

Summary
Hyperpolarization-activated cyclic nucleotide-regulated (HCN) channels, which generate the Ih current, mediate a number of important brain functions. The HCN1 isoform regulates dendritic integration in cortical pyramidal neurons, and provides an inhibitory constraint on both working memory in prefrontal cortex and spatial learning and memory in the hippocampus. Altered expression of HCN1 following seizures may contribute to the development of temporal lobe epilepsy. Yet the regulatory networks and pathways governing HCN channel expression and function in the brain are largely unknown. Here we report the presence of nine alternative N-terminal splice forms of the brain-specific cytoplasmic protein TRIP8b and demonstrate the differential effects of six isoforms to downregulate or upregulate HCN1 surface expression. Furthermore, we find that all TRIP8b isoforms inhibit channel opening by shifting activation to more negative potentials. TRIP8b thus functions as an auxiliary subunit that provides a mechanism for the dynamic regulation of HCN1 channel expression and function.
Introduction
The hyperpolarization-activated cation current (Ih), encoded by the HCN1-4 gene family, regulates the electrical activity of a wide array of neurons (for review, Robinson and Siegelbaum, 2003). In cortical layer V and hippocampal CA1 pyramidal neurons, Ih channels are predominantly composed of HCN1 subunits that are targeted to the distal regions of the apical dendrites (Santoro et al., 1997; Magee, 1998; Lorincz et al., 2002). At these sites, Ih inhibits the integration of excitatory inputs, dendritic excitability and the induction of long-term synaptic plasticity (Stuart and Spruston, 1998; Magee, 1999; Williams and Stuart, 2000; Nolan et al., 2004; Tsay et al., 2007; George et al., 2009). Such effects may underlie the behavioral role of HCN1 as an inhibitory constraint on hippocampal-dependent and prefrontal cortex-dependent forms of learning and memory (Nolan et al., 2004; Wang et al., 2007).
Complementary to the regulatory action of HCN1 channels on neural activity, both physiological and pathological patterns of neuronal activity regulate the expression of HCN1. Stimuli that induce long-term potentiation or long-term depression produce, respectively, an upregulation (Fan et al., 2005) or downregulation (Brager and Johnston, 2007) of Ih and HCN1 expression, providing a homeostatic scaling mechanism that offsets changes in synaptic efficacy by controlling dendritic excitability. In contrast, maladaptive downregulation of HCN1 expression following seizures enhances dendritic excitability and may contribute to the development of temporal lobe epilepsy (Chen et al., 2001; Brewster et al., 2002, 2005; Shah et al., 2004; Jung et al, 2007; Shin et al, 2008). These observations suggest that powerful regulatory mechanisms must govern the ongoing expression, function and localization of HCN channels in the brain.
One widespread mechanism for regulating channel expression, function and trafficking is through the association of the pore-forming channel subunits with auxiliary subunits (Yu et al., 2005). Auxiliary subunits for HCN channels could provide a molecular mechanism for their activity-dependent regulation and might explain why the properties of native Ih in neurons differ from the properties of Ih formed by recombinant HCN subunits in heterologous cells (e.g. Pedarzani and Storm, 1995; Magee, 1998; Gasparini and DiFrancesco, 1999; Santoro et al., 2000; Franz et al., 2000; Chen et al., 2001a). Such subunits could also help regulate the subcellular trafficking of the HCN channels. However, despite their potential importance, the existence and function of HCN channel auxiliary subunits in the brain remain poorly understood.
We previously used a yeast two-hybrid screen to identify potential molecular partners of HCN1 and isolated a brain-specific ∼68 kD cytoplasmic protein, TRIP8b, that binds strongly to HCN channels and is tightly co-localized with HCN1 in the distal dendrites of neocortical and hippocampal pyramidal neurons (Santoro et al., 2004). An analysis of cDNA isolates reveals that the single TRIP8b gene is subject to extensive alternative splicing that affects the extreme N-terminus of the protein (Santoro et al., 2004), Thus, the TRIP8b gene encodes a family of N-terminal splice variants that differ in their initial 7-112 amino acids, whereas the subsequent 560 residues are identical in all isoforms. The TRIP8b splice variant that we previously characterized exerts a powerful effect on HCN channel trafficking. Overexpression of this splice variant in either heterologous expression systems or cultured hippocampal pyramidal neurons causes a dramatic, almost complete downregulation of surface expression of HCN1 or HCN2 (Santoro et al., 2004).
Here, we document the presence in the brain of at least nine alternatively spliced TRIP8b isoforms and show that different isoforms have differential effects on HCN1 surface expression. Importantly, one isoform leads to a marked upregulation of HCN1 surface expression and Ih density in heterologous cells, cultured hippocampal neurons, and CA1 pyramidal neurons in the intact brain. Moreover, all TRIP8b isoforms examined exert an inhibitory effect on HCN1 gating, shifting activation to more negative voltages and slowing the rate of channel opening. These results provide strong evidence that TRIP8b is an auxiliary subunit of HCN1 channels ideally poised to mediate the dynamic modulation of channel expression and function in discrete regions of the brain.
Results
Multiple splice variants of TRIP8b are expressed in the mouse brain
As we previously reported, a comparison of the various GenBank entries for TRIP8b orthologs suggests that the N-terminal domain of this protein undergoes alternative splicing (Santoro et al., 2004). To address the nature and extent of this diversity, we performed an extensive reverse transcription-polymerase chain reaction (RT-PCR) analysis of mRNA isolated from mouse brain, using TRIP8b exon-specific primers. Such analysis revealed the existence of 9 different splice variants of TRIP8b (Figure 1A; Supplementary Figure 1; Table 1). All variants include one of two alternative start codons, each encoded in a separate exon (exons 1a and 1b), followed by different combinations of the three subsequent exons (exons 2,3,4). No variations are found in the ensuing exons (exons 5-16), which encode the bulk of the protein. As we previously reported, the C-terminal half of TRIP8b (encoded by exons 10-16) contains six tetratricopeptide repeats (TPR), which constitute a conserved protein-protein binding motif (Blatch and Lassle, 1999) that interacts with the C-terminal “SNL” tripeptide sequence in HCN1, 2 and 4 (Santoro et al, 2004; see Supplementary Figure 1). Given the large intervening distance (over 50 kilobases) between the end of exon 1a and the start of exon 1b, two alternative promoters may drive expression of the TRIP8b gene.
Figure 1.
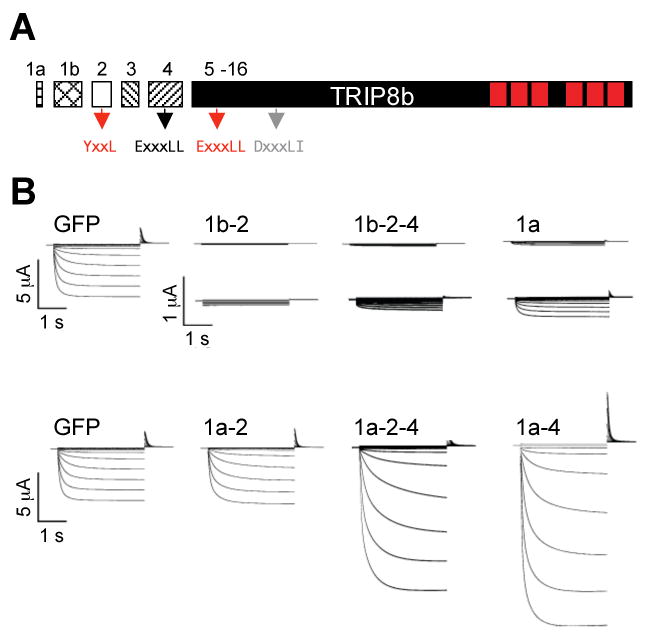
Different TRIP8b splice isoforms differentially regulate HCN1 current density.
(A) Schematic representation of exons comprising the open reading frame of TRIP8b. Exons 1a through 4 are variably spliced. Arrows show locations and identity of putative adaptin-binding consensus sequences. Tetratricopeptide repeats (TPR) shown as red rectangles.
(B) Two microelectrode voltage-clamp current traces from Xenopus oocytes co-injected with cRNA encoding HCN1 together with GFP (control) or one of six TRIP8b splice isoforms (labeled). Currents were elicited by a 3 second voltage steps from a holding potential of -30 mV to a range of test potentials between +5 and -95 mV in increments of 10 mV, followed by a depolarizing step to 0 mV. Middle row of traces show currents for HCN1 coexpressed with TRIP8b(1b-2), TRIP8b(1b-2-4) and TRIP8b(1a) currents at an expanded current scale (note scale bars). HCN1 plus GFP traces on top and bottom rows are identical to aid in comparisons.
Table 1.
Relative abundance of TRIP8b splice variants expressed in mouse brain and effects of six major variants on HCN1 channel expression and gating. Splice form mRNA abundance was assessed by quantitative real-time PCR analysis as a percentage of total TRIP8b mRNA. The indicated range of values describes the variability observed between different mRNA/cDNA preparations. ΔV1/2 values refer to the difference in the midpoint of activation observed between HCN1 channels expressed without or with the indicated TRIP8b splice variant (see Figure 4 and text). TBD, to be determined.
| TRIP8b Isoform | Effect on HCN1 gating (ΔV1/2) | Effect on HCN1 expression | % TRIP8b mRNA |
|---|---|---|---|
| 1a | -13.4 mV | ∼10-fold reduction | ∼25-30% |
| 1a-2 | -9.3 mV | no effect | <10% |
| 1a-2-3-4 | TBD | TBD | <1% |
| 1a-2-4 | -9.7 mV | ∼5-fold increase | <10% |
| 1a-3-4 | TBD | TBD | <5% |
| 1a-4 | -11.6 mV | ∼6-fold increase | ∼30-40% |
| 1b-2 | -10.7 mV | >100-fold reduction | ∼10-15% |
| 1b-2-3-4 | TBD | TBD | <1% |
| 1b-2-4 | -14.7 mV | >50-fold reduction | ∼10-15% |
To determine the relative expression levels of individual TRIP8b isoforms, we performed quantitative real-time PCR analysis on mouse brain mRNA, using primers spanning specific exon-exon boundaries (see Experimental Procedures; Supplementary Table 1). The abundance of each of the splice forms was determined as a percentage of total TRIP8b mRNA, the latter measured using primer pairs that define amplicons within the constant domain (exons 5-16). Our results indicate that TRIP8b(1a-4) is the predominant TRIP8b mRNA species in the brain, accounting for ∼30-40% of total TRIP8b mRNA. TRIP8b(1b-2), the splice variant described in our original study, accounts for ∼10-15% of total brain TRIP8b mRNA. The second most abundant species is TRIP8b(1a), representing ∼25-30% of total TRIP8b mRNA. Although it is not possible to draw a direct inference as to the relative levels of protein expression of the different TRIP8b isoforms from the mRNA data, Western blot analysis of brain tissue using an antibody that recognizes all splice variants reveals two prominent bands (Supplementary Figure 2). Consistent with the results from the quantitative PCR analysis, the most abundant TRIP8b protein species co-migrates with TRIP8b(1a-4) whereas the second TRIP8b protein species co-migrates with TRIP8b(1a). The relative abundance of all 9 TRIP8b splice forms identified by real-time PCR analysis is presented in Table 1, along with a summary of the effects of the major TRIP8b isoforms on HCN1 expression and gating, which we present in detail below.
Alternative splice forms of TRIP8b differentially regulate HCN1 channel expression
Sequence analysis reveals the presence of consensus protein trafficking motifs in distinct TRIP8b exons, suggesting that the various isoforms may exert specific effects on HCN1 trafficking and surface expression (Supplementary Figure 1). Indeed, we have previously shown that TRIP8b(1b-2) induces the near-complete internalization of HCN1 from the plasma membrane, resulting in an accumulation of channel protein in intracellular vesicles, a subset of which can be identified as early endosomes (Santoro et al., 2004). Consistent with this effect, TRIP8b(1b-2) contains a potential tyrosine-based trafficking motif (YXXØ) in exon 2. In addition, dileucine-based ([DE]XXXL[LI]) trafficking motifs are found in exons 4, 5 and 6. These signals are known to recruit distinct sets of sorting machinery, including Adaptor Protein complexes (AP-1, AP-2, and AP-3) that bind both clathrin and the YXXØ and [DE]XXXL[LI] signaling sequences of cargo proteins (Bonifacino and Traub, 2003).
Do different TRIP8b isoforms have distinct effects on HCN1 expression? To address this question, we co-injected cRNA encoding HCN1 with cRNA encoding each of six TRIP8b splice variants into Xenopus oocytes and performed two-microelectrode voltage clamp analysis of HCN1 current density. Maximal tail current amplitude, as determined by a fit of the Boltzmann equation to tail current activation curves, was dramatically affected by five of the six alternative splice forms. As described previously, TRIP8b(1b-2) essentially abolishes Ih current expression, reducing it to undetectable levels (>100-fold reduction) (Figure 1B, 2). We now find that TRIP8b(1b-2-4) produces a similarly dramatic reduction in Ih. TRIP8b(1a) also decreases expression of HCN1, although the reduction in current density is ∼10-fold, significantly less than the effect of TRIP8b(1b-2) or TRIP8b(1b-2-4). In contrast, TRIP8b(1a-2) has no effect on Ih levels. Strikingly, TRIP8b(1a-4) and TRIP8b(1a-2-4) produce up to a ∼6-fold increase in Ih current density (Figure 1B, 2).
Figure 2.
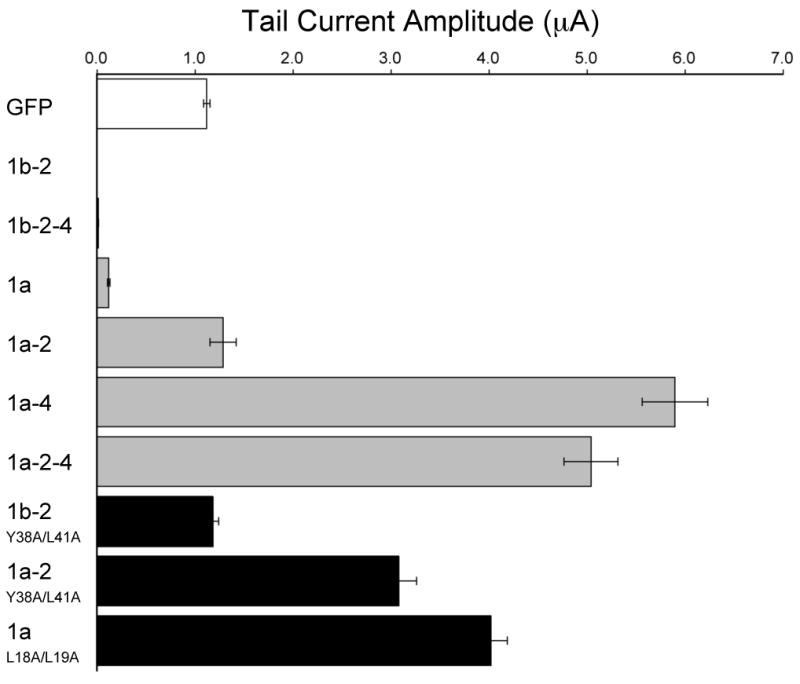
Effects of wild-type and mutant TRIP8b isoforms on HCN1 current density.
Population data showing maximal tail current amplitudes for HCN1 coexpressed with GFP (open bar), six wild-type TRIP8b isoforms (grey bars) or three selected TRIP8b isoforms with mutations in putative adaptin-binding sites (black bars) (see Experimental Procedures for details of tail current amplitude measurements). Mean current ± s.e.m. (n= number of experiments) for each pair of expressed constructs is as follows. HCN1 + GFP: 1.12 ± 0.03 μA (n=140). HCN1 + TRIP8b(1b-2): undetectable current (<0.005 μA; n=36). HCN1 + TRIP8b(1b-2-4): 0.02 ± 0.00 μA (n=21). HCN1 + TRIP8b(1a): 0.12 ± 0.01 μA (n=29). HCN1 + TRIP8b(1a-2): 1.29 ± 0.13 μA (n=16). HCN1 + TRIP8b(1a-4): 5.90 ± 0.33 μA (n=30). HCN1 + TRIP8b(1a-2-4): 5.04 ± 0.27 μA (n=35). HCN1 + TRIP8b(1b-2)Y38AL41A: 1.18 ± 0.06 μA (n=15). HCN1 + TRIP8b(1a-2)Y14AL17A: 3.08 ± 0.18 μA(n=15). HCN1 + TRIP8b(1a)L18AL19A: 4.02 ± 0.17 μA (n=13).
Both the downregulation and upregulation of HCN1 currents represent specific effects since current levels expressed by a variety of other channels, including CNGA1 (a channel closely related to the HCN channels), KAT1 and Kv1.2, are largely unaffected by coexpression with the TRIP8b isoforms (Santoro et al., 2004 and Supplementary Figure 3). The only significant effect seen with any of the TRIP8b isoforms on channels other that HCN1 was a very small (∼15%) increase in Kv1.2 current amplitude upon coexpression with TRIP8b(1a-4)).
The upregulation or downregulation of Ih density produced by these newly characterized TRIP8b isoforms appear to be caused largely by changes in channel surface expression, similar to our previous finding that TRIP8b(1b-2) promotes endocytosis of surface HCN1 channels. Thus, coexpression in Xenopus oocytes of TRIP8b(1a) with HCN1 channels tagged using green fluorescence protein (GFP-HCN1) results in a dramatic redistribution of the labeling to intracellular puncta, similar to what we previously observed for TRIP8b(1b-2) (Santoro et al., 2004; Supplementary Figure 4). In contrast, TRIP8b(1a-4) increases the level of GFP-HCN1 associated with the dendritic plasma membrane when over-expressed in neurons in vivo (see below, Figure 7).
Importance of consensus Adaptor Protein binding motifs in TRIP8b for HCN1 channel trafficking
To directly test whether the YXXØ motif in exon 2 is a critical determinant of the effects of certain TRIP8b isoforms on HCN1 trafficking, we introduced point mutations that disrupt this consensus site. Consistent with our hypothesis, the double point mutation Y38A/L41A in the YXXØ motif blocks the ability of TRIP8b(1b-2) to downregulate Ih (Figure 2). Interestingly, the same double point mutation in the background of TRIP8b(1a-2) causes this isoform, which normally has no effect on channel surface expression, to increase Ih current density ∼3-fold above baseline levels (Figure 2). This suggests that the lack of effect of wild-type TRIP8b(1a-2) on Ih levels may result from opposing influences of distinct sequences to enhance HCN1 expression and promote channel internalization.
In addition to the YXXØ motif in exon 2, other regions of TRIP8b must also contribute to the downregulation of channel expression because TRIP8b(1a), which lacks exon 2, also exerts a strong inhibitory effect on HCN1 surface expression. We therefore explored the role of the two dileucine-based trafficking motifs ([DE]XXXL[LI]) present within the invariant region of TRIP8b (exons 5,6; Figure 1A; Supplementary Figure 1). Mutation of the first dileucine motif (ESRPLL to ESRPAA; TRIP8b(1a)L18A/L19A) abolished the ability of TRIP8b(1a) to downregulate Ih density and also revealed a latent stimulatory effect of this isoform, which now increased current levels ∼4-fold above baseline (Figure 2). In contrast, mutation of the second motif (DGSDLI to DGSDAA; TRIP8b(1a)L87A/I88A) had no effect on the ability of TRIP8b(1a) to downregulate Ih (data not shown). A quadruple mutant that disrupted both dileucine motifs (L18A/L19A/L87A/L88A) behaved similarly to the L18A/L19A mutant (data not shown).
HCN1, TRIP8b and AP-2 form a macromolecular complex in vivo
The finding that the YXXL motif in exon 2 and EXXXLL motif in exon 5 are critical for the ability of distinct isoforms of TRIP8b to reduce HCN1 surface expression strongly supports a model in which TRIP8b acts as a bridge protein, recruiting AP-2 adaptors to TRIP8b/HCN1 channel complexes that enable the formation of clathrin coated pits and subsequent channel endocytosis. Consistent with this hypothesis, we find that HCN1 and TRIP8b are indeed tightly associated in the brain. Thus, an anti-TRIP8b antibody co-immunoprecipitates up to 50% of HCN1 present in solubilized membrane protein extracts of mouse forebrain cortex (Figure 3A). Given the limits of the precipitation reaction, these results suggest that the majority of HCN1 protein is normally associated with TRIP8b in the forebrain cortex.
Figure 3.
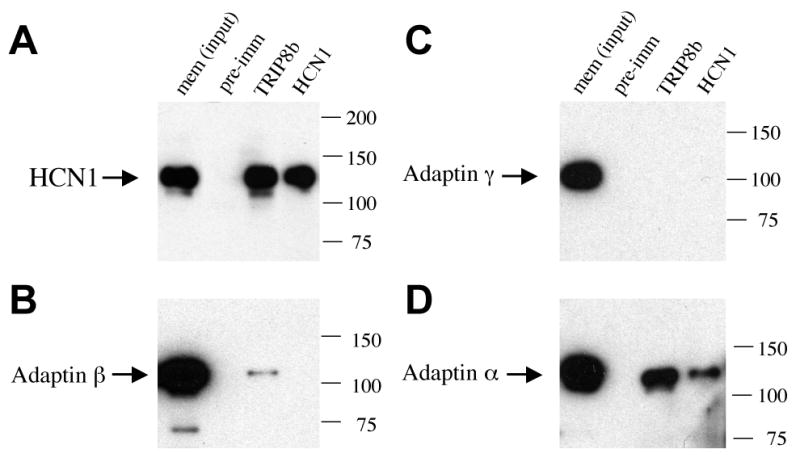
Association of TRIP8b with HCN1 and Adaptor Protein 2 (AP-2) in forebrain.
Western blots of total protein from solubilized membrane extracts of adult mouse forebrain cortex (input lane) or proteins immunoprecipitated using pre-immune serum (pre-imm lane), an anti-TRIP8b antibody (TRIP8b lane) or an anti-HCN1 antibody (HCN1 lane). Gels were probed with antibodies to HCN1 (A), adaptin β (B), adaptin γ (C) or adaptin α (D). In (A), input sample contained 50% of extract subjected to immunoprecipitation reactions. Note that TRIP8b antibody co-precipitates a large fraction of total HCN1. In (B), input sample contained 10% of extract subjected to immunoprecipitation reactions, and in (C,D) it contained 5% of extract. TRIP8b antibody co-precipitates adaptin β (weak signal) and adaptin α, but not adaptin γ. Anti-HCN1 antibody co-precipitates adaptin α. Molecular weight marker positions are indicated to the right of each panel.
Next we examined whether AP trafficking proteins are, in fact, associated with the HCN1/TRIP8b complexes. When the brain proteins co-immunoprecipitated with anti-TRIP8b antibodies were probed with antibodies directed to adaptin β (recognizing the β subunit of both adaptor proteins AP-1 and AP-2), we detected a weak but specific positive signal (Figure 3B). Further probing of the complexes with antibodies directed to adaptin γ (the γ subunit specific to AP-1) and adaptin α (the α subunit specific to AP-2) revealed that the in vivo association with TRIP8b was specific for AP-2, with no detectable levels of AP-1 complexes (Figure 3C, 3D).
TRIP8b inhibits HCN1 channel opening
Many auxiliary subunits alter channel gating in addition to regulating trafficking. We speculated that the interaction of TRIP8b with HCN1 might account for certain discrepancies between the properties of native and heterologously expressed Ih. For example, the voltage-dependent opening of native Ih in hippocampal CA1 neurons, which express mainly HCN1 and HCN2 subunits, occurs at significantly more negative potentials than does that of recombinant heteromeric channels formed from these same subunits (e.g. see Magee, 1998; Chen et al., 2001a). Similarly, the efficacy of cAMP to facilitate the opening of Ih in CA1 neurons (Pedarzani and Storm, 1995; Gasparini and DiFrancesco, 1999; Franz et al., 2000) is considerably less than that seen with the recombinant HCN1/HCN2 heteromers (Chen et al., 2001a).
To address the possibility that TRIP8b alters HCN1 channel gating, we compared the voltage-dependence of channel activation using a two-microelectrode voltage clamp of intact oocytes when HCN1 is coexpressed with GFP or with different TRIP8b isoforms. In contrast to the differential effects of the TRIP8b isoforms on HCN1 trafficking, all splice variants tested exert a similar inhibitory effect on HCN1 opening, shifting the voltage at which HCN1 channels are half-activated (V1/2) by ∼10 mV towards more negative potentials (Figure 4C; Table 1). Although the near complete downregulation of HCN1 current with TRIP8b(1b-2) prevented us from examining its effects on channel gating, the TRIP8b(1b-2)Y38A/L41A exon 2 mutant, which does not downregulate HCN1, also exerts a hyperpolarizing shift in HCN1 gating. Furthermore, all TRIP8b isoforms examined significantly slow the kinetics of channel opening and speed the kinetics of channel closing (Figure 4D). The mean V1/2 ± s.e.m. values for HCN1 coexpressed with either GFP (as a control) or a TRIP8b splice variant were as follows: HCN1 + GFP, -61.7 ± 0.2 mV (n=140); HCN1 + TRIP8b(1a-4): -73.3 ± 0.6 mV (n=16); HCN1 + TRIP8b(1a-2): -71.0 ± 0.5 mV (n=16); HCN1 + TRIP8b(1b-2)Y38A/L41A, -72.4 ± 0.5 mV (n=15); HCN1 + TRIP8b(1b-2-4), -76.4 ± 0.7 mV (n=9); HCN1 + TRIP8b(1a-2-4), -71.4 ± 0.3 mV (n=35); HCN1 + TRIP8b(1a), -75.1 ± 0.6 mV (n=29).
Figure 4.
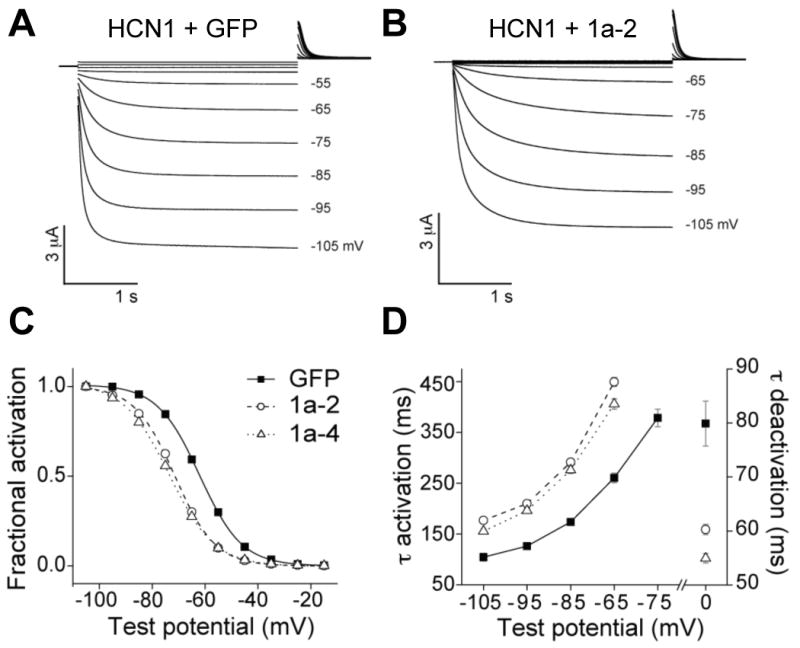
Coexpression with TRIP8b inhibits the opening of HCN1 channels.
(A) Two microelectrode voltage-clamp current traces from oocytes coinjected with cRNA encoding HCN1 and GFP. Three second voltage steps applied from a holding potential of -30 mV to a range of test potentials between +5 mV and -105 mV in increments of 10 mV, followed by a depolarizing step to 0 mV.
(B) Two microelectrode voltage-clamp current records from oocytes coinjected with cRNA encoding HCN1 and TRIP8b(1a-2). Protocol as above.
(C) Normalized tail current activation curves with fits of Boltzmann equation for HCN1 expressed with GFP (squares), TRIP8b(1a-2) (circles) or TRIP8b(1a-4) (triangles). Mean values for V1/2 ± s.e.m. (n) were as follows. HCN1 + GFP: -61.7 ± 0.2 mV (n=140); HCN1 + TRIP8b(1a-4): -73.3 ± 0.6 mV (n=16); HCN1 + TRIP8b(1a-2): -71.0 ± 0.5 mV (n=16) (P<10-4; ANOVA, Tukey HSD test). Note that there is no significant difference in the midpoint of activation of HCN1 channels in the presence or absence of GFP (HCN1 alone, V1/2 = -62.8 ± 0.5 mV, n=19).
(D) Time constants of current activation during hyperpolarizing steps to indicated voltages and time constant of deactivation during step to 0 mV (right axis).
Intriguingly, the changes in HCN1 channel gating observed with TRIP8b are the opposite of the effects of cAMP, which shifts the V1/2 to more positive potentials, speeds the kinetics of channel activation and slows the time course of deactivation. Previous studies from our laboratory indicate that basal levels of cAMP in oocytes are sufficient to facilitate the opening of HCN channels to more positive potentials (Chen et al., 2001a; Wang et al., 2002). Thus, TRIP8b might inhibit channel opening by antagonizing the facilitatory action of endogenous levels of cAMP.
To address this possibility, we examined the effects of TRIP8b on the gating of two mutant HCN1 channels in which a conserved arginine residue (R538) in the cyclic nucleotide binding domain was mutated to either alanine (R538A) or glutamate (R538E). These mutations have no effect on channel gating in the absence of cAMP but cause a 20-fold (R538A) or 2000-fold (R538E) decrease in the affinity of the channel for cAMP, preventing basal levels of cAMP from enhancing channel opening (Chen et al., 2001a; Zhou and Siegelbaum, 2007). Thus, if TRIP8b inhibits HCN1 gating by antagonizing the facilitatory action of basal levels of cAMP, then we expect that the gating of the two mutant channels should be unaffected by the presence of TRIP8b.
Under the conditions of our experiments, we find that the voltage-dependent opening of HCN1R538A and HCN1R538E in the absence of TRIP8b is shifted to more negative potentials by ∼10-15 mV relative to wild-type HCN1 (Figure 5A), reflecting the blockade of the normal facilitatory effect of basal cAMP (note: this 10-15 mV shift is greater than the 7 mV shift reported previously by Chen et al., 2001a, due to differences in extracellular recording solutions; see Experimental Procedures). Importantly, the two mutant channels show no change in gating upon coexpression of TRIP8b (Figure 5A). The simplest interpretation of these results is that TRIP8b does indeed inhibit HCN1 gating by antagonizing the actions of basal levels of cAMP. However, these findings do not rule out a more complex scheme, in which TRIP8b inhibits channel gating at a second site that is independent of cAMP action.
Figure 5.
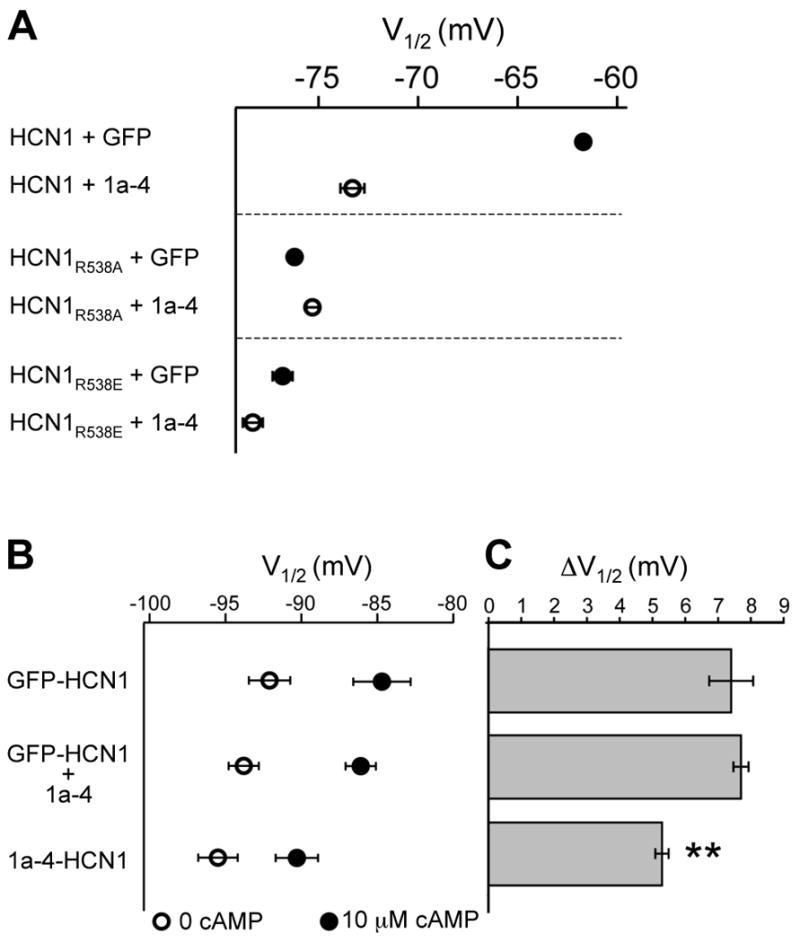
Effects of TRIP8b on the modulation of HCN1 channel gating by cAMP in intact oocytes and cell-free inside-out patches.
(A) In intact oocytes, TRIP8b(1a-4) coexpression shifts the gating of wild-type HCN1 to more negative voltages but does not alter gating of two HCN1 mutants with decreased affinity for cAMP. Oocytes were co-injected with cRNAs encoding wild-type or mutant HCN1 and either GFP or TRIP8b(1a-4). Symbols plot mean V1/2 values in absence (filled symbols) or presence (open symbols) of TRIP8b(1a-4). Error bars show s.e.m. Mean values for V1/2 ± s.e.m. (n) as follows. HCN1 + GFP: -61.7 ± 0.2 mV (n=140); HCN1 + TRIP8b(1a-4): -73.3 ± 0.6 mV (n=16); HCN1R538A + GFP: -76.2 ± 0.3 mV (n=27); HCN1R538A + TRIP8b(1a-4): -75.3 ± 0.3 (n=27); HCN1R538E + GFP: -76.8 ± 0.5 mV (n=18); HCN1R538E + TRIP8b(1a-4): -78.3 ± 0.5 mV (n=21).
(B) In cell-free patches, channels from oocytes expressing a TRIP8b(1a-4)-HCN1 fusion protein show a reduced response to cAMP compared to channels from oocytes expressing either a GFP-HCN1 fusion protein or coexpressing GFP-HCN1 with TRIP8b(1a-4). Patches were held at -30 mV and 3 sec hyperpolarizing steps applied to a series of test potentials from -40 mV to -130 mV. Tail currents were measured upon return to -30 mV. Open symbols show V1/2 in absence of cAMP and filled symbols show V1/2 in presence of a saturating concentration of cAMP (10 μM). Error bars show s.e.m. Mean ± s.e.m (n) values for V1/2 in the absence of cAMP are as follows. GFP-HCN1: -92.1 ± 1.4 mV (n=6); GFP-HCN1 + TRIP8(1a-4): -93.8 ± 1.0 mV (n=8); TRIP8b(1a-4)-HCN1: -95.5 ± 1.3 mV (n=7). There is no significant difference in the V1/2 values of the three populations in the absence of cAMP (P>0.15; ANOVA, Tukey HSD test). There is also no significant difference between the V1/2 values of the GFP-HCN1 fusion protein with or without TRIP8b(1a-4) coexpression in the presence of cAMP. Note that TRIP8b(1a-4) also caused no change in the V1/2 when coexpressed with wild-type HCN1, either in the absence (P>0.9, ANOVA, Tukey HSD test) or presence of cAMP (P> 0.8, ANOVA, Tukey HSD test; HCN1, n=12, HCN1 + TRIP8b(1a-4), n=14).
(C) Effect of 10 μM cAMP measured as difference in V1/2 (ΔV1/2) in the presence and absence of cAMP. Bars show mean ΔV1/2 values; error bars show s.e.m. Mean ΔV1/2 ± s.e.m. values are as follows. GFP-HCN1: 7.4 ± 0.7 mV (n=6); GFP-HCN1 + TRIP8(1a-4): 7.7 ± 0.2 mV (n=8): TRIP8b(1a-4)-HCN1, 5.3 ± 0.2 mV (n=7). The ΔV1/2 for TRIP8b(1a-4)-HCN1 was significantly less than that for GFP-HCN1 or for GFP-HCN1 coexpressed with TRIP8b(1a-4) (P<0.003; ANOVA, Tukey HSD test).
To test more directly the idea that TRIP8b inhibits the action of cAMP, we examined the effects on HCN1 gating when cAMP is applied to the internal surface of the channels in cell-free patches in the absence or presence of TRIP8b. According to the cAMP hypothesis, TRIP8b should have no effect on the gating of HCN1 in the cell-free patches in the absence of cAMP, but should reduce the facilitatory effect of bath-applied cAMP. In accord with the first prediction, the V1/2 of HCN1 channel activation in the absence of cAMP is identical in the absence or presence of TRIP8b(1a-4). However, contrary to the second prediction, TRIP8b coexpression does not alter the response of the channel to cAMP in the cell-free patches. Thus, the maximal shift in the V1/2 of channel activation in response to a saturating concentration (10 μM) of cAMP (7-8 mV) is not altered when HCN1 is coexpressed with TRIP8b (Figure 5B). This indicates that TRIP8b does not alter cAMP efficacy. Moreover, TRIP8b appears to have no effect on the affinity of the channel for cAMP, as the voltage shift in response to a sub-saturating concentration of cAMP that is approximately equal to the EC50 (100 nM) is also unchanged by TRIP8b coexpression (data not shown).
One possible reason why TRIP8b(1a-4) might inhibit the action of cAMP in intact oocytes but fail to antagonize the action of cAMP in inside-out patches is that TRIP8b dissociates from HCN1 upon patch excision. To test this idea we constructed a fusion protein in which the C-terminus of TRIP8b(1a-4) is attached to the N-terminus of HCN1. The TRIP8b-HCN1 fusion protein forms functional hyperpolarization-activated channels whose V1/2 in intact oocytes (TRIP8b-HCN1, V1/2 = -81.1 ± 0.4 mV, n=10) is ∼15 mV more negative than the V1/2 of channels formed by a GFP-HCN1 fusion protein (GFP-HCN1, V1/2 = -64.5 ± 0.2 mV, n=14). This ∼15 mV shift is comparable to the shift we observe when TRIP8b(1a-4) and HCN1 are coexpressed as separate proteins (see Figures 4, 5A, and Table 1), indicating that TRIP8b is able to exert its full inhibitory effect as a fusion protein.
In cell-free patches, GFP-HCN1 and TRIP8b(1a-4)-HCN1 show a similar voltage-dependence of gating in the absence of cAMP, with no significant difference in their V1/2 values (Figure 5B). However, the maximal shift in the V1/2 of the TRIP8b-HCN1 fusion protein in response to a saturating concentration of cAMP is now significantly reduced compared to the maximal response of GFP-HCN1 (Figure 5B, 5C). Such results support the view that TRIP8b does indeed antagonize the ability of cAMP to shift HCN1 channel opening to more positive potentials and that the interaction of TRIP8b with HCN1 may be weakened following patch excision.
TRIP8b(1a-4) enhances endogenous Ih current density and inhibits its gating in hippocampal neurons in vitro
If TRIP8b is indeed an HCN channel auxiliary subunit, we would expect that it should alter the gating and exert isoform-specific effects on the expression of native Ih in neurons, similar to its effects on HCN1 in heterologous systems. We previously reported that TRIP8b(1b-2) drastically reduces endogenous Ih current levels when transfected into hippocampal pyramidal neurons in dissociated cell culture (Santoro et al., 2004). Here we asked whether TRIP8b(1a-4) can increase Ih current density and shift activation to more negative voltages in cultured hippocampal neurons, similar to its effects in Xenopus oocytes.
Whole-cell recordings were obtained from neurons infected with a lentivirus expressing either GFP or GFP-tagged TRIP8b(1a-4). We found that TRIP8b(1a-4) produces a ∼2.5-fold increase in Ih current density, an ∼8.5 mV negative shift in the midpoint of Ih activation, and an increase in the time constant of Ih activation at all voltages tested (Figure 6), effects similar to those observed in the oocytes. When combined with our previous results that Ih in hippocampal neurons is downregulated by TRIP8b(1b-2), these data verify the ability of distinct TRIP8b isoforms to either upregulate or downregulate the expression of Ih and to produce an inhibitory effect on the opening of native HCN channels in neurons.
Figure 6.
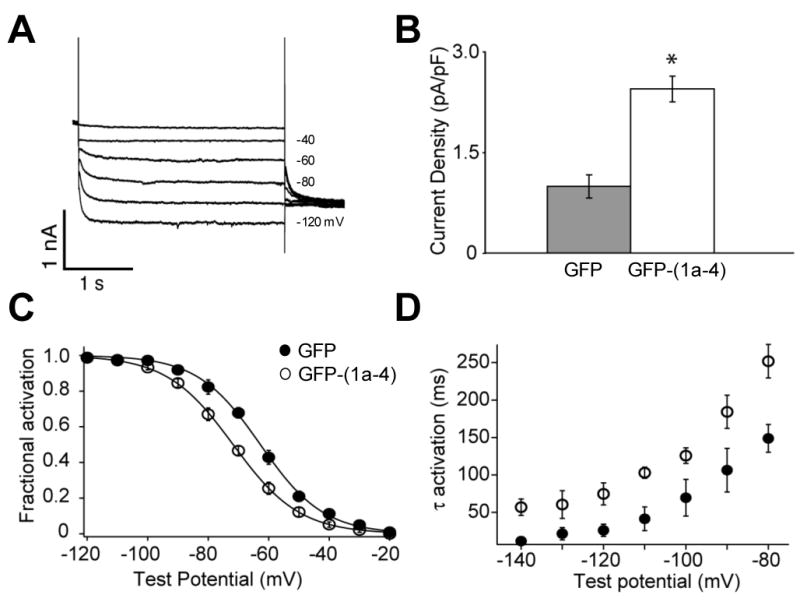
Over-expression of TRIP8b (1a-4) in cultured hippocampal neurons alters the expression and gating properties of endogenous Ih.
(A) Ih current traces from a whole-cell voltage-clamp recording of a cultured hippocampal pyramidal neuron over-expressing GFP-TRIP8b(1a-4). Currents were elicited by a series of 3-second hyperpolarizing voltage steps from a holding potential of -65 mV to a range of test potentials between -20 mV and -120 mV in increments of 10 mV, followed by a step to -100 mV. Every second trace is displayed.
(B) TRIP8b(1a-4) enhances maximal Ih tail current density (normalized by capacitance). Mean density ± s.e.m. (n) as follows. GFP: 0.99 ± 0.17 pA/PF (n=8); GFP-TRIP8b(1a-4): 2.44 ± 0.2 pA/PF (n=6), P<0.001 (t-test).
(C) TRIP8b(1a-4) shifts native Ih activation to more negative potentials. Tail current activation curves fit by Boltzmann equation for group data. Mean V1/2 values ± s.e.m. (n) as follows. GFP: -63.7 ± 1.4 mV (n=8); GFP-TRIP8b(1a-4): -72.12 ± 0.7 mV (n=6); P<0.001, t-test.
(D) TRIP8b(1a-4) slows the time course of Ih activation. Single exponential time constants plotted as function of voltage during the hyperpolarizing steps.
Effects of TRIP8b(1a-4) and TRIP8b(1b-2) over-expression in hippocampal CA1 pyramidal neurons in vivo
To determine whether TRIP8b isoforms can differentially regulate HCN1 expression in neurons in the intact hippocampus, we injected lentivirus expressing GFP-TRIP8b(1a-4), GFP-TRIP8b(1b-2), or GFP alone into the CA1 region of adult mice in vivo. This method achieves stable protein expression that persists for several weeks. Surprisingly, none of the TRIP8b constructs alter the expression or distribution of endogenous HCN1 channels in wild-type mice, as assayed by HCN1 immunofluorescence, even 3-4 weeks after viral injection and despite the strong expression of the lentivirus-driven TRIP8b proteins (data not shown).
We reasoned that virally expressed TRIP8b may fail to regulate HCN1 channels in adult brain if the channels preexist in stable complexes with endogenous TRIP8b. We therefore examined the effects of TRIP8b using co-injection of independent lentivirus vectors that express a GFP-HCN1 fusion protein and HA-tagged TRIP8b in the background of HCN1 knockout mice (Nolan et al., 2004), to ensure that both HCN1 and TRIP8b proteins would be synthesized simultaneously. Indeed, this approach reveals dramatic, isoform-dependent effects of TRIP8b on the expression and trafficking of the GFP-HCN1 fusion protein (Figure 7).
Figure 7.
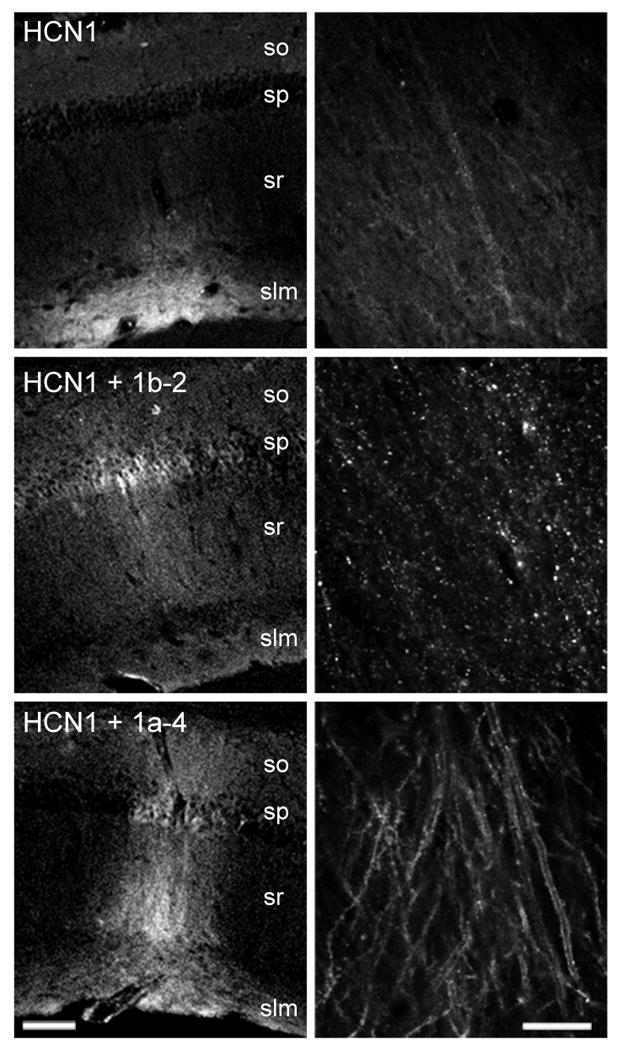
Over-expression of TRIP8b(1b-2) and TRIP8b(1a-4) in hippocampal CA1 neurons in vivo affects the intracellular distribution of HCN1. Lentiviruses driving expression of a GFP-HCN1 fusion protein were stereotaxically injected in vivo in the CA1 region of the hippocampus of HCN1 knockout mice, along with a second lentivirus driving expression of dsRed2 (top), HA-TRIP8b(1b-2) (middle), or HA-TRIP8b(1a-4) (bottom). Confocal images from thin, fixed hippocampal slices show direct GFP fluorescence, as a marker of injected GFP-HCN1 channel protein expression and localization. so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; slm, stratum lacunosum-moleculare. Left, low magnification images (scale bar, 100 μm). Right, high magnification images at the level of stratum radiatum (scale bar 20 μm). Notice the railroad-track pattern of intense GFP-HCN1 signal upon coexpression with TRIP8b(1a-4) representing enhanced membrane signal in CA1 pyramidal neuron apical dendrites as observed in confocal optical cross-section. Also note the appearance of intracellular puncta upon coexpression with TRIP8b(1b-2). Representative images from the following total number of experiments (n, number of independent injection sites): GFP-HCN1 + dsRed2, n=35 (12 animals); GFP-HCN1 + HA-TRIP8b(1a-4), n=16 (5 animals); GFP-HCN1 + HA-TRIP8b(1b-2), n=6 (2 animals).
When GFP-HCN1 is coexpressed with the soluble fluorescent protein dsRed2 (as a control), we observe a high level of expression of channels that are efficiently targeted to the distal portion of the apical dendrites, identical to the distribution of endogenous HCN1 protein normally observed in wild-type mice (e.g. Santoro et al., 1997, 2004). Coexpression of GFP-HCN1 with TRIP8b(1a-4) strongly enhances the overall expression of the GFP-HCN1 fusion construct, with intense GFP-HCN1 labeling of apical dendrites as well as enhanced labeling of basal dendrites and soma (Figure 7). The enhanced railroad-track pattern of the GFP-HCN1 signal in the CA1 apical dendrites is strongly suggestive of increased HCN1 channel expression in the dendritic surface membrane viewed in a confocal cross-section along the longitudinal axis of the dendrite (Figure 7, bottom right). In stark contrast, coexpression of GFP-HCN1 with TRIP8b(1b-2) results in the sequestering of the GFP-HCN1 channel into intracellular puncta, visible both at the level of the soma and apical dendrites (Figure 7, middle right), consistent with our previous results in heterologous cells (Santoro et al., 2004). Thus, TRIP8b exerts similar isoform-dependent actions on HCN1 channel expression in Xenopus oocytes, isolated hippocampal neurons in vitro, and CA1 pyramidal neurons in vivo.
Discussion
This study demonstrates that TRIP8b is an auxiliary subunit that associates with HCN1 channels in the brain, thus regulating channel surface expression and gating. We identify nine different TRIP8b splice variants in brain and demonstrate the differential effects of six of these isoforms on HCN1 surface expression. TRIP8b(1b-2) and TRIP8b(1b-2-4) cause a near complete loss of surface expression; TRIP8b(1a) causes a smaller but still marked ∼10-fold decrease in HCN1 current density; TRIP8b(1a-2) has no effect on surface expression; and TRIP8b(1a-4) and TRIP8b(1a-2-4) cause up to a ∼6-fold increase in surface expression. In distinction to the differential effects of TRIP8b isoforms on channel expression, all splice variants examined exert a similar action to inhibit HCN1 channel gating, which involves a shift in the voltage-dependence of channel opening to more negative potentials, a slowing of the rate of activation upon hyperpolarization, and a speeding of the rate of deactivation upon depolarization.
The isoform-specific effects of TRIP8b on channel surface expression and the invariant inhibitory action on channel opening are also observed with native Ih in cultured hippocampal pyramidal neurons. Moreover, TRIP8b isoforms also differentially regulate surface expression of HCN1 in hippocampal CA1 pyramidal neurons in vivo. These results, together with the robust biochemical association between HCN1 and TRIP8b in vivo (Figure 3) and the tight co-localization of the two proteins in the distal dendrites of cortical pyramidal neurons (Santoro et al., 2004), strongly suggest that TRIP8b plays an important role in the regulation of Ih in the brain.
Trafficking motifs within TRIP8b
Specific sequences within the N-terminus of the TRIP8b protein mediate the inhibitory effects on HCN1 surface expression, most likely by recruiting AP-2 adaptor complexes that promote channel internalization through clathrin-mediated endocytosis (Bonifacino and Traub, 2003). Our mutagenesis experiments establish the role of the YXXL and EXXXLL consensus AP-binding site motifs, present in exon 2 and exon 5, respectively, in the action of distinct TRIP8b isoforms to decrease the surface expression of HCN1 channels. This interpretation is supported by the recent identification of AP-2 subunits and clathrin binding to immobilized recombinant TRIP8b following affinity chromatography of brain extracts (Popova et al., 2008). Interestingly, the effects of the trafficking motifs present within specific TRIP8b exons on HCN1 channel expression are not autonomous but appear to be sensitive to neighboring protein sequence. Thus, although TRIP8b(1a-2) contains the YXXL motif in exon 2, this isoform has little effect on HCN1 expression levels. Moreover, whereas the EXXXLL motif in exon 5 is present in all TRIP8b isoforms, this motif induces a downregulation of HCN1 current density only in the background of TRIP8b(1a).
Although we have not identified specific sequences that mediate the upregulation of HCN1 current density by TRIP8b(1a-4) and TRIP8b(1a-2-4), upregulation may be a latent property of all TRIP8b isoforms, rather than a specific effect of exon 4. Thus, despite the presence of exon 4, TRIP8b(1b-2-4) causes a near-complete reduction in surface expression, similar to the effect of TRIP8b(1b-2). Moreover, TRIP8b(1a) carrying mutations in the EXXXLL motif of exon 5 or TRIP8b(1a-2) carrying mutations in the YXXL motif of exon 2 cause a significant increase in HCN1 current density above baseline levels, despite the absence of exon 4.
Upregulation of current density by wild-type TRIP8b(1a-4), TRIP8b(1a-2-4) and the mutated isoforms is likely to be caused by an increase in HCN1 surface expression rather than an effect on channel gating as all TRIP8b isoforms have an inhibitory effect on channel opening. Moreover, in vivo expression of TRIP8b(1a-4) produces a marked increase in the level of GFP-tagged HCN1 protein associated with the dendritic plasma membrane (Figure 7). Whether the positive effect of TRIP8b on HCN1 surface expression is due to increased protein translation, protein stabilization, or enhanced trafficking to the surface membrane remains to be determined.
Mechanism of the effects of TRIP8b on HCN1 gating
The facilitation of HCN channel opening by the direct binding of cAMP provides the best known means of regulating HCN channel function (Robinson and Siegelbaum, 2003; Craven and Zagotta, 2006). In cell-free patches, submicromolar concentrations of cAMP shift the gating of HCN channels to more positive voltages. This high sensitivity of HCN channels to ligand enables their opening in intact cells to be enhanced by low basal levels of cAMP (Chen et al., 2001a; Wang et al., 2002). Our results suggest that the inhibitory effects of TRIP8b on HCN1 channel opening in intact cells may be due to an antagonism of these facilitatory effects of basal levels of cAMP as we find that TRIP8b does not alter the gating of mutant HCN1 channels (R538A or R538E) that do not bind cAMP with high affinity. However, this simple view is not supported by our finding that, in cell-free patches, the response of HCN1 channels to direct application of cAMP is unaffected by coexpression TRIP8b. This negative result may be due, in part, to the failure of TRIP8b to remain associated with HCN1 channels in cell-free patches as a TRIP8b-HCN1 fusion protein does exhibit a reduced response to cAMP.
In contrast to our findings with HCN1, a recent study demonstrates that HCN2 channels in cell-free patches do show a reduced response to cAMP when coexpressed with TRIP8b as independent proteins (Zolles et al., 2009, this issue of Neuron). This may indicate that HCN2 and TRIP8b form a more stable complex than do HCN1 and TRIP8b. Nonetheless, our findings with HCN1, and preliminary results with HCN2 (Lei Hu, unpublished data) indicate that the extent to which TRIP8b reduces the maximal voltage shift produced by a saturating concentration of cAMP in cell-free patches, either when TRIP8b is coexpressed with HCN2 or fused to HCN1, is significantly less than the extent to which TRIP8b shifts the voltage-dependent gating of either HCN1 or HCN2 in intact cells. This quantitative discrepancy between whole-cell and cell-free results may indicate that the ability of TRIP8b to inhibit HCN channel opening, either by antagonizing the action of cAMP or through some other mechanism, requires an intracellular factor that is lost upon patch excision.
Potential roles of TRIP8b in regulating HCN1 expression and function in vivo
To date, our understanding of the mechanisms that modulate HCN channel function in vivo is limited. The opposing effects of different TRIP8b isoforms to upregulate or down-regulate levels of HCN1 expression provide an attractive molecular mechanism to mediate the reported up- and downregulation of Ih and HCN1 levels as a function of neural activity. For example, in CA1 neurons relatively weak patterns of neuronal activation that induce long-term depression of excitatory synaptic transmission down-regulate Ih, producing a homeostatic enhancement in dendritic integration and excitability (Brager and Johnston, 2007). In contrast, a very strong pattern of neuronal activity associated with the induction of long-term potentiation upregulates Ih, producing a homeostatic inhibitory effect on dendritic integration (Fan et al., 2005; Campanac et al., 2008). Slightly weaker patterns of activity that induce a moderate amount of long-term potentiation lead to a downregulation of Ih, representing a complementary excitatory effect to enhance dendritic integration (Wang et al., 2003; Campanac et al., 2008).
In entorhinal cortex and hippocampal CA1 pyramidal neurons, sites of high TRIP8b expression, intense levels of neuronal activity during seizures lead to a long-lasting downregulation of Ih current density, which increases neuronal excitability and may contribute to the development of epilepsy (Shah et al., 2004; Shin et al., 2008). Interestingly, this latter effect on Ih is independent of long-term changes in HCN protein levels. An increase in the expression of TRIP8b splice variants that cause HCN1 internalization could produce such a long-term downregulation of Ih current density without altering total protein levels. Recent findings indicate that dendrites are not only endowed with the ability to initiate regulated, local mRNA translation, but also possess RNA splicing capability (Glanzer et al., 2005). Thus, the hypothesis that neuronal activity might control the alternative splicing of TRIP8b isoforms, potentially on a local level, represents an intriguing area of investigation.
Changes in the relative mRNA or protein levels of individual TRIP8b splice variants due to alterations in transcription, RNA processing or translation provide a potential means for regulating Ih on a relatively slow time scale, requiring minutes to hours to manifest. However, it is also possible that TRIP8b might mediate effects on a more rapid time scale due to changes in protein phosphorylation. Activation of different protein kinases, for example CaMKII or PKC, underlies the rapid, activity-dependent modulation of Ih in hippocampal slices in response to different patterns of neuronal activity (Fan et al., 2005; Brager and Johnston, 2007). Based on sequence analysis, several potential serine/threonine phosphorylation sites are present on the different TRIP8b splice variants. These sites could modulate the binding of TRIP8b to trafficking factors or to HCN1 itself, thus providing a mechanism to achieve rapid decreases (or increases) in net Ih density.
In hippocampal CA1 neurons Ih is likely to be comprised of both HCN1 and HCN2 (Santoro et al., 2000). Although here we focus on the effects of TRIP8b on HCN1, we previously found that TRIP8b(1b-2) produces a potent downregulation in the surface expression of HCN2, which also contains the C-terminal SNL tripeptide-TRIP8b binding site (Santoro et al., 2004). As discussed above, a recent study (Zolles et al., 2009, this issue of Neuron) and our preliminary results (Lei Hu, unpublished data) indicate that the gating of HCN2 is regulated by TRIP8b in a manner similar to that of HCN1. However, our preliminary results and those of Zolles et al. (2009) indicate that TRIP8b isoforms may produce differential effects on the surface expression of HCN2 compared to HCN1. Interestingly, discordant changes in the expression of HCN1 versus HCN2 subunits as a result of neuronal activity have been described in a number of paradigms (e.g. Brewster et al., 2002, 2005; Fan et al., 2005; Jung et al., 2007; Shin et al., 2008). In particular, following kainate-induced status epilepticus, there is a specific downregulation and redistribution of HCN1 protein from the distal dendrites to the soma of hippocampal pyramidal neurons; in contrast, the level and distribution of HCN2 subunits are unaltered (Shin et al., 2008). Furthermore, there is a decrease in the amount of TRIP8b associated with HCN1, but not HCN2. Differences in the nature and effect of HCN channel interactions with TRIP8b could thus provide the molecular means to achieve such subunit-specific modulation.
In addition to regulating HCN1 expression, the strong influence of TRIP8b on HCN1 gating in intact cells is likely to modulate the behavior of Ih in vivo. Interestingly, we find that the voltage-dependence of Ih in cultured dissociated hippocampal neurons from newborn animals, which express low levels of TRIP8b, is significantly more positive than that of Ih in CA1 neurons from adult mice, where high endogenous levels of TRIP8b are present (R.A. Piskorowski, unpublished results). We find here that over-expression of TRIP8b in the cultured neurons shifts the voltage-dependence of Ih activation to more negative potentials, more closely matching the voltage-dependence of Ih in CA1 neurons from adults. Of further interest, the activation of Ih in CA1 distal apical dendrites is shifted by ∼6 mV to more negative voltages than those required to activate Ih in more proximal regions of the dendrite (Magee, 1998), an effect that might be explained by a more prominent association of TRIP8b with HCN1 channels in the distal dendrites, where TRIP8b and HCN1 co-localization is tightest (Santoro et al., 2004). The high levels of TRIP8b in neurons may also explain why the effect of cAMP on native Ih in CA1 pyramidal neurons (Pedarzani and Storm, 1995; Gasparini and DiFrancesco, 1999; Franz et al., 2000) is much less than the effect of cAMP on HCN1/HCN2 heteromers in heterologous cells (Chen et al., 2001a).
In conclusion, our findings strongly suggest that TRIP8b is a critical factor in regulating the gating and trafficking of HCN channel subunits in the brain to dynamically control neuronal activity. Moreover, such TRIP8b/HCN channel interactions are likely to have important consequences for the modulation of neural function both in physiological states as well as in disease.
Experimental procedures
Plasmids, molecular biology and heterologous expression
Oocyte expression
All constructs were cloned into plasmids pGHE or pGH19, linearized, and transcribed into cRNA using T7 polymerase (MessageMachine, Ambion) as described (Santoro et al., 2004). TRIP8b cDNA constructs were generated by reverse transcription-PCR (RT-PCR) from polyA+ RNA from adult mouse brain (see below). Site-directed mutagenesis was performed using QuickChange Mutagenesis kit (Stratagene), and polymerase chain reaction (PCR) cloning. Oocytes were injected with 50 nl of cRNA solution each, at a constant concentration of 0.5 mg/ml for HCN channel constructs and 0.2 mg/ml for GFP or TRIP8b constructs.
Lentivirus expression
The lentiviral expression vector containing the CaMKII promoter, pFCK(0.4)GW was kindly provided by Pavel Osten (Max Planck Institute, Heidelberg; Dittgen et al., 2004). Each lentiviral expression plasmid was transfected into HEK293FT cells with virus packaging plasmids pVSVg and pΔ8.9. After 24-48 hours the culture media containing assembled viruses was filtered, placed over a 20% sucrose cushion and spun at 26000 rpm for 3 h to purify the virus. The pellet was resuspended in sterile saline and stored at -80 °C. Virus titers were measured using high-density cultured hippocampal neurons. For in vivo delivery, virus was concentrated to 107 IU/ml in sterile saline and stereotaxically injected into the hippocampal CA1 region of adult mice (age 3-9 months) as described (Zakharenko et al., 2003). HCN1 knockout animals (genotype HCN1-/-) were generated and bred as described (Nolan et al., 2003). For each animal, four injections were performed: dorsal region of left and right hippocampus, ventral region of left and right hippocampus.
Real-time PCR
PolyA+ RNA from adult mouse brain (Clontech) was used to generate first strand cDNA using random hexamer priming (Superscript, Invitrogen). Standard curves were generated using plasmids encoding the full-length sequence for each TRIP8b variant, added in 4-fold increments (15.6 fg, 62.5 fg, 250 fg, 1000 fg per reaction). A negative control without DNA was run in all experiments, and reactions in which contaminating DNA copy number exceeded 10% of the 15.6 fg titration point were eliminated. cDNA was added to each reaction in 4-fold increments (corresponding to 1 ng, 4 ng, 16 ng of initial polyA+ RNA). For each isoform, two sets of primers were used: an isoform specific set (spanning the exon-exon junctions corresponding to that splice form) and a reference set amplifying a sequence in the common region of TRIP8b mRNA (Supplementary Table 1). For each determination, both sets were run against the same reference plasmid DNA, as well as against the cDNA sample, and values obtained for cDNA samples plotted against the standard curve. Accordingly, the copy number of each splice variant is expressed as a fraction of the total TRIP8b mRNA copy number, with both values determined using the same reference plasmid DNA to correct for errors due to variability in the purity of DNA. Only primer pairs yielding the same reaction efficiency when tested against reference plasmid DNA or cDNA were used. All PCR experiments were performed in a GeneAmp 5700 Sequence Detector from Applied Biosystems (Foster City, CA). Samples were run in triplicate using SYBR Green PCR Master Mix (Applied Biosystems). Reaction products were run on a 2% agarose gel to check for expected amplicon size. In each case, a dissociation (melting) curve was evaluated for the presence of a single final product.
Oocyte electrophysiology
Two-microelectrode voltage-clamp recordings were obtained 3 d after cRNA injection, using a Warner Instruments OC-725C amplifier. Data were digitized and acquired with an ITC-16 interface (Instrutech), filtered at 1 KHz and sampled at 2 KHz, and analyzed using Pulse software (HEKA). Microelectrodes filled with 3 M KCl had resistances of 0.5-2.0 MΩ. Oocytes were bathed in extracellular solution containing (in mM): 94 NaCl, 4 KCl, 10 Hepes, and 2 MgCl2, pH 7.5. This solution is more physiological than the isotonic KCl solution used in some of our previous studies, and results in some differences in HCN1 V1/2 values compared to prior work (e.g. Chen et al., 2001a). Three-second-long voltage steps were applied in 10 mV increments from a holding potential of -30 mV. Peak tail-current amplitudes were measured at 0 mV after the decay of the capacitive transient, and tail-current I-V curves fitted using the Boltzmann equation: I(V) = A1 + A2/{1 + exp[(V-V1/2)/s]} in which A1 is the offset caused by holding current, A2 is the maximal tail current amplitude, V is the test pulse voltage, V1/2 is the midpoint voltage of activation, and s is the slope (in mV). Time constants of Ih activation and deactivation were obtained by fitting a single exponential function, I=I0exp(-t/τ), following an initial delay, to current activation time courses during the hyperpolarizing steps and to tail current decay following the step to 0 mV, respectively.
For inside-out patch clamp measurements, recordings were obtained 3-5 d after cRNA injections. Data were acquired, filtered and analyzed as described above. The pipette solution contained (in mM): 97 KCl, 1 NaCl, 10 HEPES, and 1 MgCl2, 1.8 CaCl2, pH 7.4 with KOH; the bath solution contained (in mM): 107 KCl, 5 NaCl, 10 HEPES, 1 MgCl2, and 1 EGTA, pH 7.3 with KOH. The Ag-AgCl ground wire was connected to the bath solution by a 3 M KCl agar bridge electrode. Three-second long hyperpolarizing voltages were applied in 10 mV step increments from a holding potential of -30 mV. Tail currents were measured at -30 mV. All recordings were obtained at room temperature (22-24 °C).
Brain extract analysis and immunohistochemistry
Brain tissue was homogenized in ice cold 10 mM Tris-HCl (pH7.4), 320 mM sucrose, 5 mM EDTA, with protease inhibitor cocktail (CompleteMini, Roche) in a glass-teflon-type homogenizer, centrifuged for 5 min at 3000g, the supernatant collected and spun for 20 min at 100,000 g. The pellet was resuspended in homogenization buffer (as above) including 150 mM NaCl and 1% Triton, and protein concentration determined using Micro BCA reagent (Pierce). Immunoprecipitation was carried out on 100 μg of solubilized membrane extract in 400 μl of homogenization buffer including 150 mM NaCl and 0.2% Triton, overnight at 4 °C. Samples were eluted directly in 2× Laemmli buffer, and Western blotting was performed as described (Nolan et al., 2003). Primary antibodies: rat monoclonal anti-HCN1 (7C3), gift of F. Müller and B. Kaupp; rabbit polyclonal anti-TRIP8b (794, produced in our laboratory, see below); mouse monoclonals anti-adaptin α, β and γ (all BD Biosciences). Secondary antibodies: HRP-anti-rat conjugate, HRP-anti-mouse conjugate (Jackson ImmunoResearch), HRP-anti-rabbit conjugate (Cell Signaling). Protein bands were visualized by chemiluminescence using SuperSignal reagent (Pierce).
For immunohistochemistry, animals were perfused with ice-cold 1× PBS followed by 4% paraformaldehyde in 1× PBS, 40 μm slices cut with a vibratome, and permeabilized in PBS+0.1% Triton, followed by incubation in blocking solution (PBS+3% normal goat serum). Primary antibody incubation was carried out in blocking solution overnight at 4 °C. Antibodies used were: rat monoclonal anti-HCN1 (7C3), rat monoclonal anti-HA (Roche), rabbit anti-TRIP8b (794). Rhodamine-red conjugated goat anti-rat and Cy5-conjugated goat anti-rabbit secondary antibodies were from Jackson ImmunoResearch; GFP was visualized by direct fluorescence. Slices were mounted with Gel/Mount (Biomeda, CA), and fluorescence imaging performed on an inverted laser scanning confocal microscope (BioRad MRC 1000).
For antibody generation, a fragment corresponding to amino acids 1-176 of TRIP8b(1a-4) was subcloned into vector pGEX-4T-1 (Pharmacia) and gluthatione-S-transferase (GST-)TRIP8b fusion protein purified as described (Santoro et al., 1997, 2004). Polyclonal antibodies were generated in rabbit (Covance), and serum 794 tested for specificity by Western blot analysis using protein extracts from Xenopus oocytes expressing each of the cloned TRIP8b isoforms. The serum showed comparable affinity for all isoforms, and recognized two prominent protein species in extracts from mouse brain membranes (see Supplemental Figure 2).
Hippocampal cultures and electrophysiology
The hippocampus was dissected from the brains of P0-P1 rats, digested with papain, washed with protease inhibitor, then triturated, and neurons counted and plated onto differentiated primary astrocyte feeder layers. Neurons were grown in Neurobasal media supplemented with B27, glutamine and antibiotics. Cultures were infected with lentivirus expressing either GFP or a GFP-TRIP8b fusion construct under the control of a 0.4 kb fragment of the CaMKII promoter (vector pFCK(0.4)GW, see above).
Hippocampal cultures were used for whole-cell recordings 7-17 days after infection. Whole-cell patch recordings were obtained from pyramidal-shaped neurons using 2-3 MΩ borosilicate glass pipettes. Intracellular solution was (in mM): KMethylsulfate (130), KCl (10), HEPES (10), NaCl(4), EGTA (0.1), MgATP (4), Na2GTP (0.3), Phosphocreatine (10). The external bath solution contained (in mM): 145 NaCl, 3 KCl, 10 HEPES, 3 CaCl2, 8 glucose, 2 MgCl2 (pH adjusted to 7.30 with NaOH). Only recordings with uncompensated whole-cell series resistance below 15 MΩ were used for analysis. Series resistance compensation of 80-90% was used. To eliminate spontaneous activity, 2 μM tetrodotoxin (TTX) was added to the bath solution after establishing that a cell displayed the expected action potential shape and firing pattern for a pyramidal neuron. All data were obtained in pairs, with one GFP-expressing neuron matched with each GFP-TRIP8b expressing neuron, as described (Santoro et. al 2004).
Supplementary Material
Acknowledgments
This work was partially supported by grants NS36658 and MH80745 from NIH to S.A.S, by a Fellowship from the Italian Academy for Advanced Studies in America (B.S.) and a Research Grant from the Epilepsy Foundation (B.S.). We thank Nikolaj Klöcker, Bernd Fakler and colleagues for sharing with us their unpublished data that first suggested that TRIP8b inhibits the facilitatory action of cAMP on HCN2 channels. We are grateful to Catherine Smith, Alexander Kushnir, and Jayeeta Basu for their valuable contributions during the course of this study, and to John Riley for his excellent technical assistance. We thank Pavel Osten for his kind gift of plasmid pFCK(0.4)GW, and Frank Müller for generously providing the HCN1-7C3 monoclonal antibody.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blatch GL, Lassle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Brager DH, Johnston D. Plasticity of intrinsic excitability during long-term depression is mediated through mGluR-dependent changes in I(h) in hippocampal CA1 pyramidal neurons. J Neurosci. 2007;27:13926–13937. doi: 10.1523/JNEUROSCI.3520-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster A, Bender RA, Chen Y, Dube C, Eghbal-Ahmadi M, Baram TZ. Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J Neurosci. 2002;22:4591–4599. doi: 10.1523/JNEUROSCI.22-11-04591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster AL, Bernard JA, Gall CM, Baram TZ. Formation of heteromeric hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in the hippocampus is regulated by developmental seizures. Neurobiol Dis. 2005;19:200–207. doi: 10.1016/j.nbd.2004.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanac E, Daoudal G, Ankri N, Debanne D. Downregulation of dendritic Ih in CA1 pyramidal neurons after LTP. J Neurosci. 2008;28:8635–8643. doi: 10.1523/JNEUROSCI.1411-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med. 2001;7:331–337. doi: 10.1038/85480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol. 2001a;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven KB, Zagotta WN. CNG and HCN channels: two peas, one pod. Annu Rev Physiol. 2006;68:375–401. doi: 10.1146/annurev.physiol.68.040104.134728. [DOI] [PubMed] [Google Scholar]
- Dittgen T, Nimmerjahn A, Komai S, Licznerski P, Waters J, Margrie TW, Helmchen F, Denk W, Brecht M, Osten P. Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc Natl Acad Sci U S A. 2004;101:18206–18211. doi: 10.1073/pnas.0407976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h) Nat Neurosci. 2005;8:1542–1551. doi: 10.1038/nn1568. [DOI] [PubMed] [Google Scholar]
- Franz O, Liss B, Neu A, Roeper J. Single-cell mRNA expression of HCN1 correlates with a fast gating phenotype of hyperpolarization-activated cyclic nucleotide-gated ion channels (Ih) in central neurons. Eur J Neurosci. 2000;12:2685–2693. doi: 10.1046/j.1460-9568.2000.00151.x. [DOI] [PubMed] [Google Scholar]
- Gasparini S, DiFrancesco D. Action of serotonin on the hyperpolarization-activated cation current (Ih) in rat CA1 hippocampal neurons. Eur J Neurosci. 1999;11:3093–3100. doi: 10.1046/j.1460-9568.1999.00728.x. [DOI] [PubMed] [Google Scholar]
- George MS, Abbott LF, Siegelbaum SA. Hyperpolarization-activated cation channels inhibit inhibit EPSPs by interactions with M-type K+ channels. Nat Neurosci. 2009;12:577–584. doi: 10.1038/nn.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzer J, Miyashiro KY, Sul JY, Barrett L, Belt B, Haydon P, Eberwine J. RNA splicing capability of live neuronal dendrites. Proc Natl Acad Sci. 2005;102:16859–16864. doi: 10.1073/pnas.0503783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Jones TD, Lugo JN, Jr, Sheerin AH, Miller JW, D'Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lörincz A, Notomi T, Tamás G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002;5:1185–1193. doi: 10.1038/nn962. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:508–514. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, Morozov A. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell. 2003;115:551–564. doi: 10.1016/s0092-8674(03)00884-5. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsaki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. Protein kinase A-independent modulation of ion channels in the brain by cyclic AMP. Proc Natl Acad Sci U S A. 1995;92:11716–11720. doi: 10.1073/pnas.92.25.11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova NV, Plotnikov AN, Ziganshin RKh, Deyev IE, Petrenko AG. Analysis of proteins interacting with TRIP8b adapter. Biochemistry (Mosc) 2008;73:644–651. doi: 10.1134/s0006297908060035. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Santoro B, Grant SG, Bartsch D, Kandel ER. Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci U S A. 1997;94:14815–14820. doi: 10.1073/pnas.94.26.14815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Wainger BJ, Siegelbaum SA. Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J Neurosci. 2004;24:10750–10762. doi: 10.1523/JNEUROSCI.3300-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Brager D, Jaramillo TC, Johnston D, Chetkovich DM. Mislocalization of h channel subunits underlies h channelopathy in temporal lobe epilepsy. Neurobiol Dis. 2008;32:26–36. doi: 10.1016/j.nbd.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Spruston N. Determinants of voltage attenuation in neocortical pyramidal neuron dendrites. J Neurosci. 1998;18:3501–3510. doi: 10.1523/JNEUROSCI.18-10-03501.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsay D, Dudman JT, Siegelbaum SA. HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron. 2007;56:947–953. doi: 10.1016/j.neuron.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chen S, Nolan MF, Siegelbaum SA. Activity-dependent regulation of HCN pacemaker channels by cyclic AMP: signaling through dynamic allosteric coupling. Neuron. 2002;36:451–461. doi: 10.1016/s0896-6273(02)00968-6. [DOI] [PubMed] [Google Scholar]
- Wang Z, Xu NL, Wu CP, Duan S, Poo MM. Bidirectional changes in spatial dendritic integration accompanying long-term synaptic modifications. Neuron. 2003;37:463–472. doi: 10.1016/s0896-6273(02)01189-3. [DOI] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Site independence of EPSP time course is mediated by dendritic I(h) in neocortical pyramidal neurons. J Neurophysiol. 2000;83:3177–3182. doi: 10.1152/jn.2000.83.5.3177. [DOI] [PubMed] [Google Scholar]
- Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA. Overview of Molecular Relationships in the Voltage-Gated Ion Channel Superfamily. Pharmacol Rev. 2005;57:387–395. doi: 10.1124/pr.57.4.13. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron. 2003;39:975–990. doi: 10.1016/s0896-6273(03)00543-9. [DOI] [PubMed] [Google Scholar]
- Zhou L, Siegelbaum SA. Gating of HCN channels by cyclic nucleotides: residue contacts that underlie ligand binding, selectivity, and efficacy. Structure. 2007;15:655–670. doi: 10.1016/j.str.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolles G, Wenzel D, Bildl W, Schulte U, Hofmann A, Muller C, Thumfart JO, Vlachos A, Deller T, Pfeifer A, Flesichmann BK, Roeper J, Fakler B, Klöcker N. Association with the auxiliary subunit PEX5R/Trip8b controls responsiveness of HCN channels to cAMP and adrenergic stimulation. Neuron. 2009 doi: 10.1016/j.neuron.2009.05.008. in press. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.