~515 to ~475 nm), reflecting the pH-induced transformation of the natively unfolded α-synuclein to the partially folded compact conformation with solvent exposed hydrophobic clusters. Hydrodynamic methods revealed that this partially folded conformation is characterized by substantially decreased hydrodynamic dimensions (RS = 27.9 ± 0.4 Å and Rg = 30 ± 1 Å versus RS = 31.8±0.4 Å and Rg = 40 ± 1 Å measured at pH 7.5). Furthermore, the profile of the Kratky plot at neutral pH was typical for a random coil conformation, whereas that at pH 3 showed changes consistent with the development of the beginnings of a tightly packed core (Fig. 2D). This means that protonation of α-synuclein results in transformation of the natively unfolded protein into a partially folded and more compact conformation with a significant amount of ordered secondary structure, increased affinity for ANS and the beginnings of a tightly packed core, all hallmarks of the partially folded intermediate [108, 114]. Comparable structural changes were induced in α-synuclein by high temperatures [108, 114].
Fig 2.

Structural properties and conformational behavior of human α-synuclein.
A. Far-UV CD spectra measured under different conditions.
B. FTIR spectra measured for natively unfolded, partially folded and fibrilar forms of α-synuclein.
C. ANS spectra under different conditions.
D. Kratky plot representation of the results of small angle X-ray scattering analysis of α-synuclein at different experimental conditions.
The pH dependence of [θ]222 is shown in Fig 3A. There is little change in the far-UV CD spectrum between pH ~9.0 and ~5.5. However, a decrease in pH from 5.5 to 3.0 results in a ~2-fold increase in negative intensity in the vicinity of 220 nm, and a further decrease in pH is accompanied by a reversal in the spectral intensity. Fig. 3A shows that the pH-induced changes in the far-UV CD spectrum of α-synuclein are completely reversible (compare open and solid symbols) and are independent of protein concentration (at least in the range of 0.1–1.5 mg/ml, compare circles and squares). These observations are consistent with the assumption that the pH-induced increase in structure of α-synuclein represents an intramolecular process and not self-association [108, 114]. Fig. 3A further shows that the pH-induced structural transition observed by far-UV CD coincided with that detected by changes in ANS fluorescence. The position of the transition (between pH 5.5 and 3.0) indicates that protonation of one or more carboxylates is responsible for the structural change.
Fig. 3.

Conformational behavior human α-synuclein.
A. pH-Induced folding of α-synuclein.
B. Temperature-induced folding of the nativley unfolded α-synuclein.
Fig. 3B represents the temperature-dependence of [θ]222 and shows that increase in temperature induced formation of secondary structure in α-synuclein [108, 114]. The major spectral changes occurred over the range of 3 to 50°C. Further heating lead to a less pronounced effect. Interestingly, Fig. 3B shows that the structural changes induced in α-synuclein by heating were completely reversible (cf. open and filled symbols). These data indicate that high temperatures induce a reversible transition of α-synuclein to a partially folded intermediate. This intermediate has a similar CD spectrum to that induced by low pH (see Fig. 2A).
Summarizing, α-synuclein, is unstructured under conditions of neutral pH, but does not represent a random coil. It has some residual structure (at least a region with a preference for helical conformation [115]), leading to partial compaction [108, 114]. Either a decrease in pH, or an increase in temperature, transformed α-synuclein into a partially folded conformation. This partially folded conformation resembles the pre-molten globule state, an intermediate, preceding the molten globule in the refolding of globular proteins [13, 32, 33]. The structure-forming effects of low pH were attributed to minimization of the large net negative charge present at neutral pH, thereby decreasing intramolecular charge-charge repulsion and permitting hydrophobic-driven collapse to the partially folded intermediate. The effect of elevated temperatures was attributed to increased strength of the hydrophobic interaction at higher temperatures, leading to a stronger hydrophobic driving force for folding [108, 114, 119]. This illustrates “turned out” response to the changes in the environment typical for the natively unstructured proteins, which, unlike “normal” globular proteins, gain rather than lose ordered structure at extreme pH and high temperatures [114].
Conformational behavior of α-synuclein: A protein-chameleon concept
It has been shown that the PD-related mutations A30P and P53T do not affect the conformational behavior of α-synuclein and structural transitions induced in these three proteins by a decrease in pH or an increase in temperature or methanol concentration were shown to be indistinguishable [120, 121]. Likewise, wild type α-synuclein, and the A30P and A53T mutants may be transformed into the partially folded intermediate state by decreasing the pH or increasing the temperature [120, 121]. Importantly, the structure of this intermediate state was shown to be independent of the mutations. Thus, the monomeric forms of WT, A30P and A53T α-synucleins exhibit identical structural properties and conformational behavior [120, 121].
Similarly, the analysis of conformational behavior of different members of the synuclein family revealed that they possess a comparable response to the changes in their environment. In fact, although far-UV CD spectra of α-, β-, and γ-synucleins were slightly different at neutral pH, all three proteins possessed almost identical far-UV CD spectra at acidic pH suggesting that they adopt a partially folded intermediate with comparable degree of folding [116]. This hypothesis was further confirmed by the results of gel-filtration analysis, which showed that although β-synuclein was slightly more extended than α- and γ-synucleins at neutral pH, all three proteins possessed the same degree of compaction in acidic solutions [116].
An important characteristic of the α-synuclein primary structure, which is likely related to its functional activity, is seven imperfect repeats within the first 95 residues, resulting in a variation in hydrophobicity [122–124] with a strictly conserved periodicity of 11 [123]. Such a periodicity is characteristic of the amphipathic lipid-binding α-helical domains of apolipoproteins [122, 123], which have been extensively studied and assigned to subclasses according to their unique structural and functional properties [125, 126]. These seven imperfect 11-residue repeat sequences were predicted to form five amphipathic helices on the amino-terminal half of human α-synuclein [127–129], with helices 1–4 predicted to associate with lipid vesicles [130, 131], whereas helix 5 being likely responsible for protein–protein interactions [128]. It has been pointed out that α-synuclein shares the defining properties of the class A2 lipid-binding helix, distinguished by clustered basic residues at the polar-apolar interface, positioned ±100° from the center of apolar face; predominance of lysines relative to arginines among these basic residues; and several glutamate residues at the polar surface [125, 126, 131]. In agreement with these structural features, α-synuclein was shown to bind specifically to synthetic vesicles containing acidic phospholipids [128, 131]. This binding was shown to be accompanied by a dramatic increase in α-helix content [128, 131] and was attributed to the formation of two curved α-helices (Val3-Val37 and Lys45-Thr92) connected by a well ordered, extended linker [132], whereas the acidic, glutamate-rich C-terminal region (Asp98-Ala140) was shown to behave as a highly mobile tail; i.e., it remained unstructured even in the presence of membranes [129, 132]. Recently it has been established that C-terminal tail of the protein can gain protease-insensitive conformation when the micelle bound α-synuclein is exposed to calcium [133].
Conformational behavior of α-synuclein under the variety of environments was extensively analyzed. This analysis revealed that structure of α-synuclein is extremely sensitive to the environment and can be easily modified. Intriguingly, extended natively unfolded conformation was shown to be effectively stabilized via the methionine oxidation [134–136]. It has been shown that under the mild oxidative conditions (1–2% H2O2) all four methionines of α-synuclein, Met1, Met5, Met116, and Met127, located outside the repeat-containing region, are successfully oxidized to the methionine sulfoxides [135]. The oxidized form of α-synuclein was shown to be more unfolded than non-oxidized protein as manifested by the larger contribution of unordered structure to both FTIR and far-UV CD spectra [135], and by detectable decrease in the α-synuclein-MetO compactness [134]. This was attributed to the decreased hydrophobicity of oxidized methionine leading to a decrease in the overall hydrophobicity of the protein. Given the decrease in hydrophobicity, it was not a big surprise that the oxidized protein was less prone to oligomerize and aggregate, being substantially non-amyloidogenic, and even able to inhibit the fibrillation of non-modified α-synuclein [135].
It has been shown that this protein adopts pre-molten globule-like partially folded conformation not only at low pH [108] and high temperature [108], but under the variety of conditions, including the presence of low concentrations of organic solvents [137] and TMAO [138], the presence of different metal ions [139], various salts [140], several common pesticides/herbicides [141–143], heparin and other glycosoaminoglycans [144], some polycations [145], or as a result of a spontaneous oligomerization both in vitro and in vivo [146]. Furthermore, the addition of different alcohols was shown to increase the content of ordered secondary structure in α-synuclein [137]. Interestingly, the structural transformations induced by high solvent concentrations were dependent on the type of alcohol, with simple alcohols inducing a β-sheet-enriched conformation whereas fluorinated alcohols promoting α-helix-rich species [137]. Interestingly, both α-helical and β-structural species were shown to be initially monomeric, but underwent association over longer times, and β-rich rich conformations were strongly prone to form amorphous aggregates [137]. Oligomeric α-helical globular species potentially possessing rigid tertiary structure were induced in α-synuclein by high concentrations of TMAO [138].
Besides these monomeric conformations, α-synuclein is able to form morphologically different oligomers and aggregates. For example, the prolonged incubation of this protein at different temperatures resulted in a temperature-dependent, progressive aggregation, with dimers being formed first [146]. This temperature-modulated oligomerization was shown to be accompanied by small but reproducible increase in the ordered secondary structure content. Interestingly, the trapped oligomeric conformation was structurally similar to the pre-molten globule-like partially folded monomeric confomer induced by low pH or high temperature [146]. Therefore, it has been concluded that the partially folded pre-molten globule-like conformation of α-synuclein can be stabilized as the protein undergoes a highly selective self-assembly process during prolonged incubation at elevated temperatures [146]. The formation of oxidative dimers and higher-order oligomers with dityrosine cross-links in α-synuclein under the conditions of oxidative stress was also reported [147].
In addition to covalent and non-covalent dimers, α-synuclein was shown to form a series of morphologically different soluble oligomers. There are four tyrosines, Tyr39, Tyr125, Tyr132, and Tyr135, in α-synuclein, which were shown to be easily nitrated under the appropriate conditions in vitro [148]. It has been established that nitrated α-synuclein remains assembled into the oligomeric spheroids even after incubation for a very prolonged time [148]. The formation of several oligomeric “protofibrilar” species with different morphologies were detected by atomic force microscopy at early fibrillation stages of α-synuclein [24, 25, 149–152]. The first-formed α-synuclein protofibrils appeared to be predominantly spherical with heights varying between 2.5 and 4.2 nm [151, 152]. Under the appropriate conditions these spherical oligomers were shown to convert into the annular structures (doughnuts) [152]. In addition to the completed rings, doughnuts, the existence of partially formed rings (crescents) has been observed [152]. The formation of both doughnuts and fibrils was shown to require initial formation of spherical, β-structure enriched, α-synuclein oligomers. However, the subsequent assembly processes seem to require different conditions. Importantly, the doughnuts were not observed once spherical oligomers have disappeared and α-synuclein was converted to fibrils [152]. Based on these observations it has been suggested that the doughnuts are not on the direct monomer-to-fibril pathway, but must “reopen” to be converted to fibrils [152].
The incubation of the spherical α-synuclein oligomers with brain-derived membranes was shown to produce pore-like annular protofibrils too [152]. It has been also reported that incubation of α-synuclein with different metals for one day at 4° gave rise to three different classes of oligomers, where Cu2+, Fe3+ and Ni2+ yielded 0.8–4 nm spherical particles, similar to α-synuclein incubated without metal ions, Mg2+, Cd2+ and Zn2+ gave larger, 5–8 nm spherical oligomers, whereas Co2+ and Cd2+ frequent annular (doughnut-like) oligomers, 70–90 nm in diameter with Ca2+ and 22–30 nm in diameter with Co2+ [153].
Finally, α-synuclein was shown to assemble into large insoluble aggregates of two distinctive morphologies – amorphous aggregates and fibrils. The appearance of the particular type of the insoluble aggregate is determined by the environmental conditions. For example, α-synuclein precipitated from solutions containing high concentrations of simple alcohols predominantly in a form of amorphous aggregates. In many other cases, the major insoluble species were amyloid-like fibrils. In a few cases, the successful partitioning between these two pathways has been observed and α-synuclein was present in both fibrillar and amorphous forms simultaneously.
Data overviewed above indicate that α-synuclein possesses a remarkable conformational plasticity, being able to adopt structurally unrelated conformations including the substantially unfolded “basic” state, an amyloidogenic partially folded conformation, and different α-helical or β-structural species folded to a different degree, both monomeric and oligomeric [114, 116]. Furthermore, it might form several morphologically different types of aggregates, including oligomers (spheres or doughnuts), amorphous aggregates, and or amyloid-like fibrils [114, 116]. Based on this astonishing conformational behavior the concept of a protein-chameleon was proposed, according to which the structure of α-synuclein to a dramatic degree depends on the environment: the choice between all the mentioned above conformations is determined by the peculiarities of protein surroundings [114].
Fig. 4 provides a physical explanation for this phenomenal chameleon behavior of α-synuclein. Here the hypothetical folding-energy landscape of a typical globular protein (Fig. 4A) is compared with that of a natively unfolded protein (Fig. 4B). Note both landscapes are depicted schematically in one-dimensional cross-section. Fig. 4A shows that the folding landscape of a globular protein is characterized by a deep energy minimum, thus resembling a funnel [1, 154, 155]. It has been proposed that this folding landscape profile determines the ability of a globular protein to fold into a unique conformation, its native state, as a protein sequence possessing fast folding must satisfy two essential requirements: (1) thermodynamic stability meaning the existence of a deep global minimum in the energy landscape and (2) kinetic accessibility meaning the existence of a basin of attraction sloping toward that minimum [154]. Contrarily to a globular protein, the ‘topology’ of the landscape of a natively unfolded α-synuclein is characterized by numerous local energy minima, due to which this protein tend to behave as a highly frustrated system without any stable well-folded conformation (Fig. 4B). This folding landscape profile determines the conformational plasticity of α-synuclein (and other natively unfolded proteins) and also provides some clues on how this protein can specifically interact with so many ligands of so different nature (membrane, lipids, other proteins, metal ions, small organic molecules, etc.). If the interaction with a particular binding partner affects the α-synuclein folding landscape making some energy minima deeper and some energy barriers higher (see Figs. 4C1–3), then this protein would fold on a template-dependent manner gaining a specific structure needed to form a given complex.
Fig. 4.

A diagram showing the folding energy landscapes of a typical globular protein (A) [1] and of a typical natively unfolded protein in the absence (B) or presence of different binding partners (C). These landscapes are depicted schematically in one-dimensional cross-section.
Partial Folding to an Amyloidogenic Conformation Is Crucial for α-Synuclein Fibrillation
The amyloidogenesis of a-synuclein in vitro has been studied extensively (for recent reviews see [108, 114, 156]). Although α-synuclein is a natively unfolded protein, it forms fibrils of highly organized secondary structure. For example, the FTIR spectrum of α-synuclein fibrils shows the major contribution from β-sheet (see Fig. 2B). Furthermore, X-ray diffraction analysis of α-synuclein fibrils showed the characteristic pattern of a cross β-sheet structure in which the β-strands lie perpendicular to the long fiber axis, typical of all amyloid fibrils [157]. Electron microscopy analysis indicates that human α-synuclein filaments are typically 6–9 nm in width and several microns long. Atomic force microscopy images of α-synuclein fibrillation reveal three different fibrillar species, corresponding to protofilaments, protofibrils and mature fibrils. Clearly, within the fibril α-synuclein cannot be present as an extended linear polymer, since in a fully extended conformation the maximal linear dimension of a polypeptide with n residues is n × 3.63 Å [158]. This gives ~51 nm for α-synuclein, which is at least 5 times greater than the diameter of the filament. Thus, α-synuclein must be folded within fibril. It has been shown that the decrease in pH or an increase in temperature induces partial folding of α-synuclein. In contrast to an unfolded protein, a partially folded intermediate is anticipated to have contiguous hydrophobic patches on its surface, which are likely to foster self-association, and hence potentially fibrillation.
The histological dye Thioflavin T, ThT, is widely used for the detection of amyloid fibrils due to a number of characteristic spectral changes [159–163]. Fig. 5 represents time-dependent changes in the ThT fluorescence during the process of α-synuclein fibril formation as a function of pH at 37°C (Fig. 5A) or as a function of temperature at pH 7.5 (Fig. 5B). The kinetic changes in the ThT fluorescence intensity at 482 nm are described by characteristic sigmoidal curves, with an initial lag phase, a subsequent exponential growth phase, and a final equilibrium phase. Such curves are consistent with a nucleation-dependent polymerisation model, in which the lag corresponds to the nucleation phase and the exponential part to fibril growth (elongation) [164–167].
Fig. 5.

Effect of pH (A) and tempreature (B) on fibrillation of human α-synuclein.
Fig. 5 shows that both decreasing the pH and increasing the temperature result in a very substantial acceleration of the kinetics of α-synuclein fibrillation [108, 114]. pH has similar effects on both the lag time and the elongation rate [108, 114]. Furthermore, the pH- and temperature-induced changes in the α-synuclein fibrillation kinetics were shown to be coincident with the pH- and temperature-driven structural transformations [108, 114]. In other words an excellent correlation between intramolecular conformational change and fibril formation has been established, suggesting that the partially structured conformation is a key amyloidogenic intermediate on the fibril-forming pathway [108, 114]. The results were consistent with the minimum scheme for α-synuclein fibrillation, where the first step is partial folding to this amyloidogenic conformation, the second step is the formation of nucleus and the final stage is the fibril formation [108, 114]. From this model the two key kinetic steps are the structural transformation leading to the aggregation-prone conformation, and the nucleus formation [108, 114]. Therefore, factors that shift the equilibrium in favor of the amyloidogenic partially folded conformation will facilitate fibril formation, as observed. It has been also assumed that fibrillation of α-synuclein, leading to Lewy Body formation and Parkinson’s and related Lewy Body diseases, as well as development of other synucleinopathies, may arise from various factors that would significantly populate or increase the concentration of this monomeric but aggregation-competent form [108, 114].
An increase in protein concentration obviously increases the absolute concentration of the amyloidogenic conformation. Therefore, high protein concentration is predicted to accelerate fibrillation, as is in fact observed in vitro [108, 114] and in the PD patient with the α-synuclein gene triplication [83–85]. The data suggest that the key partially folded conformation, once formed, oligomerizes rapidly to form fibrils. This reflects an important aspect of the kinetics of nucleus formation, where the appearance of the partially folded aggregation-prone conformation represents the rate-limiting step in α-synuclein nucleation. This partially folded intermediate was shown to be stabilized by numerous factors [114]. This includes point mutation [120, 121], high temperatures [108], low pH [108], the presence of several common pesticides and herbicides [141–143], or metal ions [139, 142], or at moderate concentrations of trimethylamine-N-oxide [138], or other organic solvents [137]. Under all these conditions α-synuclein was shown to undergo significantly enhanced fibrillation. In contrast, fibril formation was considerably slowed or inhibited under conditions favoring formation of more folded conformations [137, 138], or by stabilization of the fully unfolded form, e.g. by oxidation of its methionines [135] or by stabilization of off-pathway oligomers via nitration of tyrosines [148]. Importantly, all the conditions favoring the aggregation-prone partially folded conformation accelerated both nucleation and elongation stages of fibril assembly. This means that the pre-molten globule-like partially folded intermediate is likely involved in the formation of fibril nucleus and in the subsequent propagation of fibrils.
Interestingly, α-synuclein mutants associated with the early-onset of PD have been shown to be more aggregation prone in vitro than the wild type protein [120, 121, 130, 149–151, 168]. However, neither the natively unfolded, nor the partially folded intermediate conformations, were affected by the familial PD point mutations [120, 121]. Based on these observations it has been concluded that the effect of the enhanced aggregation of mutants is attributed to the increased propensity of their partially folded intermediates to aggregate, rather than to any changes in the monomeric natively unfolded species [120, 121].
It has been shown that molecular crowding modeled by high concentrations of different polymers (proteins, polysaccharides and polyethyleneglycols) dramatically accelerated α-synuclein fibrillation in vitro [169, 170]. The stimulation was observed in the presence of high concentrations of both charged and neutral polymers, namely proteins, polysaccharides and polyethylene glycols, and the magnitude of the accelerating effect depended on the nature of the polymer, its length and concentration [169]. The results suggested that the major factor responsible for the accelerated fibrillation under crowded conditions was the excluded volume, which favored self-association of α-synuclein due to the effectively increased protein concentration [169].
It is worth noting that the formation of amyloid-like fibrils is not the only pathological outcome of protein deposition diseases and in several disorders (as well as in numerous in vitro experiments) protein deposits are composed of the amorphous aggregates, cloud-like inclusions without defined structure, as in light chain deposition disease. Similarly, soluble oligomers represent another alternative final product of the aggregation process. The physiological significance of the oligomers is that they may be the toxic species. The choice between three aggregation pathways, fibrillation, amorphous aggregate formation or oligomerization, is determined by the amino acid sequence (could be modified by mutations) and by the peculiarities of protein environment.
AMYLOIDOGENESIS OF NATIVELY UNFOLDED PROTEINS INVOLVED IN VARIOUS CONFORMATIONAL DISEASES
Tables 1 and 2 show that many of the known amyloidogenic proteins are natively unfolded. It is reasonable to assume that such proteins are well suited for amyloidogenesis, as they lack significant secondary and tertiary structure and do not have much of the specific intra-chain interactions. In the absence of such conformational constraints they would be expected to be substantially more conformationally motile, and thus able to polymerize more readily than tightly packed globular proteins. However, this is not always the case and many natively unfolded proteins form fibrils in vitro with the same rates to those of ordered globular proteins (i.e., within a time frame of a few hours to several days and weeks). The delay in the amyloidogenesis of these proteins was attributed to the requirement for considerable structural rearrangement within the unfolded polypeptide chain. In fact, substantial evidence suggests that the earliest stage of fibrillation of these proteins is their partial folding. The following section considers illustrative examples of fibril formation of natively unfolded proteins.
β- and γ-Synucleins
Synucleins belong to a family of closely related presynaptic proteins that arise from three distinct genes, described currently only in vertebrates [171]. This family includes: α-synuclein, which also known as the non-amyloid component precursor protein, NACP, or synelfin [124, 172, 173]; β-synuclein, also referred to as phosphoneuro-protein 14 or PNP14 [173–175] and γ-synuclein, also known as breast cancer-specific gene 1 or BCSG1 and persyn [176–179].
Human β-synuclein is a 134-aa neuronal protein showing 78% identity to α-synuclein. The α- and β-synucleins share a conserved C-terminus with three identically placed tyrosine residues. However, β-synuclein is missing 11 residues within the specific NAC region [122, 180]. The activity of β-synuclein may be regulated by phosphorylation [174]. This protein, like α-synuclein, is expressed predominantly in the brain, however, in contrast to α-synuclein, β-synuclein is distributed more uniformly throughout the brain [181, 182]. Besides the central nervous system β-Synuclein was also found in Sertoli cells of the testis [183, 184], whereas α-synuclein was found in platelets [185].
The third member of the human synuclein family is the 127-aa γ-synuclein, which shares 60% similarity with α-synuclein at the amino acid sequence level [122, 180]. This protein is specifically lacks the tyrosine rich C-terminal signature of α- and β-synucleins [122]. γ-Synuclein is abundant in spinal cord and sensory ganglia [177, 178]. Interestingly, this protein is more widely distributed within the neuronal cytoplasm than α-and β-synucleins, being present throughout the cell body and axons [178]. It was also found in metastatic breast cancer tissue [177] and epidermis [186].
The deposition of α-synuclein has been implicated in the pathogenesis of various synucleinopathies (see above). It has recently been established that in addition to the traditional α-synuclein-containing LBs and LNs, the development of PD and DLB is accompanied by appearance of novel α-, β- and γ-synuclein-positive lesions at the axon terminals of hippocampus [187]. These pathological vesicular-like lesions located at the presynaptic axon terminals in the hippocampal dentate, hilar, and CA2/3 regions have been co-stained by antibodies to α- and β-synucleins, whereas antibodies to γ-synuclein detect previously unrecognized axonal spheroid-like inclusions in the hippocampal dentate molecular layer [187]. This broadens the concept of neurodegenerative “synucleinopathies” by implicating β- and γ-synucleins, in addition to α-synuclein, in the onset/progression of these two diseases. Additionally, abnormal expression of γ-synuclein has recently been reported in some breast tumors [177]. Using Northern blots and in situ hybridization it has been shown that a high percentage of malignant breast tumors, but not benign breast tumors or normal breast tissue, express γ-synuclein mRNA [177]. In addition, a direct link between γ-synuclein over-expression and increased invasiveness of breast tumor cells has been demonstrated [186].
As it has been already mentioned, human β- and γ-synucleins, being 78% and 60% identical to α-synuclein, preserve some characteristic features of α-synuclein, while missing others. Therefore, they serve as good models for the analysis focused on the clarification of structural outputs of sequence variability on the structure and behavior of a natively unfolded protein. Structural properties of the members of synuclein family have been compared using several physico-chemical methods [116]. It has been established that all three proteins showed far-UV CD spectra typical of an unfolded polypeptide chain. Interestingly, α-and γ-synucleins possessed almost indistinguishable spectra, whereas the far UV-CD spectrum of β-synuclein showed a slightly increased degree of disorder. The increased unfoldedness of β-synuclein was further confirmed by hydrodynamic studies performed by size-exclusion chromatography and SAXS. In fact, size-exclusion chromatographic analysis showed that β-synuclein was slightly more extended than α-and γ-synucleins: the RS of β-synuclein was typical of a completely unfolded polypeptide chain, while α- and γ-synucleins were more compact than expected for a random coil [116]. This emphasized the importance of the NAC region to maintain the residual partially collapsed structure in α- and γ-synucleins. SAXS analysis further confirmed this conclusion. Guinier analysis of the scattering data shows that the synucleins are characterized by rather different Rg values at neutral pH. The observed Rg value for α-synuclein at neutral pH (40±1 Å) is smaller than that estimated for a random coil conformation for a protein of this size (52 Å), indicating that the natively unfolded conformation of this protein is more compact than a random coil. On the other hand, the observed Rg value for β-synuclein (49±1 Å) matches that expected for a completely unfolded polypeptide chain of this length (51 Å), which indicates the random coil conformation for this protein. γ-Synuclein had a very large Rg (61±1 Å) under the conditions studied. This may be due to the very significant asymmetry of this protein or because of its self-association. Analysis of the SAXS forward-scattering intensity values, I(0) (which is proportional to the molecular weight of the molecule), confirmed that the large Rg is due to association. In fact, the I(0) value for γ-synuclein is more than twice that of for α- and β-synucleins [116]. Finally, the Kratky plots showed that the α- and β-synucleins do not have a well-developed globular structure, whereas γ-synuclein showed a characteristic maximum at low angles, indicating the presence of some globular structure in the oligomer [116].
The analysis of conformational behavior of different members of the synuclein family revealed that they possess a comparable response to the changes in their environment. In fact, although far-UV CD spectra of α-, β-, and γ-synucleins were slightly different at neutral pH, all three proteins possessed almost identical far-UV CD spectra at acidic pH suggesting that they adopt a partially folded intermediate with comparable degree of folding [116]. This hypothesis was further confirmed by the results of gel-filtration analysis, which showed that although β-synuclein was slightly more extended than α- and γ-synucleins at neutral pH, all three proteins possessed the same degree of compaction in acidic solutions [116].
Summarizing, conformational analysis revealed that α-, β-, and γ-synucleins are natively unfolded under physiological conditions in vitro, and are able to adopt comparable partially folded conformations at acidic pH or at high temperature [116]. Although both α- and γ-synucleins were shown to form fibrils, β-synuclein did not fibrillate, being incubated under the same conditions [116]. However, even non-amyloidogenic β-synuclein can be forced to fibrillate in the presence of some metals (Zn2+, Pb2+, and Cu2+) [188]. This metal-induced fibrillation of β-synuclein was further accelerated by the addition of GAGs and high concentrations of crowding agents. Furthermore, β-Synuclein was shown to undergo fast oligomerization and fibrillation in the presence of pesticides, whereas the addition of low concentrations of organic solvents induced the formation of amorphous aggregates [188]. It has been shown that β-synuclein did not fibrillate in crowded environments or in the presence of glycosaminoglycans (i.e., under the conditions known to induce very fast fibrillation of α-synuclein) when metals were not present in media [188]. Analysis of the fibrillation of both β- and γ-synucleins revealed that similar to α-synuclein these proteins have to adopt a partially folded amyloidogenic conformation to aggregate successfully [116, 188].
Intriguingly, the addition of either β- or γ-synuclein in a 1:1 molar ratio to α-synuclein solution substantially increased the duration of the lag-time and dramatically reduced the elongation rate of α-synuclein fibrillation [116]. Fibrillation was completely inhibited at a 4:1 molar excess of β- or γ-synuclein over α-synuclein [116]. β-Synuclein inhibited α-synuclein aggregation in an animal models too [189]. This suggests that β- and γ-synucleins may act as regulators of α-synuclein fibrillation in vivo, potentially acting as chaperones. Therefore, one possible factor in the etiology of PD would be a decrease in the levels of β- or γ-synucleins [116].
Amylin (Islet Amyloid Polypeptide, IAAP) and Type II Diabetes
In addition to insulin, pancreatic islet β-cells produce a polypeptide called amylin or islet amyloid polypeptide, IAAP [190]. Amylin has several functions associated with the normal regulation of energy metabolism. Dysfunction of amylin due to mutation and/or amyloid fibril formation has been associated with the development of non-insulin-dependent diabetes mellitus (NIDDM), also known as type II diabetes [191–193]. Type-II diabetes is characterized by chronic insulin resistance and progressive decline in pancreatic β-cell function. One of the most common pathological features of type-II diabetes is the deposition of amyloid fibrils in the islets of Langerhans of the pancreas [194–197]. Human IAPP or amylin, is the major protein component of these amyloid deposits [190, 198]. Amylin is an unstructured peptide hormone of 37 amino acid residues. The natively unfolded nature of this peptide was established using far-UV CD spectroscopy [192, 199, 200] and electron paramagnetic resonance spectroscopy [201]. For example, eight spin-labeled derivatives of IAPP were analyzed using electron paramagnetic resonance spectroscopy. In solution, all eight derivatives gave rise to electron paramagnetic resonance spectra with sharp lines indicative of rapid motion on the sub-nanosecond time scale, which are consistent with a rapidly tumbling and highly dynamic peptide [201]. Human amylin and its 8–37 fragment were shown to form fibrils under physiological conditions. The process of polymerization is relatively fast (lag-times were 100 and 50 min for full-length amylin and its 8–37 fragment, respectively) and results in the appearance of typical amyloid fibrils [200]. Interestingly, both peptides showed formation of a partially folded (pre-molten globule-like) intermediate early in the fibrillation process. It takes ~90 min for full-length amylin to form such an intermediate, whereas this period was almost half as long for the truncated peptide, showing excellent agreement with the fibrillation lag-times [200].
Amyloid β-Protein and Alzheimer’s Disease
Alzheimer’s disease (AD) is the most prevalent age-dependent dementia, which is characterized pathologically by the accumulation of extracellular amyloid deposits, senile plaques, in the cerebral cortex and vasculature and of intracellular neurofibrillary tangles (paired helical filaments, PHFs). Amyloid deposits contain the amyloid β-protein (Aβ), which is a 40–42 residue peptide, produced by endoproteolytic cleavage of the amyloid β-protein precursor (APP). PHFs are assembled from the protein tau (see below). Many lines of evidence support the crucial role of Aβ in AD. The Aβ peptide becomes neurotoxic to cortical cell cultures when aggregated as amyloid-like β-strand structures [202–204], Aβ protofibrils [205], or other aggregates such as Aβ derived diffusible ligands that kill mature neurons at nanomolar concentrations and cause neurological dysfunction in the hippocampus [206]. The two major Aβ peptides are the 40-residue Aβ1–40 and the 42-residue Aβ1–42, which differ in the absence or presence of two extra C-terminal residues (Ile41-Ala42). The N-terminal (residues 1–28) residues comprise a hydrophilic domain with a high proportion of charged residues (46%), whereas the C-terminal domain (residues 29–40 or 29–42) is completely hydrophobic and is presumably associated with the cell membrane of APP. Although the Aβ1–40 and Aβ1–42 peptides are ubiquitous in biological fluids of humans (at an approximate ratio of 9:1), it is thought that the longer Aβ1–42 is more pathogenic, due to its higher quantities in the amyloid plaques of sporadic AD cases, its even higher quantities in patients afflicted with early-onset AD [207, 208], and because of the greater in vitro tendency of the Aβ1–42 to aggregate and precipitate as amyloid [209, 210]. Fibrillation of Aβ is associated with the development of the cascade of neuropathogenetic events, ending with the appearance of cognitive and behavioral features typical of AD. Aβ appears to be unfolded at the beginning of the fibrillation under physiological conditions. NMR studies have shown that monomers of Aβ(1–40), or Aβ(1–42) possess no α-helical or β-sheet structure [211], i.e. they exist predominately as random extended chains. Partial refolding to the pre-molten globule-like conformation has been detected at the earliest stages of Aβ fibrillation [211].
Tau-Protein in Alzheimer’s Disease
Tau, a microtubule assembly protein isolated from brain microtubules, represents a family of isoforms, which migrate as close bands of 55–62 kDa in SDS gel electrophoresis. Heterogeneity is explained in part by alternative mRNA splicing leading to the appearance of three or four repeats in the C-terminal region [212, 213]. Post-translational phosphorylation of tau is an additional source of microheterogeneity [214]. In vitro, tau binds to microtubules, promotes microtubule assembly, and affects the dynamic instability of individual microtubules [215–219]. In situ, tau is highly enriched in the axons [220]. In living cells and brain tissue, tau protein has been estimated as comprising 0.025–0.25% of total protein [221, 222]. On the basis of its in vitro activity and its distribution, it is believed that tau regulates the organization of neuronal microtubules. Interest in tau dramatically increased with the discovery of its aggregation in neuronal cells in the progress of Alzheimer’s disease and various other neurodegenerative disorders, especially frontotemporal dementia [223, 224]. In these cases specific tau-containing neurofibrillary tangles (paired helical filaments) are formed [224]. Filaments isolated from end-stage AD are particularly well characterized and consist of all six full-length tau isoforms extensively phosphorylated and organized into twisted paired helical filaments (PHFs) and nontwisted straight filaments (SFs) [225, 226]. Hyperphosphorylation was shown to be a common characteristic of pathological tau [227]. Hyperphosphorylated tau isolated from patients with AD was shown to be unable to bind to microtubules and promote microtubule assembly. However, both of these activities were restored after enzymatic dephosphorylation of tau protein [228–231].
During brain development, tau is phosphorylated at many residues, including sites phosphorylated with GSK-3β, cdk 5, and MAPK [232]. In vitro, tau can be phosphorylated on multiple sites by several kinases (for a review, see [233]). Most of the in vitro phosphorylation sites of tau are located within the microtubule interacting region (repeat domain) and sequences flanking the repeat domain. Many of these sites are also phosphorylated in PHF-tau [234, 235]. In fact, 10 major phosphorylation sites have been identified in tau isolated from PHFs from patients with AD [234]. All of these sites are located in regions flanking tau’s repeat domain and constitute recognition sites for several AD diagnostic antibodies, which may point to an important role for these phosphorylation sites for AD pathogenesis. Hyperphosphorylation was shown to be accompanied by the transformation from the unfolded state of tau into a partially folded conformation [236, 237], accelerating dramatically the self-assembly of this protein into paired helical filaments in vitro [229, 238]. To analyze the potential role of tau hyperphosphorylation in tauopathies, mutated tau proteins have been produced, in which all 10 serine/threonine residues known to be highly phosphorylated in PHF-tau were substituted for negatively charged residues, thus producing a model for a defined and permanent hyperphosphorylation-like state of tau protein [239]. It has been demonstrated that, like hyperphosphorylation, glutamate substitutions induce compact structure elements and SDS-resistant conformational domains in tau protein, as well as lead to the dramatic acceleration of its fibrillation [239].
Prior the aggregation, tau protein was shown to be in a mostly random coil structure. This conclusion followed from the conformational analysis of this protein by circular dichroism, Fourier transform infrared, X-ray scattering and biochemical assays [240]. Analysis of the primary structure reveals a very low content of hydrophobic amino acids and a high content of charged residues, which was sufficient to explain the lack of folding [240]. Analysis of the hydrodynamic radii confirms a mostly random coil structure of various tau isoforms and tau domains. However, the protein was further unfolded by high concentrations of strong denaturant GdmCl, indicating the presence of some residual structure in this protein. This conclusion was supported by a FRET-based approach where the distances between different domains of tau were determined. The combined data show that tau is mostly disordered and flexible but tends to assume a hairpin-like overall fold which may be important in the transition to a pathological aggregate [240].
Intriguingly, purified recombinant tau isoforms do not detectably aggregate over days of incubation under physiological conditions. However, aggregation and fibrillization of tau protein can be greatly accelerated under near-physiological conditions in vitro by the addition of anionic surfactants [241]. Based on the detailed analysis of tau fibrillation in the presence of anionic inducers using a set of spectroscopic techniques (circular dichroism spectroscopy and reactivity with thioflavin S and 8-anilino-1-naphthalenesulfonic acid fluorescent probes) it has been established that the inducer stabilized a monomeric partially folded species with the structural characteristics of a pre-molten globule state [242]. The stabilization of this intermediate was sufficient to trigger the fibrilliation of full-length tau protein [94, 95, 223–225, 227–230, 242–245].
ABri Peptide and Familial British Dementia
The ABri is a 34 residue peptide that is the major component of amyloid deposits in familial British dementia (FBD), which is an autosomal dominant disorder with onset at around the fifth decade of life and full penetrance by age 60 characterized by the presence of amyloid deposits in cerebral blood vessels and brain parenchyma that coexist with neurofibrillary tangles in limbic areas [246]. FBD patients develop progressive dementia, spasticity, and cerebellar ataxia. The protein subunit (termed ABri) is an example of an amyloid molecule created de novo by the abolishment of the stop codon in its precursor, a protein comprised of 266 amino acid residues (BRI-266) that is codified by a single gene, BRI, located on the long arm of chromosome 13 [247, 248]. The FBD has a single nucleotide change (TGA→AGA, codon 267) that results in an arginine residue substitution for the stop codon in the wild-type precursor molecule and a longer open reading frame of 277 amino acid residues (BRI-277 instead of BRI-266). The ABri amyloid peptide is formed by the 34 C-terminal amino acid residues of the mutant precursor protein BRI-277, presumably generated from furin-like processing [249]. Thus, the point mutation at the stop codon of BRI results in the generation of the 34 residue ABri peptide (instead of the shorter 23 residue WT peptide), which is deposited as amyloid fibrils causing neuronal dysfunction and dementia [250]. It has been emphasized that athough FBD and AD share almost identical neurofibrillar pathology and neuronal loss that co-localize with amyloid deposits, the primary sequences of the amyloid proteins (ABri and Aβ) differ. Therefore, ABri and Aβ amyloid deposition in the brain can trigger similar neuropathological changes (neuronal loss and dementia) and thus may be a key event in the initiation of neurodegeneration [250].
Using far-UV CD and NMR spectroscopy it has been recently established that ABri is in the random coil-like conformation at slightly acidic pH [250]. The solution pH was shown to play an important role in promoting the amyloid-like β-sheet structure and the characteristic fibril morphology of ABri and this protein forms amyloid fibrils at pH 4.9 with no distinct fibril morphology being observed at neutral and slightly basic pH (pH 7.1–8.3), except for smaller spherical aggregates that gradually disappeared and assembled into larger amorphous aggregates [250]. It has been also pointed out that at pH 4.9 the ABri undergoes relatively slow β-aggregation, where it is possible for fibril formation to occur, similar to the behavior of the amyloid Aβ peptide [250].
ADan in Familial Danish Dementia
Familial Danish dementia is a neurodegenerative disorder linked to a genetic defect in the BRI2 gene. Similar to FBD, familial Danish dementia (FDD) results form the genetic alterations in the this gene and the deposited amyloid protein, ADan, is the C-terminal proteolytic fragment of a genetically altered BRI2 precursor molecule [251]. The amyloid peptides ABri and ADan originate as a result of two different genetic defects at, or immediately before, the BRI2 stop codon with a common final outcome in both diseases: regardless of the nucleotide changes, the ordinarily occurring stop codon is either non-existent (in FBD) or out of frame (in FDD) causing the genesis of an extended precursor featuring a C-terminal piece that does not exist in normal conditions (reviewed in [252]). ABri and ADan are released by a furin-like proteolytic processing. Both these peptides are 34-residues-long, which share 100% homology on the first 22 residues, a completely different 12 amino acid C-terminus and have no sequence identity to any other known amyloid protein. Despite the structural differences among the corresponding amyloid subunits FDD and FBD show striking clinical and neuropathological similarities with AD, including the presence of neurofibrillary tangles, parenchymal amyloid and pre-amyloid deposits and cerebral amyloid angiopathy (CAA) co-localizing with inflammatory markers, reactive microglia and activation products of the complement system (reviewed in [252]). Structural analysis revealed that similar to Aβ and ABri, ADan is a typical natively unfolded protein, which is characterized by a random coil structure in a wide pH range and is prone to form fibrils in a pH-dependent manner [253].
Prion Protein and Prion Diseases
Prion diseases, collectively referred to as the transmissible spongiform encephalopathies (TSEs), are caused by the pathological deposition of the prion protein in its aggregated form and are characterized by unique infectious prion particles. TSEs include Creutzfeldt-Jakob disease, scrapie, bovine spongiform encephalopathy (BSE) and chronic wasting disease of mule deer and elk [254]. The most important aspect is the transmission of prion protein aggregates from one individual or species to another, causing prion diseases. The characteristic pathological features of TSEs are spongiform degeneration of the brain and accumulation of the abnormal, protease-resistant prion protein isoform in the central nervous system, which sometimes forms amyloid-like plaques.
Native prion protein (PrPC) is attached to the extracellular plasma membrane surface by a glycosylphosphatidylinositol lipid anchor and undergoes endocytosis. The N-terminal region of about 100 amino acids in PrPC (from amino acid 23 to 126) is largely unstructured in the isolated molecule in solution [255]. The C-terminal domain is folded into a largely α-helical conformation (three α-helices and a short antiparallel β-sheet) and stabilized by a single disulphide bond linking helices 2 and 3 [256]. The central event in the pathogenesis of prion diseases is a major conformational change of the C-terminal region of the prion protein (PrP) from an α-helical (PrPC) to a β-sheet-rich isoform (PrPSc), and PrPSc propagates itself by causing the conversion of PrPC to PrPSc. Although unstructured in the isolated molecule, the N-terminal region contains tight binding sites for Cu2+ ions and acquires structure following copper binding [257, 258]. Two pathological GSS-like mutations, Y145Stop and Q160Stop, result in C-terminal truncated isoforms. The truncation occurs just after the central region from amino acid 90 to 145, which was shown to be converted into β-sheet as a result of the PrPC to PrPSc conversion [259, 260]. In contrast to the full-length prion protein, the Y145Stop mutant was shown to aggregate under native nondenaturing rather neutral pH conditions [261].
Investigations of the steps required for prion propagation and neurodegeneration in transgenic mice expressing chimeric mouse–hamster–mouse or mouse–human–mouse PrP transgenics indicated that the last 50 residues in the disordered N-terminal region play a particularly important role in the interaction of PrPC with PrPSc leading to the conversion of the former to the latter [262, 263]. Those residues are largely unordered or weakly helical in the full-length PrPC [264, 265], but are predicted to be β-structure in PrPSc [266]. These observations emphasize a crucial role of the disordered N-terminal region in the modulation of prion protein aggregation. Several kinetics studies have revealed the existence of partially folded intermediates for the prion protein [266–268], and is reasonable to assume that fibrillation requires partial unfolding of the C-terminal domain prior to self-association.
In an elegant study, structural properties and aggregation propensities of the Y145Stop and Q160Stop mutants were analyzed by a variety of biophysical techniques [269]. It has been shown that although both proteins are substantially disordered, a continuous stretch of positive secondary chemical NMR shifts was found for residues 144–154 in Q160Stop protein, indicative of helical structure. This clearly demonstrated that although the vast majority of a polypeptide chain is substantially disordered, a significantly populated helix 1 is present in human Q160Stop protein [269]. Q160Stop protein was shown to fibrillate faster than shorter Y145Stop variant. Intriguingly, helix 1 was not converted to the β-sheet during the protein aggregation. Based on the results of this analysis it has been concluded that the highly charged helix 1 is involved in the aggregation of Q160Stop protein likely via the formation of intermolecular salt bridges [269].
Polyglutamine Repeat Diseases: Huntingtin, Ataxin-1, Androgen Receptor and Atrophin-1
Currently there are at least eight known hereditary diseases, including Huntington’s, Kennedy disease (spinal and bulbar muscular atrophy, SBMA), spinocerebellar ataxia type 1 (SCA1), dentatorubral-pallidoluysian atrophy (DRPLA), spinocerebellar ataxia type 2 (SCA2), Machado-Joseph disease (MJD/SCA3), SCA6, and SCA7, in all of which the expansion of a CAG repeat in the gene leads to neurodegeneration [270, 271]. These diseases are accompanied by the progressive death of neurons, with insoluble, granular, and fibrous deposits being found in the cell nuclei of the affected neurons. The neurotoxicity in these diseases is due to the expansion of the (CAG)N-encoded polyglutamine (polyGln) repeat, which lead to the formation of amyloid fibrils and neuronal death. In Huntington’s diseases, the CAG repeat that encodes the polyQ region is part of exon 1 in the 3,140-residue huntingtin protein [272]. The polyQ repeat varies between 16 and 37 residues in healthy individuals, and individuals who are afflicted by disease have repeats of >38 residues.
The age of onset and the severity of the progression of spinocerebellar ataxia type-1 (SCA1), an autosomal-dominant neurodegenerative disorder characterized by ataxia and progressive motor deterioration, are directly correlated with the length of polyQ segment in ataxin-1, a nuclear protein of ~800 residues [273–275]. When the number of glutamine residues in the polyQ tract exceeds a threshold (39–44 glutamine residues), ataxin-1 aggregates with granular or fibrillar morphologies accumulate intranuclearly and eventually lead to cell death [276, 277].
Kennedy disease (also known as spinal and bulbar muscular atrophy, SBMA) is linked to the expansion of a Gln-rich segment in the androgen receptor [278]; healthy individuals have a 15- to 31-residue polyQ segment, and individuals who are afflicted with the disease have 40–62 Gln residues. Intriguingly, in the human androgen receptor there are three polyglutamine repeats ranging in size from five to 22 residues, stretches of seven prolines and five alanines, and a polyglycine repeat of 24 residues. Polymorphisms in the length of the largest polyglutamine and the polyglycine repeats of the androgen receptor have been associated with a number of clinical disorders, including prostate cancer, benign prostatic hyperplasia, male infertility and rheumatoid arthritis [279]. Circular dichroism analysis of a region of the androgen receptor N-terminal domain lacking the largest polyglutamine stretch, but containing the remaining repeats, showed that it lacked stable secondary structure in aqueous solution [279].
The onset of the dentatorubral-pallidoluysian atrophy (DRPLA), another progressive neurodegenerative disorder characterized by a distinctive pathology in the cerebellar and pallidal outflow pathways, is inversely correlated with the polyQ repeat size in the corresponding DRPLA protein, a product of the gene on chromosome 12p [280]. The repeat size varied from 7–23 in normal individuals and was expanded to 49–75 in DRPLA patients.
The mechanistic hypothesis linking CAG repeat expansion to toxicity involves the tendency of longer polyGln sequences, regardless of protein context, to form insoluble aggregates [281–289]. To help evaluate various possible mechanisms, the biophysical properties of a series of simple polyglutamine peptides have been analyzed. The circular dichroism spectra of poly(Gln) peptides with repeat lengths of 5, 15, 28 and 44 residues were shown to be nearly identical and were consistent with a high degree of random coil structure, suggesting that the length-dependence of disease is not related to a conformational change in the monomeric states of expanded poly(Gln) sequences [287] In contrast, there was a dramatic acceleration in the spontaneous formation of ordered, amyloid-like aggregates for poly(Gln) peptides with repeat lengths of greater than 37 residues. Several studies have revealed the existence of partially folded intermediates of polyglutamine-repeat proteins as key species in fibrillation [288, 290, 291], and these are assumed to be precursors of oligomeric aggregates [287].
Nuclear Poly(A) Binding Protein and Oculopharyngeal Muscular Dystrophy
The nuclear poly(A) binding protein (PABPN1) stimulates polyadenylation and controls the length of poly(A) tails [292, 293]. PABPN1 is a 33-kD protein containing a single RNA binding domain (residues 161–258) consisting of a eukaryotic RNA recognition motif that is flanked by an acidic N and a basic C terminus. At the N-terminus, PABPN1 has a natural 12 Ala stretch that is interrupted after the tenth Ala residue by a single Gly residue. Trinucleotide expansions result in an extension of the wild-type 10 Ala sequence before the Gly residue to a maximum of 17 Ala residues. Individuals carrying the extensions develop the disease oculopharyngeal muscular dystrophy (OPMD) that is characterized by swallowing difficulties, eyelid drooping, and limb weakness [294]. Hystopathologically, OPMD is characterized by intranuclear inclusions of palisade-like structures in muscle fibers [295], with PABPN1 being the major constituent of these nuclear inclusions [296]. Far-UV CD analysis revealed that both full length protein and its N-terminal fragment (residues 1–125) are almost completely unfolded in solution. The low content of secondary structural elements at the termini of PABPN1 might be due to the high content of Pro and Glu (20 Pro and 24 Glu within 125 residues) in the N terminus and the highly positively charged C terminus (11 Arg within 40 residues) [297]. Intriguingly, the N-terminal highly amyloidogenic fragment of the PABPN1 with the extended polyalanine stretch showed some gain in secondary structure and possessed spectral properties typical of the pre-molten globule conformation [297].
Calcitonin and Medullary Thyroid Cancer
Medullary thyroid cancer (MTC) is a nonepithelial, neuroendocrine tumor with a more aggressive clinical behavior than differentiated thyroid cancer [298]. MTC comprises 5–10% of all thyroid cancers and arises from the parafollicular or C-cells of neuroendocrine origin, which produce the hormone calcitonin and make up 1% of thyroid cells. C-cells are found throughout the thyroid gland but are concentrated in the posterior upper third of the lateral lobes, which is where most MTC are found [299, 300]. MTC occurs as a sporadic form or less commonly, as a hereditary form, as part of multiple endocrine neoplasia syndromes types 2A (MEN 2A) and 2B (MEN 2B) [300]. Histologically, MTC is characterized by uniform polygonal cells with central nuclei and finely granular eosinophilic cytoplasm. Stromal amyloid-like deposits are frequently found in MTC representing the distinguishing feature of this disease [300]. The major component of these deposits is calcitonin, a 32-residue polypeptide hormone with an N-terminal disulphide bridge (between residues 1 and 7) and a C-terminal proline amide residue [301, 302]. Besides being involved in MTC, calcitonin is known to cause a rapid, but short-lived, decline in calcium and phosphate levels in the blood by promoting the incorporation of these ions into bone [303]. This activity has lead to the use of calcitonin in the treatment of conditions such as osteoporosis and Paget’s disease, as well as malignancy-caused hypercalcemia and musculoskeletal pain [303–305]. Therefore, calcitonin is considered as a bioactive peptide, which unfortunately has limited pharmaceutical potential due to a high tendency to aggregate. In addition to mentioned above high tendency to self-associate in the form of amyloid fibrils in patients with MCT, in vitro preparations of calcitonin designed for patient administration experience a serious problem during the production, storage, and administration [306].
The far-UV CD spectra revealed that in aqueous solution, the calcitonin showed little evidence of adopting an ordered conformation [307, 308], whereas in 100% trifluoroethanol a dominant α-helix component was clearly observed [307]. Conformations of human calcitonin in several solvents have been studied by solution NMR spectroscopy. In TFE-H2O mixtures, calcitonin forms α-helical structure between the residues 9 and 21 [309]. In DMSO-H2O, the central region (residues 16–21) of calcitonin might form a short double-stranded antiparallel β-sheet [310]. It was suggested that human calcitonin exhibits an amphiphilic nature when it forms an α-helix [311]. However, in D2O, human calcitonin was shown to be in a random coil conformation as determined from the 1H chemical shift data [312]. Subsequent studies by two-dimensional NOESY and CD measurements indicated that it adopts an extended conformation with high flexibility in aqueous solution. Recently, α-helical conformation was reported to be present in the central region of human calcitonin in aqueous acidic solution, although it is shorter than that in TFE-H2O [313]. During the fibril formation in the acetic acid solution (pH 3.3), local conformational transitions from an α-helix to a β-sheet structure at the central region and from a random coil to a β-sheet at the C terminus region were shown to occur simultaneously [314]. It was reported that the rate of human calcitonin fibril formation at neutral pH is much faster than that at acidic pH [314]. Intriguingly, far-UV CD spectra of human calcitonin measured at neutral pH reflected the presence of more ordered secondary structure than at acidic pH, as it followed from the decreased signal at 200 nm and an increased negative ellipticity around 220 nm [315]. Therefore, human calcitonin is likely in the pre-molten globule-like conformation at the conditions favoring amyloid fibril formation.
Gelsolin and Finnish-Type Familial Amyloidosis
Finnish-type familial gelsolin amyloidosis is an autosomal dominantly inherited neurodegenerative disease where homozygotes generally do not survive past their thirties. Extensive deposition of mutant gelsolin fragments as amyloid in the basement membrane of the skin, blood vessel walls, central nervous system, and the ocular system causes cutis laxa, cranial neuropathy, and corneal lattice dystrophy [316–318]. The mutations (D187N/Y) causing gelsolin amyloidosis disrupts Ca2+ binding within domain 2 which makes it susceptible to aberrant cleavage by furin yielding a secreted 68 kDa fragment referred to as C68 [319, 320]. The C68 fragment is further cleaved in at least two positions by metalloprotease(s) producing 8 (residues A173-M243) and 5 kDa (residues A173-R225) gelsolin fragments which compose the amyloid deposits in patients [321, 322]. Far-UV CD analysis of the amyloidogenic A173-M243 fragment of the D187N gelsolin revealed that this protein being substantially unfolded possesses structural features typical of the pre-molten globule [323].
FIBRILLATION OF NATIVELY UNFOLDED PROTEINS UNRELATED TO HUMAN CONFORMATIONAL DISEASES
Table 2 lists natively unfolded proteins that are known to be able to form amyloid-like fibrils, but are not related to conformational disease. In addition to these proteins, a large set of short peptides, both disease-related and non-disease-related were shown to form fibrils under physiological conditions in vitro. Although the majority of those peptides are unfolded in aqueous solutions, they will not be considered in this chapter, as they definitely consitite an attractive target for the specialized review.
Yeast Prions
There are at least two natural genetic elements, the [PSI+] and [URE3] factors, in the budding yeast Saccharomyces cerevisiae that exhibit non-Mendelian inheritance but can be “cured” by treatment of the cells with low concentrations of the protein denaturant guanidine hydrochloride [324]. The [PSI+] factor is a self-perpetuating, conformationally altered form of a cellular protein [325–327]. The inheritance of [PSI+] from mother to daughter cells is based on the transmission of conformational information from ordered nonfunctional Sup35p aggregates (amyloid-like fibrils) to the soluble, functional form of Sup35p, a subunit of the polypeptide chain release complex that is essential for translation termination [328, 329]. In vitro, the prion-determining region of Sup35p, NM, is initially disordered but slowly converts to a partially folded structure and then fibrils [330–332]. In particular, factors such as elevated temperatures, chemical chaperones and certain mutations that increased the amount of structure increased the amount of aggregation [332]. The acceleration of Sup35p fibril formation is determined by the acceleration of slow conformational changes rather than by providing stable nuclei. Strikingly, inhibitory mutations map exclusively within a short glutamine/asparagine-rich region of Sup35p, and all but one occur at polar residues. Even after replacement of this region with polyglutamine, Sup35p retains its ability to form fibrils [327]. This suggests similarities between the prion-like propagation of [PSI+] and polyglutamine-mediated pathogenesis of several neurodegenerative diseases (see above).
The [URE3] element is another factor of S. cerevisiae that propagates by a prion-like mechanism and corresponds to the loss of function of the cellular protein Ure2 [333–335]. The N-terminal region of the protein is flexible and unstructured, while its C-terminal region is compact and folded [336]. The overexpression of full-length Ure2p in wild-type S. cerevisiae strains induced a 20- to 200-fold increase in the frequency with which [URE3] arose. On the other hand, expression of just the N-terminal 65 residues of Ure2p increased the frequency of [URE3] induction 6000-fold [336–338]. These observations emphasize a key role of the disordered N-terminal domain in the fibrillation of Ure2 protein. A dimeric intermediate is populated transiently during refolding and populated under conditions correlating with fibril formation. Interestingly, the native dimer and dimeric intermediate are significantly more stable than either of their monomeric counterparts. Stabilization of the native state disfavors amyloid nucleation [339].
Prothymosin α
This is a very acidic protein, containing ~50% aspartic and glutamic acid, no aromatic or cysteine residues, and very few large hydrophobic aliphatic amino acids [340]. Because of these features, prothymosin α adopts a random coil-like conformation with no regular secondary structure at neutral pH [340, 341]. However, at acidic pH prothymosin α folds into a partially folded pre-molten globule-like conformation [341]. Interestingly, it has been recently shown that at low pH (below pH 3, i.e. under conditions favoring the formation of the partially folded conformation [341]), prothymosin α is capable of relatively fast formation (lag time ~100 min) of regular elongated fibrils with a flat ribbon-like structure 4–5 nm in height and 12–13 nm in width [342].
Apolipoprotein C-II
Human apolipoprotein C-II is a plasma protein consisting of 79 amino acid residues, whose function is to activate lipoprotein lipase. The structure of apoC-II in the presence of the lipid mimetic, SDS, reveals three regions of well defined amphipathic α-helix with loosely defined intervening regions that may reflect flexible hinge regions [343]. In the absence of lipid or detergent, human apolipoprotein C-II lacks ordered structure and forms amyloid ribbons after incubation for several days [344–348]. Fibril formation was dramatically accelerated by the addition of phospholipids in sub-micellar concentrations, which were shown to induce partial folding of the protein into a pre-molten globule-like intermediate [344]. In contrast, the fibrillation of apolipoprotein C-II was completely inhibited in the presence of micellar phospholipids; i.e., under conditions favoring α-helical conformation [344].
Core Histones
The bovine core histones are natively unfolded proteins in solutions of low ionic strength, due to their high net positive charge at pH 7.5 [349]. Analysis of the structural properties of core histones as a function of pH, ionic strength and organic solvent concentration demonstrated a correlation between conformational change and propensity to aggregate. Overall, the histones are able to adopt at least five different relatively stable conformations: the intrinsically unstructured form (pH 2.0, low protein concentration, low ionic strength); a fully-unfolded conformation (6 M urea); partially folded α-helical dimers (low ionic strength, pH 2.0, high protein concentrations); helical monomers (low ionic strength, pH 2.0, 10% TFE) and molten globule-like α-helical oligomers (pH 2.0, high salt) [349]. Under most of the conditions studied the histones formed amyloid fibrils with typical morphology as seen by electron microscopy. In contrast to most aggregation/amyloidogenic systems, the kinetics of fibrillation showed an inverse dependence on histone concentration, which has been attributed to partitioning to a faster pathway leading to non-fibrillar self-associated aggregates at higher protein concentrations. In keeping with the hypothesis that partial folding is required of intrinsically disordered proteins for aggregation, the rate of fibril formation correlated with conditions favoring the stabilization of the partially folded conformation [349].
Carboxymethylated α-Lactabumin
α-Lactalbumin (α-LA) is a small acidic Ca2+-binding protein involved in the regulation of lactose biosynthesis as a component of lactose synthetase [350, 351]. α-LA possesses a single Ca2+-binding site and is frequently used as a model Ca2+-binding protein distinct from the EF-hand family [352, 353]. In addition, α-LA is very attractive for studies of partially folded conformations because at either acidic pH, moderate guanidinium chloride concentrations, or elevated temperatures (apo form) it adopts the classic molten globule state [353–355]. Interestingly, it has been found that some associative forms of α-LA can induce apoptosis in tumor cells [356, 357]. αLA is known to adopt different partially folded conformations under mildly denaturing conditions [353–355, 358–360]. In addition, disulfide bridges were shown to play a very important role in the stabilization of α-LA structure [361–365]. For example, when two of the four disulfides were reduced, α-LA retained approximately half of the secondary and tertiary structure of the native protein [361, 362], the 1SS-α-LA form of α-LA was already significantly unfolded, showing only approximately one-third of the normal far-UV CD signal [366], and the fully reduced form had little tertiary structure but still retained some amount of secondary structure [363]. It has been also pointed out that the 1SS-α-LA form was characterized by conformational behavior typical of the pre-molten globule state and this form of α-LA possessed the highest rate of fibril formation in vitro [366].
GAGA Factor
GAGA factor (GAF) is as a transcription activator that binds to (GA)n-rich sites in the ultrabithorax and engrailed promoters [367, 368], regulating a number of Drosophila housekeeping genes, including histone H3/H4 [369], and the heat shock genes hsp26 [370] and hsp70 [371]. GAF was shown to be essential for homeotic gene expression [372]. There are two major isoforms of GAF, proteins of 519 and 581 amino acid residues. Both isoforms share the same N-terminal part, but have distinct glutamine-rich C termini of different lengths, which are glutamine-rich (23 % in the 519 residue and 42 % in the 581 residue form) [373]. Structural and fibrillation properties of the truncated GAF forms, GAF1 (residues 137–519, which include DNA-binding domain (310–372) and glutamine-rich domain (446–519) and GAF2 (residues 137–443 that do not contain glutamine-rich domain) were analyzed. It has been shown that both of these forms are significantly unfolded in solution. However, only GAF1 form containing the glutamine-rich region was able to form the amyloid-like fibrils in vitro in a concentration-dependent manner: at high concentrations, fibrils appeared almost instantly, whereas at low concentrations a large lag time for fibril formation was observed [373].
Caseins
Casein micelles are large colloidal aggregates whose light-scattering properties determine the milk opacity. Casein is a heterogeneous phosphoprotein comprising four distinct gene products, αS1-, αS2-, β-, and κ-caseins, which have been classified as intrinsically disordered proteins [38, 118, 374], because they are extremely flexible, essentially unfolded, and have relatively little secondary or tertiary structure under physiological conditions [38, 374]. In fact, tryptophan emission parameters (intensity and wavelength of emission maximum) and CD spectra showed that at neutral and alkaline pH the caseins exist predominantly in random coil conformation [375]. It has been shown recently that κ-casein is prone to form amyloid-like fibrils and this process is effectively inhibited by αS- and β-caseins [376].
Merozoite Surface Protein 2 (MSP2)
Merozoite surface protein 2 (MSP2) is one of the most abundant proteins on the merozoite surface of Plasmodium falciparum, which proved to be a promising anti-malaria vaccine target [377, 378]. MSP2 is a glycosyl-phosphatidylinositol anchored protein of mass approximately 30 kDa [379]. It has conserved N- and C-terminal domains of 25 and 74 residues, respectively, but is highly polymorphic in the central variable region [380]. MSP2 is largely disordered in solution, but has a propensity to form amyloid-like fibrils under physiological conditions. The N-terminal conserved region (MSP21–25) represents a part of the protease-resistant core of these fibrils [381].
Apo Cytochrome c552
The c-type cytochromes are electron-transfer proteins that contain a haem prosthetic group which is covalently attached through thioether linkages to two cysteine residues that are in a CXXCH motif in the protein [382]. Structural properties and amyloidogenic behavior of the mutated cytochrome c552 from Hydogenobacter thermophilus in which the two cysteine residues involved in covalent linkages to the haem (Cys 11 and Cys 14) were replaced with alanine residues was analyzed [383]. The holo-protein was shown to be highly helical, whereas the apo-form of the C11A/C14A cytochrome c552 has a CD spectrum characteristic of a mainly random coil conformation with approximately 14% α-helical secondary structure. This partially folded pre-molten globule-like conformation of the C11A/C14A cytochrome c552 possessed pronounced amyloidogenic properties [383].
SH3 Domain at Acidic pH
The SH3 domain (60–85 residues) is a small protein module with a simple fold found in many proteins that mediates specific protein-protein interactions within the cell [384, 385]. At neutral pH, PI3-SH3 (84 residues) is characterized by a well defined structure (a β-barrel composed of two perpendicular, antiparallel β-sheets of three and two strands [386–389]), the monomeric A state stabilized at acidic pH is substantially unfolded [390]. In fact, it has been shown that the A form possesses circular dichroism in the far- and near-UV regions and 1H NMR spectra typical of essentially unfolded conformation which is able to interact effectively with ANS. NMR diffusion measurements indicate, however, that the effective hydrodynamic radius of the A state is only 23% higher than that of the native protein and is 20% lower than that of the protein unfolded by high concentrations of GdmCl. At these conditions favoring the pre-molten globule-like conformation, PI3-SH3 was shown to aggregate readily into amyloid fibrils [390].
CONCLUSIONS
Natively unfolded proteins represent a high fraction of naturally occurring amyloidogenic proteins. This is most likely due to the ease with which they can form the β-sheet topology required in amyloid fibrils, in contrast to folded globular proteins, for which the native conformation places major constraints for the required topological rearrangement. Antipodal to tightly folded globular proteins which require substantial unfolding to become amyloidogenic, the first, and critical, step in the aggregation of intrinsically disordered proteins or polypeptides is partial folding. This results in a partially folded conformation with hydrophobic surface patches that favors self-association. As with partially folded intermediates formed during the folding/unfolding of globular proteins, such aggregate-prone conformations can polymerize to form fibrillar or amorphous aggregates, or soluble oligomers.
Acknowledgments
This work was supported by grant from the National Institutes of Health LM007688-0A1 and by the Programs of the Russian Academy of Sciences for the “Molecular and cellular biology” and “Fundamental science for medicine”.
References
- 1.Onuchic JN, Nymeyer H, Garcia AE, Chahine J, Socci ND. The energy landscape theory of protein folding: insights into folding mechanisms and scenarios. Adv Protein Chem. 2000;53:87–152. doi: 10.1016/s0065-3233(00)53003-4. [DOI] [PubMed] [Google Scholar]
- 2.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 3.Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 4.Bellotti V, Mangione P, Stoppini M. Biological activity and pathological implications of misfolded proteins. Cell Mol Life Sci. 1999;55:977–991. doi: 10.1007/s000180050348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uversky VN, Talapatra A, Gillespie JR, Fink AL. Protein Deposits As the Molecular Basis of Amyloidosis. II. Localized Amyloidosis and Neurodegenerative Disordres. Med Sci Monitor. 1999;5:1238–1254. [Google Scholar]
- 6.Uversky VN, Talapatra A, Gillespie JR, Fink AL. Protein Deposits As the Molecular Basis of Amyloidosis. I. Systemic Amyloidoses. Med Sci Monitor. 1999;5:1001–1012. [Google Scholar]
- 7.Rochet JC, Lansbury PT., Jr Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol. 2000;10:60–68. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 8.Uversky VN, Fink AL. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 9.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 10.Shirahama T, Benson MD, Cohen AS, Tanaka A. Fibrillar assemblage of variable segments of immunoglobulin light chains: an electron microscopic study. J Immunol. 1973;110:21–30. [PubMed] [Google Scholar]
- 11.Shirahama T, Cohen AS. High-resolution electron microscopic analysis of the amyloid fibril. J Cell Biol. 1967;33:679–708. doi: 10.1083/jcb.33.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jimenez JL, Guijarro JI, Orlova E, Zurdo J, Dobson CM, Sunde M, et al. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. Embo J. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uversky VN. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell Mol Life Sci. 2003;60:1852–1871. doi: 10.1007/s00018-003-3096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, et al. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci U S A. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zerovnik E. Amyloid-fibril formation. Proposed mechanisms and relevance to conformational disease. Eur J Biochem. 2002;269:3362–3371. doi: 10.1046/j.1432-1033.2002.03024.x. [DOI] [PubMed] [Google Scholar]
- 16.Lansbury PT., Jr Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc Natl Acad Sci U S A. 1999;96:3342–3344. doi: 10.1073/pnas.96.7.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9–23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 18.Dobson CM. The structural basis of protein folding and its links with human disease. Philos Trans R Soc Lond B Biol Sci. 2001;356:133–145. doi: 10.1098/rstb.2000.0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khurana R, Gillespie JR, Talapatra A, Minert LJ, Ionescu-Zanetti C, Millett I, et al. Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry. 2001;40:3525–3535. doi: 10.1021/bi001782b. [DOI] [PubMed] [Google Scholar]
- 20.Smith DP, Jones S, Serpell LC, Sunde M, Radford SE. A systematic investigation into the effect of protein destabilisation on beta 2-microglobulin amyloid formation. J Mol Biol. 2003;330:943–954. doi: 10.1016/s0022-2836(03)00687-9. [DOI] [PubMed] [Google Scholar]
- 21.Selkoe DJ. Alzheimer’s disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 22.Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, et al. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc Natl Acad Sci U S A. 2002;99:13990–13995. doi: 10.1073/pnas.222433299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, et al. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 25.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 26.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 27.Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- 28.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 29.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 30.Daughdrill GW, Pielak GJ, Uversky VN, Cortese MS, Dunker AK. Natively disordered proteins. In: Buchner J, Kiefhaber T, editors. Handbook of Protein Folding. Weinheim, Germany: Wiley-VCH, Verlag GmbH & Co. KGaA; 2005. pp. 271–353. [Google Scholar]
- 31.Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Uversky VN, Ptitsyn OB. “Partly folded” state, a new equilibrium state of protein molecules: four-state guanidinium chloride-induced unfolding of beta-lactamase at low temperature. Biochemistry. 1994;33:2782–2791. doi: 10.1021/bi00176a006. [DOI] [PubMed] [Google Scholar]
- 33.Uversky VN, Ptitsyn OB. Further evidence on the equilibrium “pre-molten globule state”: four-state guanidinium chloride-induced unfolding of carbonic anhydrase B at low temperature. J Mol Biol. 1996;255:215–228. doi: 10.1006/jmbi.1996.0018. [DOI] [PubMed] [Google Scholar]
- 34.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 35.Williams RM, Obradovic Z, Mathura V, Braun W, Garner EC, Young J, et al. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput. 2001:89–100. doi: 10.1142/9789814447362_0010. [DOI] [PubMed] [Google Scholar]
- 36.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Inform Ser Workshop Genome Inform. 1999;10:30–40. [PubMed] [Google Scholar]
- 38.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, Rost B. NORSp: Predictions of long regions without regular secondary structure. Nucleic Acids Res. 2003;31:3833–3835. doi: 10.1093/nar/gkg515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Linding R, Russell RB, Neduva V, Gibson TJ. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003;31:3701–3708. doi: 10.1093/nar/gkg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, et al. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- 43.Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 44.Jones DT, Ward JJ. Prediction of disordered regions in proteins from position specific score matrices. Proteins. 2003;53 (Suppl 6):573–578. doi: 10.1002/prot.10528. [DOI] [PubMed] [Google Scholar]
- 45.Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–2139. doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- 46.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 47.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 48.Dunker AK, Brown CJ, Obradovic Z. Identification and functions of usefully disordered proteins. Adv Protein Chem. 2002;62:25–49. doi: 10.1016/s0065-3233(02)62004-2. [DOI] [PubMed] [Google Scholar]
- 49.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. Febs J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 50.Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, et al. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998:473–484. [PubMed] [Google Scholar]
- 51.Dunker AK, Obradovic Z. The protein trinity--linking function and disorder. Nat Biotechnol. 2001;19:805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 52.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 53.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 54.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 55.Tompa P, Csermely P. The role of structural disorder in the function of RNA and protein chaperones. Faseb J. 2004;18:1169–1175. doi: 10.1096/fj.04-1584rev. [DOI] [PubMed] [Google Scholar]
- 56.Tompa P, Szasz C, Buday L. Structural disorder throws new light on moonlighting. Trends Biochem Sci. 2005;30:484–489. doi: 10.1016/j.tibs.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 57.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 59.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, et al. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–1898. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, et al. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–1916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, et al. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18:343–384. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 63.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- 64.Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44:1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 65.Goedert M. Filamentous nerve cell inclusions in neurodegenerative diseases: tauopathies and alpha-synucleinopathies. Philos Trans R Soc Lond B Biol Sci. 1999;354:1101–1118. doi: 10.1098/rstb.1999.0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spillantini MG, Goedert M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci. 2000;920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x. [DOI] [PubMed] [Google Scholar]
- 67.Trojanowski JQ, Lee VM. Parkinson’s disease and related alpha-synucleinopathies are brain amyloidoses. Ann N Y Acad Sci. 2003;991:107–110. doi: 10.1111/j.1749-6632.2003.tb07468.x. [DOI] [PubMed] [Google Scholar]
- 68.Galvin JE, Lee VM, Trojanowski JQ. Synucleinopathies: clinical and pathological implications. Arch Neurol. 2001;58:186–190. doi: 10.1001/archneur.58.2.186. [DOI] [PubMed] [Google Scholar]
- 69.Marti MJ, Tolosa E, Campdelacreu J. Clinical overview of the synucleinopathies. Mov Disord. 2003;18 (Suppl 6):S21–27. doi: 10.1002/mds.10559. [DOI] [PubMed] [Google Scholar]
- 70.Norris EH, Giasson BI, Lee VM. Alpha-synuclein: normal function and role in neurodegenerative diseases. Curr Top Dev Biol. 2004;60:17–54. doi: 10.1016/S0070-2153(04)60002-0. [DOI] [PubMed] [Google Scholar]
- 71.Jellinger KA. Neuropathological spectrum of synucleinopathies. Mov Disord. 2003;18 (Suppl 6):S2–12. doi: 10.1002/mds.10557. [DOI] [PubMed] [Google Scholar]
- 72.Dev KK, Hofele K, Barbieri S, Buchman VL, van der Putten H. Part II: alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology. 2003;45:14–44. doi: 10.1016/s0028-3908(03)00140-0. [DOI] [PubMed] [Google Scholar]
- 73.Duda JE, Lee VM, Trojanowski JQ. Neuropathology of synuclein aggregates. J Neurosci Res. 2000;61:121–127. doi: 10.1002/1097-4547(20000715)61:2<121::AID-JNR1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 74.Mitra K, Gangopadhaya PK, Das SK. Parkinsonism plus syndrome--a review. Neurol India. 2003;51:183–188. [PubMed] [Google Scholar]
- 75.Dickson DW. Alpha-synuclein and the Lewy body disorders. Curr Opin Neurol. 2001;14:423–432. doi: 10.1097/00019052-200108000-00001. [DOI] [PubMed] [Google Scholar]
- 76.Goedert M. Parkinson’s disease and other alpha-synucleinopathies. Clin Chem Lab Med. 2001;39:308–312. doi: 10.1515/CCLM.2001.047. [DOI] [PubMed] [Google Scholar]
- 77.Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson’s disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- 78.Forno LS. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 79.Lewy FH. Paralysis Agitans. Pathologische Anatomie. In: Lewandowski M, editor. Handbuch der Neurologie. Berlin: Springer; 1912. pp. 920–933. [Google Scholar]
- 80.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 81.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 82.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 83.Singleton A, Gwinn-Hardy K, Sharabi Y, Li ST, Holmes C, Dendi R, et al. Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain. 2004;127:768–772. doi: 10.1093/brain/awh081. [DOI] [PubMed] [Google Scholar]
- 84.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 85.Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 86.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 87.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Galpern WR, Lang AE. Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol. 2006;59:449–458. doi: 10.1002/ana.20819. [DOI] [PubMed] [Google Scholar]
- 89.Helmer C, Joly P, Letenneur L, Commenges D, Dartigues JF. Mortality with dementia: results from a French prospective community-based cohort. Am J Epidemiol. 2001;154:642–648. doi: 10.1093/aje/154.7.642. [DOI] [PubMed] [Google Scholar]
- 90.Aronson MK, Ooi WL, Geva DL, Masur D, Blau A, Frishman W. Dementia. Age-dependent incidence, prevalence, and mortality in the old old. Arch Intern Med. 1991;151:989–992. doi: 10.1001/archinte.151.5.989. [DOI] [PubMed] [Google Scholar]
- 91.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 92.Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- 93.Clark CM, Ewbank D, Lee VM-Y, Trojanowski JQ. Molecular pathology of Alzheimer’s disease: neuronal cytoskeletal abnormalities. In: Growdon JH, Rossor MN, editors. The dementias. Vol. 19 of Blue books of practical neurology. Boston: Butterworth–Heinemann; 1998. pp. 285–304. [Google Scholar]
- 94.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Braak H, Braak E, Grundke-Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986;65:351–355. doi: 10.1016/0304-3940(86)90288-0. [DOI] [PubMed] [Google Scholar]
- 96.Arai Y, Yamazaki M, Mori O, Muramatsu H, Asano G, Katayama Y. Alpha-synuclein-positive structures in cases with sporadic Alzheimer’s disease: morphology and its relationship to tau aggregation. Brain Res. 2001;888:287–296. doi: 10.1016/s0006-8993(00)03082-1. [DOI] [PubMed] [Google Scholar]
- 97.Lippa CF, Schmidt ML, Lee VM, Trojanowski JQ. Antibodies to alpha-synuclein detect Lewy bodies in many Down’s syndrome brains with Alzheimer’s disease. Ann Neurol. 1999;45:353–357. doi: 10.1002/1531-8249(199903)45:3<353::aid-ana11>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 98.Marui W, Iseki E, Ueda K, Kosaka K. Occurrence of human alpha-synuclein immunoreactive neurons with neurofibrillary tangle formation in the limbic areas of patients with Alzheimer’s disease. J Neurol Sci. 2000;174:81–84. doi: 10.1016/s0022-510x(99)00327-5. [DOI] [PubMed] [Google Scholar]
- 99.Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome) J Neurol Sci. 1989;94:79–100. doi: 10.1016/0022-510x(89)90219-0. [DOI] [PubMed] [Google Scholar]
- 100.Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- 101.Wakabayashi K, Takahashi H. Cellular pathology in multiple system atrophy. Neuropathology. 2006;26:338–345. doi: 10.1111/j.1440-1789.2006.00713.x. [DOI] [PubMed] [Google Scholar]
- 102.Taylor TD, Litt M, Kramer P, Pandolfo M, Angelini L, Nardocci N, et al. Homozygosity mapping of Hallervorden-Spatz syndrome to chromosome 20p12.3-p13. Nat Genet. 1996;14:479–481. doi: 10.1038/ng1296-479. [DOI] [PubMed] [Google Scholar]
- 103.Malandrini A, Cesaretti S, Mulinari M, Palmeri S, Fabrizi GM, Villanova M, et al. Acanthocytosis, retinitis pigmentosa, pallidal degeneration. Report of two cases without serum lipid abnormalities. J Neurol Sci. 1996;140:129–131. doi: 10.1016/0022-510x(96)00155-4. [DOI] [PubMed] [Google Scholar]
- 104.Sugiyama H, Hainfellner JA, Schmid-Siegel B, Budka H. Neuroaxonal dystrophy combined with diffuse Lewy body disease in a young adult. Clin Neuropathol. 1993;12:147–152. [PubMed] [Google Scholar]
- 105.Swaiman KF. Hallervorden-Spatz syndrome and brain iron metabolism. Arch Neurol. 1991;48:1285–1293. doi: 10.1001/archneur.1991.00530240091029. [DOI] [PubMed] [Google Scholar]
- 106.Dooling EC, Schoene WC, Richardson EP., Jr Hallervorden-Spatz syndrome. Arch Neurol. 1974;30:70–83. doi: 10.1001/archneur.1974.00490310072012. [DOI] [PubMed] [Google Scholar]
- 107.Jankovic J, Kirkpatrick JB, Blomquist KA, Langlais PJ, Bird ED. Late-onset Hallervorden-Spatz disease presenting as familial parkinsonism. Neurology. 1985;35:227–234. doi: 10.1212/wnl.35.2.227. [DOI] [PubMed] [Google Scholar]
- 108.Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 109.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 110.Bracken C, Iakoucheva LM, Romero PR, Dunker AK. Combining prediction, computation and experiment for the characterization of protein disorder. Curr Opin Struct Biol. 2004;14:570–576. doi: 10.1016/j.sbi.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 111.Romero P, Obradovic Z, Dunker AK. Natively disordered proteins : functions and predictions. Appl Bioinformatics. 2004;3:105–113. doi: 10.2165/00822942-200403020-00005. [DOI] [PubMed] [Google Scholar]
- 112.Obradovic Z, Peng K, Vucetic S, Radivojac P, Dunker AK. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins. 2005;61 (Suppl 7):176–182. doi: 10.1002/prot.20735. [DOI] [PubMed] [Google Scholar]
- 113.Yang ZR, Thomson R, McNeil P, Esnouf RM. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics. 2005;21:3369–3376. doi: 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
- 114.Uversky VN. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn. 2003;21:211–234. doi: 10.1080/07391102.2003.10506918. [DOI] [PubMed] [Google Scholar]
- 115.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 116.Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, et al. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem. 2002;277:11970–11978. doi: 10.1074/jbc.M109541200. [DOI] [PubMed] [Google Scholar]
- 117.Morar AS, Olteanu A, Young GB, Pielak GJ. Solvent-induced collapse of alpha-synuclein and acid-denatured cytochrome c. Protein Sci. 2001;10:2195–2199. doi: 10.1110/ps.24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Syme CD, Blanch EW, Holt C, Jakes R, Goedert M, Hecht L, et al. A Raman optical activity study of rheomorphism in caseins, synucleins and tau. New insight into the structure and behaviour of natively unfolded proteins. Eur J Biochem. 2002;269:148–156. doi: 10.1046/j.0014-2956.2001.02633.x. [DOI] [PubMed] [Google Scholar]
- 119.Uversky VN, Fink AL. Biophysical properties of human alpha-synuclein and its role in Parkinson’s disease. In: Pandalai SG, editor. Recent Research Developments in Proteins. Kerala, India: Transworld Research Network; 2002. pp. 153–186. [Google Scholar]
- 120.Li J, Uversky VN, Fink AL. Conformational behavior of human alpha-synuclein is modulated by familial Parkinson’s disease point mutations A30P and A53T. Neurotoxicology. 2002;23:553–567. doi: 10.1016/s0161-813x(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 121.Li J, Uversky VN, Fink AL. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry. 2001;40:11604–11613. doi: 10.1021/bi010616g. [DOI] [PubMed] [Google Scholar]
- 122.Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21:249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- 123.George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 124.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Segrest JP, De Loof H, Dohlman JG, Brouillette CG, Anantharamaiah GM. Amphipathic helix motif: classes and properties. Proteins. 1990;8:103–117. doi: 10.1002/prot.340080202. [DOI] [PubMed] [Google Scholar]
- 126.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 127.Bisaglia M, Schievano E, Caporale A, Peggion E, Mammi S. The 11-mer repeats of human alpha-synuclein in vesicle interactions and lipid composition discrimination: a cooperative role. Biopolymers. 2006;84:310–316. doi: 10.1002/bip.20440. [DOI] [PubMed] [Google Scholar]
- 128.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 129.Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc Natl Acad Sci U S A. 2004;101:8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Biere AL, Wood SJ, Wypych J, Steavenson S, Jiang Y, Anafi D, et al. Parkinson’s disease-associated alpha-synuclein is more fibrillogenic than beta- and gamma-synuclein and cannot cross-seed its homologs. J Biol Chem. 2000;275:34574–34579. doi: 10.1074/jbc.M005514200. [DOI] [PubMed] [Google Scholar]
- 131.Perrin RJ, Woods WS, Clayton DF, George JM. Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J Biol Chem. 2000;275:34393–34398. doi: 10.1074/jbc.M004851200. [DOI] [PubMed] [Google Scholar]
- 132.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 133.de Laureto PP, Tosatto L, Frare E, Marin O, Uversky VN, Fontana A. Conformational properties of the SDS-bound state of alpha-synuclein [robed by limited proteolysis: Unexpected rigidity of the acidic C-terminal tail. Biochemistry. 2006;45:11523–11531. doi: 10.1021/bi052614s. [DOI] [PubMed] [Google Scholar]
- 134.Yamin G, Glaser CB, Uversky VN, Fink AL. Certain metals trigger fibrillation of methionine-oxidized alpha-synuclein. J Biol Chem. 2003;278:27630–27635. doi: 10.1074/jbc.M303302200. [DOI] [PubMed] [Google Scholar]
- 135.Uversky VN, Yamin G, Souillac PO, Goers J, Glaser CB, Fink AL. Methionine oxidation inhibits fibrillation of human alpha-synuclein in vitro. FEBS Lett. 2002;517:239–244. doi: 10.1016/s0014-5793(02)02638-8. [DOI] [PubMed] [Google Scholar]
- 136.Glaser CB, Yamin G, Uversky VN, Fink AL. Methionine oxidation, alpha-synuclein and Parkinson’s disease. Biochim Biophys Acta. 2005;1703:157–169. doi: 10.1016/j.bbapap.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 137.Munishkina LA, Phelan C, Uversky VN, Fink AL. Conformational behavior and aggregation of alpha-synuclein in organic solvents: modeling the effects of membranes. Biochemistry. 2003;42:2720–2730. doi: 10.1021/bi027166s. [DOI] [PubMed] [Google Scholar]
- 138.Uversky VN, Li J, Fink AL. Trimethylamine-N-oxide-induced folding of alpha-synuclein. FEBS Lett. 2001;509:31–35. doi: 10.1016/s0014-5793(01)03121-0. [DOI] [PubMed] [Google Scholar]
- 139.Uversky VN, Li J, Fink AL. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem. 2001;276:44284–44296. doi: 10.1074/jbc.M105343200. [DOI] [PubMed] [Google Scholar]
- 140.Munishkina LA, Henriques J, Uversky VN, Fink AL. Role of protein-water interactions and electrostatics in alpha-synuclein fibril formation. Biochemistry. 2004;43:3289–3300. doi: 10.1021/bi034938r. [DOI] [PubMed] [Google Scholar]
- 141.Uversky VN, Li J, Fink AL. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson’s disease. FEBS Lett. 2001;500:105–108. doi: 10.1016/s0014-5793(01)02597-2. [DOI] [PubMed] [Google Scholar]
- 142.Uversky VN, Li J, Bower K, Fink AL. Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: implications for Parkinson’s disease. Neurotoxicology. 2002;23:527–536. doi: 10.1016/s0161-813x(02)00067-0. [DOI] [PubMed] [Google Scholar]
- 143.Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- 144.Cohlberg JA, Li J, Uversky VN, Fink AL. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry. 2002;41:1502–1511. doi: 10.1021/bi011711s. [DOI] [PubMed] [Google Scholar]
- 145.Goers J, Uversky VN, Fink AL. Polycation-induced oligomerization and accelerated fibrillation of human alpha-synuclein in vitro. Protein Sci. 2003;12:702–707. doi: 10.1110/ps.0230903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Uversky VN, Lee HJ, Li J, Fink AL, Lee SJ. Stabilization of partially folded conformation during alpha-synuclein oligomerization in both purified and cytosolic preparations. J Biol Chem. 2001;276:43495–43498. doi: 10.1074/jbc.C100551200. [DOI] [PubMed] [Google Scholar]
- 147.Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- 148.Yamin G, Uversky VN, Fink AL. Nitration inhibits fibrillation of human alpha-synuclein in vitro by formation of soluble oligomers. FEBS Lett. 2003;542:147–152. doi: 10.1016/s0014-5793(03)00367-3. [DOI] [PubMed] [Google Scholar]
- 149.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 150.Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 151.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ding TT, Lee SJ, Rochet JC, Lansbury PT., Jr Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry. 2002;41:10209–10217. doi: 10.1021/bi020139h. [DOI] [PubMed] [Google Scholar]
- 153.Lowe R, Pountney DL, Jensen PH, Gai WP, Voelcker NH. Calcium(II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci. 2004;13:3245–3252. doi: 10.1110/ps.04879704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 155.Nymeyer H, Socci ND, Onuchic JN. Landscape approaches for determining the ensemble of folding transition states: success and failure hinge on the degree of frustration. Proc Natl Acad Sci U S A. 2000;97:634–639. doi: 10.1073/pnas.97.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Uversky VN. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04764.x. In press. [DOI] [PubMed] [Google Scholar]
- 157.Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Creighton TE. Proteins. Structures and Molecular Properties. New York: W. H. Freeman and Company; 1993. [Google Scholar]
- 159.Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal Biochem. 1989;177:244–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 160.Naiki H, Higuchi K, Matsushima K, Shimada A, Chen WH, Hosokawa M, et al. Fluorometric examination of tissue amyloid fibrils in murine senile amyloidosis: use of the fluorescent indicator, thioflavine T. Lab Invest. 1990;62:768–773. [PubMed] [Google Scholar]
- 161.Levine H., 3rd Soluble multimeric Alzheimer beta(1–40) pre-amyloid complexes in dilute solution. Neurobiol Aging. 1995;16:755–764. doi: 10.1016/0197-4580(95)00052-g. [DOI] [PubMed] [Google Scholar]
- 162.Stsiapura VI, Maskevich AA, Kuzmitsky VA, Turoverov KK, Kuznetsova IM. Computational study of thioflavin T torsional relaxation in the excited state. J Phys Chem A. 2007;111:4829–4835. doi: 10.1021/jp070590o. [DOI] [PubMed] [Google Scholar]
- 163.Maskevich AA, Stsiapura VI, Kuzmitsky VA, Kuznetsova IM, Povarova OI, Uversky VN, et al. Spectral properties of thioflavin T in solvents with different dielectric properties and in a fibril-incorporated form. J Proteome Res. 2007;6:1392–1401. doi: 10.1021/pr0605567. [DOI] [PubMed] [Google Scholar]
- 164.Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J Biol Chem. 1999;274:19509–19512. doi: 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]
- 165.Jarrett JT, Lansbury PT., Jr Amyloid fibril formation requires a chemically discriminating nucleation event: studies of an amyloidogenic sequence from the bacterial protein OsmB. Biochemistry. 1992;31:12345–12352. doi: 10.1021/bi00164a008. [DOI] [PubMed] [Google Scholar]
- 166.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 167.Lomakin A, Teplow DB, Kirschner DA, Benedek GB. Kinetic theory of fibrillogenesis of amyloid beta-protein. Proc Natl Acad Sci U S A. 1997;94:7942–7947. doi: 10.1073/pnas.94.15.7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J Biol Chem. 1999;274:9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 169.Uversky VN, E MC, Bower KS, Li J, Fink AL. Accelerated alpha-synuclein fibrillation in crowded milieu. FEBS Lett. 2002;515:99–103. doi: 10.1016/s0014-5793(02)02446-8. [DOI] [PubMed] [Google Scholar]
- 170.Shtilerman MD, Ding TT, Lansbury PT., Jr Molecular crowding accelerates fibrillization of alpha-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson’s disease? Biochemistry. 2002;41:3855–3860. doi: 10.1021/bi0120906. [DOI] [PubMed] [Google Scholar]
- 171.Clayton DF, George JM. Synucleins in synaptic plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:120–129. [PubMed] [Google Scholar]
- 172.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 174.Nakajo S, Tsukada K, Omata K, Nakamura Y, Nakaya K. A new brain-specific 14-kDa protein is a phosphoprotein. Its complete amino acid sequence and evidence for phosphorylation. Eur J Biochem. 1993;217:1057–1063. doi: 10.1111/j.1432-1033.1993.tb18337.x. [DOI] [PubMed] [Google Scholar]
- 175.Tobe T, Nakajo S, Tanaka A, Mitoya A, Omata K, Nakaya K, et al. Cloning and characterization of the cDNA encoding a novel brain-specific 14-kDa protein. J Neurochem. 1992;59:1624–1629. doi: 10.1111/j.1471-4159.1992.tb10991.x. [DOI] [PubMed] [Google Scholar]
- 176.Ji H, Liu YE, Jia T, Wang M, Liu J, Xiao G, et al. Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing. Cancer Res. 1997;57:759–764. [PubMed] [Google Scholar]
- 177.Ninkina NN, Alimova-Kost MV, Paterson JW, Delaney L, Cohen BB, Imreh S, et al. Organization, expression and polymorphism of the human persyn gene. Hum Mol Genet. 1998;7:1417–1424. doi: 10.1093/hmg/7.9.1417. [DOI] [PubMed] [Google Scholar]
- 178.Buchman VL, Hunter HJ, Pinon LG, Thompson J, Privalova EM, Ninkina NN, et al. Persyn, a member of the synuclein family, has a distinct pattern of expression in the developing nervous system. J Neurosci. 1998;18:9335–9341. doi: 10.1523/JNEUROSCI.18-22-09335.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lavedan C, Leroy E, Dehejia A, Buchholtz S, Dutra A, Nussbaum RL, et al. Identification, localization and characterization of the human gamma-synuclein gene. Hum Genet. 1998;103:106–112. doi: 10.1007/s004390050792. [DOI] [PubMed] [Google Scholar]
- 180.Lucking CB, Brice A. Alpha-synuclein and Parkinson’s disease. Cell Mol Life Sci. 2000;57:1894–1908. doi: 10.1007/PL00000671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shibayama-Imazu T, Okahashi I, Omata K, Nakajo S, Ochiai H, Nakai Y, et al. Cell and tissue distribution and developmental change of neuron specific 14 kDa protein (phosphoneuroprotein 14) Brain Res. 1993;622:17–25. doi: 10.1016/0006-8993(93)90796-p. [DOI] [PubMed] [Google Scholar]
- 182.Nakajo S, Shioda S, Nakai Y, Nakaya K. Localization of phosphoneuroprotein 14 (PNP 14) and its mRNA expression in rat brain determined by immunocytochemistry and in situ hybridization. Brain Res Mol Brain Res. 1994;27:81–86. doi: 10.1016/0169-328x(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 183.Nakajo S, Tsukada K, Kameyama H, Furuyama Y, Nakaya K. Distribution of phosphoneuroprotein 14 (PNP 14) in vertebrates: its levels as determined by enzyme immunoassay. Brain Res. 1996;741:180–184. doi: 10.1016/s0006-8993(96)00914-6. [DOI] [PubMed] [Google Scholar]
- 184.Shibayama-Imazu T, Ogane K, Hasegawa Y, Nakajo S, Shioda S, Ochiai H, et al. Distribution of PNP 14 (beta-synuclein) in neuroendocrine tissues: localization in Sertoli cells. Mol Reprod Dev. 1998;50:163–169. doi: 10.1002/(SICI)1098-2795(199806)50:2<163::AID-MRD6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 185.Hashimoto M, Yoshimoto M, Sisk A, Hsu LJ, Sundsmo M, Kittel A, et al. NACP, a synaptic protein involved in Alzheimer’s disease, is differentially regulated during megakaryocyte differentiation. Biochem Biophys Res Commun. 1997;237:611–616. doi: 10.1006/bbrc.1997.6978. [DOI] [PubMed] [Google Scholar]
- 186.Ninkina NN, Privalova EM, Pinon LG, Davies AM, Buchman VL. Developmentally regulated expression of persyn, a member of the synuclein family, in skin. Exp Cell Res. 1999;246:308–311. doi: 10.1006/excr.1998.4292. [DOI] [PubMed] [Google Scholar]
- 187.Galvin JE, Uryu K, Lee VM, Trojanowski JQ. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci U S A. 1999;96:13450–13455. doi: 10.1073/pnas.96.23.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yamin G, Munishkina LA, Karymov MA, Lyubchenko YL, Uversky VN, Fink AL. Forcing the non-amyloidogenic beta-synuclein to fibrillate. J Mol Biol. 2005 doi: 10.1021/bi048778a. In press. [DOI] [PubMed] [Google Scholar]
- 189.Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001;32:213–223. doi: 10.1016/s0896-6273(01)00462-7. [DOI] [PubMed] [Google Scholar]
- 190.Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci U S A. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jaikaran ET, Clark A. Islet amyloid and type 2 diabetes: from molecular misfolding to islet pathophysiology. Biochim Biophys Acta. 2001;1537:179–203. doi: 10.1016/s0925-4439(01)00078-3. [DOI] [PubMed] [Google Scholar]
- 192.Jaikaran ET, Higham CE, Serpell LC, Zurdo J, Gross M, Clark A, et al. Identification of a novel human islet amyloid polypeptide beta-sheet domain and factors influencing fibrillogenesis. J Mol Biol. 2001;308:515–525. doi: 10.1006/jmbi.2001.4593. [DOI] [PubMed] [Google Scholar]
- 193.Higham CE, Jaikaran ET, Fraser PE, Gross M, Clark A. Preparation of synthetic human islet amyloid polypeptide (IAPP) in a stable conformation to enable study of conversion to amyloid-like fibrils. FEBS Lett. 2000;470:55–60. doi: 10.1016/s0014-5793(00)01287-4. [DOI] [PubMed] [Google Scholar]
- 194.Rocken C, Linke RP, Saeger W. Immunohistology of islet amyloid polypeptide in diabetes mellitus: semi-quantitative studies in a post-mortem series. Virchows Arch A Pathol Anat Histopathol. 1992;421:339–344. doi: 10.1007/BF01660981. [DOI] [PubMed] [Google Scholar]
- 195.Schneider HM, Storkel S, Will W. [Amyloid of islets of Langerhans and its relation to diabetes mellitus (author’s transl)] Dtsch Med Wochenschr. 1980;105:1143–1147. doi: 10.1055/s-2008-1070828. [DOI] [PubMed] [Google Scholar]
- 196.Kahn SE, Andrikopoulos S, Verchere CB. Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes. 1999;48:241–253. doi: 10.2337/diabetes.48.2.241. [DOI] [PubMed] [Google Scholar]
- 197.Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9:151–159. [PubMed] [Google Scholar]
- 198.Westermark P, Wernstedt C, Wilander E, Hayden DW, O’Brien TD, Johnson KH. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci U S A. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yoon S, Welsh WJ. Rapid assessment of contact-dependent secondary structure propensity: relevance to amyloidogenic sequences. Proteins. 2005;60:110–117. doi: 10.1002/prot.20477. [DOI] [PubMed] [Google Scholar]
- 200.Goldsbury C, Goldie K, Pellaud J, Seelig J, Frey P, Muller SA, et al. Amyloid fibril formation from full-length and fragments of amylin. J Struct Biol. 2000;130:352–362. doi: 10.1006/jsbi.2000.4268. [DOI] [PubMed] [Google Scholar]
- 201.Jayasinghe SA, Langen R. Identifying structural features of fibrillar islet amyloid polypeptide using site-directed spin labeling. J Biol Chem. 2004;279:48420–48425. doi: 10.1074/jbc.M406853200. [DOI] [PubMed] [Google Scholar]
- 202.Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, et al. Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol Pharmacol. 1994;45:373–379. [PubMed] [Google Scholar]
- 203.Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 204.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 205.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, et al. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 207.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Jr, Younkin LH, et al. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43) J Biol Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 208.Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 209.Barrow CJ, Zagorski MG. Solution structures of beta peptide and its constituent fragments: relation to amyloid deposition. Science. 1991;253:179–182. doi: 10.1126/science.1853202. [DOI] [PubMed] [Google Scholar]
- 210.Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 211.Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J Mol Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 212.Himmler A. Structure of the bovine tau gene: alternatively spliced transcripts generate a protein family. Mol Cell Biol. 1989;9:1389–1396. doi: 10.1128/mcb.9.4.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Himmler A, Drechsel D, Kirschner MW, Martin DW., Jr Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9:1381–1388. doi: 10.1128/mcb.9.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kenessey A, Yen SH. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. 1993;629:40–46. doi: 10.1016/0006-8993(93)90478-6. [DOI] [PubMed] [Google Scholar]
- 215.Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977;116:207–225. doi: 10.1016/0022-2836(77)90213-3. [DOI] [PubMed] [Google Scholar]
- 216.Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977;116:227–247. doi: 10.1016/0022-2836(77)90214-5. [DOI] [PubMed] [Google Scholar]
- 217.Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Brandt R, Lee G. The balance between tau protein’s microtubule growth and nucleation activities: implications for the formation of axonal microtubules. J Neurochem. 1993;61:997–1005. doi: 10.1111/j.1471-4159.1993.tb03613.x. [DOI] [PubMed] [Google Scholar]
- 219.Brandt R, Lee G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. J Biol Chem. 1993;268:3414–3419. [PubMed] [Google Scholar]
- 220.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Drubin DG, Feinstein SC, Shooter EM, Kirschner MW. Nerve growth factor-induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J Cell Biol. 1985;101:1799–1807. doi: 10.1083/jcb.101.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Khatoon S, Grundke-Iqbal I, Iqbal K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem. 1992;59:750–753. doi: 10.1111/j.1471-4159.1992.tb09432.x. [DOI] [PubMed] [Google Scholar]
- 223.Crowther RA, Goedert M. Abnormal tau-containing filaments in neurodegenerative diseases. J Struct Biol. 2000;130:271–279. doi: 10.1006/jsbi.2000.4270. [DOI] [PubMed] [Google Scholar]
- 224.Delacourte A, Buee L. Normal and pathological Tau proteins as factors for microtubule assembly. Int Rev Cytol. 1997;171:167–224. doi: 10.1016/s0074-7696(08)62588-7. [DOI] [PubMed] [Google Scholar]
- 225.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 226.Ruben GC, Iqbal K, Grundke-Iqbal I, Wisniewski HM, Ciardelli TL, Johnson JE., Jr The microtubule-associated protein tau forms a triple-stranded left-hand helical polymer. J Biol Chem. 1991;266:22019–22027. [PubMed] [Google Scholar]
- 227.Vulliet R, Halloran SM, Braun RK, Smith AJ, Lee G. Proline-directed phosphorylation of human Tau protein. J Biol Chem. 1992;267:22570–22574. [PubMed] [Google Scholar]
- 228.Lu Q, Wood JG. Functional studies of Alzheimer’s disease tau protein. J Neurosci. 1993;13:508–515. doi: 10.1523/JNEUROSCI.13-02-00508.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 230.Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Iqbal K, Zaidi T, Bancher C, Grundke-Iqbal I. Alzheimer paired helical filaments. Restoration of the biological activity by dephosphorylation. FEBS Lett. 1994;349:104–108. doi: 10.1016/0014-5793(94)00650-4. [DOI] [PubMed] [Google Scholar]
- 232.Watanabe A, Hasegawa M, Suzuki M, Takio K, Morishima-Kawashima M, Titani K, et al. In vivo phosphorylation sites in fetal and adult rat tau. J Biol Chem. 1993;268:25712–25717. [PubMed] [Google Scholar]
- 233.Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323 ( Pt 3):577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Watanabe A, et al. Hyperphosphorylation of tau in PHF. Neurobiol Aging. 1995;16:365–371. doi: 10.1016/0197-4580(95)00027-c. discussion 371–380. [DOI] [PubMed] [Google Scholar]
- 235.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, et al. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. 1995;270:823–829. doi: 10.1074/jbc.270.2.823. [DOI] [PubMed] [Google Scholar]
- 236.Uversky VN, Winter S, Galzitskaya OV, Kittler L, Lober G. Hyperphosphorylation induces structural modification of tau-protein. FEBS Lett. 1998;439:21–25. doi: 10.1016/s0014-5793(98)01303-9. [DOI] [PubMed] [Google Scholar]
- 237.Hagestedt T, Lichtenberg B, Wille H, Mandelkow EM, Mandelkow E. Tau protein becomes long and stiff upon phosphorylation: correlation between paracrystalline structure and degree of phosphorylation. J Cell Biol. 1989;109:1643–1651. doi: 10.1083/jcb.109.4.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A. 2001;98:6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Eidenmuller J, Fath T, Hellwig A, Reed J, Sontag E, Brandt R. Structural and functional implications of tau hyperphosphorylation: information from phosphorylation-mimicking mutated tau proteins. Biochemistry. 2000;39:13166–13175. doi: 10.1021/bi001290z. [DOI] [PubMed] [Google Scholar]
- 240.von Bergen M, Barghorn S, Jeganathan S, Mandelkow EM, Mandelkow E. Spectroscopic approaches to the conformation of tau protein in solution and in paired helical filaments. Neurodegener Dis. 2006;3:197–206. doi: 10.1159/000095257. [DOI] [PubMed] [Google Scholar]
- 241.Chirita CN, Necula M, Kuret J. Anionic micelles and vesicles induce tau fibrillization in vitro. J Biol Chem. 2003;278:25644–25650. doi: 10.1074/jbc.M301663200. [DOI] [PubMed] [Google Scholar]
- 242.Chirita CN, Congdon EE, Yin H, Kuret J. Triggers of full-length tau aggregation: a role for partially folded intermediates. Biochemistry. 2005;44:5862–5872. doi: 10.1021/bi0500123. [DOI] [PubMed] [Google Scholar]
- 243.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 244.Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al. Defective brain microtubule assembly in Alzheimer’s disease. Lancet. 1986;2:421–426. doi: 10.1016/s0140-6736(86)92134-3. [DOI] [PubMed] [Google Scholar]
- 245.Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res. 1996;38:200–208. doi: 10.1016/0169-328x(95)00316-k. [DOI] [PubMed] [Google Scholar]
- 246.Plant GT, Revesz T, Barnard RO, Harding AE, Gautier-Smith PC. Familial cerebral amyloid angiopathy with nonneuritic amyloid plaque formation. Brain. 1990;113 ( Pt 3):721–747. doi: 10.1093/brain/113.3.721. [DOI] [PubMed] [Google Scholar]
- 247.Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, et al. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999;399:776–781. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- 248.Ghiso JA, Holton J, Miravalle L, Calero M, Lashley T, Vidal R, et al. Systemic amyloid deposits in familial British dementia. J Biol Chem. 2001;276:43909–43914. doi: 10.1074/jbc.M105956200. [DOI] [PubMed] [Google Scholar]
- 249.Kim SH, Wang R, Gordon DJ, Bass J, Steiner DF, Lynn DG, et al. Furin mediates enhanced production of fibrillogenic ABri peptides in familial British dementia. Nat Neurosci. 1999;2:984–988. doi: 10.1038/14783. [DOI] [PubMed] [Google Scholar]
- 250.Srinivasan R, Jones EM, Liu K, Ghiso J, Marchant RE, Zagorski MG. pH-dependent amyloid and protofibril formation by the ABri peptide of familial British dementia. J Mol Biol. 2003;333:1003–1023. doi: 10.1016/j.jmb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 251.Vidal R, Revesz T, Rostagno A, Kim E, Holton JL, Bek T, et al. A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc Natl Acad Sci U S A. 2000;97:4920–4925. doi: 10.1073/pnas.080076097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Rostagno A, Tomidokoro Y, Lashley T, Ng D, Plant G, Holton J, et al. Chromosome 13 dementias. Cell Mol Life Sci. 2005;62:1814–1825. doi: 10.1007/s00018-005-5092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Surolia I, Reddy GB, Sinha S. Hierarchy and the mechanism of fibril formation in ADan peptides. J Neurochem. 2006;99:537–548. doi: 10.1111/j.1471-4159.2006.04072.x. [DOI] [PubMed] [Google Scholar]
- 254.Aronoff-Spencer E, Burns CS, Avdievich NI, Gerfen GJ, Peisach J, Antholine WE, et al. Identification of the Cu2+ binding sites in the N-terminal domain of the prion protein by EPR and CD spectroscopy. Biochemistry. 2000;39:13760–13771. doi: 10.1021/bi001472t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Riek R, Hornemann S, Wider G, Glockshuber R, Wuthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231) FEBS Lett. 1997;413:282–288. doi: 10.1016/s0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 256.Burns CS, Aronoff-Spencer E, Dunham CM, Lario P, Avdievich NI, Antholine WE, et al. Molecular features of the copper binding sites in the octarepeat domain of the prion protein. Biochemistry. 2002;41:3991–4001. doi: 10.1021/bi011922x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Wildegger G, Liemann S, Glockshuber R. Extremely rapid folding of the C-terminal domain of the prion protein without kinetic intermediates. Nat Struct Biol. 1999;6:550–553. doi: 10.1038/9323. [DOI] [PubMed] [Google Scholar]
- 258.Hosszu LL, Baxter NJ, Jackson GS, Power A, Clarke AR, Waltho JP, et al. Structural mobility of the human prion protein probed by backbone hydrogen exchange. Nat Struct Biol. 1999;6:740–743. doi: 10.1038/11507. [DOI] [PubMed] [Google Scholar]
- 259.Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, Bastidas RB, et al. A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol. 1997;273:614–622. doi: 10.1006/jmbi.1997.1328. [DOI] [PubMed] [Google Scholar]
- 260.Vanik DL, Surewicz KA, Surewicz WK. Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol Cell. 2004;14:139–145. doi: 10.1016/s1097-2765(04)00155-8. [DOI] [PubMed] [Google Scholar]
- 261.Kundu B, Maiti NR, Jones EM, Surewicz KA, Vanik DL, Surewicz WK. Nucleation-dependent conformational conversion of the Y145Stop variant of human prion protein: structural clues for prion propagation. Proc Natl Acad Sci U S A. 2003;100:12069–12074. doi: 10.1073/pnas.2033281100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Scott M, Groth D, Foster D, Torchia M, Yang SL, DeArmond SJ, et al. Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell. 1993;73:979–988. doi: 10.1016/0092-8674(93)90275-u. [DOI] [PubMed] [Google Scholar]
- 263.Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 264.Donne DG, Viles JH, Groth D, Mehlhorn I, James TL, Cohen FE, et al. Structure of the recombinant full-length hamster prion protein PrP(29–231): the N terminus is highly flexible. Proc Natl Acad Sci U S A. 1997;94:13452–13457. doi: 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.James TL, Liu H, Ulyanov NB, Farr-Jones S, Zhang H, Donne DG, et al. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci U S A. 1997;94:10086–10091. doi: 10.1073/pnas.94.19.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998;93:337–348. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- 267.Apetri AC, Surewicz WK. Kinetic intermediate in the folding of human prion protein. J Biol Chem. 2002;277:44589–44592. doi: 10.1074/jbc.C200507200. [DOI] [PubMed] [Google Scholar]
- 268.Martins SM, Chapeaurouge A, Ferreira ST. Folding intermediates of the prion protein stabilized by hydrostatic pressure and low temperature. J Biol Chem. 2003;278:50449–50455. doi: 10.1074/jbc.M307354200. [DOI] [PubMed] [Google Scholar]
- 269.Watzlawik J, Skora L, Frense D, Griesinger C, Zweckstetter M, Schulz-Schaeffer WJ, et al. Prion protein helix1 promotes aggregation but is not converted into beta-sheet. J Biol Chem. 2006;281:30242–30250. doi: 10.1074/jbc.m605141200. [DOI] [PubMed] [Google Scholar]
- 270.Cummings CJ, Zoghbi HY. Trinucleotide repeats: mechanisms and pathophysiology. Annu Rev Genomics Hum Genet. 2000;1:281–328. doi: 10.1146/annurev.genom.1.1.281. [DOI] [PubMed] [Google Scholar]
- 271.Cummings CJ, Zoghbi HY. Fourteen and counting: unraveling trinucleotide repeat diseases. Hum Mol Genet. 2000;9:909–916. doi: 10.1093/hmg/9.6.909. [DOI] [PubMed] [Google Scholar]
- 272.Perutz MF. Glutamine repeats and inherited neurodegenerative diseases: molecular aspects. Curr Opin Struct Biol. 1996;6:848–858. doi: 10.1016/s0959-440x(96)80016-9. [DOI] [PubMed] [Google Scholar]
- 273.Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron. 2002;35:819–822. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 274.Bates G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet. 2003;361:1642–1644. doi: 10.1016/S0140-6736(03)13304-1. [DOI] [PubMed] [Google Scholar]
- 275.Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 276.Yue S, Serra HG, Zoghbi HY, Orr HT. The spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract. Hum Mol Genet. 2001;10:25–30. doi: 10.1093/hmg/10.1.25. [DOI] [PubMed] [Google Scholar]
- 277.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 278.Gusella J, MacDonald M. No post-genetics era in human disease research. Nat Rev Genet. 2002;3:72–79. doi: 10.1038/nrg706. [DOI] [PubMed] [Google Scholar]
- 279.McEwan IJ. Structural and functional alterations in the androgen receptor in spinal bulbar muscular atrophy. Biochem Soc Trans. 2001;29:222–227. doi: 10.1042/0300-5127:0290222. [DOI] [PubMed] [Google Scholar]
- 280.Nagafuchi S, Yanagisawa H, Ohsaki E, Shirayama T, Tadokoro K, Inoue T, et al. Structure and expression of the gene responsible for the triplet repeat disorder, dentatorubral and pallidoluysian atrophy (DRPLA) Nat Genet. 1994;8:177–182. doi: 10.1038/ng1094-177. [DOI] [PubMed] [Google Scholar]
- 281.Zoghbi HY, Orr HT. Polyglutamine diseases: protein cleavage and aggregation. Curr Opin Neurobiol. 1999;9:566–570. doi: 10.1016/S0959-4388(99)00013-6. [DOI] [PubMed] [Google Scholar]
- 282.Ross CA, Wood JD, Schilling G, Peters MF, Nucifora FC, Jr, Cooper JK, et al. Polyglutamine pathogenesis. Philos Trans R Soc Lond B Biol Sci. 1999;354:1005–1011. doi: 10.1098/rstb.1999.0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Preisinger E, Jordan BM, Kazantsev A, Housman D. Evidence for a recruitment and sequestration mechanism in Huntington’s disease. Philos Trans R Soc Lond B Biol Sci. 1999;354:1029–1034. doi: 10.1098/rstb.1999.0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wanker EE. Protein aggregation and pathogenesis of Huntington’s disease: mechanisms and correlations. Biol Chem. 2000;381:937–942. doi: 10.1515/BC.2000.114. [DOI] [PubMed] [Google Scholar]
- 285.McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, et al. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- 286.McCampbell A, Fischbeck KH. Polyglutamine and CBP: fatal attraction? Nat Med. 2001;7:528–530. doi: 10.1038/87842. [DOI] [PubMed] [Google Scholar]
- 287.Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol. 2001;311:173–182. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 288.Chen S, Berthelier V, Hamilton JB, O’Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 289.Perutz MF, Pope BJ, Owen D, Wanker EE, Scherzinger E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid beta-peptide of amyloid plaques. Proc Natl Acad Sci U S A. 2002;99:5596–5600. doi: 10.1073/pnas.042681599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Chow MK, Paulson HL, Bottomley SP. Destabilization of a non-pathological variant of ataxin-3 results in fibrillogenesis via a partially folded intermediate: a model for misfolding in polyglutamine disease. J Mol Biol. 2004;335:333–341. doi: 10.1016/j.jmb.2003.08.064. [DOI] [PubMed] [Google Scholar]
- 291.Poirier MA, Li H, Macosko J, Cai S, Amzel M, Ross CA. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem. 2002;277:41032–41037. doi: 10.1074/jbc.M205809200. [DOI] [PubMed] [Google Scholar]
- 292.Wahle E. A novel poly(A)-binding protein acts as a specificity factor in the second phase of messenger RNA polyadenylation. Cell. 1991;66:759–768. doi: 10.1016/0092-8674(91)90119-j. [DOI] [PubMed] [Google Scholar]
- 293.Wahle E, Ruegsegger U. 3′-End processing of pre-mRNA in eukaryotes. FEMS Microbiol Rev. 1999;23:277–295. doi: 10.1111/j.1574-6976.1999.tb00400.x. [DOI] [PubMed] [Google Scholar]
- 294.Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- 295.Tome FM, Chateau D, Helbling-Leclerc A, Fardeau M. Morphological changes in muscle fibers in oculopharyngeal muscular dystrophy. Neuromuscul Disord. 1997;7 (Suppl 1):S63–69. doi: 10.1016/s0960-8966(97)00085-0. [DOI] [PubMed] [Google Scholar]
- 296.Calado A, Tome FM, Brais B, Rouleau GA, Kuhn U, Wahle E, et al. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum Mol Genet. 2000;9:2321–2328. doi: 10.1093/oxfordjournals.hmg.a018924. [DOI] [PubMed] [Google Scholar]
- 297.Scheuermann T, Schulz B, Blume A, Wahle E, Rudolph R, Schwarz E. Trinucleotide expansions leading to an extended poly-L-alanine segment in the poly (A) binding protein PABPN1 cause fibril formation. Protein Sci. 2003;12:2685–2692. doi: 10.1110/ps.03214703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Clark JR, Fridman TR, Odell MJ, Brierley J, Walfish PG, Freeman JL. Prognostic variables and calcitonin in medullary thyroid cancer. Laryngoscope. 2005;115:1445–1450. doi: 10.1097/01.mlg.0000168114.90852.a6. [DOI] [PubMed] [Google Scholar]
- 299.Ogilvie JB, Kebebew E. Indication and timing of thyroid surgery for patients with hereditary medullary thyroid cancer syndromes. J Natl Compr Canc Netw. 2006;4:139–147. doi: 10.6004/jnccn.2006.0014. [DOI] [PubMed] [Google Scholar]
- 300.Fialkowski EA, Moley JF. Current approaches to medullary thyroid carcinoma, sporadic and familial. J Surg Oncol. 2006;94:737–747. doi: 10.1002/jso.20690. [DOI] [PubMed] [Google Scholar]
- 301.Sletten K, Westermark P, Natvig JB. Characterization of amyloid fibril proteins from medullary carcinoma of the thyroid. J Exp Med. 1976;143:993–998. doi: 10.1084/jem.143.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Silver MM, Hearn SA, Lines LD, Troster M. Calcitonin and chromogranin A localization in medullary carcinoma of the thyroid by immunoelectron microscopy. J Histochem Cytochem. 1988;36:1031–1036. doi: 10.1177/36.8.3392392. [DOI] [PubMed] [Google Scholar]
- 303.Silverman SL. Calcitonin. Am J Med Sci. 1997;313:13–16. doi: 10.1097/00000441-199701000-00003. [DOI] [PubMed] [Google Scholar]
- 304.Schneider D, Hofmann MT, Peterson JA. Diagnosis and treatment of Paget’s disease of bone. Am Fam Physician. 2002;65:2069–2072. [PubMed] [Google Scholar]
- 305.Mehta NM, Malootian A, Gilligan JP. Calcitonin for osteoporosis and bone pain. Curr Pharm Des. 2003;9:2659–2676. doi: 10.2174/1381612033453622. [DOI] [PubMed] [Google Scholar]
- 306.Arvinte T, Cudd A, Drake AF. The structure and mechanism of formation of human calcitonin fibrils. J Biol Chem. 1993;268:6415–6422. [PubMed] [Google Scholar]
- 307.Siligardi G, Samori B, Melandri S, Visconti M, Drake AF. Correlations between biological activities and conformational properties for human, salmon, eel, porcine calcitonins and Elcatonin elucidated by CD spectroscopy. Eur J Biochem. 1994;221:1117–1125. doi: 10.1111/j.1432-1033.1994.tb18832.x. [DOI] [PubMed] [Google Scholar]
- 308.Gaudiano MC, Colone M, Bombelli C, Chistolini P, Valvo L, Diociaiuti M. Early stages of salmon calcitonin aggregation: effect induced by ageing and oxidation processes in water and in the presence of model membranes. Biochim Biophys Acta. 2005;1750:134–145. doi: 10.1016/j.bbapap.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 309.Doi M, Kobayashi Y, Kyogoku Y, Takimoto M, Goda K. In: River JE, Marshall GR, editors. Structure study of human calcitonin; 11th Proc Am Pept Symp; 1990; July 9–14, 1989; La Jolla, California. 1990. [Google Scholar]
- 310.Motta A, Temussi PA, Wunsch E, Bovermann G. A 1H NMR study of human calcitonin in solution. Biochemistry. 1991;30:2364–2371. doi: 10.1021/bi00223a010. [DOI] [PubMed] [Google Scholar]
- 311.Epand RM, Epand RF, Orlowski RC, Schlueter RJ, Boni LT, Hui SW. Amphipathic helix and its relationship to the interaction of calcitonin with phospholipids. Biochemistry. 1983;22:5074–5084. doi: 10.1021/bi00291a005. [DOI] [PubMed] [Google Scholar]
- 312.Wüthrich K. NMR in biological research: Peptides and proteins. Amsterdam, Holland: North-Holland Publishing Company; 1976. [Google Scholar]
- 313.Jeon YH, Kanaori K, Takashima H, Kosiba T, Nosaka YA. Comparative studies on human calcitonin fibrillation in aqueous and trifluoroethanol solution by proton NMR spectroscopy. Proc ICMRBS XVIII; 1998; Tokyo. 1998. p. 61. [Google Scholar]
- 314.Kamihira M, Naito A, Tuzi S, Nosaka AY, Saito H. Conformational transitions and fibrillation mechanism of human calcitonin as studied by high-resolution solid-state 13C NMR. Protein Sci. 2000;9:867–877. doi: 10.1110/ps.9.5.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kamihira M, Oshiro Y, Tuzi S, Nosaka AY, Saito H, Naito A. Effect of electrostatic interaction on fibril formation of human calcitonin as studied by high resolution solid state 13C NMR. J Biol Chem. 2003;278:2859–2865. doi: 10.1074/jbc.M205285200. [DOI] [PubMed] [Google Scholar]
- 316.Kiuru S. Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid. 1998;5:55–66. doi: 10.3109/13506129809007291. [DOI] [PubMed] [Google Scholar]
- 317.Kiuru-Enari S, Somer H, Seppalainen AM, Notkola IL, Haltia M. Neuromuscular pathology in hereditary gelsolin amyloidosis. J Neuropathol Exp Neurol. 2002;61:565–571. doi: 10.1093/jnen/61.6.565. [DOI] [PubMed] [Google Scholar]
- 318.Kiuru-Enari S, Keski-Oja J, Haltia M. Cutis laxa in hereditary gelsolin amyloidosis. Br J Dermatol. 2005;152:250–257. doi: 10.1111/j.1365-2133.2004.06276.x. [DOI] [PubMed] [Google Scholar]
- 319.Chen CD, Huff ME, Matteson J, Page L, Phillips R, Kelly JW, et al. Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca(2+) stabilization. Embo J. 2001;20:6277–6287. doi: 10.1093/emboj/20.22.6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Kangas H, Seidah NG, Paunio T. Role of proprotein convertases in the pathogenic processing of the amyloidosis-associated form of secretory gelsolin. Amyloid. 2002;9:83–87. [PubMed] [Google Scholar]
- 321.de la Chapelle A, Tolvanen R, Boysen G, Santavy J, Bleeker-Wagemakers L, Maury CP, et al. Gelsolin-derived familial amyloidosis caused by asparagine or tyrosine substitution for aspartic acid at residue 187. Nat Genet. 1992;2:157–160. doi: 10.1038/ng1092-157. [DOI] [PubMed] [Google Scholar]
- 322.Maury CP. Gelsolin-related amyloidosis. Identification of the amyloid protein in Finnish hereditary amyloidosis as a fragment of variant gelsolin. J Clin Invest. 1991;87:1195–1199. doi: 10.1172/JCI115118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Suk JY, Zhang F, Balch WE, Linhardt RJ, Kelly JW. Heparin accelerates gelsolin amyloidogenesis. Biochemistry. 2006;45:2234–2242. doi: 10.1021/bi0519295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Lindquist S. Mad cows meet psi-chotic yeast: the expansion of the prion hypothesis. Cell. 1997;89:495–498. doi: 10.1016/s0092-8674(00)80231-7. [DOI] [PubMed] [Google Scholar]
- 325.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 326.Paushkin SV, Kushnirov VV, Smirnov VN, Ter-Avanesyan MD. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. Embo J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 327.DePace AH, Santoso A, Hillner P, Weissman JS. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell. 1998;93:1241–1252. doi: 10.1016/s0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 328.Serio TR, Lindquist SL. [PSI+]: an epigenetic modulator of translation termination efficiency. Annu Rev Cell Dev Biol. 1999;15:661–703. doi: 10.1146/annurev.cellbio.15.1.661. [DOI] [PubMed] [Google Scholar]
- 329.Serio TR, Cashikar AG, Moslehi JJ, Kowal AS, Lindquist SL. Yeast prion [psi +] and its determinant, Sup35p. Methods Enzymol. 1999;309:649–673. doi: 10.1016/s0076-6879(99)09043-6. [DOI] [PubMed] [Google Scholar]
- 330.Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 331.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 332.Scheibel T, Lindquist SL. The role of conformational flexibility in prion propagation and maintenance for Sup35p. Nat Struct Biol. 2001;8:958–962. doi: 10.1038/nsb1101-958. [DOI] [PubMed] [Google Scholar]
- 333.Taylor KL, Wickner RB. [URE3] and [PSI]: prions of Saccharomyces cerevisiae. Contrib Microbiol. 2001;7:21–31. doi: 10.1159/000060373. [DOI] [PubMed] [Google Scholar]
- 334.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 335.Thual C, Bousset L, Komar AA, Walter S, Buchner J, Cullin C, et al. Stability, folding, dimerization, and assembly properties of the yeast prion Ure2p. Biochemistry. 2001;40:1764–1773. doi: 10.1021/bi001916l. [DOI] [PubMed] [Google Scholar]
- 336.Masison DC, Wickner RB. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science. 1995;270:93–95. doi: 10.1126/science.270.5233.93. [DOI] [PubMed] [Google Scholar]
- 337.Masison DC, Maddelein ML, Wickner RB. The prion model for [URE3] of yeast: spontaneous generation and requirements for propagation. Proc Natl Acad Sci U S A. 1997;94:12503–12508. doi: 10.1073/pnas.94.23.12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Thual C, Komar AA, Bousset L, Fernandez-Bellot E, Cullin C, Melki R. Structural characterization of Saccharomyces cerevisiae prion-like protein Ure2. J Biol Chem. 1999;274:13666–13674. doi: 10.1074/jbc.274.19.13666. [DOI] [PubMed] [Google Scholar]
- 339.Zhu L, Zhang XJ, Wang LY, Zhou JM, Perrett S. Relationship between stability of folding intermediates and amyloid formation for the yeast prion Ure2p: a quantitative analysis of the effects of pH and buffer system. J Mol Biol. 2003;328:235–254. doi: 10.1016/s0022-2836(03)00249-3. [DOI] [PubMed] [Google Scholar]
- 340.Gast K, Damaschun H, Eckert K, Schulze-Forster K, Maurer HR, Muller-Frohne M, et al. Prothymosin alpha: a biologically active protein with random coil conformation. Biochemistry. 1995;34:13211–13218. doi: 10.1021/bi00040a037. [DOI] [PubMed] [Google Scholar]
- 341.Uversky VN, Gillespie JR, Millett IS, Khodyakova AV, Vasiliev AM, Chernovskaya TV, et al. Natively unfolded human prothymosin alpha adopts partially folded collapsed conformation at acidic pH. Biochemistry. 1999;38:15009–15016. doi: 10.1021/bi990752+. [DOI] [PubMed] [Google Scholar]
- 342.Pavlov NA, Cherny DI, Heim G, Jovin TM, Subramaniam V. Amyloid fibrils from the mammalian protein prothymosin alpha. FEBS Lett. 2002;517:37–40. doi: 10.1016/s0014-5793(02)02572-3. [DOI] [PubMed] [Google Scholar]
- 343.MacRaild CA, Hatters DM, Howlett GJ, Gooley PR. NMR structure of human apolipoprotein C-II in the presence of sodium dodecyl sulfate. Biochemistry. 2001;40:5414–5421. doi: 10.1021/bi002821m. [DOI] [PubMed] [Google Scholar]
- 344.Hatters DM, Lawrence LJ, Howlett GJ. Sub-micellar phospholipid accelerates amyloid formation by apolipoprotein C-II. FEBS Lett. 2001;494:220–224. doi: 10.1016/s0014-5793(01)02355-9. [DOI] [PubMed] [Google Scholar]
- 345.Hatters DM, Lindner RA, Carver JA, Howlett GJ. The molecular chaperone, alpha-crystallin, inhibits amyloid formation by apolipoprotein C-II. J Biol Chem. 2001;276:33755–33761. doi: 10.1074/jbc.M105285200. [DOI] [PubMed] [Google Scholar]
- 346.Hatters DM, MacPhee CE, Lawrence LJ, Sawyer WH, Howlett GJ. Human apolipoprotein C-II forms twisted amyloid ribbons and closed loops. Biochemistry. 2000;39:8276–8283. doi: 10.1021/bi000002w. [DOI] [PubMed] [Google Scholar]
- 347.Hatters DM, Minton AP, Howlett GJ. Macromolecular crowding accelerates amyloid formation by human apolipoprotein C-II. J Biol Chem. 2002;277:7824–7830. doi: 10.1074/jbc.M110429200. [DOI] [PubMed] [Google Scholar]
- 348.Hatters DM, Howlett GJ. The structural basis for amyloid formation by plasma apolipoproteins: a review. Eur Biophys J. 2002;31:2–8. doi: 10.1007/s002490100172. [DOI] [PubMed] [Google Scholar]
- 349.Munishkina LA, Fink AL, Uversky VN. Conformational prerequisites for formation of amyloid fibrils from histones. J Mol Biol. 2004;342:1305–1324. doi: 10.1016/j.jmb.2004.06.094. [DOI] [PubMed] [Google Scholar]
- 350.Hill RL, Brew K. Lactose synthetase. Adv Enzymol Relat Areas Mol Biol. 1975;43:411–490. doi: 10.1002/9780470122884.ch5. [DOI] [PubMed] [Google Scholar]
- 351.Permyakov EA, Berliner LJ. alpha-Lactalbumin: structure and function. FEBS Lett. 2000;473:269–274. doi: 10.1016/s0014-5793(00)01546-5. [DOI] [PubMed] [Google Scholar]
- 352.Hiraoka Y, Segawa T, Kuwajima K, Sugai S, Murai N. alpha-Lactalbumin: a calcium metalloprotein. Biochem Biophys Res Commun. 1980;95:1098–1104. doi: 10.1016/0006-291x(80)91585-5. [DOI] [PubMed] [Google Scholar]
- 353.Permyakov EA, Yarmolenko VV, Kalinichenko LP, Morozova LA, Burstein EA. Calcium binding to alpha-lactalbumin: structural rearrangement and association constant evaluation by means of intrinsic protein fluorescence changes. Biochem Biophys Res Commun. 1981;100:191–197. doi: 10.1016/s0006-291x(81)80081-2. [DOI] [PubMed] [Google Scholar]
- 354.Dolgikh DA, Gilmanshin RI, Brazhnikov EV, Bychkova VE, Semisotnov GV, Venyaminov S, et al. Alpha-Lactalbumin: compact state with fluctuating tertiary structure? FEBS Lett. 1981;136:311–315. doi: 10.1016/0014-5793(81)80642-4. [DOI] [PubMed] [Google Scholar]
- 355.Kuwajima K. The molten globule state of alpha-lactalbumin. Faseb J. 1996;10:102–109. doi: 10.1096/fasebj.10.1.8566530. [DOI] [PubMed] [Google Scholar]
- 356.Hakansson A, Zhivotovsky B, Orrenius S, Sabharwal H, Svanborg C. Apoptosis induced by a human milk protein. Proc Natl Acad Sci U S A. 1995;92:8064–8068. doi: 10.1073/pnas.92.17.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Svensson M, Sabharwal H, Hakansson A, Mossberg AK, Lipniunas P, Leffler H, et al. Molecular characterization of alpha-lactalbumin folding variants that induce apoptosis in tumor cells. J Biol Chem. 1999;274:6388–6396. doi: 10.1074/jbc.274.10.6388. [DOI] [PubMed] [Google Scholar]
- 358.Veprintsev DB, Narayan M, Permyakov SE, Uversky VN, Brooks CL, Cherskaya AM, et al. Fine tuning the N-terminus of a calcium binding protein: alpha-lactalbumin. Proteins. 1999;37:65–72. doi: 10.1002/(sici)1097-0134(19991001)37:1<65::aid-prot7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 359.Veprintsev DB, Permyakov SE, Permyakov EA, Rogov VV, Cawthern KM, Berliner LJ. Cooperative thermal transitions of bovine and human apo-alpha-lactalbumins: evidence for a new intermediate state. FEBS Lett. 1997;412:625–628. doi: 10.1016/s0014-5793(97)00841-7. [DOI] [PubMed] [Google Scholar]
- 360.Gussakovsky EE, Haas E. Two steps in the transition between the native and acid states of bovine alpha-lactalbumin detected by circular polarization of luminescence: evidence for a premolten globule state? Protein Sci. 1995;4:2319–2326. doi: 10.1002/pro.5560041109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Takeda K, Ogawa K, Ohara M, Hamada S, Moriyama Y. Conformational changes of alpha-lactalbumin induced by the stepwise reduction of its disulfide bridges: the effect of the disulfide bridges on the structural stability of the protein in sodium dodecyl sulfate solution. J Protein Chem. 1995;14:679–684. doi: 10.1007/BF01886906. [DOI] [PubMed] [Google Scholar]
- 362.Hendrix TM, Griko Y, Privalov P. Energetics of structural domains in alpha-lactalbumin. Protein Sci. 1996;5:923–931. doi: 10.1002/pro.5560050514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Okazaki A, Katsumata K, Kuwajima K. Hydrogen-exchange kinetics of reduced alpha-lactalbumin bound to the chaperonin GroEL. J Biochem (Tokyo) 1997;121:534–541. doi: 10.1093/oxfordjournals.jbchem.a021619. [DOI] [PubMed] [Google Scholar]
- 364.Redfield C, Schulman BA, Milhollen MA, Kim PS, Dobson CM. Alpha-lactalbumin forms a compact molten globule in the absence of disulfide bonds. Nat Struct Biol. 1999;6:948–952. doi: 10.1038/13318. [DOI] [PubMed] [Google Scholar]
- 365.Luo Y, Baldwin RL. The 28–111 disulfide bond constrains the alpha-lactalbumin molten globule and weakens its cooperativity of folding. Proc Natl Acad Sci U S A. 1999;96:11283–11287. doi: 10.1073/pnas.96.20.11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Goers J, Permyakov SE, Permyakov EA, Uversky VN, Fink AL. Conformational prerequisites for alpha-lactalbumin fibrillation. Biochemistry. 2002;41:12546–12551. doi: 10.1021/bi0262698. [DOI] [PubMed] [Google Scholar]
- 367.Biggin MD, Tjian R. Transcription factors that activate the Ultrabithorax promoter in developmentally staged extracts. Cell. 1988;53:699–711. doi: 10.1016/0092-8674(88)90088-8. [DOI] [PubMed] [Google Scholar]
- 368.Soeller WC, Poole SJ, Kornberg T. In vitro transcription of the Drosophila engrailed gene. Genes Dev. 1988;2:68–81. doi: 10.1101/gad.2.1.68. [DOI] [PubMed] [Google Scholar]
- 369.Gilmour DS, Thomas GH, Elgin SC. Drosophila nuclear proteins bind to regions of alternating C and T residues in gene promoters. Science. 1989;245:1487–1490. doi: 10.1126/science.2781290. [DOI] [PubMed] [Google Scholar]
- 370.Glaser RL, Thomas GH, Siegfried E, Elgin SC, Lis JT. Optimal heat-induced expression of the Drosophila hsp26 gene requires a promoter sequence containing (CT)n.(GA)n repeats. J Mol Biol. 1990;211:751–761. doi: 10.1016/0022-2836(90)90075-W. [DOI] [PubMed] [Google Scholar]
- 371.Lee H, Kraus KW, Wolfner MF, Lis JT. DNA sequence requirements for generating paused polymerase at the start of hsp70. Genes Dev. 1992;6:284–295. doi: 10.1101/gad.6.2.284. [DOI] [PubMed] [Google Scholar]
- 372.Wilkins RC, Lis JT. Dynamics of potentiation and activation: GAGA factor and its role in heat shock gene regulation. Nucleic Acids Res. 1997;25:3963–3968. doi: 10.1093/nar/25.20.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Agianian B, Leonard K, Bonte E, Van der Zandt H, Becker PB, Tucker PA. The glutamine-rich domain of the Drosophila GAGA factor is necessary for amyloid fibre formation in vitro, but not for chromatin remodelling. J Mol Biol. 1999;285:527–544. doi: 10.1006/jmbi.1998.2355. [DOI] [PubMed] [Google Scholar]
- 374.Farrell HM, Jr, Qi PX, Uversky VN. New views of protein structure: Applications to the caseins. Protein structure and functionality. In: Fishman M, Qi PX, editors. Advances in Biopolymers: Molecules, Clusters, Networks and Interactions. Washington, DC: American Chemical Society Publishers; 2006. pp. 52–70. [Google Scholar]
- 375.Chakraborty A, Basak S. pH-induced structural transitions of caseins. J Photochem Photobiol B. 2007;87:191–199. doi: 10.1016/j.jphotobiol.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 376.Thorn DC, Meehan S, Sunde M, Rekas A, Gras SL, MacPhee CE, et al. Amyloid fibril formation by bovine milk kappa-casein and its inhibition by the molecular chaperones alphaS- and beta-casein. Biochemistry. 2005;44:17027–17036. doi: 10.1021/bi051352r. [DOI] [PubMed] [Google Scholar]
- 377.Genton B, Al-Yaman F, Betuela I, Anders RF, Saul A, Baea K, et al. Safety and immunogenicity of a three-component blood-stage malaria vaccine (MSP1, MSP2, RESA) against Plasmodium falciparum in Papua New Guinean children. Vaccine. 2003;22:30–41. doi: 10.1016/s0264-410x(03)00536-x. [DOI] [PubMed] [Google Scholar]
- 378.Genton B, Anders RF, Alpers MP, Reeder JC. The malaria vaccine development program in Papua New Guinea. Trends Parasitol. 2003;19:264–270. doi: 10.1016/s1471-4922(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 379.Genton B, Betuela I, Felger I, Al-Yaman F, Anders RF, Saul A, et al. A recombinant blood-stage malaria vaccine reduces Plasmodium falciparum density and exerts selective pressure on parasite populations in a phase 1–2b trial in Papua New Guinea. J Infect Dis. 2002;185:820–827. doi: 10.1086/339342. [DOI] [PubMed] [Google Scholar]
- 380.Felger I, Steiger S, Hatz C, Smith T, Beck HP. Antigenic cross-reactivity between different alleles of the Plasmodium falciparum merozoite surface protein 2. Parasite Immunol. 2003;25:531–543. doi: 10.1111/j.0141-9838.2004.00664.x. [DOI] [PubMed] [Google Scholar]
- 381.Low A, Chandrashekaran IR, Adda CG, Yao S, Sabo JK, Zhang X, et al. Merozoite surface protein 2 (MSP2) of Plasmodium falciparum: Expression, structure, dynamics and fibril formation of the conserved N-terminal domain. Biopolymers. 2007 doi: 10.1002/bip.20764. [DOI] [PubMed] [Google Scholar]
- 382.Pettigrew GW, Moore GR. Cytochromes c: Evolutionary, Structural and Physiochemical Aspects. Berlin: Springer; 1990. [Google Scholar]
- 383.Pertinhez TA, Bouchard M, Tomlinson EJ, Wain R, Ferguson SJ, Dobson CM, et al. Amyloid fibril formation by a helical cytochrome. FEBS Lett. 2001;495:184–186. doi: 10.1016/s0014-5793(01)02384-5. [DOI] [PubMed] [Google Scholar]
- 384.Musacchio A, Wilmanns M, Saraste M. Structure and function of the SH3 domain. Prog Biophys Mol Biol. 1994;61:283–297. doi: 10.1016/0079-6107(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 385.Morton CJ, Campbell ID. SH3 domains. Molecular ‘Velcro’. Curr Biol. 1994;4:615–617. doi: 10.1016/s0960-9822(00)00134-2. [DOI] [PubMed] [Google Scholar]
- 386.Booker GW, Gout I, Downing AK, Driscoll PC, Boyd J, Waterfield MD, et al. Solution structure and ligand-binding site of the SH3 domain of the p85 alpha subunit of phosphatidylinositol 3-kinase. Cell. 1993;73:813–822. doi: 10.1016/0092-8674(93)90259-s. [DOI] [PubMed] [Google Scholar]
- 387.Koyama S, Yu H, Dalgarno DC, Shin TB, Zydowsky LD, Schreiber SL. 1H and 15N assignments and secondary structure of the PI3K SH3 domain. FEBS Lett. 1993;324:93–98. doi: 10.1016/0014-5793(93)81539-c. [DOI] [PubMed] [Google Scholar]
- 388.Koyama S, Yu H, Dalgarno DC, Shin TB, Zydowsky LD, Schreiber SL. Structure of the PI3K SH3 domain and analysis of the SH3 family. Cell. 1993;72:945–952. doi: 10.1016/0092-8674(93)90582-b. [DOI] [PubMed] [Google Scholar]
- 389.Liang J, Chen JK, Schreiber ST, Clardy J. Crystal structure of P13K SH3 domain at 20 angstroms resolution. J Mol Biol. 1996;257:632–643. doi: 10.1006/jmbi.1996.0190. [DOI] [PubMed] [Google Scholar]
- 390.Guijarro JI, Sunde M, Jones JA, Campbell ID, Dobson CM. Amyloid fibril formation by an SH3 domain. Proc Natl Acad Sci U S A. 1998;95:4224–4228. doi: 10.1073/pnas.95.8.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]