

Summary
p110α (PIK3CA) is the most frequently mutated kinase in human cancer, and numerous drugs targeting this kinase are currently in pre-clinical development or early stage clinical trials. Clinical resistance to protein kinase inhibitors frequently results from point mutations that block drug binding; similar mutations in p110α are likely, but currently none have been reported. Using a S. cerevisiae screen against a structurally diverse panel of PI3K inhibitors, we have identified a potential hotspot for resistance mutations (I800), a drug-sensitizing mutation (L814C), and a surprising lack of resistance mutations at the “gatekeeper” residue. Our analysis further reveals that clinical resistance to these drugs may be attenuated by using multi-targeted inhibitors that simultaneously inhibit additional PI3K pathway members.
Significance
Point mutations that block drug binding are likely to be a major mechanism of clinical resistance to PI3K-targeted cancer therapy. Here we report resistance mutations in the oncogenic PI3K isoform p110α, as well as a drug-sensitizing mutation that will be useful for chemical genetic studies. This study anticipates p110α mutations that are likely to emerge against PI3K-targeted drugs, and identifies inhibitor classes that can overcome these resistance mutations. Our experiments in mammalian cells show that multi-targeted inhibitors with additional PI3K pathway targets are less susceptible to drug resistance than selective PI3K inhibitors. The screening protocol described here is applicable to several other drug targets that inhibit S. cerevisiae growth in addition to p110α.
Introduction
p110α is a class IA phosphoinositide 3-kinase (PI3K) that phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) at the D-3 position of its inositol ring, generating phosphatidylinositol-3,4,5-trisphosphate (PIP3) (Fruman et al., 1998). PIP3 recruits downstream signaling proteins such as Akt to the plasma membrane, leading to increased growth, proliferation, motility, and cell survival (Cantley, 2002).
p110α has attracted considerable interest as a drug target (Hennessy et al., 2005; Luo et al., 2003; Stephens et al., 2005) since its identification as an oncogene (Chang et al., 1997) and the discovery of activating point mutations in its encoding gene PIK3CA (Samuels et al., 2004), as it is the most frequently mutated oncogene in breast (27%) and endometrial (22%) cancers (frequencies obtained from the Wellcome Trust Sanger Institute Cancer Genome Project, http://www.sanger.ac.uk/genetics/CGP). In the absence of p110α mutation, other PI3K pathway members, including Receptor Tyrosine Kinases (RTKs), Ras, PTEN, and Akt are frequently mutated instead (Brugge et al., 2007; Cully et al., 2006; Shaw and Cantley, 2006), highlighting the critical role PI3K signaling plays in oncogenesis and tumor maintenance (Gupta et al., 2007; Lim and Counter, 2005). More than 80% of reported p110α mutations are confined to the helical domain mutations E542K and E545K, and the kinase domain mutation H1047R (Samuels et al., 2004).
These mutants exhibit increased catalytic activity (Ikenoue et al., 2005; Samuels et al., 2004) and are capable of transforming a number of cell lines in tissue culture and xenograft models (Ikenoue et al., 2005; Isakoff et al., 2005; Kang et al., 2005; Samuels et al., 2005; Zhao et al., 2005). Mounting evidence for p110α’s importance in cancer has spurred the development of PI3K-targeted inhibitors, several of which are now entering clinical trials as cancer therapy (Marone et al., 2008; Ward and Finan, 2003).
As these targeted PI3K inhibitors progress through clinical trials, it is likely that drug resistance will emerge in cancer patients as it does to targeted protein kinase inhibitors (Daub et al., 2004). The most frequent mechanism of resistance to protein kinase inhibitors in cancer therapy is point mutation of the target kinase that blocks drug binding (Daub et al., 2004; Gorre et al., 2001; Mahon et al., 2000; Shah et al., 2002; Shah and Sawyers, 2003), and mutation at one position in particular is frequently observed across the protein kinase family, including BCR-Abl (T315I), c-Kit (T670I), PDGFR (T674I), and EGFR (T790M) (Daub et al., 2004). This position is termed the gatekeeper because it controls access to a large hydrophobic pocket in which most kinase inhibitors bind (Bishop et al., 2000; Liu et al., 1998). Recent crystal structures of inhibitor-bound p110γ reveal a similar large hydrophobic pocket, termed the affinity pocket, which is occupied by the most potent PI3K inhibitors (Knight et al., 2006). Structure-activity relationship (SAR) data (Knight et al., 2006) and the crystal structure of p110α (Huang et al., 2007) indicate that this affinity pocket is conserved in all p110 isoforms, and structural alignment reveals that Ile848 occupies a position in p110α that is structurally analogous to the gatekeeper residue in protein kinases (Walker et al., 1999). This suggests the possibility that drug resistance in p110α might arise as it often does in protein kinases, by mutation at the gatekeeper residue Ile848 or at other residues lining the affinity pocket. Although PI3Ks share significant sequence and structural homology with the protein kinase family, the level of homology is not high enough to confidently predict resistance mutations in p110α from the corresponding mutations in protein kinases.
The identification and characterization of protein kinase resistance mutations from cancer patients has led to the development of effective second generation inhibitors (Carter et al., 2005; Quintas-Cardama et al., 2007; Shah et al., 2004). Remarkably, mutagenic screens in mammalian tissue culture are able to reproduce these resistance mutations in vitro (Azam et al., 2003; Engelman et al., 2006; von Bubnoff et al., 2005). These studies are typically performed with some knowledge of resistance mutations in the target kinase that have already been identified in cancer patients. In this study we sought to identify p110α resistance mutations in vitro before they occur in cancer patients, with no prior knowledge of clinical resistance in the PI3K family.
The structural diversity of PI3K inhibitors in clinical development (Marone et al., 2008) suggests that a different spectrum of resistance mutations may arise against each inhibitor. In order to screen several PI3K inhibitors simultaneously, we used a PI3K expression system in the budding yeast S. cerevisiae, which allows an unlimited number of drug conditions to be screened via replica plating or robotic pinning. Unlike previous mutagenic screens in mammalian tissue culture which rely on oncogenic transformation by the target kinase (Azam et al., 2003; Engelman et al., 2006; von Bubnoff et al., 2005), our screen is based on growth inhibition induced by overexpression of membrane-localized p110α (Rodriguez-Escudero et al., 2005; Tugendreich et al., 2001). p110α activity severely inhibits growth in S. cerevisiae because it has no endogenous p110 homolog and therefore does not produce or degrade endogenous PIP3 (Odorizzi et al., 2000). Previous studies have shown that p110α-induced growth inhibition can be partially reversed by the low potency PI3K inhibitor LY294002 (Rodriguez-Escudero et al., 2005); in this study we used a structurally diverse panel of high potency PI3K inhibitors that completely rescue S. cerevisiae growth. Drug-resistant p110α mutants were identified by continued growth inhibition at drug concentrations that rescue growth from wild-type p110α.
An unexpected benefit of the yeast screen was the ability to identify drug sensitization in addition to drug resistance, by enhanced growth rescue at low inhibitor concentrations that do not rescue growth from wild-type p110α. Currently there is no truly selective p110α inhibitor available, so the drug-sensitized mutants we identified will be valuable tools for the study of p110α’s role in biological processes that rely on PI3K signaling, from oncogenesis and tumor maintenance (Gupta et al., 2007; Lim and Counter, 2005) to processes as varied as neuronal development and animal behavior, viral replication, and stem cell self-renewal (Cosker and Eickholt, 2007; Hale and Randall, 2007; Kwon et al., 2006; Paling et al., 2004; Rodgers and Theibert, 2002). Additional benefits to screening with yeast are the speed and simplicity of yeast culture in comparison to mammalian tissue culture, the straightforward recovery of plasmid DNA from screen hits for DNA sequencing, and the ability to screen inhibitors that would confound mammalian transformation assays by their off-target effects (Fan et al., 2006).
To determine whether p110α is susceptible to drug-resistant mutations in a similar manner to protein kinases, and to search for drug-sensitized p110α mutants, we mutagenized residues that line the affinity pocket and screened against a structurally diverse panel of PI3K inhibitors. Using a high-throughput, parallel approach with robotic pinning, we generated a measure of catalytic activity and an inhibition profile for every clone in our mutant library, allowing for a detailed structure-function analysis of the p110α affinity pocket. Potential drug-resistant and drug-sensitizing mutations identified with the yeast screen were further characterized in mammalian systems to rule out artifacts from heterologous yeast expression: by kinase assays following expression in the human cell line HEK-293T, and by transformation assays with the human breast epithelial cell line MCF10A.
Results
PI3K activity inhibits growth in S. cerevisiae, and can be rescued by selective PI3K inhibitors
In order to use PI3K-induced toxicity to study p110α inhibition in S. cerevisiae, we generated high copy (2μ) plasmids that express p110α-CAAX under control of a GAL1 promoter, and transformed them into a wild-type strain AFS92 and a drug-permeablized strain YRP1 (Δerg6, Δpdr5, Δsnq2) (Gray et al., 1998). When grown on galactose, both strains show greater than 1000-fold growth inhibition compared to empty vector and kinase-dead controls (Fig. 1A). PI3K-induced growth inhibition is more severe in the YRP1 strain than in the AFS92 strain, and there is no difference in growth inhibition between wild-type p110α-CAAX and the oncogenic H1047R mutant (Fig. 1A). The PI3K inhibitor PI-103 completely rescues growth in the drug-permeablized YRP1 strain, but only partially rescues growth in the wild-type AFS92 strain (Fig. 1A). Growth rescue by several other PI3K inhibitors is more efficient in YRP1 than in AFS92, (data not shown), and therefore the YRP1 strain was chosen for use in all further experiments.
Figure 1. Rescue of PI3K-induced growth inhibition in S. cerevisiae by selective PI3K inhibitors.
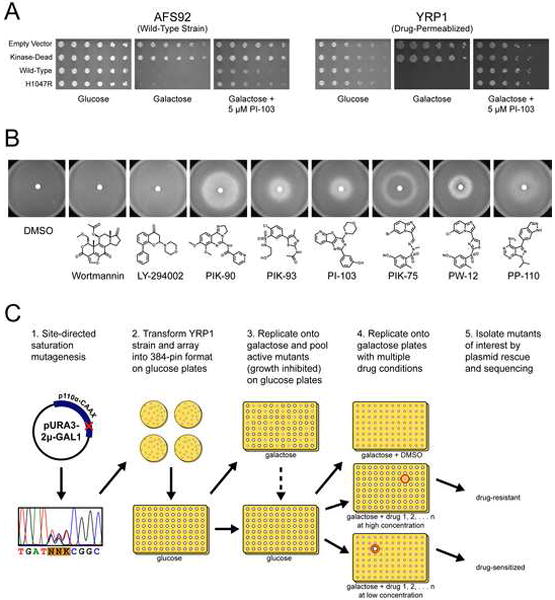
A: Three-fold serial dilutions of AFS92 and YRP1 yeast strains containing the pURA3-2μ-GAL1 plasmid expressing the indicated p110α-CAAX mutants were spotted onto agar plates of SD −URA media containing glucose, galactose, or galactose with 5 μM PI-103. All plates were incubated at 30°C for 2 days except the YRP1 strains grown on galactose, which were incubated for 6 days.
B: Reverse halo assays with YRP1-pURA3-2μ-GAL1-p110αH1047R-CAAX grown on SD −URA +Galactose media, each plate spotted with 10 μl of a 10 mM DMSO stock of PI3K inhibitor.
C: Schematic overview of the S. cerevisiae growth inhibition screen used to identify drug-resistant and drug-sensitized p110α mutants.
It was desirable to include as many diverse PI3K inhibitors as possible in the planned screen of p110α mutants, in order to identify pan-inhibitor resistance mutations as well as inhibitor-specific mutations, and to give the best chance of identifying a mutant-inhibitor pair that confers drug sensitivity. To test all PI3K inhibitors for compatibility with the yeast assay format, we used a variation on the traditional halo assay which we term “reverse halo assay.” In this assay, a PI3K inhibitor is spotted onto a cellulose disc in the middle of a lawn of yeast, and instead of inhibiting growth as in a traditional halo assay, the PI3K inhibitor rescues growth, creating a “growth halo” of healthy yeast. The diameter and intensity of this halo depends on the inhibitor’s potency, stability, diffusion rate, and lack of S. cerevisiae toxicity.
Two non-selective PI3K inhibitors, wortmannin and LY294002, did not produce growth halos in the reverse halo assay (Fig. 1B). This is not surprising because wortmannin inhibits the essential S. cerevisiae kinase STT4 at low nM concentrations (Cutler et al., 1997), while LY294002 is only a μM inhibitor of p110α and other PI3K family members (Brunn et al., 1996; Vlahos et al., 1994). The imido-pyrazine PIK-75 produced only a thin ring of growth (Fig. 1B), suggesting an exceedingly narrow dose window in which a mutagenic screen could be performed. Five structurally diverse p110α inhibitors, PIK-90, PIK-93, PI-103, PW-12, and PP-110 produced growth halos of various size and intensity (Fig. 1B), and therefore were selected for use in subsequent mutagenic screens.
Figure 1C illustrates how p110α-induced growth inhibition in S. cerevisiae was used to screen for drug resistance and sensitization. Residues of interest were subject to saturation mutagenesis with randomized primers, and the resulting libraries were transformed into the permeablized yeast strain YRP1. Individual colonies were picked and arrayed into 384-pin format and then replicated with a robotic pinner onto glucose and galactose-containing media to determine the relative colony size, and therefore PI3K activity, of each mutant clone. Active mutants were picked and arrayed onto new 384-pin format plates and then replicated onto multiple PI3K inhibitor plates to screen for drug resistance and sensitization.
Mutation of the p110α gatekeeper residue Ile848
Initially we focused on the p110α gatekeeper residue, because in protein kinases this position is the most frequent site of drug-resistant mutations (Daub et al., 2004). Sequence alignment of p110α with protein kinases reveals Ile848 to be its gatekeeper residue (Fig. 2A), which is positioned in the active site similarly to the gatekeeper residue in protein kinases (Fig. 2B). Using site-directed mutagenesis with randomized primers, we mutated Ile848 to all 20 amino acids in the low copy plasmid pURA3-CEN/ARS-GAL1-p110αH1047R-CAAX. These plasmids were transformed into YRP1, and the resulting strains were grown on either glucose or galactose to determine the relative PI3K activity of each mutant (Fig. 2C). With the exception of mild growth inhibition by the conservative mutations I848L, I848S, and I848V, the other 16 p110α mutants did not inhibit yeast growth (Fig. 2C), suggesting these mutants are catalytically inactive or unstable when expressed in yeast. This result corroborates and extends previous work showing loss of catalytic activity with the I848A and I848G mutations (Alaimo et al., 2005). The inability of p110α to tolerate non-conservative mutations at the gatekeeper position is reflected in the evolutionary conservation of lipid kinases at this position. Only Ile, Leu, Met and Val are found at the gatekeeper position in the PI3K family, and only Ile is found among the p110 isoforms, while a greater diversity of amino acid side chains is observed in the protein kinase family (Fig. 2D).
Figure 2. p110α gatekeeper mutants have reduced catalytic activity.
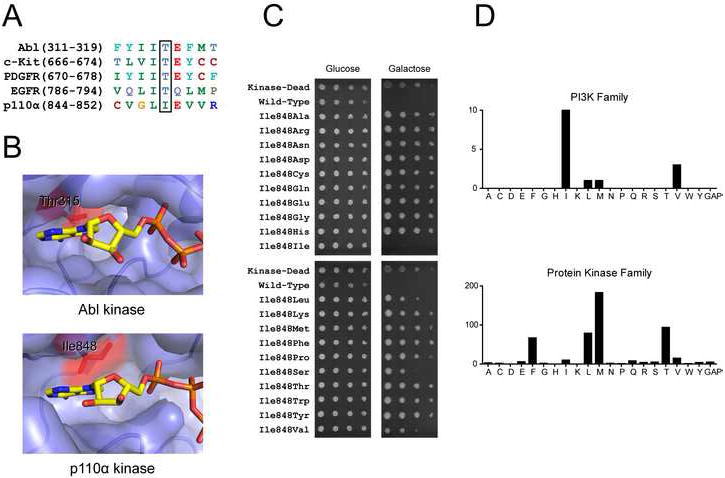
A: Sequence alignment of p110α with protein kinases that display clinical resistance mutations at the gatekeeper position
B: ATP binding sites of Abl (PDB code 2G1T) and p110α (PDB code 2RD0). ATP from p110γ co-crystal (PDB code 1E8X) was overlaid onto the apo p110α structure by structural alignment. The gatekeeper residue is colored red and labeled in both structures.
C: Six-fold serial dilutions of YRP1-pURA3-2μ-GAL1-p110αH1047R-CAAX strains with the indicated p110α mutations were spotted onto agar plates of SD −URA media containing either glucose or galactose. The plates were incubated at 30°C for 2 days (glucose) and 6 days (galactose).
D: Distribution of residues at the gatekeeper position in the human PI3K and protein kinase families.
A mutagenic screen of the p110α affinity pocket against a diverse panel of PI3K inhibitors
Minimal growth inhibition by p110α gatekeeper mutants in the low copy vector made screening for drug resistance impossible, so the high copy plasmid pURA3-2μ-GAL1-p110αH1047R-CAAX was used in all further experiments. Ile848 appeared relatively intolerant to mutation (Fig. 2C), so the screen was expanded to include seven additional residues surrounding the p110α affinity pocket. These residues were chosen because the affinity pocket is occupied by most potent PI3K inhibitors, but not by ATP (Knight et al., 2006) (Fig. 3A), making it a likely site for drug-resistant mutations. Ile800, Leu807, Leu814, Tyr836, Gly837, Cys838, and Ile848 were chosen based on proximity to the affinity pocket, and lack of interaction with the catalytic Lys802, DFG motif (responsible for Mg2+ coordination), or ATP (Fig 3B). One additional residue outside the affinity pocket, Ser854, was chosen due to possible inhibitor-specific H-bonds. These eight residues are highly conserved in the PI3K family and almost 100% identical among the p110 isoforms (Fig. 3C).
Figure 3. Affinity pocket residues chosen for saturation mutagenesis.
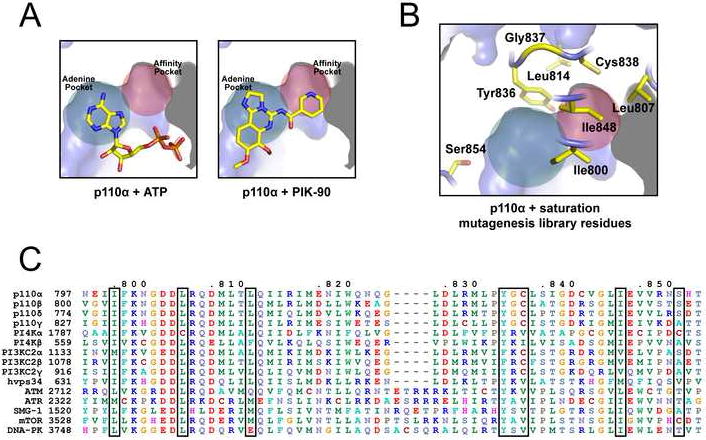
A: p110α active site, with the adenine pocket colored green and the affinity pocket colored red. The p110α apo structure (PDB code 2RD0) was overlaid with ATP (PDB code 1E8X) and PIK-90 (PDB code 2CHX) from p110γ co-crystal structures.
B: p110α affinity pocket residues chosen for saturation mutagenesis, shown on the p110α structure (PDB code 2RD0).
C: Sequence alignment of the human PI3K family. Affinity pocket residues chosen for saturation mutagenesis are indicated by boxes.
The chosen residues were mutagenized and screened against PI3K inhibitors as described in Figure 1C. Each residue was subject to saturation mutagenesis with randomized NNK primers, and the resulting eight libraries (Fig. S1) were transformed into the yeast strain YRP1. 384 colonies from each NNK library were picked and arrayed onto 384-pin format plates, and then replicated onto glucose and galactose plates to determine the PI3K activity of each clone by colony size, measured with Cellprofiler image analysis software (Fig. 4A). Active mutants with relative colony sizes less than 70% of wild-type were picked and arrayed onto new 384-pin format plates, and then replicated onto multiple PI3K inhibitor containing plates (Table S4) to screen for drug resistance and sensitization. PIK-85 was used in place of PIK-90 for all screens; it is identical to PIK-90 except for an enol group in place of PIK-90’s amide. An array of pooled active clones replicated onto either DMSO or 5 μM PI-103 is shown in Fig. 4B. Potential drug-resistant and drug-sensitized clones identified on the 384-pin inhibitor plates were validated by serial dilution analysis (Fig. S2). Serial dilutions of the drug-resistant mutant I800M and the drug-sensitized mutant L814C are shown in Fig. 4C.
Figure 4. Growth inhibition screen of p110α mutant libraries in S. cerevisiae.
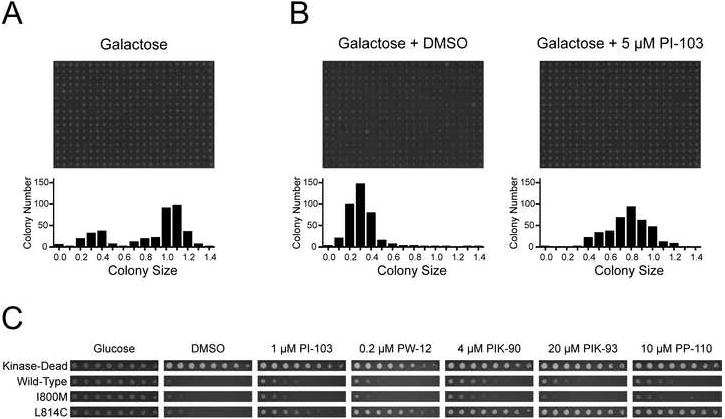
A: YRP1-pURA3-2μ-GAL1-p110αH1047R-CAAX mutant library pinned onto SD −URA +Galactose and grown for 5 days at 30°C, and the distribution of colony sizes for that plate, relative to the same array plated on SD −URA +Glucose and grown for 3 days at 30°C (not shown).
B: Representative image of pooled active clones from YRP1-pURA3-2μ-GAL1-p110αH1047R-CAAX mutant libraries, pinned onto SD −URA +Galactose with either DMSO control or 5 μM PI-103 and grown for 5 days at 30°C, and the corresponding distributions of colony sizes, relative to the same array plated on SD −URA +Glucose and grown for 3 days at 30°C (not shown).
C: Ten-fold serial dilutions of YRP1-pURA3-2μ-GAL1-p110αH1047R-CAAX strains with the indicated p110α mutations spotted onto SD −URA +Galactose media containing the indicated PI3K inhibitors and grown for 5 days at 30°C.
Characterization of p110α mutants by mammalian expression and in vitro kinase assay
Drug-resistant and drug-sensitized mutations identified by the yeast screen require validation in a mammalian context, to confirm that their altered activities and inhibitor sensitivities are not due to membrane localization by the C-terminal CAAX box motif, lack of a p85 binding partner, or other artifacts of S. cerevisiae expression. Mutations of interest were introduced into Myc-tagged p110α-H1047R, and then expressed in the human cell line HEK-293T for purification and enzymatic assay. Most of the mutants tested have slightly reduced activity (Fig. 5A): I800L and I800M show approximately 2-fold decreases in PI3K activity, while L814C’s activity is significantly reduced. In vitro IC50 values were determined for all p110α mutants against the inhibitors used in the yeast screen, as well as BEZ-235, which we obtained after the screen was performed. Mutation at Ile800 confers the greatest drug resistance, with both I800L and I800M showing approximately 5-10 fold decreases in inhibitor potency for PIK-90, PIK-93, PI-103, and PP-110, although I800L is sensitized to the compounds PW-12 and BEZ-235 (Fig. 5B). Mutation at Leu814 confers the greatest drug sensitization, with the L814C mutant sensitized more than 10-fold to PIK-90 and PP-110, and approximately 100-fold to PIK-93 (Fig. 5B).
Figure 5. Characterization of yeast screen hits by mammalian expression and in vitro kinase assay.
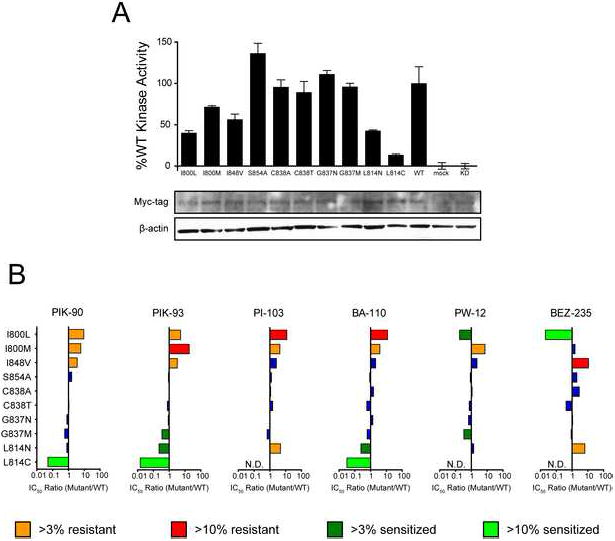
A: The indicated Myc-tagged p110αH1047R mutants were immunoprecipitated from HEK-293T cells and assayed for PI3K activity. Data are represented as mean ± SEM.
B: In vitro IC50 values were determined for each p110α mutant against the indicated PI3K inhibitors at 10 μM ATP. Changes in inhibitor sensitivity are shown as the ratio of mutant IC50 / WT IC50. The label N.D. (not determined) indicates that the data from the performed dose response experiment were insufficient to generate an IC50 curve.
I800L, I800M, and L814C mutants transform MCF10A cells, and confer resistance or sensitization to PI3K inhibitors
The oncogenic potential of the most drug-resistant and drug-sensitized p110α mutants were assessed in the untransformed breast cell line MCF10A, which requires Epidermal Growth Factor (EGF) for growth under normal tissue culture conditions, but can be transformed to EGF-independent growth by p110α-H1047R (Isakoff et al., 2005). I800L, I800M, L814C, wild-type, and kinase-dead (K802R) mutations were made in the retroviral plasmid pMIG-p110α-H1047R, and then transduced into MCF10A cells. The pMIG vector contains an internal ribosome entry site (IRES)-GFP sequence: infected MCF10A cell lines were all FACS sorted to greater than 95% GFP positive for use in further experiments.
p110α-expressing MCF10A cell lines were cultured in media without EGF to determine whether the drug-resistant and drug-sensitized mutant p110α-H1047Rs could support EGF-independent growth, and to monitor changes in PI3K inhibitor sensitivity for each mutant. The I800L and I800M mutant cell lines grow at the same rate as p110α-H1047R, while the L814C mutation caused a small but consistent decrease in growth (Fig. 6A). I800L and I800M confer resistance to all inhibitors, with the exception that I800L is sensitized to PW-12 and BEZ-235 (Fig. 6B). PI3K inhibitor resistance is most pronounced with the selective PI3K inhibitors PIK-90 and PIK-93, and less dramatic against the multi-targeted PI3K inhibitors PI-103 and BEZ-235 (mTOR), PW-12 (multiple protein kinases), and PP-110 (receptor tyrosine kinases) (Fig. 6B) (Fan et al., 2006; Hayakawa et al., 2007; Hayakawa et al., 2007; Knight et al., 2006; Raynaud et al., 2007; Stauffer et al., 2008) (B.A. and K.M.S., submitted manuscript). Similarly, L814C confers strong sensitization of cell growth to PIK-90 and PIK-93, moderate sensitization to PW-12 and PP-110, and minimal or no sensitization to PI-103 and BEZ-235 (Figure 6C), again likely due to the selectivity of PIK-90 and PIK-93, and the relative promiscuity of PW-12, PP-110, PI-103, and BEZ-235. As a reference, inhibitory values for the PI3K inhibitors against p110α and selected off-target protein kinases are shown in Table S1.
Figure 6. Yeast screen hits I800L, I800M, and L814C confer EGF-independent growth to MCF10A cells, and maintain altered inhibitor sensitivities in vivo.
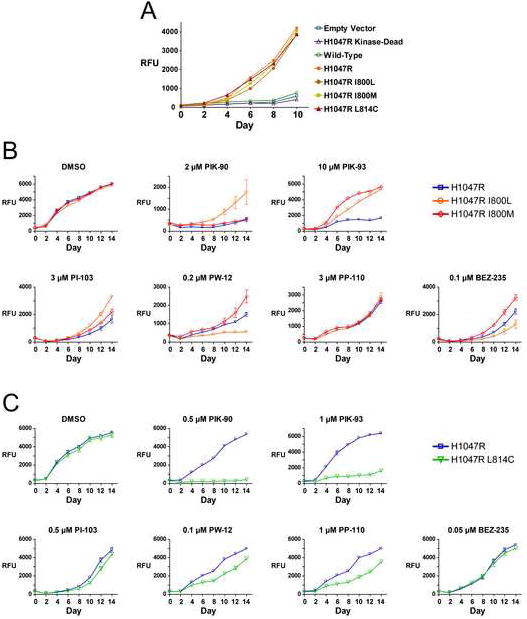
A: MCF10A cell lines expressing the indicated p110α mutants were cultured in growth media lacking EGF, and monitored for growth by Alamar Blue assay. Data are represented as mean ± SEM.
B and C: Growth of the I800L, I800M, and L814C MCF10A cell lines in media lacking EGF was monitored as in Fig. 6A in the presence of the indicated PI3K inhibitors. Data are represented as mean ± SEM.
I800L, I800M, and L814C mutants retain the ability to induce EGF-independent Akt phosphorylation in MCF10A cells
Phosphorylation of Akt residues Thr308 and Ser473 was monitored in the I800L, I800M, and L814C-expressing MCF10A cell lines to determine whether these p110α mutants retain the ability to activate the canonical downstream PI3K signaling pathway. The I800L and I800M mutations to p110α-H1047R produce high phospho-Akt levels comparable to p110α-H1047R after 24 hours EGF starvation, but L814C gives reduced, although still detectable levels (Fig. 7A). Phospho-Akt can be fully recovered in all cell lines by one hour treatment with EGF-containing media, and this stimulation can be blocked in all lines by 30 μM PI-103 (Fig. 7A). GFP levels, which are coupled to p110α expression by an IRES promoter, are greatly reduced in all p110α-expressing cell lines, and p110α levels are not highly elevated in comparison to the empty vector control (Fig. 7A). This suggests that MCF10A cells cannot tolerate overexpression of p110α-H1047R, and shows that expression near endogenous levels is sufficient for Akt activation and transformation to EGF independence.
Figure 7. Yeast screen hits I800L, I800M, and L814C produce EGF-independent Akt phosphorylation in MCF10A cells, which maintains altered inhibitor sensitivities.
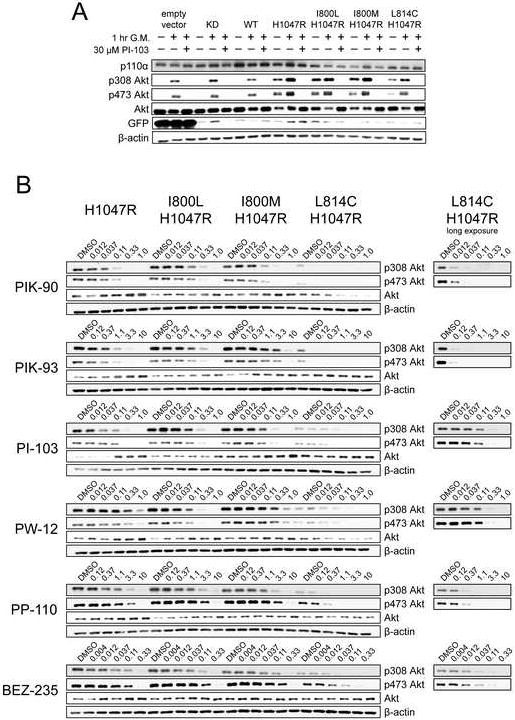
A: MCF10A cell lines expressing the indicated p110α mutants were cultured in growth media lacking EGF for 24 hours, and then treated with the indicated combinations of normal growth media (G. M.) and 30 μM PI-103. After 1 hour, the cells were lysed and subject to western blot analysis with the indicated antibodies.
B: The indicated MCF10A cell lines were cultured in growth media lacking EGF for 24 hours, and then treated for 1 hour with serial dilutions of the indicated PI3K inhibitors, after which the cells were lysed and subjected to western blot analysis with the indicated antibodies. The PI3K inhibitor concentrations are indicated in μM.
The I800L, I800M, and L814C cell lines were treated with PI3K inhibitors in serial dilution to determine how each mutation affects the inhibitor sensitivity of phospho-Akt levels. Similar to the MCF10A growth curves, the most striking resistance and sensitization occurs with the selective PI3K inhibitors PIK-90 and PIK-93, most likely because these results are not confounded by off-target effects. I800L is approximately 3-fold resistant to PIK-90 and PIK-93, I800M is approximately 10-fold resistant to PIK-93, and L814C is approximately 5-fold and 30-fold sensitized to PIK-90 and PIK-93 respectively (Fig. 7B), consistent with in vitro kinase assays and the MCF10A growth curves. Significant resistance is not observed against the multi-targeted inhibitors PI-103, PW-12, PP-110 and BEZ-235, but sensitization is seen with I800L to PW-12 and L814C to PW-12, PP-110, and BEZ-235 (Fig. 7B), again consistent with in vitro kinase assays and the MCF10A growth curves. Slight resistance and sensitization are observed with the I800L mutant to the dual PI3K/mTOR inhibitors PI-103 and BEZ-235 respectively. This trend is seen in phospho-308 Akt but not phospho-473, most likely because mTOR is the kinase that phosphorylates Ser473 in these cells (Sarbassov et al., 2005).
Tolerance to mutation in the p110α affinity pocket
In order to determine how tolerant each p110α affinity pocket residue is to mutation, we compared the colony size distributions generated for each residue in the course of our mutagenic screen, as seen in Figure 4A. All mutant libraries display a peak of relative colony sizes close to 1.0, corresponding to no growth inhibition and therefore kinase-dead mutants (Fig. 8A). The majority of mutant libraries also display a second peak of smaller colony sizes, corresponding to growth inhibition and therefore PI3K activity (Fig. 8A). The tolerance to mutation for each residue was quantified by Σ(1-x)2, where x equals relative colony size. The resulting values were converted into heat map color scale and displayed on p110α (Fig 8B). The I800, I807, L814, and G837 residues are most tolerant to mutation, C837, I848, and S854 are less tolerant to mutation, and Y836 appears to be completely intolerant to mutation, with even the conservative mutation Y836F causing a substantial loss of kinase activity (Fig. S2). Intolerance to mutation at Tyr 836 corroborates previous results with p110α (Alaimo et al., 2005). Recently published work has shown that the mutation corresponding to Y836M in PI4KIIIβ produces an active kinase that is resistant to Wortmannin and PIK-93, and the same mutation in PI4KIIIα kills all catalytic activity (Balla et al., 2008). It is currently unclear why this mutation is tolerated in PI4KIIIβ but abolishes kinase activity in PI4KIIIα and p110α. Further experiments with additional PI3K family members may better reveal the sequence determinants for mutation tolerance and inhibitor sensitivity.
Figure 8. Tolerance to mutation in the p110α affinity pocket.
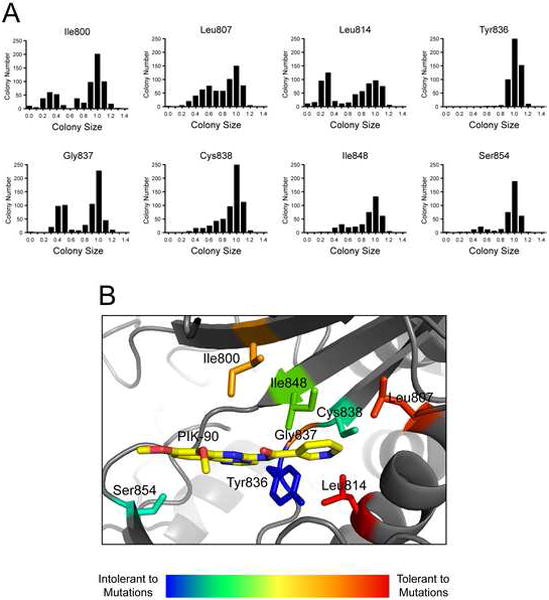
A: Colony size distributions for 384 colony arrays of the indicated p110α mutant libraries, grown in the YRP1 strain on SD −URA +Galactose media as described in Fig. 4A.
B: Tolerance to mutation as calculated by Σ(1-x)2 from the distributions in Fig. 8A, where x equals relative colony size. These values were converted into heat map values and are shown on the p110α crystal structure (PDB code 2RD0) with the PI3K inhibitor PIK-90 from the p110γ co-crystal structure (PDB code 2CHX) overlaid by structural alignment.
Discussion
We describe here the development of a resistance screen based on PI3K-induced growth inhibition of S. cerevisiae. Unlike resistance screens based on oncogenic transformation of mammalian cells, this screen allows for the identification of drug-sensitized mutants in addition to drug-resistant mutants, and allows for detailed structure-function analysis of the drug target, because activity levels and inhibitor sensitivities are determined for every clone in the mutant library. Previous studies have used growth inhibition in S. cerevisiae by heterologous expression of a mammalian drug target to screen a compound library for potent inhibitors (Boschelli et al., 2001), but to our knowledge such a screen has never been used to identify drug-resistant and drug-sensitizing mutations in the target protein. In addition to p110α, several other drug targets inhibit S. cerevisiae growth, including Akt1, PDK1, p38 kinase, Src, RhoA, RhoC, PARP-1, and HIV Protease (Brugge et al., 1987; Tugendreich et al., 2001), and the methods described here should be broadly applicable for the identification of drug-resistant and drug-sensitized mutations in these proteins as well.
Our results in S. cerevisiae indicate that the p110α gatekeeper residue Ile848 will not be a hotspot for inhibitor resistance in p110α as it is in the protein kinase family (Figs. 2C and 8A). While protein kinases can tolerate dramatic mutations at the gatekeeper position, from smaller, space-creating mutations that sensitize protein kinases to analog-specific inhibitors (Bishop et al., 2000), or larger, steric clash-inducing mutations that confer inhibitor resistance (Daub et al., 2004), it appears that p110α cannot tolerate significant mutations at this position. This suggests the possibility that the PI3K fold is less tolerant of mutations than the protein kinase fold in this region, which may reduce the clinical development of resistance to p110α-targeted drugs, although a more complete mutagenic screen of p110α would be required to prove this conclusively.
In contrast to Ile848, the residue Ile800 is tolerant to mutations (Fig. 8A), two of which confer drug resistance: I800L and I800M. The resistance conferred by these mutations is smaller than observed for the T315I gatekeeper mutant of BCR-Abl, but comparable to most other mutations that confer clinical resistance to imatinib (Shah et al., 2002; von Bubnoff et al., 2005). This residue is a potential hotspot for clinical resistance to PI3K inhibitors; fortunately our screen has identified PW-12 and BEZ-235 as inhibitors that potently target the I800L mutant. The fact that I800L confers resistance to all other inhibitors, but sensitizes p110α to PW-12 and BEZ-235 suggests that these inhibitors share a similar binding mode that differs from other scaffolds near residue Ile800. There are several inhibitor-bound PI3K structures available, but the co-crystal structures of PW-12 and BEZ-235 have not been reported, so it is difficult to rationalize these trends. Interestingly, the residue corresponding to Ile800 in mTOR is also leucine, and mTOR is potently targeted by BEZ-235.
The drug-sensitizing mutation L814C will be a valuable tool to study the effect of p110α inhibition in various biological systems, because there is no selective p110α inhibitor currently available. One possible concern with the L814C mutation is that its “sensitizing” effect may simply be due to the loss of enzymatic activity (Fig. 5A), but in vitro kinase assays reveal large shifts in IC50 values (Fig. 5B), and yeast serial dilution analysis (Fig. 4C) and MCF10A transformation assays (Fig. 6A) indicate that this mutant behaves similarly to wild-type in a cellular context, although it does show reduced Akt phosphorylation (Fig. 7B).
Transferring the L814C, I800L, and I800M mutations to other members of the PI3K family presents an excellent opportunity to study the cellular roles of each p110 isoform by specific inhibition or drug resistance. I800 and L814 are absolutely conserved among the p110 isoforms, and highly conserved among the rest of the PI3K family, so the likelihood of successfully generating drug-resistant and drug-sensitized mutants for each family member appears high. Concerning the loss of activity caused by gatekeeper mutations in p110α, it will be interesting to test whether this trend holds for other PI3K family members, or is unique to p110α.
The advent of high throughput screening and the great success of imatinib led the pharmaceutical industry to focus on highly specific kinase inhibitors, but currently a multi-targeted approach is gaining acceptance (Branca, 2005; Jimeno and Hidalgo, 2006). Inhibiting multiple targets can increase efficacy (Fan et al., 2006) and theoretically should decrease the likelihood of drug resistance, although no study to our knowledge has conclusively shown a reduced likelihood of clinical resistance with multi-targeted vs. highly specific drugs. Our results with the MCF10A cell line (Figs. 6, 7, S3) show that multi-targeted inhibitors are not as susceptible to drug resistance by mutation of the single target p110α, especially when they target additional kinases in the PI3K signaling pathway.
PI-103 and BEZ-235 block PI3K signaling downstream of p110α by inhibiting mTOR, and PP-110 blocks PI3K signaling upstream of p110α by inhibiting RTKs. The ability of these three inhibitors to block p110αH1047R-dependent MCF10A growth is largely unaffected by p110α resistance mutations, suggesting that mutation of additional targets is required to confer drug resistance. The possibility of accumulating multiple resistance mutations in a single cancer cell seems unlikely, but slight growth advantages at each step may increase the odds of successive mutations, especially during prolonged treatment. The rate and probability of resistance mutation accumulation will be an important area of study for multi-targeted drugs in cancer therapy, accessible by careful monitoring of clinical trials as well as mammalian tissue culture screens.
By focusing on a small subset of p110α residues that line the affinity pocket, we have identified a potential resistance hotspot at Ile800 that confers 5-30 fold resistance to most PI3K inhibitors. While this discovery should help guide the design of second-generation PI3K inhibitors, a more complete mutagenic screen of p110α will be necessary to uncover the full spectrum of resistance mutations.
Experimental Procedures
Plasmids
Yeast Expression Plasmids: A C-terminal myristoylation sequence (CAAX box) was added to human p110α-H1047R by three rounds of PCR amplification with the following primers: 1) hp110α-FM and hp110α-CRM, 2) hp110α-FL and hp110α-CRL. (All oligonucleotide primers used for cloning are described in Table S2). The resulting fragment was cloned into a high copy (2μ), URA3 yeast expression vector with a GAL1 promoter by gap repair/homologous recombination to create the vector pURA3-2μ-GAL1-p110αH1047R-CAAX. Wild-type and kinase-dead plasmids were created by site-directed mutagenesis with the following primers: hp110αR1047H-F, hp110αR1047H-R, hp110αK802R-F, and hp110αK802R-R.
Mammalian Expression Plasmids: An N-terminal Myc tag was added to human p110α-H1047R by PCR with the following primers: p110α-BamHI-NtermMyc-F and p110α-EcoRV-R. The resulting fragment was digested with BamHI and EcoRV, and then ligated into the mammalian expression vector pcDNA3 to create pcDNA3-Myc-p110α-H1047R. Several point mutations to this vector were created by site-directed mutagenesis.
Retroviral Plasmids: Human p110αH1047R was PCR amplified with the primers hp110αF-XhoI and hp110αR-HpaI, and the resulting fragment was digested with XhoI and HpaI and then ligated into the IRES-GFP retroviral vector pMIG to create pMIG-p110α-H1047R. Several p110α point mutations to this vector were made by site-directed mutagenesis.
Murine Ecotropic Pseudotyping Vector: pcDNA3-EcoR plasmid DNA encoding the murine ecotropic receptor EcoR/MCAT-1 was a generous gift from Jeffrey Henise.
Yeast Strains and Media
The S. cerevisiae strains YRP1 (Δerg6, Δpdr5, Δsnq2) and AFS92 were used for all experiments. Strains were grown at 30°C on SD -URA +Glucose or +Galactose. PI3K inhibitors were added to media in DMSO at 1:100. YRP1 was transformed with plasmid DNA as described in Supplemental Data.
Reverse Halo Assay
Log phase YRP1-pURA3-2μ-GAL1-p110αH1047R-CAAX cultures grown in SD −URA +Glucose were washed three times with water and then spread into a lawn on SD −URA +Galactose agarose plates at approximately 106 cells per plate. After drying, a small cellulose disc was placed in the middle of each plate, and then 10 μl of a DMSO inhibitor stock was spotted onto the cellulose disc. The plates were incubated at 30°C for 5-7 days before imaging.
PI3K Inhibitors
BEZ-235 was a generous gift from Yi Liu. All other inhibitors used in this study were synthesized following previously reported protocols (Knight et al., 2006; Stauffer et al., 2008) (B.A. and K.M.S., submitted manuscript). In all tissue culture experiments, DMSO inhibitor stocks were used at 1:1000.
Library Construction
The yeast expression vector pURA3-2μ-GAL1-p110αH1047R-CAAX was mutagenized at the residues I800, L807, L814, Y836, G837, C838, and S854 by quickchange PCR with degenerate NNK primers (Table S3), where N = 25% A, 25% C, 25% G, 25% T, and K = 50% G, 50% T. The resulting PCR reactions were purified with a Qiagen PCR Cleanup kit, DpnI digested for 1:30 hours, and then re-purified. 5 μl of each purified, digested PCR product was transformed into TOP-10 One Shot chemically competent E. coli (Invitrogen) and plated onto a single 10 cm plate of LB media with carbenicillin antibiotic, yielding 1-4 × 103 colonies per transformation. Colonies were grown for 2 days at 37°C and then pooled by scraping, transferred to 1.5ml tubes, and spun down to pellets of approximately 0.5 ml. Plasmid DNA was isolated from each pellet with the Qiagen Miniprep kit and verified by restriction digest and DNA sequencing (Fig. S1).
Screening and Image Analysis
p110α mutant libraries were transformed into YRP1 by electroporation and plated onto SD -URA +Glucose media. 384 colonies from each library were arrayed by hand and then replicated with a Virtek colony arrayer (Waterloo, Ontario, Canada) to obtain uniformly sized colonies. The arrays were further replicated onto two media conditions: 1) SD -URA +Galactose media with added PI3K inhibitor or DMSO alone, and 2) SD -URA +Glucose media. The plates were incubated at 30°C until the colonies had grown sufficiently (2-7 days depending on strain and media conditions) and then photographed. Colony size was calculated with Cellprofiler image analysis software, available at www.cellprofiler.org, and each SD -URA +Galactose colony size value was divided by the corresponding SD -URA +Glucose colony size to normalize for variation in pinning efficiency.
In Vitro PI3K Assays
pcDNA3-p110α plasmid DNA was transfected into HEK-293T cells with Lipofectamine 2000 (Invitrogen). After 48 hours the cells were trypsinized, washed with PBS, and pelleted for storage at -80°C. Pellets were lysed by vortexing in PI3K lysis buffer (50 mM Tris (pH 7.4), 300 mM NaCl, 5 mM EDTA, 0.02% NaN3, 1% Triton X-100, protease inhibitor cocktail tablets (Roche), 8 mM sodium orthovanadate, 83 μM PMSF, 1X Phosphatase Inhibitor Cocktails 1 and 2 (Sigma)) and then immunoprecipitated by overnight incubation with Anti-c-Myc Agarose Affinity Gel (Sigma-Aldrich). The immunoprecipitates were washed with the following buffers: twice with buffer A (PBS, 1 mM EDTA, 1% Triton X-100), twice with buffer B (100 mM Tris pH 7.4, 500 mM LiCl, 1 mM EDTA), twice with buffer C (50 mM Tris pH 7.4, 100 mM NaCl), twice with PBS, and then assayed for PI3K activity in 96 well format essentially as described (Knight et al., 2007). Briefly, immunoprecipitated Myc-p110α was incubated “on bead” with shaking at 25°C with 100 μg/ml phosphatidylinositol, 10 μCi γ32P-ATP, 10 μM ATP, 1 mg/ml BSA, 25 mM HEPES pH 7.4, 10 mM MgCl2, 1:50 DMSO ± PI3K inhibitor. After 1 hour, 4 μl of each reaction was spotted onto a dry nitrocellulose membrane pre-rinsed with wash solution (1 M NaCl, 1% H3PO4). After the spots dried, the membrane was washed five times with wash solution, dried with a heat lamp, and exposed on a phosphor screen overnight. The phosphor screen was then scanned with a Typhoon phosphorimager (GE Healthcare) and the resulting data was quantified with the MATLAB script Spot (Knight et al., 2007).
Mammalian Cell Lines and Cell Culture
MCF10A cells (ATCC) were cultured at 37°C, 5% CO2 in MCF10A growth media (1:1 DMEM:F-12 supplemented with 5% filtered, heat inactivated horse serum, 20 ng/ml EGF, 100 μg/ml hydrocortisone, 1 ng/ml cholera toxin, 10 μg/ml insulin, and penicillin/streptomycin) as described (Debnath et al., 2003).
Generation of p110α-expressing MCF10A cell lines
Mutant p110α expressing MCF10A lines were created by retroviral infection. Ecotropic p110α viral stocks were made by transfecting pMIG-p110α plasmid DNA into the Phoenix Eco cell line. Retroviral supernatants were collected at 48, 72, 96, and 120 hours, spun at 500 RPM for 5 minutes at 4°C, and stored in 0.5 ml aliquots at -80°C. Human MCF10A cells were pseudotyped for infection with murine ecotropic virus by transient transfection with pcDNA3-EcoR/MCAT1. Transfection was performed by nucleofection (Amaxa Nucleofector) following the manufacturer’s instructions, and the transfected cells were plated into 6-well plates. 24 hours after nucleofection with pcDNA3-EcoR/MCAT1, the MCF10A cells were infected with thawed p110α viral stocks for 12 hours, switched to growth media for 6 hours, and then expanded into 75cm2 flasks. After 3 days, the infected cells were FACS sorted for GFP expression.
MCF10A Western Blots
40-70% confluent MCF10A cultures were starved for 24 hours with MCF10A media lacking EGF and insulin, and then treated for 60 minutes with PI3K inhibitors or a DMSO control. After 60 minutes, the cells were lysed with PI3K lysis buffer and subject to western blotting with antibodies purchased from Santa Cruz (anti-GFP) and Cell Signaling (all other antibodies) as directed by the supplier.
MCF10A Transformation Assays
MCF10A cell lines were assayed for oncogenic transformation by their ability to grow in media lacking EGF. Cells were seeded into black sided, clear bottom, 96-well plates at 2 × 103 cells per well in 100 μl growth media. After 2 days, the cells were washed with PBS and then switched to 100 μl growth media lacking EGF. Cell proliferation was monitored every 2 days by incubation with resazurin (alamar blue). A stock solution of 120 μg/ml resazurin in PBS was added to the cells at 1:20, incubated for 2 hours at 37°C, 15 minutes at room temperature, and then assayed for fluorescence with an excitation wavelength of 520 nm and an emission wavelength of 590 nm. After fluorescence measurement, the resazurin containing media was replaced with 100 μl fresh growth media lacking EGF and the cells were returned to 37°C, 5% CO2.
Structural Analysis
Visualization and structural alignment of x-ray crystal structures was performed with the Pymol Molecular Graphics System, available at http://www.pymol.org.
Supplementary Material
Acknowledgments
We are grateful to Karl Kulcher for the YRP1 strain; Yi Liu at Intellikine for the inhibitor BEZ-235; Dorothea Fiedler, Assen Roguev, and Nevan Krogan for assistance with the colony arrayer; Sohye Kang and Peter Vogt for p110αH1047R containing plasmids; Brandon Tavshanjian for assistance with generation of the MCF10A cell lines, and Jeffrey Henise and Jack Taunton for the pcDNA3-EcoR/MCAT1 vector. This work was supported by the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alaimo PJ, Knight ZA, Shokat KM. Targeting the gatekeeper residue in phosphoinositide 3-kinases. Bioorg Med Chem. 2005;13:2825–2836. doi: 10.1016/j.bmc.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112:831–843. doi: 10.1016/s0092-8674(03)00190-9. [DOI] [PubMed] [Google Scholar]
- Balla A, Tuymetova G, Toth B, Szentpetery Z, Zhao X, Knight ZA, Shokat K, Steinbach PJ, Balla T. Design of drug-resistant alleles of type-III phosphatidylinositol 4-kinases using mutagenesis and molecular modeling. Biochemistry. 2008;47:1599–1607. doi: 10.1021/bi7017927. [DOI] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Boschelli DH, Wang YD, Ye F, Wu B, Zhang N, Dutia M, Powell DW, Wissner A, Arndt K, Weber JM, Boschelli F. Synthesis and Src kinase inhibitory activity of a series of 4-phenylamino-3-quinolinecarbonitriles. J Med Chem. 2001;44:822–833. doi: 10.1021/jm000420z. [DOI] [PubMed] [Google Scholar]
- Branca MA. Multi-kinase inhibitors create buzz at ASCO. Nat Biotechnol. 2005;23:639. doi: 10.1038/nbt0605-639. [DOI] [PubMed] [Google Scholar]
- Brugge J, Hung MC, Mills GB. A new mutational AKTivation in the PI3K pathway. Cancer Cell. 2007;12:104–107. doi: 10.1016/j.ccr.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Brugge JS, Jarosik G, Andersen J, Queral-Lustig A, Fedor-Chaiken M, Broach JR. Expression of Rous sarcoma virus transforming protein pp60v-src in Saccharomyces cerevisiae cells. Mol Cell Biol. 1987;7:2180–2187. doi: 10.1128/mcb.7.6.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC, Jr, Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. Embo J. 1996;15:5256–5267. [PMC free article] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, 3rd, Edeen PT, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci U S A. 2005;102:11011–11016. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HW, Aoki M, Fruman D, Auger KR, Bellacosa A, Tsichlis PN, Cantley LC, Roberts TM, Vogt PK. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 1997;276:1848–1850. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- Cosker KE, Eickholt BJ. Phosphoinositide 3-kinase signalling events controlling axonal morphogenesis. Biochem Soc Trans. 2007;35:207–210. doi: 10.1042/BST0350207. [DOI] [PubMed] [Google Scholar]
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- Cutler NS, Heitman J, Cardenas ME. STT4 is an essential phosphatidylinositol 4-kinase that is a target of wortmannin in Saccharomyces cerevisiae. J Biol Chem. 1997;272:27671–27677. doi: 10.1074/jbc.272.44.27671. [DOI] [PubMed] [Google Scholar]
- Daub H, Specht K, Ullrich A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat Rev Drug Discov. 2004;3:1001–1010. doi: 10.1038/nrd1579. [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, Naumov GN, Yeap BY, Jarrell E, Sun J, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, Shokat KM, Weiss WA. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–349. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- Gray NS, Wodicka L, Thunnissen AM, Norman TC, Kwon S, Espinoza FH, Morgan DO, Barnes G, LeClerc S, Meijer L, et al. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science. 1998;281:533–538. doi: 10.1126/science.281.5376.533. [DOI] [PubMed] [Google Scholar]
- Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, Nye E, Stamp G, Alitalo K, Downward J. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Hale BG, Randall RE. PI3K signalling during influenza A virus infections. Biochem Soc Trans. 2007;35:186–187. doi: 10.1042/BST0350186. [DOI] [PubMed] [Google Scholar]
- Hayakawa M, Kaizawa H, Kawaguchi K, Ishikawa N, Koizumi T, Ohishi T, Yamano M, Okada M, Ohta M, Tsukamoto S, et al. Synthesis and biological evaluation of imidazo[1,2-a]pyridine derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem. 2007;15:403–412. doi: 10.1016/j.bmc.2006.09.047. [DOI] [PubMed] [Google Scholar]
- Hayakawa M, Kaizawa H, Moritomo H, Koizumi T, Ohishi T, Yamano M, Okada M, Ohta M, Tsukamoto S, Raynaud FI, et al. Synthesis and biological evaluation of pyrido[3’,2’:4,5]furo[3,2-d]pyrimidine derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem Lett. 2007;17:2438–2442. doi: 10.1016/j.bmcl.2007.02.032. [DOI] [PubMed] [Google Scholar]
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–1748. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562–4567. doi: 10.1158/0008-5472.CAN-04-4114. [DOI] [PubMed] [Google Scholar]
- Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- Jimeno A, Hidalgo M. Multitargeted therapy: can promiscuity be praised in an era of political correctness? Crit Rev Oncol Hematol. 2006;59:150–158. doi: 10.1016/j.critrevonc.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Feldman ME, Balla A, Balla T, Shokat KM. A membrane capture assay for lipid kinase activity. Nat Protoc. 2007;2:2459–2466. doi: 10.1038/nprot.2007.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, Counter CM. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell. 2005;8:381–392. doi: 10.1016/j.ccr.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Liu Y, Shah K, Yang F, Witucki L, Shokat KM. A molecular gate which controls unnatural ATP analogue recognition by the tyrosine kinase v-Src. Bioorg Med Chem. 1998;6:1219–1226. doi: 10.1016/s0968-0896(98)00099-6. [DOI] [PubMed] [Google Scholar]
- Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- Mahon FX, Deininger MW, Schultheis B, Chabrol J, Reiffers J, Goldman JM, Melo JV. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96:1070–1079. [PubMed] [Google Scholar]
- Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Odorizzi G, Babst M, Emr SD. Phosphoinositide signaling and the regulation of membrane trafficking in yeast. Trends Biochem Sci. 2000;25:229–235. doi: 10.1016/s0968-0004(00)01543-7. [DOI] [PubMed] [Google Scholar]
- Paling NR, Wheadon H, Bone HK, Welham MJ. Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J Biol Chem. 2004;279:48063–48070. doi: 10.1074/jbc.M406467200. [DOI] [PubMed] [Google Scholar]
- Quintas-Cardama A, Kantarjian H, Cortes J. Flying under the radar: the new wave of BCR-ABL inhibitors. Nat Rev Drug Discov. 2007;6:834–848. doi: 10.1038/nrd2324. [DOI] [PubMed] [Google Scholar]
- Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, Henley A, Di-Stefano F, Ahmad Z, Guillard S, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–5850. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- Rodgers EE, Theibert AB. Functions of PI 3-kinase in development of the nervous system. Int J Dev Neurosci. 2002;20:187–197. doi: 10.1016/s0736-5748(02)00047-3. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Escudero I, Roelants FM, Thorner J, Nombela C, Molina M, Cid VJ. Reconstitution of the mammalian PI3K/PTEN/Akt pathway in yeast. Biochem J. 2005;390:613–623. doi: 10.1042/BJ20050574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y, Diaz LA, Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–573. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- Shah NP, Sawyers CL. Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene. 2003;22:7389–7395. doi: 10.1038/sj.onc.1206942. [DOI] [PubMed] [Google Scholar]
- Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Stauffer F, Maira SM, Furet P, Garcia-Echeverria C. Imidazo[4,5-c]quinolines as inhibitors of the PI3K/PKB-pathway. Bioorg Med Chem Lett. 2008;18:1027–1030. doi: 10.1016/j.bmcl.2007.12.018. [DOI] [PubMed] [Google Scholar]
- Stephens L, Williams R, Hawkins P. Phosphoinositide 3-kinases as drug targets in cancer. Curr Opin Pharmacol. 2005;5:357–365. doi: 10.1016/j.coph.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Tugendreich S, Perkins E, Couto J, Barthmaier P, Sun D, Tang S, Tulac S, Nguyen A, Yeh E, Mays A, et al. A streamlined process to phenotypically profile heterologous cDNAs in parallel using yeast cell-based assays. Genome Res. 2001;11:1899–1912. doi: 10.1101/gr.191601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- von Bubnoff N, Veach DR, van der Kuip H, Aulitzky WE, Sanger J, Seipel P, Bornmann WG, Peschel C, Clarkson B, Duyster J. A cell-based screen for resistance of Bcr-Abl-positive leukemia identifies the mutation pattern for PD166326, an alternative Abl kinase inhibitor. Blood. 2005;105:1652–1659. doi: 10.1182/blood-2004-06-2445. [DOI] [PubMed] [Google Scholar]
- Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- Ward SG, Finan P. Isoform-specific phosphoinositide 3-kinase inhibitors as therapeutic agents. Curr Opin Pharmacol. 2003;3:426–434. doi: 10.1016/s1471-4892(03)00078-x. [DOI] [PubMed] [Google Scholar]
- Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102:18443–18448. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.