

Abstract
The isotype switch defect in CD40−/− mice is corrected by wild-type (WT) CD40 transgene, but not by a mutant CD40 transgene that does not bind tumor necrosis factor receptor-associated factors (TRAF) 2 and 3. To define the individual roles of TRAF2 and TRAF3 in CD40 activation of B cells, we introduced mutant CD40 transgenes that selectively lack the ability to bind TRAF2 (ΔTR2), TRAF3 (ΔTR3) or both (ΔTR2,3) into B cells of CD40−/− mice. Serum IgG1 and IgE levels, IgG1 antibody response to sub-optimal doses of the T cell-dependent antigen keyhole limpet hemocyanin, germinal center formation, CD40-mediated proliferation, isotype switching and activation of the non-canonical NF-κB pathway were partially diminished in ΔTR2 and ΔTR3 mice and virtually absent in ΔTR2,3 mice. These results suggest that TRAF2 and TRAF3 can each independently mediate class switch recombination (CSR) driven by CD40, but both are required for optimal CD40-driven isotype switching.
Keywords: B cells, IgE, isotype switching
Introduction
The tumor necrosis factor (TNF) receptor family member CD40 expressed on all mature B cells mediates T cell-dependent (TD) Ig class switching and synergizes with cytokines to induce class switch recombination (CSR) in vitro (1). Thus, deficiency of CD40 or of its ligand, CD40L, expressed on activated T cells results in inability of the B cells to undergo class switching from IgM to IgG, IgA and IgE in response to TD antigens (2).
The intracellular (IC) domain of CD40 binds to TNF receptor-associated factor (TRAF) molecules, which play an important role in CD40 signaling. Structural studies have shown that the IC domain of CD40 assumes a hairpin configuration and that ligand binding results in the assembly of CD40 trimers, which recruit TRAF proteins (3, 4). The IC domain of CD40 contains a TRAF6-binding site with a core KxxPxE motif, which is conserved in human and mouse CD40 (5). Downstream of the TRAF6 site, there is a conserved PXQXT sequence [amino acids (a.a.) 250–254 in huCD40 and 251–255 in muCD40], which is essential for binding to TRAF2 and TRAF3 (5–7). The threonine residue in this sequence makes contact with residues in the C-terminal end of CD40, which is important for the hairpin configuration (3, 4). Mutation of this threonine residue to alanine drastically reduces the binding of both TRAF2 and TRAF3 to CD40, probably by disrupting its hairpin structure (8, 9). In addition, TRAF2 and TRAF3 individually bind to distinct residues in the IC domain of CD40. It has been shown that mutation of the P250 residue in the PXQXT motif of huCD40 to glycine strongly reduces TRAF2 binding without a significant effect on TRAF3 binding (10). Conversely, mutation of the Q263 residue in huCD40 to alanine, as well as deletion of Q263 and E264, strongly reduce TRAF3 binding without a significant effect on TRAF2 binding (11).
A number of studies have examined the role of TRAF molecules in CD40-mediated B cell activation by reconstituting B cells of mice deficient in CD40 with mutated CD40 transgenes. Using this approach, we and others have shown that CD40-mediated CSR was fully restored in mice reconstituted with a wild-type (WT) CD40 transgene (12–14). Mice reconstituted with a CD40 transgene that selectively lost the capacity to bind TRAF6 had normal CSR. In contrast, CSR was severely impaired in mice bearing a mutant T255A CD40 transgene, which has lost the ability to bind TRAF2 and TRAF3, and was abolished in mice bearing a mutant transgene that fails to bind all three TRAF molecules. These results indicate that binding to TRAF2 and/or TRAF3 is essential for CD40-driven CSR. Based on studies in which TRAF3 was over-expressed in B cell lines, it has been suggested that TRAF3 inhibits CD40-mediated signaling in B cells (15–17). Nevertheless, in two studies that examined up-regulation of IL-4-driven Cγ1 and Cε germ line transcript (GLT) expression, TRAF3 was found to be important for CD40 up-regulation of these transcripts (18, 19). However, the individual roles of TRAF2 and TRAF3 in CD40-driven CSR remain unknown. To address this question, we have generated mice whose B cells express CD40 transgenes that selectively lack the ability to bind TRAF2, TRAF3 or both. We show that TRAF2 and TRAF2 can each independently mediate CSR driven by CD40.
Materials and methods
Generation of CD40−/− mice with CD40 transgenes
PCR-generated CD40 WT and mutated gene products (Fig. 1A) were cloned in the pBSVE6BK vector containing an Igμ enhancer and Ig heavy chain (IgVH) promoter and used to generate founder mice as previously described (12). Mice were used at 8–12 weeks of age according to the guidelines of the Animal Care Committee of Children's Hospital.
Fig. 1.
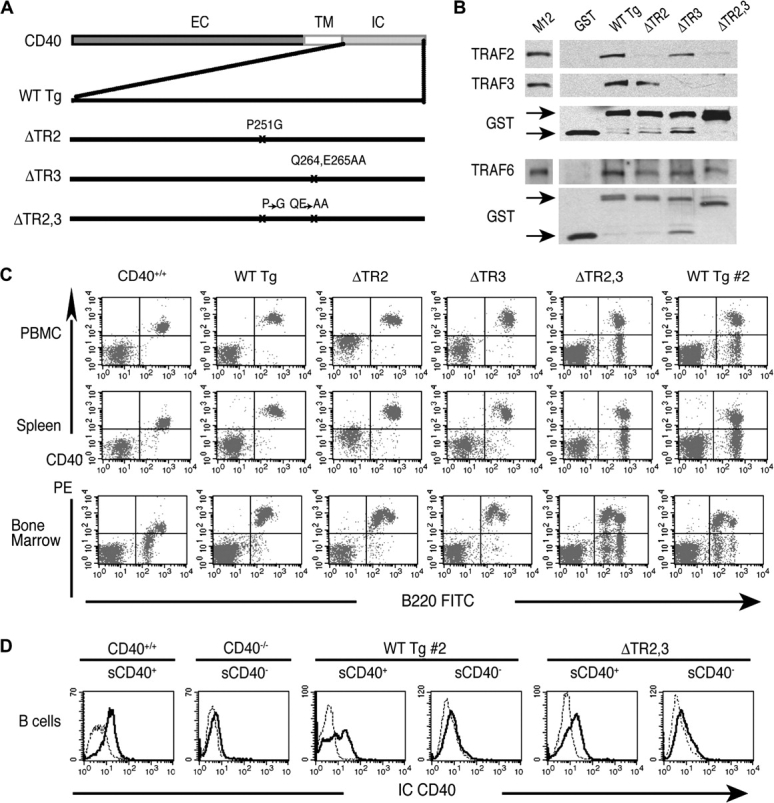
Characterization of the CD40 mutants. (A) Schematic representation of murine WT CD40 and mutant transgenes: WT (WT Tg), P251G (ΔTR2), QE264/265AA (ΔTR3) and P251G,QE264/265AA (ΔTR2,3). (B) Association of TRAF proteins with GST-CD40 mutants in M12 B cell lysates. M12 cell lysates were used as controls. (C) Representative FACS analysis of CD40 surface expression in PBMC, spleen and BM of CD40 transgenic mice. (D) Representative FACS analysis of CD40 IC staining (IC CD40, solid line) or isotype control (dotted line) in surface (s) CD40+ or CD40− B cells isolated from WT Tg #2 and ΔTR2,3 mice by two step sorting: purified B cells obtained by using MACS CD43 beads were stained with anti-mouse CD40–PE and were sorted into CD40+ and CD40− B cells using MACS anti-PE MicroBeads. Purified B cells from CD40+/+ and CD40−/− mice were used as controls. EC, extracellular domain; TM, transmembrane domain.
Flow cytometry analysis
Single-cell suspensions were stained and analyzed on a FACSCalibur cytometer (Becton Dickinson, Mountain View, CA, USA) using FITC or PE-conjugated mAbs to CD3, CD4 CD8α, B220, CD40, IgM, rat IgG2a and hamster IgG (PharMingen, San Diego, CA, USA) as previously described (12). Annexin V–FITC (Biovision Inc., Mountain view, CA, USA) staining was performed as per the manufacturer's instructions. IC staining of CD40 was performed on purified B cells post-permeabilization using the FIX& PERM cell Permeabilization Kit (Invitrogen, Camarillo, CA, USA) according to the manufacturer's instructions. The antibody used was goat anti-mouse CD40 (T-20, Santa Cruz Biotechnology, Santa Cruz, CA, USA) followed by donkey anti-goat IgG FITC (Abcam, Cambridge, MA, USA). For isotype control, purified goat IgG (Abcam) was used.
Co-precipitation of TRAF proteins with CD40-GST fusion proteins
IC regions corresponding to WT and mutant CD40 were amplified from transgenic constructs by PCR as previously described (12). Preparation of glutathione-S-transferase (GST)-fusion proteins, M12 B cell lysates, pull-down assays and western blotting using rabbit anti-mouse TRAF2 (MBL International, Woburn, MA, USA), mouse mAb to TRAF3 and rabbit anti-mouse TRAF6 (Santa Cruz Biotechnology) were previously described (12). Binding of TRAF proteins to GST-CD40 fusion proteins was evaluated by densitometry scanning of the TRAF and GST bands. The ratio of TRAF band to the corresponding GST band in mutants versus WT Tg CD40 was then calculated.
Serum Ig levels and antibody responses to TD antigen
Igs were assayed by ELISA (12). Keyhole limpet hemocyanin (KLH) immunization on days 0 and 14 and analysis of anti-KLH antibody responses were described previously (12). KLH antibody levels were measured by ELISA, using anti-KLH-specific standards (PharMingen) to determine IgM, IgG1 and IgG3 concentrations (12). Optical density at 405 nm of serum dilutions (1:450), which is in the linear part of the titration curve, were used for IgE.
Germinal center formation
Frozen spleen tissue sections were stained with biotin-labeled peanut agglutinin (Vector Laboratories, Burlingame, CA, USA) as previously described (12). Germinal centers (GCs) were counted in a blinded fashion.
RT-PCR
RNA was extracted from cultured splenic B cells on day 4 and RT-PCR for Cε GLT, activation-induced cytidine deaminase (AID), Iμ-Cε mature transcripts and β2 microglobulin (β2 m) was performed as described previously (12). Various dilutions were used in both reactions to ensure that the products measured were in the linear range. The densitometric analysis of the scanned bands was evaluated using the National Institutes of Health Image program 1.63f. Relative expression levels normalized to β2 m levels were calculated as a ratio of the value of each transcript to the corresponding β2 m value.
Proliferation and IgE synthesis of B cells
CD40+ B cells were purified from spleen cell suspensions by magnetic sorting using MACS CD43 or MACS anti-PE MicroBeads Kits (Miltenyi Biotec, Auburn, CA, USA) according to the manufacturer's instructions. Purified cells were >95% B220+ CD40+ by FACS analysis. Proliferation of CD40+ B cells (0.5 × 106 ml−1) in response to hamster IgM anti-mouse CD40 (0.1–1 μg ml−1, HM40-3, PharMingen) and LPS (10 μg ml−1; Sigma, Saint Louis, MO, USA) was measured after 3 days using [3H]-thymidine as in ref. (12). For Ig synthesis, B cells were stimulated with IL-4 (50 ng ml−1, R&D Systems, Minneapolis, MN, USA) plus anti-CD40 (0.1–1 μg ml−1) or LPS (10 μg ml−1) for 6 days and supernatants were assayed for IgE and IgG1 by ELISA (12).
Surface expression of CD23, CD54 and CD86
B cells (>90% B220+) were stimulated with sCD40L (1:20 dilution of supernatants from muCD40L:muCD8-transfected J558L cells), control supernatants (1:20 dilution of J558L cells transfected with empty plasmid) or LPS (10 μg ml−1) and double stained for CD40 and CD23, CD54 or CD86 (PharMingen) as previously described (12).
Activation of NF-κB and MAP kinase phosphorylation
CD40+ splenic B cells were rested for 1 h and then stimulated with anti-CD40 (1 μg ml−1). Cell lysates were probed with anti-phospho-IκBα, anti-IκBα (Cell Signaling, Danvers, MA, USA) and actin (Chemicon International, Temecula, CA, USA). Nuclear extracts were prepared from cells stimulated for 16 h using a Nuclear Extract Kit (Active Motif, Carlsbad, CA, USA) and probed with anti-p52 and poly ADP-ribose polymerase 1 (Santa Cruz Biotechnology). Activated MAP kinases were detected in lysates of cells stimulated for 5 and 10 min by immunoblotting with phospho-p38 (Cell Signaling) and phospho-SAPK/c-Jun-N-terminal kinase (JNK) (Biosource, Camarillo, CA, USA) and membranes were reprobed with kinase-specific antibody to p38 (Cell Signaling).
Results
Reconstitution of CD40−/− mice with CD40 transgenes
WT and mutant murine CD40 constructs were generated as illustrated in Fig. 1(A). CD40 P251G, designated ΔTR2, carries a P251G point mutation, which corresponds to the P250G mutation in human CD40 that has been reported to destroy TRAF2, but not TRAF3, binding (10). CD40 QE264/265AA, designated ΔTR3, carries the point mutation Q264A, which corresponds to the Q263A mutation in human CD40 that destroys TRAF3, but not TRAF2, binding (11). In addition, the deletion of Q263 and E264 in huCD40, that correspond to Q264 and E265 in muCD40, severely impairs TRAF3, but not TRAF2, binding (11). CD40 P251G,QE264/265AA, designated ΔTR2,3, carries both mutations present in ΔTR2 and ΔTR3.
To confirm that the muCD40 mutants lack the expected associations with TRAF2 and TRAF3, GST-WT CD40 and mutant fusion proteins were examined for their capacity to bind TRAF proteins in lysates of the murine B cell line M12, which expresses TRAF2, TRAF3 and TRAF6. As expected, GST-CD40 WT, but not GST, associated with TRAF2, TRAF3 and TRAF6 (Fig. 1B). In contrast, GST-ΔTR2 showed severely impaired association with TRAF2 but retained normal ability to associate with TRAF3. Conversely, GST-ΔTR3 showed severely impaired association with TRAF3 but retained normal ability to associate with TRAF2. In three experiments, densitometry scanning revealed that the amount of TRAF2 that associated with GST-ΔTR2 was 3.3 ±0.6% (mean ± standard error) of that associated with GST-WT CD40 and that the amount of TRAF3 that associated with GST-ΔTR3 was 3.0 ± 0.9% of that associated with GST-WT CD40. GST-ΔTR2,3 showed barely detectable association with TRAF2 and TRAF3 that amounted to, respectively, 1.71 ± 0.04 and 0.54 ± 0.40% of their association with GST-WT CD40. All three mutant GST-CD40 fusion proteins bound TRAF6.
Constructs containing WT CD40 and all three CD40 mutant cDNAs were used to create transgenic mice, which were bred on the CD40−/− background. Lines that expressed CD40 only on B220+ cells were selected for study. For each of the constructs, at least two transgenic lines derived from separate founders were studied and similar results were obtained. FACS analysis revealed that CD40 was expressed with comparable intensity on all peripheral B cells in spleen and blood from mice reconstituted with WT (WT Tg mice), ΔTR2 (ΔTR2 mice) and ΔTR3 (ΔTR3 mice) transgenes (Fig. 1C). All three lines of ΔTR2,3 mice examined expressed CD40 on only a fraction of the B cells. The two ΔTR2,3 lines chosen for study expressed CD40 on 50–60% of peripheral B cells in spleen and blood. As a control for ΔTR2,3 mice, we used a previously generated line of CD40−/− mice reconstituted with WT CD40 transgene (WT Tg #2) that expressed CD40 on a comparable percentage of B cells and in comparable intensity to ΔTR2,3 mice (12, Fig. 1C). B cells from ΔTR2,3 mice and WT Tg #2 mice that did not express surface CD40 showed no detectable presence of CD40 protein intracellularly, ruling out a defect in CD40 trafficking in these cells (Fig. 1D). WT Tg #2 mice behaved similarly to WT Tg mice that expressed CD40 on all B cells with regard to serum Ig levels, antibody responses and in vitro CD40-driven isotype switching and signaling by CD40 sorted cells (data not shown). Therefore, for simplicity, we present data only on the WT Tg mice that express CD40 on all B cells.
Bone marrow (BM) from all transgenic mice lines had normal cellularity and normal percentage and expression profile of IgM+ and B220+ cells (data not shown). In normal BM, CD40 is highly expressed on B220high cells but is absent or poorly expressed on B220low cells. In contrast, in all transgenic lines, both B220low and B220high BM cells expressed high levels of CD40 (Fig. 1C). This likely reflects the fact that the EμVH promoter, used to drive transgene expression, is active at earlier stages of B cell development than the endogenous CD40 gene promoter, which is poorly active in pre-B cells and immature B cells (20).
Spleens were normal in size and cellularity in all transgenic mice. The numbers of CD3+ and B220+ cells and the expression of IgM on B220+ cells in the spleens of all transgenic mice were similar to those of CD40+/+ mice (data not shown). Furthermore, the distribution of transitional T1 cells, marginal zone B cells and follicular B cells in the spleen was comparable between CD40+/+, CD40−/− and all four strains of CD40 transgenic mice (data not shown). This suggests that introduction of the transgenes did not interfere with B cell development. The thymus was normal in size, architecture and numbers of CD4+, CD8+ and CD3+ cells in all the transgenic lines (data not shown).
Serum Ig levels
CD40−/− mice have severely decreased serum IgG1 and IgE levels and normal IgM and IgG3 levels (21, 22). As previously reported, introduction of the CD40 WT transgene in the CD40−/− background resulted in normalization of serum IgG1 and IgE levels (Fig. 2). Reconstitution with ΔTR2 and ΔTR3 restored serum IgG1 and IgE levels to a substantial degree, but in both strains, serum IgG1 and IgE levels remained significantly lower than those in WT Tg mice. IgE levels in ΔTR3 mice were significantly lower than in ΔTR2 mice. Reconstitution with ΔTR2,3 partially restored serum IgG1 and IgE levels, consistent with previous findings in mice reconstituted with a T255A mutant transgene that is severely impaired in TRAF2 and TRAF3 binding (12). None of the transgenes had a significant effect on serum IgM or IgG3 levels.
Fig. 2.

Serum Igs in CD40 transgenic mice. Serum Ig levels from non-immunized 12-week-old CD40−/− mice reconstituted with CD40 transgenes, CD40+/+ littermates and CD40−/− mice. Student’s t test was used for statistical analysis: *P < 0.05, **P < 0.01, ***P < 0.001.
Antibody response and GC formation to the TD antigen KLH
As previously reported, CD40−/− mice make normal IgM and IgG3, but no detectable IgG1 or IgE, antibody responses to KLH (12, 22). Mice reconstituted with WT Tg mounted IgG1 and IgE antibody responses in response to immunization with 400 μg KLH that were comparable to those of CD40+/+ mice. The IgG1 and IgE anti-KLH responses of ΔTR2 and ΔTR3 mice were comparable to those of mice reconstituted with WT Tg (Fig. 3A). In contrast, ΔTR2,3 mice had significantly diminished IgG1 and IgE antibody responses. However, unlike the case of CD40−/− mice, these responses were clearly measurable as previously observed in mice reconstituted with the T255A mutant (12). None of the transgenes had a significant effect on IgM or IgG3 anti-KLH responses.
Fig. 3.
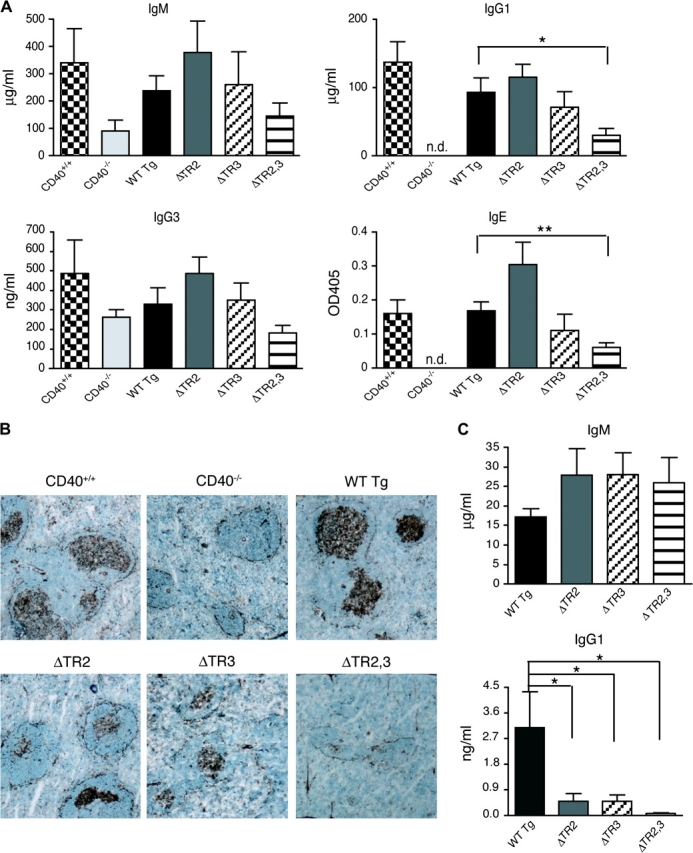
Antibody response and GC formation in response to TD antigen. (A) IgM, IgG1, IgG3 and IgE KLH-specific antibody responses to immunization with 400 μg KLH (ND = not detectable). (B) GCs in representative spleen sections (×40) from mice immunized with KLH examined for peanut agglutinin binding. (C) IgM and IgG1 KLH-specific antibody responses to immunization with 100 μg KLH. Statistical analysis was performed as in Fig. 2.
Spleens from KLH-immunized CD40+/+ mice revealed the presence of multiple GCs (Fig. 3B). As previously reported (21, 22), GCs were absent from spleens of non-immunized mice (data not shown) and from spleens of immunized CD40−/− mice (Fig. 3B). Mice reconstituted with WT Tg had prominent GCs in their spleens with numbers similar to those found in CD40+/+ mice. ΔTR2 and ΔTR3 mice had fewer GCs, which were less developed than GCs of control WT Tg mice. GCs were virtually absent from spleens of ΔTR2,3 mice (Fig. 3B).
Because of the decreased size of GCs in ΔTR2 and ΔTR3 mice, we examined the response of CD40 transgenic mice to immunization with a 4-fold lower dose of KLH (100 micrograms per mouse). Figure 3C shows that ΔTR2 and ΔTR3 mice made normal IgM anti-KLH responses but significantly lower IgG1 anti-KLH responses compared with WT Tg controls in response to the lower dose of KLH. The antibody response of ΔTR2,3 mice was virtually undetectable.
CD40-mediated proliferation and isotype switching in vitro
CD40 ligation in the presence of IL-4 causes isotype switching to IgG1 and IgE (23). We compared the capacity of B cells from CD40−/− mice reconstituted with WT and mutant transgenes to undergo isotype switching to IgG1 and IgE in response to anti-CD40+IL-4, using three concentrations of anti-CD40 (0.1, 0.5 and 1.0 μg ml−1). Purified B cells were used from ΔTR2 mice, ΔTR3 mice and WT Tg controls. Because ΔTR2,3 mice express CD40 on only a fraction of their B cells, CD40+ B cells were sorted from the splenic B cells of these mice and WT Tg controls. The purification procedure by itself did not activate the B cells because unstimulated sorted CD40+ B cells do neither proliferate nor secrete IgE or IgG1 (data not shown). Furthermore, comparable data were obtained using WT Tg B cells prepared by either negative sorting (CD43− cells) or positive sorting with anti-CD40 mAb (CD40+ cells). B cells from ΔTR2 and ΔTR3 mice proliferated significantly less in response to anti-CD40 (Fig. 4A) and secreted significantly less IgG1 and IgE in response to anti-CD40+IL-4 compared with B cells from WT Tg mice (Fig. 4B and C), as determined by analysis of variance analysis of the dose response curve. The proliferative defect was more pronounced at the lower concentration of anti-CD40 used (0.1 μg ml−1). This was confirmed by CFSE dilution studies (data not shown). B cells from ΔTR2,3 mice failed to proliferate and secreted no detectable amounts of IgG1 or IgE in response to anti-CD40+IL-4 (Fig. 4A–C). B cells from ΔTR2 and ΔTR3 mice did not exhibit significantly increased apoptosis and cell death, as assessed by Annexin V staining at day 3, in response to stimulation with anti-CD40 compared with B cells from CD40+/+ or WT Tg mice (Fig. 4D). However, B cells from ΔTR2,3 mice exhibited increased apoptosis and cell death in response to stimulation with anti-CD40, but not LPS, which was comparable to that observed in B cells from CD40−/− mice. The defect in proliferation and IgG1 and IgE production in B cells from mice reconstituted with mutant CD40 transgenes was not due to a general impairment in the ability of these B cells to undergo isotype switching or to respond to IL-4 because they proliferated normally and secreted normal amounts of IgE and IgG1 in response to LPS+IL-4 (Fig. 4A–C).
Fig. 4.
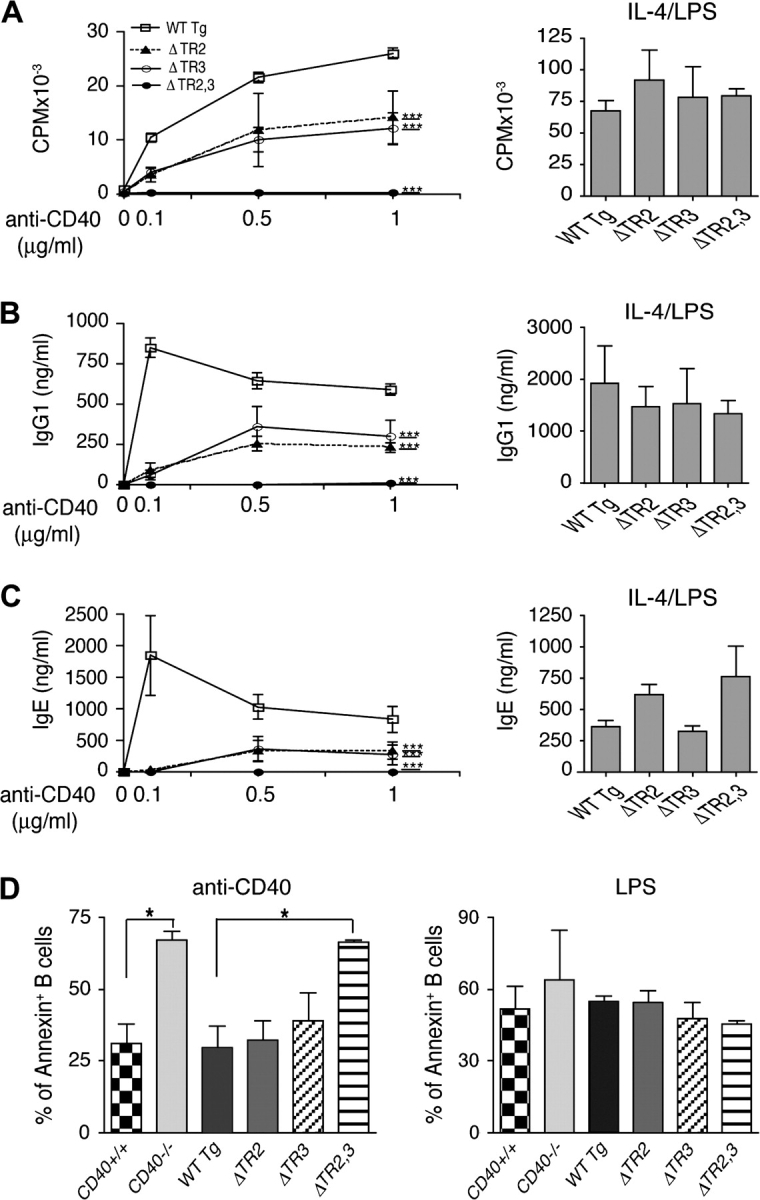
B cell proliferation, Annexin V staining and Ig synthesis in vitro. Sorted CD40+ B cells were used to examine (A) Proliferation in response to anti-CD40 or IL-4+LPS and (B) IgG1 and (C) IgE synthesis in response to IL-4 in the presence of anti-CD40 or LPS. Results shown are the mean of three experiments. Analysis was done by two-way analysis of variance: ***P < 0.001. (D) Annexin V–FITC staining of CD40+ B cells after 3d stimulation with anti-CD40 (1 μg ml−1) or LPS (10 μg ml−1) and were analyzed by FACS. Results shown are the mean of two experiments and are expressed as percentage of Annexin V+ B cells. Student’s t test was used for statistical analysis: *P < 0.05.
Molecular events in CD40-mediated isotype switching
CD40 ligation synergizes with IL-4 to drive molecular events that result in CSR. We compared the ability of CD40 to activate molecular events involved in CSR to IgE in B cells stimulated with low (0.1 μg ml−1) and high (1.0 μg ml−1) concentrations of anti-CD40 plus IL-4. These events include expression of Cε GLTs, AID and mature Iμ-Cε transcripts (24). Expression of these transcripts was estimated semi-quantitatively by calculating the ratio of expression of each transcript to that of the transcript of the house keeping gene β2 m expression (n = 3). Expression of transcripts was compared in B cells with mutated CD40 transgenes and B cells with CD40 WT transgene. In the absence of stimulation, or upon stimulation with anti-CD40 alone or IL-4 alone, none of the B cells expressed Cε GLT, AID or Iμ-Cϵ transcripts (data not shown). Figure 5(A) shows that, as previously reported (12), CD40 ligation synergized with IL-4 in inducing Cε GLT, AID and Iμ-Cε transcripts in B cells from WT Tg mice. At the lower concentration of anti-CD40, Cϵ GLT expression was diminished in ΔTR2 B cells and severely diminished in ΔTR3 B cells. At the higher concentration of anti-CD40, Cε GLT expression was normal in ΔTR2 B cells but was still decreased in ΔTR3 B cells. At the lower concentration of anti-CD40, AID expression was normal in ΔTR2 B cells but significantly decreased in ΔTR3 B cells. At the higher concentration of anti-CD40, AID expression was normal in ΔTR2 and ΔTR3 B cells. Iμ-Cε transcript expression was severely diminished in ΔTR2 and ΔTR3 B cells at the lower concentration of anti-CD40 and partially decreased at the higher concentration of anti-CD40. Cε GLT, AID or Iμ-Cε transcripts were virtually undetectable in ΔTR2,3 B cells at both concentrations of anti-CD40 (Fig. 5A and B). B cells from all mice expressed comparable amounts of Cε GLT, AID and Iμ-Cε transcripts in response to LPS+IL-4 (data not shown).
Fig. 5.

Molecular events in isotype switching. CD40+ B cells were stimulated with anti-CD40+IL-4 using two concentrations of anti-CD40 (0.1 and 1 μg ml−1) and examined for (A) expression of Cε GLT, AID and Iμ-Cε mature transcripts by RT-PCR at day 4. (B) Semi-quantitative analysis of three experiments performed. Values represent the ratio of each transcript/β2 m.
CD40-mediated up-regulation of CD23, CD54 and CD86 expression on B cells
CD40 ligation up-regulates the expression of CD23, CD54 and CD86 on B cells (1). We compared the ability of B cells from transgenic mice to up-regulate these surface antigens following CD40 ligation with sCD40L. Figure 6 shows that B cells from WT Tg mice up-regulated CD23, CD54 and CD86 expression following CD40 ligation. B cells from ΔTR2 and ΔTR3 mice were comparable to B cells from WT Tg mice in up-regulation of CD23, CD54 and CD86 in response to CD40 ligation. B cells from ΔTR2,3 mice failed to up-regulate the expression of these antigens in response to sCD40L. B cells from all transgenic lines up-regulated CD23, CD54 and CD86 expression comparably in response to LPS (Fig. 6).
Fig. 6.
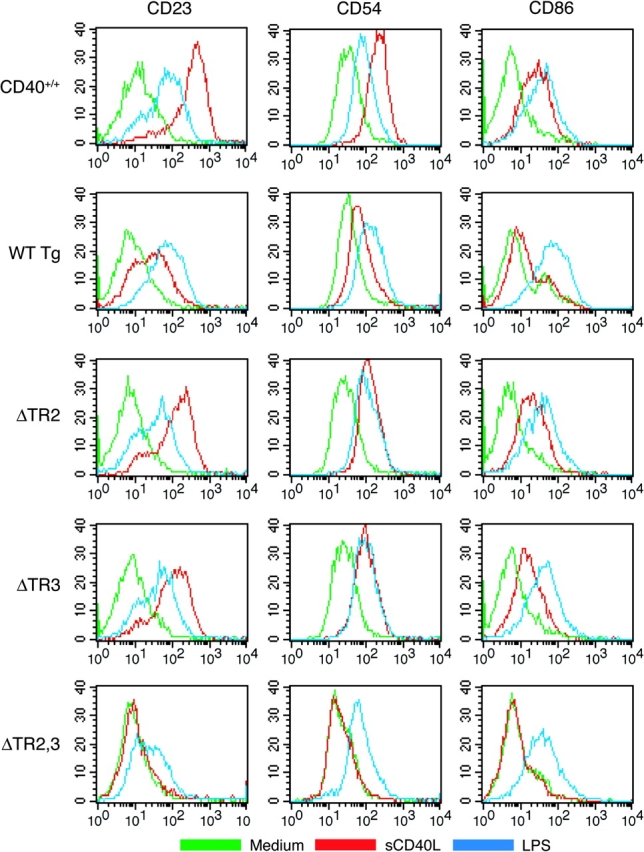
Up-regulation of CD23, CD54 and CD86 expression on B cells. Splenic B cells were left unstimulated (green) or were stimulated overnight with sCD40L supernatants of CD40L-transfected J558L cells (red) or LPS (blue) and then stained with anti-CD40–PE and FITC-conjugated mAbs to CD23, CD54 or CD86 and with the relevant isotype controls. Stimulation with control supernatants of J558L cells transfected with empty plasmid caused no up-regulation of CD23, CD54 or CD86 (data not shown). Because CD40 is expressed only on a fraction of B cells from ΔTR2,3 mice (Fig. 1C), analysis of B cells from these mice was performed by gating on CD40+ cells. Data shown are representative of three independent experiments.
CD40 signaling in B cells
CD40 ligation in B cells causes activation of NF-κB (1). This is mediated both via the canonical and non-canonical NF-κB activation pathways (25, 26). The canonical pathway involves the activation of the IκB kinase complex, which causes the phosphorylation and degradation of IκB, releasing p50 and p65 to translocate into the nucleus (27). The non-canonical pathway is activated by processing NF-κB2 precursor protein p100 to generate p52, which translocates to the nucleus (28). Figure 7(A) shows that CD40-mediated phosphorylation and degradation of IκBα in B cells from ΔTR2 and ΔTR3 mice were comparable to that in B cells from WT Tg mice and CD40+/+ WT controls. This was confirmed by scanning densitometry (n = 3, data not shown). In contrast, phosphorylation and degradation of IκBα were not detectable in B cells from ΔTR2,3 mice. Figure 7(B) shows that CD40 ligation caused the appearance of p52 in nuclear extracts from B cells of WT Tg mice. In contrast, appearance of nuclear p52 was severely diminished in nuclear extracts of B cells from ΔTR2 and ΔTR3 mice and was undetectable in B cells from ΔTR2,3 mice.
Fig. 7.

CD40 activation of NF-κB and MAP kinases in spleen B cells. Sorted CD40+ B cells were stimulated with anti-CD40 for the indicated times and examined for (A) phosphorylation and degradation of IκBα, (B) translocation of p52 in nuclear extracts after 16 h stimulation and (C) phosphorylation of p38 and JNK. Membranes were reprobed with antibodies to actin (A), poly ADP-ribose polymerase 1 (B) and anti-p38 (C) as loading controls.
CD40 ligation in B cells results in the phosphorylation and activation of the MAP kinases p38 and JNK (29). Figure 7(C) shows that CD40 ligation caused comparable phosphorylation of JNK in B cells from WT Tg mice and CD40+/+ WT controls. CD40-mediated JNK phosphorylation was diminished in ΔTR2 B cells, almost normal in ΔTR3 B cells and undetectable in ΔTR2,3 B cells. Scanning densitometry (n = 3) revealed significantly decreased JNK phosphorylation in ΔTR2 B cells at 5 min (0.28 ± 0.05-fold of WT Tg B cells, P < 0.05), but not at 10 min (0.75 ± 0.12-fold of WT Tg B cells, P > 0.05), and normal JNK phosphorylation in ΔTR3 B cells at 5 and 10 min (0.97- and 1.07-fold of WT Tg B cells, respectively). CD40 ligation caused comparable phosphorylation of p38 in B cells from CD40+/+, WT Tg, ΔTR2 and ΔTR3 mice (Fig. 7C). This was confirmed by scanning densitometry (n = 3, data not shown). In contrast, CD40 ligation caused no detectable phosphorylation of p38 in B cells from ΔTR2,3 mice (n = 3).
Discussion
Our results suggest that each of TRAF2 and TRAF3 is involved in CD40-driven CSR. Our strategy of constructing muCD40 mutants that are selectively deficient in binding TRAF2 or TRAF3 was based on data obtained with human CD40. It is thought that P250 in the PXQXT sequence of huCD40 interacts via van der Waals forces with residues F447, P449, F456 and S467 of TRAF2 (3). Furthermore, this residue points into the TRAF-binding pocket and orients properly the succeeding residues involved in hairpin formation. The P251G muCD40 mutant (ΔTR2), like the corresponding huCD40 P250G mutant (10), was drastically impaired in its ability to bind TRAF2 but retained virtually intact its ability to bind TRAF3 and TRAF6. Q263 in huCD40 makes contact with Y395 and D399 in TRAF3 (4). The QE264,265AA muCD40 mutant (ΔTR3), like the Q263A and the ΔQE263,264 huCD40 mutants (11), was drastically impaired in its ability to bind TRAF3 but retained virtually intact its ability to bind TRAF2 and TRAF6. The residual weak binding of TRAF2 and TRAF3 to the ΔTR2 and ΔTR3 mutants (<5% of that WT CD40) could have been due to incomplete loss of direct binding and/or to formation of TRAF2/3 heterodimers. The muCD40 double-mutant P251G/QE264,265AA (ΔTR2,3) had virtually no detectable binding to either TRAF2 or TRAF3 but retained virtually intact its ability to bind TRAF6. The preserved binding of TRAF3 to ΔTR2 and of TRAF2 to ΔTR3 and of TRAF6 to all mutants makes it unlikely that the selective loss of binding of TRAF proteins by these mutants is due to a global loss of structure. Nevertheless, localized loss of structure in the region where the mutation was introduced is possible.
B cells from lines of CD40−/− mice reconstituted with ΔTR2 and ΔTR3 transgenes expressed CD40 on all their B cells, whereas only 50–60% of B cells from ΔTR2,3 mice expressed CD40 (Fig. 1C). A similar finding was previously shown in the WT Tg #2 line of CD40−/− mice reconstituted with WT CD40 transgene (12). B cells that did not express the transgene on the surface showed no detectable presence of CD40 protein intracellularly, ruling out a defect in CD40 trafficking in these cells (Fig. 1D). Recently, we have also found that some lines of TACI−/− mice reconstituted with a TACI transgene under the control of the EμVH promoter used in this study also express TACI on a fraction of their B cells, while other lines express it on all B cells (our unpublished observations). Epigenetic factors that exert influences at the transgene insertion site may account for the variability of Tg expression in lines derived from different founders.
Reconstitution of B cells from CD40−/− mice with ΔTR2 and ΔTR3 transgenes partially restored serum levels of the CD40-dependent isotypes IgG1 and IgE (Fig. 2). Both transgenes reconstituted to normal the IgG1 and IgE antibody response, but only partially the GC response, to immunization with 400 μg of the TD antigen KLH (Fig. 3A and B). However, the IgG1 antibody of ΔTR2 and ΔTR3 mice to a 4-fold lower immunizing dose of KLH was significantly impaired (Fig. 3C). The ΔTR2,3 transgene reconstituted all these functions poorly. This could not simply be due to the fact that only 50–60% of B cells from ΔTR2,3 mice expressed CD40 because we have previously shown that expression of CD40 Tg on a comparable fraction of B cells from CD40−/− mice reconstituted with a WT CD40 transgene reconstituted total serum Ig levels and the TD response to KLH (12). These results suggest that TRAF2 and TRAF3 play partially overlapping roles in the in vivo antibody response to TD antigens and that both are needed for an optimal IgG1 antibody response to immunization with a lower dose of TD antigen. In vitro isotype switching to IgE and IgG1 in response to CD40/IL-4 stimulation, as well as proliferation to anti-CD40, were partially impaired in B cells from ΔTR2 and ΔTR3 mice (Fig. 4). The impairment in both isotype switching and proliferation was more marked with low concentrations of anti-CD40. These data suggest that loss of TRAF2 or TRAF3 could impair T cell-driven isotype switching under conditions of sub-optimal CD40L expression, e.g. sub-optimal TCR stimulation by limiting doses of antigens. CD40-driven isotype switching and proliferation were virtually absent in B cells from ΔTR2,3 mice, consistent with what we previously found in mice reconstituted with the T255A transgene (12).
CD40/IL-4-driven expression of Cε GLT and mature Cε transcripts was defective in B cells from ΔTR2 and ΔTR3 mice at low concentration of anti-CD40 (0.1 μg ml−1) (Fig. 5). The defect was less pronounced in ΔTR2 mice and at higher concentrations of anti-CD40. B cells from ΔTR2,3 mice were completely deficient in all CSR events. These results further support the conclusion that TRAF2 and TRAF3 play overlapping roles in CD40-dependent CSR. CD40 up-regulation of CD23, CD54 and CD86 expression in B cells was preserved in ΔTR2 and ΔTR3 mice but severely impaired in ΔTR2,3 mice (Fig. 6). The redundancy of TRAF2 and TRAF3 in CD40 up-regulation of these activation markers is not surprising given the fact that this up-regulation is dependent on activation of NF-κB and that CD40-mediated activation of the canonical NF-κB pathway was intact in B cells from ΔTR2 and ΔTR3 mice (Fig. 7A). The normal activation of the canonical NF-κB pathway in B cells from ΔTR2 mice contrasts with the impaired CD40 activation of this pathway in TRAF2−/− B cells (30). This suggests that lack of TRAF2 may have more widespread effects on the activation of B cells.
In contrast to the normal activation of the canonical NF-κB pathway, CD40 activation of the non-canonical NF-κB pathway was impaired in B cells from both ΔTR2 and ΔTR3 mice (Fig. 7B), suggesting that these two TRAF proteins play non-redundant roles in CD40-mediated activation of the non-canonical NF-κB pathway. Both TRAF2 and TRAF3 have been reported to mediate activation of the non-canonical NF-κB pathway in B cell lines (17, 31). However, TRAF2- and TRAF3-deficient B cells have increased baseline activation of this pathway (30, 32, 33), suggesting that global loss of TRAF2 or TRAF3 in B cells may have different outcomes than specific disruption of their binding to CD40. While p50 and c-Rel of the canonical NF-κB pathway are known to be important for CSR (34, 35), NF-κB-inducing kinase and the p52 partner RelB of the non-canonical NF-κB pathway also contribute to CSR and GC formation (36–38). Impaired activation of the non-canonical NF-κB pathway may contribute to the impaired CD40-driven isotype switching in ΔTR2 and ΔTR3 mice.
CD40 activation of the MAP kinase p38 was normal in B cells from ΔTR2 and ΔTR3 mice but virtually absent in B cells from ΔTR2,3 mice (Fig. 7C), indicating that TRAF2 and TRAF3 are redundant in CD40 activation of p38. CD40 activation of JNK was partially decreased in ΔTR2 B cells but not in ΔTR3 B cells and was undetectable in ΔTR2,3 B cells, suggesting that TRAF3 partially overlaps with TRAF2 in CD40 activation of JNK. Impaired JNK activation may contribute to the defective CD40-driven isotype switching in ΔTR2 mice, as JNK inhibition blocks CD40-mediated switching (39).
The fact that loss of TRAF3 binding impaired CD40-driven CSR in B cells, even slightly more than loss of TRAF2 binding, supports a role for TRAF3 in CD40-driven isotype switching. Recently, mice with B cell-specific disruption of TRAF3 were found to have increased numbers of marginal zone B cells with a corresponding increase in serum IgG3, IgG2a, IgG2b and IgA levels and in the antibody response to the type II TI antigen trinitrophenol (TNP)–Ficoll. In contrast, although these mice had also increased numbers of follicular B cells, their serum IgG1 and IgE levels and their IgG1 antibody response to the TD antigen TNP–KLH were not increased (32). This is consistent with a role for TRAF3 in the production of TD Ig isotypes. A role for TRAF3 in CSR is also consistent with the observation that the B cell- activating factor receptor and the EBV protein LMP-1, which bind exclusively or predominantly TRAF3 (31, 40), can mediate isotype switching (41, 42).
It was recently reported that a muCD40 IC domain deletion mutant that lacks a.a. 235–260 (Δ235–260) but retains the C-terminal 29 a.a. (a.a. 261–289) binds TRAF2 weakly; yet is able to reconstitute CD40-driven proliferation and isotype switching but not GC formation in CD40 null mice (43). The truncation mutant retains residues QE264,265 that are essential for TRAF3 binding. Possible TRAF3 binding to this mutant may have contributed to its relatively preserved function. In addition, the massive deletion in the IC domain of CD40 in the mutant may have resulted in conformational changes that allowed recruitment of novel signaling molecules that could have altered its function.
In summary, TRAF2 and TRAF3 can each independently mediate CSR driven by CD40, but both are required for optimal CD40-driven isotype switching.
Funding
USPHS grant (AI-31136).
Acknowledgments
We are indebted to John Manis and Michel Massaad for reading the manuscript and useful suggestions. The authors have no conflicting financial interests.
Glossary
Abbreviations
- a.a
amino acids
- AID
activation-induced cytidine deaminase
- β2 m
β2 microglobulin
- BM
bone marrow
- CD40L
CD40 ligand
- CSR
class switch recombination
- GLT
germ line transcript
- GC
germinal center
- GST
glutathione-S-transferase
- IC
intracellular
- JNK
c-Jun-N-terminal kinase
- KLH
keyhole limpet hemocyanin
- TD
T cell dependent
- TNF
tumor necrosis factor
- TNP
trinitrophenol
- TRAF
TNF receptor-associated factor
- WT
wild type
References
- 1.Banchereau J, Bazan F, Blanchard D, et al. The CD40 antigen and its ligand. Annu. Rev. Immunol. 1994;12:881. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 2.Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu. Rev. Immunol. 2004;22:307. doi: 10.1146/annurev.immunol.22.012703.104533. [DOI] [PubMed] [Google Scholar]
- 3.McWhirter SM, Pullen SS, Holton JM, Crute JJ, Kehry MR, Alber T. Crystallographic analysis of CD40 recognition and signaling by human TRAF2. Proc. Natl Acad. Sci. USA. 1999;96:8408. doi: 10.1073/pnas.96.15.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ni CZ, Welsh K, Leo E, et al. Molecular basis for CD40 signaling mediated by TRAF3. Proc. Natl Acad. Sci. USA. 2000;97:10395. doi: 10.1073/pnas.97.19.10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pullen SS, Miller HG, Everdeen DS, Dang TT, Crute JJ, Kehry MR. CD40-tumor necrosis factor receptor-associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry. 1998;37:11836. doi: 10.1021/bi981067q. [DOI] [PubMed] [Google Scholar]
- 6.Cheng G, Cleary AM, Ye Z, Hong DI, Lederman S, Baltimore D. Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science. 1995;267:1494. doi: 10.1126/science.7533327. [DOI] [PubMed] [Google Scholar]
- 7.Lee HH, Dempsey PW, Parks TP, Zhu X, Baltimore D, Cheng G. Specificities of CD40 signaling: involvement of TRAF2 in CD40-induced NF-kappaB activation and intercellular adhesion molecule-1 up-regulation. Proc. Natl Acad. Sci. USA. 1999;96:1421. doi: 10.1073/pnas.96.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pullen SS, Labadia ME, Ingraham RH, et al. High-affinity interactions of tumor necrosis factor receptor-associated factors (TRAFs) and CD40 require TRAF trimerization and CD40 multimerization. Biochemistry. 1999;38:10168. doi: 10.1021/bi9909905. [DOI] [PubMed] [Google Scholar]
- 9.Ye H, Park YC, Kreishman M, Kieff E, Wu H. The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol. Cell. 1999;4:321. doi: 10.1016/s1097-2765(00)80334-2. [DOI] [PubMed] [Google Scholar]
- 10.Pullen SS, Dang TT, Crute JJ, Kehry MR. CD40 signaling through tumor necrosis factor receptor-associated factors (TRAFs). Binding site specificity and activation of downstream pathways by distinct TRAFs. J. Biol. Chem. 1999;274:14246. doi: 10.1074/jbc.274.20.14246. [DOI] [PubMed] [Google Scholar]
- 11.Leo E, Welsh K, Matsuzawa S, et al. Differential requirements for tumor necrosis factor receptor-associated factor family proteins in CD40-mediated induction of NF-kappaB and Jun N-terminal kinase activation. J. Biol. Chem. 1999;274:22414. doi: 10.1074/jbc.274.32.22414. [DOI] [PubMed] [Google Scholar]
- 12.Jabara H, Laouini D, Tsitsikov E, et al. The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity. 2002;17:265. doi: 10.1016/s1074-7613(02)00394-1. [DOI] [PubMed] [Google Scholar]
- 13.Ahonen C, Manning E, Erickson LD, et al. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat. Immunol. 2002;3:451. doi: 10.1038/ni792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yasui T, Muraoka M, Takaoka-Shichijo Y, et al. Dissection of B cell differentiation during primary immune responses in mice with altered CD40 signals. Int. Immunol. 2002;14:319. doi: 10.1093/intimm/14.3.319. [DOI] [PubMed] [Google Scholar]
- 15.Hostager BS, Bishop GA. Cutting edge: contrasting roles of TNF receptor-associated factor 2 (TRAF2) and TRAF3 in CD40-activated B lymphocyte differentiation. J. Immunol. 1999;162:6307. [PubMed] [Google Scholar]
- 16.Hostager BS, Haxhinasto SA, Rowland SL, Bishop GA. Tumor necrosis factor receptor-associated factor 2 (TRAF2)-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J. Biol. Chem. 2003;278:45382. doi: 10.1074/jbc.M306708200. [DOI] [PubMed] [Google Scholar]
- 17.Hauer J, Puschner S, Ramakrishnan P, et al. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc. Natl Acad. Sci. USA. 2005;102:2874. doi: 10.1073/pnas.0500187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leo E, Zapata JM, Reed JC. CD40-mediated activation of Ig-Cgamma1- and Ig-cepsilon germ-line promoters involves multiple TRAF family proteins. Eur. J. Immunol. 1999;29:3908. doi: 10.1002/(SICI)1521-4141(199912)29:12<3908::AID-IMMU3908>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 19.Basaki Y, Ikizawa K, Kajiwara K, Yanagihara Y. CD40-mediated tumor necrosis factor receptor-associated factor 3 signaling upregulates IL-4-induced germline cepsilon transcription in a human B cell line. Arch. Biochem. Biophys. 2002;405:199. doi: 10.1016/s0003-9861(02)00369-7. [DOI] [PubMed] [Google Scholar]
- 20.Castigli E, Young F, Carossino AM, Alt FW, Geha RS. CD40 expression and function in murine B cell ontogeny. Int. Immunol. 1996;8:405. doi: 10.1093/intimm/8.3.405. [DOI] [PubMed] [Google Scholar]
- 21.Castigli E, Alt F, Davidson L, et al. CD40 deficient mice generated by RAG-2 deficient blastocyst complementation. Proc. Natl Acad. Sci. USA. 1994;91:12135. doi: 10.1073/pnas.91.25.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawabe T, Naka T, Yoshida K, et al. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1994;1:167. doi: 10.1016/1074-7613(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 23.Jabara HH, Fu SM, Geha RS, Vercelli D. CD40 and IgE: synergism between anti-CD40 mAb and IL-4 in the induction of IgE synthesis by highly purified human B cells. J. Exp. Med. 1990;172:1861. doi: 10.1084/jem.172.6.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manis JP, Tian M, Alt FW. Mechanism and control of class-switch recombination. Trends Immunol. 2002;23:31. doi: 10.1016/s1471-4906(01)02111-1. [DOI] [PubMed] [Google Scholar]
- 25.Tsukamoto N, Kobayashi N, Azuma S, Yamamoto T, Inoue J. Two differently regulated nuclear factor kappaB activation pathways triggered by the cytoplasmic tail of CD40. Proc. Natl Acad. Sci. USA. 1999;96:1234. doi: 10.1073/pnas.96.4.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coope HJ, Atkinson PG, Huhse B, et al. CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J. 2002;21:5375. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000;18:621. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 28.Beinke S, Ley SC. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem. J. 2004;382:393. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aicher A, Shu GL, Magaletti D, et al. Differential role for p38 mitogen-activated protein kinase in regulating CD40-induced gene expression in dendritic cells and B cells. J. Immunol. 1999;163:5786. [PubMed] [Google Scholar]
- 30.Grech AP, Amesbury M, Chan T, Gardam S, Basten A, Brink R. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-kappaB activation in mature B cells. Immunity. 2004;21:629. doi: 10.1016/j.immuni.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 31.Morrison MD, Reiley W, Zhang M, Sun SC. An atypical tumor necrosis factor (TNF) receptor-associated factor-binding motif of B cell-activating factor belonging to the TNF family (BAFF) receptor mediates induction of the noncanonical NF-kappaB signaling pathway. J. Biol. Chem. 2005;280:10018. doi: 10.1074/jbc.M413634200. [DOI] [PubMed] [Google Scholar]
- 32.Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 2007;27:253. doi: 10.1016/j.immuni.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28:391. doi: 10.1016/j.immuni.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 34.Pohl T, Gugasyan R, Grumont RJ, et al. The combined absence of NF-kappa B1 and c-Rel reveals that overlapping roles for these transcription factors in the B cell lineage are restricted to the activation and function of mature cells. Proc. Natl Acad. Sci. USA. 2002;99:4514. doi: 10.1073/pnas.072071599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zarnegar B, He JQ, Oganesyan G, Hoffmann A, Baltimore D, Cheng G. Unique CD40-mediated biological program in B cell activation requires both type 1 and type 2 NF-kappaB activation pathways. Proc. Natl Acad. Sci. USA. 2004;101:8108. doi: 10.1073/pnas.0402629101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franzoso G, Carlson L, Poljak L, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J. Exp. Med. 1998;187:147. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brady K, Fitzgerald S, Moynagh PN. Tumour-necrosis-factor-receptor-associated factor 6, NF-kappaB-inducing kinase and IkappaB kinases mediate IgE isotype switching in response to CD40. Biochem. J. 2000;3(Pt 350):735. [PMC free article] [PubMed] [Google Scholar]
- 38.Weih DS, Yilmaz ZB, Weih F. Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J. Immunol. 2001;167:1909. doi: 10.4049/jimmunol.167.4.1909. [DOI] [PubMed] [Google Scholar]
- 39.Jabara HH, Geha RS. Jun N-terminal kinase is essential for CD40-mediated IgE class switching in B cells. J. Allergy Clin. Immunol. 2005;115:856. doi: 10.1016/j.jaci.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 40.Xie P, Hostager BS, Bishop GA. Requirement for TRAF3 in signaling by LMP1 but not CD40 in B lymphocytes. J. Exp. Med. 2004;199:661. doi: 10.1084/jem.20031255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castigli E, Wilson SA, Scott S, et al. TACI and BAFF-R mediate isotype switching in B cells. J. Exp. Med. 2005;201:35. doi: 10.1084/jem.20032000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uchida J, Yasui T, Takaoka-Shichijo Y, et al. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science. 1999;286:300. doi: 10.1126/science.286.5438.300. [DOI] [PubMed] [Google Scholar]
- 43.Lu LF, Ahonen CL, Lind EF, et al. The in vivo function of a noncanonical TRAF2-binding domain in the C-terminus of CD40 in driving B-cell growth and differentiation. Blood. 2007;110:193. doi: 10.1182/blood-2006-07-038414. [DOI] [PMC free article] [PubMed] [Google Scholar]