

Abstract
Limb-girdle muscular dystrophy type 2H (LGMD2H) and sarcotubular myopathy are hereditary skeletal muscle disorders caused by mutations in TRIM32. We previously identified TRIM32 as an E3 ubiquitin ligase that binds to myosin and ubiquitinates actin. To date four TRIM32 mutations have been linked to LGMD2H, all of which occur in the C-terminal NHL domains. Unexpectedly, a fifth mutation in the B-box of TRIM32 causes a completely different, multisystemic disorder, Bardet–Biedl syndrome type 11. It is not understood how allelic mutations in TRIM32 can create such diverse phenotypic outcomes. To generate a tool for elucidating the complex in vivo functions of TRIM32, we created the first murine Trim32 knock-out model (T32KO). Histological analysis of T32KO skeletal muscles revealed mild myopathic changes. Electron microscopy showed areas with Z-line streaming and a dilated sarcotubular system with vacuoles—the latter being a prominent feature of sarcotubular myopathy. Therefore, our model replicates phenotypes of LGMD2H and sarcotubular myopathy. The level of Trim32 expression in normal mouse brain exceeds that observed in skeletal muscle by more than 100 times, as we demonstrated by real-time PCR. Intriguingly, analysis of T32KO neural tissue revealed a decreased concentration of neurofilaments and a reduction in myelinated motoraxon diameters. The axonal changes suggest a shift toward a slower motor unit type. Not surprisingly, T32KO soleus muscle expressed an elevated type I slow myosin isotype with a concomitant reduction in the type II fast myosin. These data suggest that muscular dystrophy due to TRIM32 mutations involves both neurogenic and myogenic characteristics.
INTRODUCTION
TRIM32 protein belongs to the tripartite motif (TRIM) family (1) that is currently comprised of approximately 77 members (as outlined in HUGO Gene Nomenclature Committee database, http://www.genenames.org/genefamily/trim.php). This family is defined by the presence of three linked motifs including a RING finger, a B-box and a coiled-coil domain. The domain structure of TRIM32 is shown in Supplementary Material, Figure S2C. Murine Trim32 (GenBank accession number NM_053084) and human Trim32 (GenBank accession number NM_012210) are highly homologous with 87% nucleotide and 95% amino acid identity. The similarity is even higher in conserved domains (100% identity for RING domain and 97% for NHL and B-box domains). TRIM32 has been shown to possess E3 ubiquitin ligase activity, attributable to its RING finger (2–4). E3 ubiquitin ligases are proteins that carry out post-translational modification of targets by participating in a cascade of ubiquitin-modifying reactions, which result in conjugation of a ubiquitin moiety onto a target substrate (5). Hundreds of E3 ligases have been identified, many of which have disease associations (6). Furthermore, muscle-specific ligases have been shown to participate in muscle atrophy (7).
Since its identification in 1995 (8), a pleiotropic role for TRIM32 (MIM 602290) has emerged. This protein has been implicated in diverse pathologic processes such as muscular dystrophy (9–11), cancer (3,4) and Bardet–Biedl syndrome (12) [BBS (MIM 209900)]—a multisystemic, oligogenic disorder. Moreover, TRIM32 is up-regulated in the occipital lobe of patients with Alzheimer's disease (13). At the present time, it is unclear how mutations in one protein can result in such dissimilar phenotypes as muscular dystrophy and BBS.
Mutations in Trim32 result in two distinct hereditary disorders. The first disease identified to be associated with TRIM32 mutations was limb-girdle muscular dystrophy type 2H [LGMD2H (MIM 254110)], which is a mild autosomal recessive muscular dystrophy with a variable clinical presentation. This rare skeletal muscle disorder was first described in the Manitoba Hutterites, a genetically isolated population in Canada. Affected individuals were found to carry a homozygous mutation (D487N) in one of the C-terminal NHL domains of TRIM32 [NHL repeats are conserved domains defined by amino acid sequence homologies among Ncl-1, HT2A and Lin-41 proteins (InterPro database)] (9). It was subsequently demonstrated that the identical mutation causes another muscular disease—sarcotubular myopathy (10), which was described in both Hutterite (14) and non-Hutterite (15) patients. The common mutation found in LGMD2H and sarcotubular myopathy suggests that these two conditions represent different forms of the same muscular disorder. Interestingly, the D487N mutation does not appear to disrupt the capacity of TRIM32 to perform ubiquitination, suggesting that the mutation disrupts another characteristic of the protein (2). Recently, three novel mutations in TRIM32 (R394H, T520TfsX13 and D588del) were identified in non-Hutterite European patients with LGMD (11). Similar to the previously identified D487N mutation, all these new mutations occurred in the C-terminal NHL domains of TRIM32. The onset of disease and clinical manifestations are highly variable among the different affected individuals even carrying the same D487N mutation. Phenotypic outcomes of LGMD2H and sarcotubular myopathy are primarily characterized by myopathic features (10,11,15–17). However, potentially neurogenic elements (namely neurophathic EMG components, paresthesia, paresis and hypoactive tendon reflexes) were also reported in some patients (10,11,15).
In 2006, another disease was identified to be associated with mutations in TRIM32. The homozygous P130S mutation occurs in the B-box domain of the protein and causes BBS (12). BBS is characterized by obesity, retinopathy, genito-urinary tract malformations, cognitive impairment, polydactyly, renal and cardiac anomalies, hypertension and diabetes (18). To date 12 BBS genes have been identified with Trim32 being BBS11. Although BBS genes encode for proteins which do not belong to the same functional category, mutations of these genes result in a similar pleiotropic phenotype. The precise functions of the BBS proteins have yet to be determined; however, current data support a role for many BBS proteins in function of immotile sensory (also called primary) cilia and intraflagellar transport (19). This role for TRIM32 was demonstrated in a zebrafish knockdown model (12).
Several attempts have been made to uncover molecular mechanisms underlying TRIM32's role in cancer. TRIM32 was shown to be overexpressed in skin cancer cells and to regulate NF-κB activity through ubiquitination and degradation of PIASy (protein inhibitor of STATs-signal transducers and activators of transcription) (3). By controlling PIASy stability, TRIM32 inhibits ultraviolet B (UVB)-induced keratinocyte apoptosis through the induction of NF-κB. Another report suggests a role for TRIM32 as a novel oncogene that promotes cell growth, transforming activity and cell motility (4). These authors demonstrated that TRIM32 ubiquitinates and promotes degradation of Abl-interactor 2 (Abi2)—a tumor suppressor and cell migration inhibitor.
Due to its ubiquitous expression, TRIM32 may have a number of divergent tissue-specific functions and substrates. Thus, different mutations in TRIM32 may cause alterations in its turn-over rate, subcellular localization and/or substrate specificity that could feasibly result in diverse pathological outcomes. Numerous examples can be found in the literature in which mutations in one gene cause different disorders. For example, mutations in RET protooncogene (MIM 164761) result in Hirschsprung disease [a malady of the colon (MIM 142623)], multiple endocrine neoplasia type IIA (MIM 171400) or type IIB (MIM 162300) and familial medullary thyroid carcinoma (MIM 188550) (20). Lamin A/C (MIM 150330) mutations have been linked to at least 10 degenerative heritable disorders that primarily affect muscle, adipose, bone or neural tissues and also result in premature aging syndromes (21). Several animal models are available for these conditions and serve as an invaluable aid for uncovering disease mechanisms and facilitating studies of pathogenic processes.
Here, we describe the first Trim32 knock-out (T32KO) mouse model. The T32KO model replicates human muscular dystrophy phenotypes (LGMD2H and sarcotubular myopathy). Moreover, it allowed for the uncovering of a neurogenic component caused by disruption of the Trim32 gene, suggesting a novel role for TRIM32 in the nervous system. The T32KO mouse provides an excellent model system for elucidating in vivo functions of TRIM32 and identification of future therapeutic targets.
RESULTS
Disruption of mouse Trim32 gene
To assess TRIM32s' biological role in skeletal muscle, we generated mice with TRIM32 deficiency. These mice were created using the BGA355 embryonic stem cell line (BayGenomics, San Francisco, CA, USA) carrying a gene trap insertion in the Trim32 gene within exon 2, which includes the entire open reading frame (Fig. 1A). The integration site of the β-geo cassette into Trim32 was identified by sequencing. Genotyping strategy, and PCR is shown in Figure 1A and B.
Figure 1.

Generation of Trim32-null mice. (A) Schematic of the Trim32 gene with integrated β-geo cassette. Integration site was verified by sequencing. (B) Genotyping PCR for the presence of gene trap insertion using three genotyping primers (shown by numbered arrows in A) results in production of an 800 bp band in KO, amplified from the allele with the integrated β-geo cassette (primers 1 and 3) and 300 bp band from the WT allele (primers 1 and 2). PCR from HET containing both alleles (primers 1, 2 and 3) yields both bands. (C) Schematic structure of Trim32 genetic locus on mouse chromosome 4. Mouse Trim32 gene lies within the intron flanked by exons 16 and 15 of Astn2 gene, which runs in the opposite direction. (D) RT–PCR analysis demonstrates the disruption of Trim32 after the gene trap insertion. Primers used for this analysis are represented by triangles on the map shown on (C). Trim32 PCR products are absent in T32KO, except for its most 5′ end fragment preceding the integration site of β-geo cassette. β-Actin PCR is used as cDNA control. (E) RT–PCR followed by sequencing of the products obtained after amplification from both WT and T32KO brain cDNA confirmed the preserved expression of Astn2 transcript variants 1 (GenBank accession number NM_019514) and 2 (GenBank accession number NM_207109) in T32KO. Primers Astn2Fex3 and Astn2Rex16 flank the alternatively spliced region of the Astn2 gene (exon 3 through exon 16). Lane (M): 1 kb plus DNA ladder (Invitrogen). GAPDH PCR is used as cDNA control. (F) and (G) The levels of Astn2 expression are not changed in T32KO brain and skeletal muscle as demonstrated by real-time PCR using Astn2F and Astn2R primers indicated as arrows on (C). PCR data from two representative samples for each genotype are shown on (F). Lane (–) is a negative control in which no DNA was added into the PCR. GAPDH PCR is used as cDNA control. Real-time PCR data are shown in graph (G) as average cycle threshold (Ct) for Astn2 normalized by Ct for GAPDH detected in samples from three WT and three KO animals.
The mouse Trim32 gene lies within an intron flanked by exons 16 and 15 of the Astrotactin 2 (Astn2) gene, which runs in the opposite direction on chromosome 4 (Fig. 1C). To ensure both that expression of Trim32 was disrupted and expression of Astn2 was preserved in the T32KO tissues after the gene trap insertion, we performed rigorous RT–PCR analysis. Using various pairs of primers (shown in Fig. 1C as arrowheads), we demonstrated the absence of Trim32 RNA message in the T32KO samples except for the very 5′-terminal region of Trim32 that precedes the integration site of the β-geo cassette (Fig. 1D). On the contrary, expression of the Astn2 splice isoforms in T32KO brain was not perturbed (Fig. 1E). Additionally, real-time PCR analysis demonstrated that levels of Astn2 mRNA are unchanged in T32KO tissues compared with wild-type (WT) (Fig. 1F and G). Therefore, only Trim32, and not the Astn2 gene, is disrupted in this TRIM32-deficient mouse.
To explore the possibility of whether a Trim32-β-geo fusion mRNA was generated after the gene trap insertion, another RT–PCR was carried out (Supplementary Material, Fig. S1A). Since the entire open reading frame of Trim32 is contained within a single exon, we treated RNA samples with DNase I before cDNA preparation to remove genomic DNA contamination. DNase I-treated RNA had no specific message for Trim32-β-geo fusion (Supplementary Material, Fig. S1B). β-Galactosidase immunoblotting of skeletal muscle and brain extracts further confirmed the absence of any TRIM32-β-geo fusion protein in T32KO tissues (Supplementary Material, Fig. S1C).
T32KO muscle performance
Mice heterozygous (HET) for the Trim32 null allele were expanded on the mixed 129 SvEvBrd × C57 BL/6J background. Breeding of heterozygous mice yielded a normal Mendelian ratio [22.1 ± 10.3% WT, 52.2 ± 11.5% HET and 25 ± 11.2% knock-out (KO)]. T32KO mice are born phenotypically indistinguishable from WT littermates. Homozygous KO breeders seemed to produce fewer pups than WT breeders and at a lower frequency. Body weights of T32KO males become slightly elevated (∼10%) by the age of 8 weeks compared with WT or HET; this difference persists with age (Fig. 2A). In contrast, muscle weight normalized to body weight was reduced in T32KO for both fast [tibialis anterior (TA)] and slow (soleus) muscles (Fig. 2B). This finding suggests that muscle function in T32KO mice may be impaired.
Figure 2.

Muscle performance is impaired in T32KO. (A) Comparison of body weight gain in T32KO and WT mice. Average body weight in grams ± SEM was calculated for males of each genotype and plotted as a function of age. Average body weight was ∼10% higher in T32KO after 8 weeks of age, compared with age-matched WT or HET (n = 5–15, *P < 0.05). (B) The ratio of muscle weight to body weight was calculated and expressed as the percent of WT value ± SEM for fast TA and slow (soleus) muscles. T32KO muscle weight normalized to body weight was reduced by 19 and 27% for TA and soleus muscles, respectively (n = 15, **P = 0.002, *P = 0.03). (C) Forelimb grip strength of KO (n = 21), WT (n = 26) and HET (n = 25) animals was measured using grip strength meter and expressed as percent of WT value ± SEM. Age of the tested animals: 5–9 months. Grip strength of T32KO mice was reduced by 17%, compared with WT or HET littermates (**P < 0.01 and *P < 0.05). (D) Wire hang test performance. Latency to fall was compared within genotypes as average time in seconds ± SEM, n = 11–16, age 5–9 months. Latency to fall was 1.8 times less in T32KO animals, compared with WT (*P < 0.05).
There are several tests to assess muscle performance in mice. The grip strength test is used to measure forelimb strength in mice. The average peak tension normalized to body weight was decreased in T32KO animals by 17% compared with WT or HET littermates (Fig. 2C). Muscle strength can also be tested by the use of the wire hang test. T32KO mice were less successful on the wire test, exhibiting a significantly reduced latency to fall, compared with WT littermates (Fig. 2D). Thus, muscle weight and muscle strength are impaired in T32KO animals.
Histological analysis of T32KO muscles
To further characterize T32KO skeletal muscles, we performed morphological and histochemical examination of frozen sections of muscles. All analyses were done in comparison with age-matched WT control muscles. These studies revealed a variation in myofiber size and shape in T32KO muscles with very small diameter fibers occasionally observed. Staining with anti-developmental myosin heavy chain (MYHC) did not reveal any regenerating myotubes. No inflammatory infiltrates were seen following anti-CD11b staining for immune cells. Apoptotic myonuclei were present in T32KO muscles at a low level as revealed by TUNEL staining (data not shown).
To further analyze the muscle phenotype, we utilized histochemical staining including hematoxylin–eosin, nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR), succinic dehydrogenase (SDH), non-specific esterase, modified Gomori trichrome (Fig. 3) and ATPase (data not shown). Light microscopy revealed mild dystrophic changes in T32KO muscles. Myonuclei in some fibers were displaced from the periphery to the center, which was confirmed by staining serial sections with hematoxylin and DAPI (Fig. 3A and B). The number of myofibers containing multiple centrally placed nuclei was significantly increased in T32KO muscles compared with WT (Fig. 3I and J). Fiber splitting and occasional ring fibers were apparent by hematoxylin and modified Gomori trichrome staining (Fig. 3C and D). Within a number of myofibers, fragmented areas with numerous tiny split fibers were observed (arrows in Fig. 3F). This feature was previously reported as a secondary myopathic change in neurogenic disorders (22). Some samples contained small angulated fibers (Fig. 3E and F), similar to those commonly observed following denervation; however, these fibers did not show dark staining with non-specific esterase, which is the characteristic of denervation (Fig. 3F). Another denervation/reinnervation feature, fiber type grouping, also was not an attribute of T32KO muscles as revealed by ATPase staining (data not shown). Therefore, the small fibers identified in the T32KO muscles do not appear to be a result of denervation.
Figure 3.
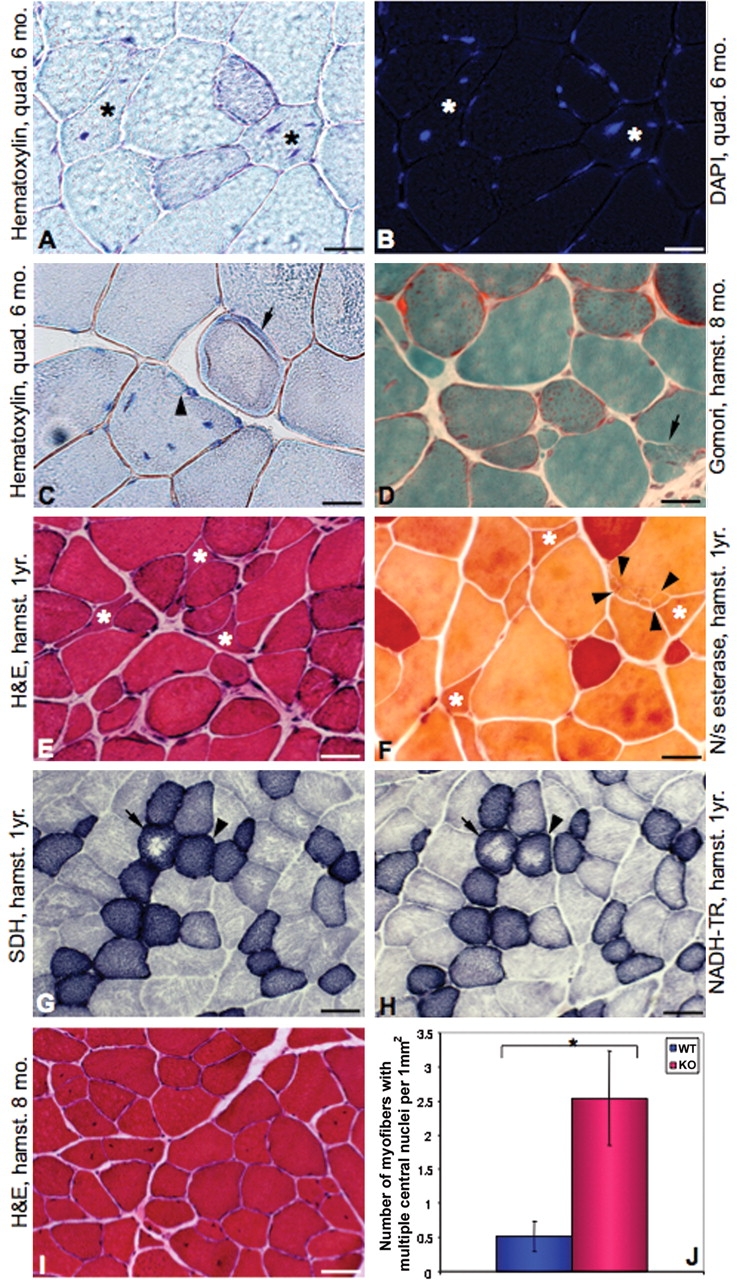
Histology of T32KO muscles. Micrographs of cross-sections of T32KO muscles. Type of muscles (quad.: quadriceps and hamstr.: hamstring group of muscles) and age of animals are specified on the micrographs. (A and B) Serial sections stained with hematoxylin (A) or DAPI (B) show internalized nuclei. Asterisks indicate fibers with multiple nuclei displaced from the periphery to the center. Scale bar is 10 µm. (C) Hematoxylin staining shows ring-fiber (arrow) and fiber splitting (arrowhead). Note mislocalized nuclei. Scale bar is 10 µm. (D) Modified Gomori trichrome staining. Arrow points to a splitting fiber. Scale bar is 10 µm. (E) Hematoxylin and eosin staining. Note fiber diameter variability and small angulated fibers (asterisks). Scale bar is 20 µm. (F) Non-specific esterase staining. Angulated fibers (asterisks) are not stained darkly in this enzymatic reaction suggesting that they are not denervated. Arrows indicate an area of the fiber that is fragmented. Scale bar is 10 µm. (G and H) Serial sections stained for SDH (G) and NADH-TR (H) enzymatic activity. Arrows point to fibers with central pallor, indicating defects in both mitochondrial and SR staining. Arrowheads indicate a fiber with a defect in SR only since SDH (mitochondrial) staining is normal. Scale bar is 40 µm. (I) Hematoxylin and eosin staining, scale bar is 40 µm. (J) Number of myofibers containing two or more centrally located nuclei was quantified in cross-sections of hamstring group of muscles from three WT and three T32KO animals (*P < 0.05).
We used NADH-TR and SDH enzymatic reactions of serial sections to differentiate between defects that are sarcoplasmic reticulum (SR) in origin versus defects that are mitochondrial in origin. NADH-TR staining reveals both SR and mitochondria, whereas SDH staining is mitochondria-specific. By means of this technique, we detected fibers with central pallor in the T32KO, where NADH-TR and/or SDH staining was reduced (Fig. 3G and H). This finding suggests defects in both SR and mitochondrial oxidative enzymes in T32KO muscles. Reduced NADH-TR and SDH enzymatic activity in muscle fibers is often observed in both myogenic and neurogenic disorders (23).
Some of these features (namely an increased number of internal nuclei, variation in fiber size, atrophic fibers dispersed between slightly hypertrophied fibers and areas with loss of NADH-TR enzyme activity) were also found in patients with LGMD2H and sarcotubular myopathy (10,15). Taken together these histological findings reveal a mild myopathy phenotype in TRIM32 null mice; however, whether they derive from a myogenic and/or a neurogenic origin cannot be discriminated based solely on histochemical staining.
Electron microscopy examination of T32KO muscles
Electron microscopy was performed to further refine the morphological analysis. Ultrastructural examination of the T32KO muscles confirmed the presence of dystrophic changes (Fig. 4). Electron micrographs showed profuse sarcotubular profiles, which were dispersed irregularly between atrophic myofibers. In addition, increased intermyofibrillar space (Fig. 4A, C and D), Z-line streaming, disorganized sarcomeres, myofibrillar degeneration (Fig. 4B and C), membranous structures as well as autophagic double-membraned vacuoles could be observed (Fig. 4D and E). Internalized nuclei were also occasionally present. Some areas had mitochondrial loss accompanied by clusters of mitochondria in adjacent areas, confirming our findings of fibers with central pallor by light microscopy. Interestingly, sarcotubular myopathy is characterized by similar features of a dilated sarcotubular system and proliferation of the T-tubule system with abundant sarcotubular profiles, vacuoles and degenerative changes in the myofibrils with Z-line streaming (10,15). These data validate the T32KO mouse as a suitable model for studying disease mechanisms of LGMD2H and sarcotubular myopathy.
Figure 4.

Electron microscopy of T32KO skeletal muscles. (A–E) Electron micrographs represent T32KO muscles (TA and solei as indicated on the figure). Note increased intermyofibrillar spaces (A) and Z-line streaming with myofibrillar degeneration and disorganized sarcomeres (B). Higher magnification shows membranous structures, dilated sarcotubular system (C, D) and autophagic double-membraned vacuoles (E). Scale bars are 2 µm for (A) and (B), 1 µm for (C) and 0.5 µm for (D) and (E). (F) Normal sarcomeric organization of WT skeletal muscle. Scale bar is 0.5 µm.
Expression of TRIM32 in skeletal muscle and brain
The creation of the T32KO mouse provided the opportunity to examine the specificity of our polyclonal anti-TRIM32 antibody (T332) that was previously described (2). As was reported by our laboratory (2) and others (9,13), TRIM32 is expressed not only in skeletal muscle, but also is highly expressed in nervous tissue. Therefore, both brain and skeletal muscle from T32KO and WT animals were subjected to western blotting with T332 antibody. As anticipated, the 80 kDa TRIM32 protein was absent in western blots using extracts from KO brains (Fig. 5A, left panel), confirming disruption of Trim32 by the gene trap insertion. However, despite the fact that Trim32 mRNA is absent from skeletal muscle of T32KO animals (Fig. 1D), T332 antibody revealed a 72 kDa-band in muscle extracts from both WT and KO animals (Fig. 5A, right panel). Previously, we attributed this band in WT skeletal muscles to the TRIM32 protein based upon the following observations: (i) the predicted molecular weight of TRIM32 is 72 kDa; (ii) the rabbit anti-peptide affinity purified T332 antibody was raised against a specific TRIM32 peptide and readily recognized recombinant TRIM32 protein and (iii) recognition of the 72 kDa band in skeletal muscle occurred only after immunization of the rabbit and was not detected on western blots probed with non-immune serum from this same rabbit. Taking into account that our RT–PCR data (Fig. 1D) showed no Trim32 mRNA in T32KO skeletal muscle, we can now conclude that T332 antibody recognizes endogenous TRIM32 protein only in brain, but not in skeletal muscle and that TRIM32 likely migrates at 80 kDa instead of the anticipated 72 kDa.
Figure 5.

TRIM32 expression in brain and skeletal muscle. (A) Western blot analysis of brain and skeletal muscle (hamstring) lysates using T332 antibody. The 80 kDa band in brain lysate is absent in T32KO, but the 72 kDa band in muscle persists in the T32KO. Anti-GAPDH staining is shown for loading control. (B) Real-time PCR data as Trim32 relative quantity normalized to GAPDH demonstrate a greater than 100-fold difference in Trim32 expression between brain and skeletal muscles (gastroc.: gastrocnemius and ham.: hamstring groups of muscles). Samples from three different WT animals were analyzed. Each sample was run in triplicate for both Trim32 and GAPDH amplifications, *P < 0.05. (C) Regular PCR using real-time PCR primers and different number of cycles is shown to visualize the real-time PCR data. Trim32 PCR band in muscles is detected only after 40 cycles of amplification, whereas in brain it can be observed after 30 cycles. GAPDH PCR used as a control shows approximately equal amount of cDNA in brain and muscle samples. M: markers (DNA ladder). Lane (–) is a negative control in which no DNA was added into the PCR.
The lack of an 80 kDa TRIM32 band in WT muscles led us to test the hypothesis that TRIM32 is expressed in muscle at a level that is below the sensitivity of detection by western blotting. To investigate the levels of Trim32 expression in brain and skeletal muscles, we used real-time PCR. These studies showed that the expression of Trim32 in brain exceeds that detected in muscle by more than 100 times (Fig. 5B and C). The observed difference in mouse tissues is in accordance with the previously published northern blot data showing much higher level of Trim32 expression in human brain, compared with human skeletal muscle (9). This high level of expression of Trim32 in brain suggests a role for the protein in the nervous system.
Identification of a protein with reduced expression in T32KO brain
We also tested several commercially available antibodies raised against different epitopes of TRIM32 on brain extracts from T32KO and WT mice (Supplementary Material, Fig. S2). Unfortunately, even though all the tested antibodies detected the recombinant protein (Supplementary Material, Fig. S2A), they failed to recognize native TRIM32 in WT brain lysates (Supplementary Material, Fig. S2B).
Intriguingly, all the antibodies tested, including our T332 antibody, recognized a band of ∼75 kDa, which was reduced in T32KO brain lysates, compared with WT (Fig. 5A and Supplementary Material, Fig. S2B). In order to ascertain the identity of this band, we immunoprecipitated the protein from brain and then analyzed it by mass spectrometry (for details, see Materials and Methods section). Thirty-one mass signals from matrix-assisted laser desorption/ionization–time of flight mass spectrometry (MALDI–TOF MS) fingerprinting were used for protein identification within the Mascot software to screen a non-redundant protein data bank (Supplementary Material, Fig. S3). The only target with a significant and high Mascot score of 174 (54% sequence coverage) was neurofilament light polypeptide (NEFL) with a predicted molecular mass of 61.5 kDa and apparent electrophoretic mobility of 68 kDa (close to that which we observed by western blotting). Moreover, this immunoprecipitated protein was soluble only in the presence of urea and thiourea or more stringent denaturing agents such as SDS or guanidine, which is in agreement with the nature of the NEFL protein, an intermediate filament from nervous tissue. Therefore, the 75 kDa band identified by TRIM32 antibodies in brain has no relation to TRIM32 and is non-specific. This situation is not unique, since in our experience many antibodies used in muscle research non-specifically stain actin—one of the most abundant muscle proteins. It is striking, however, that the intensity of the NEFL band appeared to be reduced in the T32KO brain (Fig. 5A and Supplementary Material, Fig. S2B). This finding is worthy of further exploration, given that NEFL and neurofilament medium polypeptide (NEFM) proteins control the caliber of myelinated axons (24,25).
Expression levels of neurofilaments in T32KO brain
Five major types of intermediate filaments are expressed in nervous tissue: NEFL (GenBank accession number NP_035040), NEFM (GenBank accession number NP_032717), neurofilament heavy polypeptide [NEFH (GenBank accession number NP_035034)], peripherin [PRPH (GenBank accession number NP_038667)] and α-internexin [INA (GenBank accession number NP_666212)]. Neurofilaments are major components of the neuronal cytoskeleton and are required for proper radial growth and stability of axons (26). Disruption of neurofilament genes causes loss of motor axons, hypotrophy of myelinated axons and delayed maturation of regenerating axons (24,25). Abnormal accumulation of neurofilaments have been detected in neural tissue from patients with neurodegenerative disorders (amyotrophic lateral sclerosis, Alzheimer's and Parkinson's diseases) (27). Mutations in NEFL (MIM 162280) have been reported to cause Charcot-Marie-Tooth disease type 2E (MIM 607684) and 1F (MIM 607734), which are hereditary motor and sensory neuropathies characterized by muscle atrophy and loss of touch sensation (28–31).
To explore the possibility of a neurogenic phenotype in T32KO, we assessed the protein and mRNA levels of neurofilaments in T32KO brain by western blot and real-time PCR. First, we confirmed the reduction of NEFL protein in T32KO brain using NEFL-specific antibodies (Fig. 6A and B). Levels of NEFM and NEFH were also decreased in T32KO (Fig. 6A and B). Stabilization of mRNA plays an important role in the regulation of neurofilament expression levels via elements in the 3′-UTR of the transcripts (32,33). Therefore, we tested neurofilament mRNA levels by real-time PCR. The expression of all five intermediate filaments found in nervous tissue was not changed at the mRNA level (Fig. 6C). Thus, neurofilament protein concentrations are reduced in brains from T32KO mice.
Figure 6.

Neurofilament protein concentrations are reduced in T32KO brains. (A) Western blots of T32KO brain lysates using NEFL, NEFM and NEFH-specific antibodies. GAPDH, β-actin and Ponceau S staining is shown for loading control. TRIM32 staining using T332 antibody demonstrates an 80 kDa TRIM32 band (arrowhead) in WT brain lysates, which is absent in T32KO. (B) Quantification of the blots shown in (A). Average intensity of the bands is expressed as a percent of the WT value ± SEM (n = 3, *P < 0.05). (C) Relative mRNA levels of intermediate filaments are not changed in KO brains, compared with WT, as revealed by real-time PCR. NEFL: neurofilament light polypeptide; NEFM: neurofilament medium polypeptide; NEFH: neurofilament heavy polypeptide; PRPH: peripherin and INA: α-internexin.
Reduction of diameters of myelinated axons in T32KO
Since NEFL and NEFM control axonal caliber, a change in their protein levels might affect axon calibers in T32KO. To test this hypothesis, we choose to analyze lumbar spine segment L4 ventral roots (VRs), since motor axons outgoing from this segment innervate the hind limbs, where we observed myopathic changes in the T32KO mouse model. L4 VRs exhibit many large myelinated axons that originate from motor neurons (Fig. 7A). We found that although the cross-sectional area and total number of axons in L4 VRs are preserved in the T32KO, the average diameter of the axons is significantly reduced (Fig. 7B), suggesting axonal hypotrophy rather than loss of axons. Since the axonal diameter is reduced, the density of axons per unit area is slightly increased in T32KO (Fig. 7B). The histogram in Figure 7C demonstrates two peaks corresponding to small and large axon populations, representing predominantly γ- and α-motoneurons, respectively (34). The distribution of the T32KO axon diameters is shifted to the left, compared with WT (Fig. 7C), demonstrating that the reduced average axon diameter is primarily attributable to a reduction in the diameters of large axons. Thus, T32KO mice may exhibit a neurogenic component for the pathogenesis of T32KO myopathy.
Figure 7.

Diameters of myelinated axons are reduced in T32KO. (A) Semi-thin sections of L4 VRs from WT and KO stained with toluidine blue. L4 VRs were dissected from three WT and three KO, 8.5-month-old animals. Representative micrographs are shown. Scale bar is 10 µm. Insets show low magnification images of the sections. (B) Comparison of morphometric data from KO and WT L4 VRs. Cross-sectional area of L4 VRs is represented as the mean for each genotype ± SEM. Total number of axons in L4 VRs was calculated using VR cross-sectional area and number of axons per 1000 µm2. Axon density was calculated as the number of axons per 1000 µm2. T32KO axonal density is increased by 15%, compared with WT (*P < 0.05). Average axon diameter of T32KO is reduces by 22%, compared with WT (**P < 0.0001). (C) Histogram shows axon diameter distribution in L4 VR. Axon diameters were measured in three random areas of VR containing a total of 200–240 axons. The number of axons with certain diameter was expressed as the percent of total number of axons measured in each sample (three KO and three WT). Note that the peak corresponding to large diameter axons is shifted to the left in T32KO, compared with WT (arrows). Axons with diameter more than 9.5 µm were not observed in T32KO L4 VR.
MYHC isoform composition of T32KO and WT muscles
Axon diameter defines conduction velocity (35), which in turn is reflected in the innervation pattern of different types of muscle fibers. Indeed, axons with larger diameters innervate type II (fast) muscle fibers, whereas smaller axons innervate type I (slow) muscle fibers (36). Hence, an alteration in axon diameter suggests a change in motor unit type. To test this hypothesis, we used one of the most widely accepted and reliable criterion of muscle fiber typing, the MYHC isoform composition. Adult skeletal muscle isoforms include MYHC IIa (MYH2; MIM 160740), MYHC IIb (MYH4; MIM 160742), MYHC IIx/d (MYH1; MIM 160730) and MYHC-β/slow type I (MYH7; MIM 160760). Normally, there is strong type II fiber dominance in mouse muscles with the exception of the soleus and diaphragm muscles. To determine if a reduction in axon diameters had any effect on muscle fiber types in T32KO, we analyzed the MYHC isoform composition of soleus muscles of T32KO animals by gel electrophoresis. MYHC gel electrophoresis revealed a shift in the isoform distribution in T32KO soleus muscle toward a slower phenotype (Fig. 8). WT solei were composed of ∼35–40% of MYHC type I, which is in agreement with the previously published data (37). Even though there was some variability in MYHC isoform composition between individual animals (most likely due to a mixed genetic background of the analyzed mice), the amount of total fast MYHC type II (combined IIa, IIx and IIb) was significantly decreased in T32KO solei, compared with WT. Reciprocally, the amount of slow MYHC type I was significantly increased in T32KO. Therefore, the reduction of axonal calibers in T32KO is associated with a shift toward a slower muscle fiber phenotype.
Figure 8.

Decreased axonal diameter in T32KO is associated with a shift in fiber-type-specific MYHC isoforms. (A) MYHC isoform gel electrophoresis of soleus muscle lysates. Positions of individual MYHC isoforms are indicated. Five WT and six KO muscles were analyzed. M: Benchmark protein ladder (Invitrogen). (B) Quantification of the gels shown in (A). The fraction corresponding to individual MYHC isoforms were calculated as a percent of total MYHC content ± SEM in each sample. This accounted for the differences in loading. MYHC isoforms IIa and IIx co-migrate, and therefore were quantified together. Amount of MYHC type II (fast) is decreased and amount of MYHC type I (slow) is increased in T32KO soleus muscle by ∼8%, compared with WT (*P < 0.05).
DISCUSSION
The T32KO mouse model demonstrates a myopathic phenotype with features similar to those found in patients with the muscular disorders LGMD2H and STM. Analysis of neural tissue revealed a high level of TRIM32 expression in normal mouse brain compared with muscle. Intriguingly, Trim32-null mice showed decreased concentrations of neurofilaments in brain and reduced diameters of myelinated axons in L4 VR, which were associated with a shift in fiber-type specific MYHC composition of soleus muscle toward a slower isotype. These findings suggest a neurogenic component to the muscle pathology caused by disruption of the Trim32 gene.
A relationship between TRIM32 and neurofilament proteins
A relationship between TRIM32, myosin and neurofilament proteins is emerging. A report linking another TRIM family member (called TRIM2) with neurofilaments was recently published (38). TRIM2 is able to ubiquitinate NEFL, and loss of TRIM2 in KO mice results in NEFL inclusions (38). Intriguingly, there are more than 75 TRIM family members and only three members (TRIM2, TRIM3 and TRIM32) contain NHL domains. All three are highly expressed in the nervous system and all are able to interact with different isoforms of myosin (1,2,39,40). TRIM2 and TRIM32 have confirmed E3 ubiquitin ligase activity (2–4,38). Despite the striking structural and functional similarities between TRIM 2 and 32, there are definite disparities. TRIM2 does not seem to be expressed in skeletal muscle, and the only E2 ubiquitin conjugating enzyme that works in conjunction with TRIM2 is UbcH5a (38). Even though we found that the same E2 enzyme can facilitate TRIM32 E3 ubiquitin ligase activity, the most potent E2 for TRIM32 was UbcH6 (2). Consequently, KOs of these genes cause different phenotypes in mice, although both are related to disturbances in neurofilament levels. Trim2-deficient mice show NEFL-filled axonal swellings and neurodegeneration accompanied by tremor and ataxia (38), whereas we found a reduction in neurofilament levels without the signs of neurodegeneration in T32KO. Thus, although a relationship between these proteins is emerging, its nature with regard to disease phenotypes and normal cellular processes is unclear at this time.
T32KO myopathy phenotype is compatible with neurogenic etiology
In the present study, we describe myopathic abnormalities in T32KO mice. Although, many of the changes we observed in skeletal muscles of T32KO are commonly detected in different myopathies, none of them is incompatible with a neurogenic etiology. This observation is supported by the findings of decreased neurofilament content and reduced axon diameters in L4 VR. In addition, we did not find typical features of muscular dystrophy in the T32KO muscles including inflammation, necrosis or regeneration. These signs were also absent in patients with STM (10,15). In the T32KO mouse, decreased axonal calibers were associated with a shift toward a slower motor unit type. This neurogenic alteration may also contribute to the myopathic changes found in T32KO muscles, and the observed muscle fiber type shift supports this hypothesis.
Fiber splitting is a feature of T32KO myopathy, which is considered to be a typical morphological feature of dystrophic biopsies. However, this phenomenon has also been well documented in many types of neurogenic disorders (41–45). Myogenic fiber splitting occurs due to segmental fiber necrosis and/or subendomysial regeneration from myoblasts, whereas neurogenic splitting results from stresses imposed on weakened muscles by normal weight bearing loads (22,46,47). Loss of peripherally located myonuclei in T32KO does not seem to be a sign of regeneration, since these fibers do not express regeneration markers. However, individual myofiber splits have been associated with centrally placed nuclei (46). A reduction in NADH-TR staining is also described in many myogenic and neurogenic disorders (23). Fiber-type grouping, a typical feature of neurogenic myopathies, was not observed in T32KO. Angulated fibers were found, but did not show non-specific esterase activity, characteristic of denervation/reinnervation. Therefore, our data do not indicate a denervation process in T32KO, but rather suggest other forms of plasticity affecting the entire motor unit.
Motor unit alteration in T32KO
This study identified a phenotype of reduced motor axon diameter in the absence of TRIM32. It is understood that motoneurons innervating different functional types of skeletal muscle fibers exhibit distinct physiological and morphological features. For instance, motoneurons innervating fast twitch muscle fibers (type II) exhibit a faster conduction velocity and a larger caliber of their axons compared with those that innervate slow twitch muscle fibers (type I) in the cat (36,48,49). In the present study, it is suggested that neurons in the central nervous system express a reduced concentration of neurofilament proteins, as the brain of T32KO mice demonstrates a markedly decreased expression of the light, medium and heavy neurofilament proteins. Interestingly, there is an associated reduction in the size of myelinated axons in the L4 VRs of the KO mice. The latter finding suggests that the functional properties of motoneurons may have changed in the KO mice with a slowed conduction of action potentials taking place along myelinated motor axons.
Previous studies have demonstrated that motoneurons may exert a marked influence over the contractile, biochemical and histochemical properties of skeletal muscles. For instance, feline motoneurons that normally innervate a slow twitch muscle may convert a predominantly fast muscle to a slow type when motor units are cross re-innervated (50). Chronic low-frequency activation of the motor nerve of a predominantly fast-twitch muscle may also convert its motor units to a slower phenotype in cats with slowed contractions, reduced strength, increased endurance and an associated fast-to-slow conversion of muscle histochemical features (51). Furthermore, a partial denervation of a hind-limb muscle may induce compensatory peripheral sprouting of the nerve with an increase in the motor unit size of the remaining intact motor axons (52,53). A partial denervation also may reduce motoraxon conduction velocity and prolong the action potential after-hyperpolarization half-decay time, consistent with a slowing of the motor unit twitch properties (54). Therefore, it seems possible that a decreased expression of neurofilament proteins in motoneurons of T32KO mice may result in a change of motor unit phenotype to a slower twitch type, characterized by the observed decreased motoraxon caliber and a combined decrease in type II and an increase in type I myosin isoforms in skeletal muscle. Here, there is a direct link between our findings on myelinated axons and myosin isoforms in skeletal muscle, as the studied L4 VR is a prominent contributor of motoraxons to the sciatic nerve, which innervates many hind-limb muscles, including the soleus muscle used for the analysis of the myosin expression patterns. Interestingly, there is a case report describing the muscle biopsy of one patient with sarcotubular myopathy, which revealed ‘a slight type I fiber predominance’ by histochemical ATPase staining (15). Even though reports on biopsies from patients with LGMD2H or sarcotubular myopathy are sparse and more human studies are needed, this observation supports our finding of the myosin isoform switch due to TRIM32 deficiency.
Plausible neurogenic component in LGMD2H and STM
The myopathy in T32KO replicates well the LGMD2H and STM phenotypes. The pathologic phenotype was observed only in KO animals, whereas HET and WT mice were similar to each other (e.g. in body weights and limb strength, etc.), supporting the recessive manner of inheritance and loss of function for LGMD2H and STM mutations. LGMD2H is a recessively inherited neuromuscular disorder with highly variable and slowly progressive clinical features. The histopathological changes in affected muscles from LGMD2H biopsies were described previously as being typical of a primary myopathy and similar to other muscular dystrophies (16). At the present time, it is not known whether patients with TRIM32 mutations associated with LGMD2H and STM may also exhibit an involvement of the central and peripheral nervous system. Although this clinical condition is relatively rare and reports describing humans with TRIM32 mutations have been sparse, a recent study provides an interesting case description that includes mixed neuropathic and myopathic features by electromyography and symptom presentation characterized by weakness and paresthesia in a different subject (11). Neurological examination of two patients with STM revealed a paresis in both patients (15). In addition, tendon reflexes were hypoactive or absent in several patients with STM (10,15). However, it remains unclear whether a neurogenic component may also be present in the pathogenesis of LGMD2H/STM. Future electrodiagnostic studies with detailed investigations of both motor and sensory nerve conduction properties in a larger sample of patients with the TRIM32 mutation and clinical disease may provide important added insight into the pathobiology of this disorder.
In summary, we described a new role for TRIM32 in the nervous system. The axonal changes caused by disruption of Trim32 are compatible with the observed skeletal muscle pathology, which replicates human LGMD2H and STM features. Nevertheless, since TRIM32 is expressed both in brain and skeletal muscle, capable of ubiquitinating actin and interacting with myosin (2), we would like to emphasize that the myogenic etiology of T32KO myopathy and LGMD2H/STM cannot be excluded and will be further examined. The T32KO mouse proved to be a valid model for the investigation of LGMD2H pathology and will provide a tool for future studies of in vivo functions of TRIM32 and mechanisms of disease associated with its mutations that will eventually lead to the identification of therapeutic targets.
MATERIALS AND METHODS
All experimental protocols and use of animals were conducted in accordance with the National Institute of Health Guide for Care and Use of Laboratory Animals and approved by the UCLA Institutional Animal Care and Use Committee.
Generation of Trim32-null mice
T32KO mice were generated using the BGA355 mouse embryonic stem cell line [BayGenomics (former web-site http://baygenomics.ucsf.edu/; now available at International Gene Trap Consortium, http://www.genetrap.org/cgi-bin/annotation.py?cellline=BGA355)] carrying gene trap insertion in Trim32, within exon 2 (Fig. 1A). Sequencing of the PCR product produced from BGA355 genomic DNA using Seqtag forward and β-gal reverse primers (for primer sequences see Supplementary Material, Table S1) defined the position of the integration site after nucleotide 278 starting from the ATG codon in exon 2 of the Trim32 gene. Original founder mice were 129 SvEvBrd × C57 BL/6 chimeras, which were backcrossed to C57 BL/6J WT mice to obtain germ line transmission. Heterozygotes (HET) from this cross were interbred to produce KO and WT homozygotes. All analyses were performed on interbred mice on a mixed 129 SvEvBrd × C57 BL/6J background.
PCR and genotyping
Total RNA was isolated from mouse tissues using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Genomic DNA contamination was removed by DNAase I treatment for 30 min at 37°C. To produce cDNA, 2 µg of DNA-free RNA was used for first-strand cDNA synthesis with random hexamer primers and Superscript III reverse transcriptase (Invitrogen). The resulting cDNAs were used for PCR amplification. Genotyping was performed using genomic tail DNA (Fig. 1A and B). All primers used for PCR, genotyping and real-time PCR analysis are shown in Supplementary Material, Table S1.
Real-time PCR
Primers for real-time PCR, where possible, were selected to span intron–exon junctions to exclude the possibility of amplification from genomic DNA contamination. All primers were first tested in regular PCR amplification to ensure the production of a single band in each case. All real-time reactions were performed using IQ™ SYBR Green Supermix PCR reagent and MyIQ™ Single Color Real-time PCR Detection System (Bio-Rad, Hercules, CA, USA). Optical System Software Version 1.0 (Bio-Rad) was used to analyze the results. Quantification utilized standard curves made from serial dilutions of cDNA. Data from each sample were normalized by dividing the relative quantity of target gene cDNA by the relative quantity of house-keeping gene cDNA (GAPDH) to correct for variability in cDNA concentration in the individual samples (for Fig. 5B). Data on Figures 1G and 6C are represented as the cycle threshold values (Ct) of target gene normalized by Ct of GAPDH gene. Ct values are proportional to the negative logarithm of the initial amount of input cDNA and are defined as the number of PCR cycles to reach the fluorescence threshold in each sample.
Grip strength and wire hang tests
Grip tests were performed using a grip strength meter Chatillon DFIS2 (AMETEK, Sellersville, PA, USA). The animal was placed in front of the instrument and allowed to grasp the pull bar with the forelimbs and then pulled backward by its tail in the horizontal plane. The force applied to the bar at the moment the grasp was released was recorded as the peak tension. Untrained mice were tested five times in succession without rest, and the results of the last three trials were averaged and normalized to body weight for each mouse. For the wire hang test, the mouse was placed on a suspended wire that was 2 ft from the floor. The latency to fall off the wire in seconds was scored in five trials and averaged for each mouse with the smallest value dropped from the calculation.
Muscle histochemistry
Muscles to be used for histochemistry were dissected from the mice, placed in O.C.T. Compound (Sakura Finetek, Torrance, CA, USA) and frozen in isopentane cooled in liquid nitrogen. Frozen sections were cut at 10 µm and kept frozen until use. Histology of the tissue was assessed by staining with aqueous hematoxylin or hematoxylin and eosin (H&E). Enzyme histochemical stains were also performed and included: NADH-TR, SDH, esterase-α naphtyl acetate-pararosaniline (non-specific esterase), adenosine triphosphatase alkaline and acidic (ATPase), and modified Gomori trichrome.
Electron microscopy
Muscle tissues for electron microscopy were prepared as previously described (55). Briefly, soleus and TA muscles were dissected from three WT and three KO mice and fixed with phosphate buffered 2% glutaraldehyde (Sigma-Aldrich, St Louis, MO, USA) and 2% paraformaldehyde (Ted Pella, Redding, CA, USA) fixative solution (pH 7.4) overnight. Then, the tissue was osmicated, dehydrated through 50, 70, 90 and 100% ethanol and embedded in Epon resin (Ted Pella). The tissue was cross-sectioned at 70 nm thickness with a microtome (Boeckeler Instrument, Tucson, AZ, USA). After lead contrast staining, the tissue section was examined under JEM-1200EX electron microscope (JEOL, Japan) equipped with BioScan 600W digital camera (1024 × 1024 pixels) (Gatan, Pleasanton, CA, USA). Digital Micrograph 1.2 software (Gatan) was used to generate images.
Tissue extract preparation and western blot analysis
For western blot analysis, muscles and brains were homogenized in reducing sample buffer [80 mm Tris, pH 6.8, 0.1 m dithiothreitol, 2% SDS and 10% glycerol with protease inhibitor cocktail (Sigma-Aldrich)] using a Dounce homogenizer. Forty micrograms of total protein per lane were loaded on polyacrylamide gel followed by transfer to nitrocellulose membrane.
Anti-TRIM32 antibodies used for western blotting included: T332 rabbit 1:500 (2), Aviva Systems Biology (San Diego, CA, USA) rabbit #ARP38965_T100 1:800 and #ARP31692_P050 1:1000, Lifespan Biosciences (Seattle, WA, USA) rabbit #LS-B1105 1:1000, Novus Biologicals (Littleton, CO, USA) mouse #H00022954-B01 1:500, Proteintech Group (Chicago, IL, USA) rabbit #10326-1-AP 1:500, Santa Cruz Biotechnology (Santa Cruz, CA, USA) goat N-12 #sc-49265 1:200 and T-20 #sc-49266 1:200. Other antibodies used for WB included: anti-NEFL rabbit #AB9568, anti-NEFM rabbit #AB1987 and anti-NEFH #AB1989, 1:1000 (Millipore, Billerica, MA, USA); anti-β-actin mouse #A2228 1:1000 (Sigma-Aldrich); anti-GAPDH mouse #MAB374 1:500 (Millipore) and anti-β-galactosidase rabbit #14-6773 1:400 (eBioscience, San Diego, CA, USA). Secondary antibodies conjugated with horseradish peroxidase (HRP) were purchased from Sigma-Aldrich. Specific signals were developed using ChemiGlow substrate (Alpha Innotech, San Leandro, CA, USA). Images of the blots were acquired using FlourChem FC2 Imager (Alpha Innotech). Densitometry was performed using AlphaEase FC Software Version 6.0.2 (Alpha Innotech).
Immunoprecipitation and MALDI–TOF MS
Mouse brain tissue was homogenized in RIPA buffer [50 mm Tris, pH 7.5, 150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mm EDTA, protease inhibitor cocktail (Sigma-Aldrich)] and incubated for 20 min on ice. After centrifugation for 5 min at 13 000g, the 75 kDa protein of interest remained in the pellet. The pellet was solubilized using various denaturing agents dissolved in 50 mm Tris, pH 7.5: (i) 0.1% SDS, (ii) 0.5% SDS, (iii) 1 m guanidine, (iv) 1 m urea, (v) 3 m urea or (vi) 1 m urea supplemented with 1 m thiourea. Only the last condition (50 mm Tris, pH 7.5, 1 m urea and 1 m thiourea) allowed for solubilization of the 75 kDa protein followed by successful immunoprecipitation using 2 µg of T332 antibody and protein G agarose. Immunocomplex was fractionated on SDS–PAGE and stained with SYPRO Ruby (Bio-Rad). The protein band corresponding to 75 kDa was excised from the gel and in situ digested with trypsin. The tryptic fragments were analyzed by MALDI–TOF MS using Voyager-DE STR instrument (Applied Biosystems, Foster City, CA, USA). Peptide mass fingerprinting allowed for the identification of NEFL using Mascot software [Matrix Science (Mascot search engine), http://www.matrixscience.com/]. Twenty-seven out of 31 selected mass signals matched, achieving high sequence coverage (54%) and significant Mascot score of 174 for NEFL.
Morphometric analysis of L4 VRs
L4 VR specimens were prepared as following. Three WT and three KO mice were transcardially perfused with phosphate buffered 2% glutaraldehyde and 2% paraformaldehyde fixative solution (pH 7.4). L4 VRs were dissected out and post-fixed in the same fixative solution at 4°C overnight. On the next day, the tissue was osmicated, dehydrated through 50, 70, 90 and 100% ethanol and embedded in Epon resin. The VR was cross-sectioned at 300 nm thickness with a microtome, and transferred onto glass slides, stained with 0.25% toluidine blue, and examined under a Nikon E600 light microscope (Nikon, Japan). The images were captured using a Micropublisher 5.0 megapixel digital camera (Q Imaging, Burnaby, BC, Canada).
Morphometric parameters analyzed included cross-sectional area of L4 VRs, total number of axons, axonal density (number of axons per 1000 µm2) and average axon diameter. For the axon diameter distribution histogram, diameters of 200–240 axons were measured and the quantity of axons with certain diameter was expressed as the percent of total number of axons measured in each sample. To avoid discrepancy in the measurements due to the angle at which cross-sections were obtained, the smallest diameters were utilized for oval shaped axons. The measurements were performed using light microscope Axio Imager and digital image processing software AxioVision (Carl Zeiss MicroImaging, Thornwood, NY, USA).
MYHC isoform gel electrophoresis
The procedure was previously described (56) with modifications (57). Briefly, the separating gel consisted of 30% glycerol, 8% acrylamide/bisacrylamide (50:1), 0.2 m Tris (pH 8.8), 0.1 m glycine, 0.4% SDS, 0.1% APS, 0.05% TEMED; the stacking gel was 30% glycerol, 4% acrylamide/bisacrylamide (50:1), 70 mm Tris (pH 6.8), 4 mm EDTA, 0.4% SDS, 0.1% APS and 0.05% TEMED. The composition of the upper running buffer was 300 mm Tris base, 450 mm glycine, 0.3% SDS and 0.12% β-mercaptoethanol. Upper running buffer without β-mercaptoethanol was diluted 1:6 to prepare the lower running buffer. One microgram of total muscle lysate was loaded per lane and allowed to run for 20 h at 140 V at 4°C. Gels were stained with silver staining kit (Bio-Rad).
Statistical analysis
Statistical analysis of all data was carried out by Student's t-test. Differences were considered statistically significant if the P-value was less than 0.05. Error bars on all graphs are represented by standard errors of means (SEM).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by National Institute of Arthritis, Musculoskeletal and Skin Diseases grant (R01-AR052693), the Muscular Dystrophy Association (MDA 4084) and the Adelson Medical Research Foundation.
ACKNOWLEDGEMENTS
The authors thank Jane Wen and José Guadelupe Morales III for excellent technical support, Ms Zeynep Durer for assistance with MALDI–TOF MS, Ms Birgitta Sjöstrand for excellent ultrastructural support, Dr Cristina Ghiani for guidance with quantitative PCR and Dr Irina Kramerova for valuable discussions and advice. The real-time PCR data were generated on equipment in the Mental Retardation Research Center at UCLA.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Reymond A., Meroni G., Fantozzi A., Merla G., Cairo S., Luzi L., Riganelli D., Zanaria E., Messali S., Cainarca S., et al. The tripartite motif family identifies cell compartments. EMBO J. 2001;20:2140–2151. doi: 10.1093/emboj/20.9.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kudryashova E., Kudryashov D., Kramerova I., Spencer M.J. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J. Mol. Biol. 2005;354:413–424. doi: 10.1016/j.jmb.2005.09.068. [DOI] [PubMed] [Google Scholar]
- 3.Albor A., El-Hizawi S., Horn E.J., Laederich M., Frosk P., Wrogemann K., Kulesz-Martin M. The interaction of Piasy with Trim32, an E3-ubiquitin ligase mutated in limb-girdle muscular dystrophy type 2H, promotes Piasy degradation and regulates UVB-induced keratinocyte apoptosis through NFκB. J. Biol. Chem. 2006;281:25850–25866. doi: 10.1074/jbc.M601655200. [DOI] [PubMed] [Google Scholar]
- 4.Kano S., Miyajima N., Fukuda S., Hatakeyama S. Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl-interactor 2. Cancer Res. 2008;68:5572–5580. doi: 10.1158/0008-5472.CAN-07-6231. [DOI] [PubMed] [Google Scholar]
- 5.Pickart C.M., Eddins M.J. Ubiquitin: structures, functions, mechanisms. Biochim. Biophys. Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 6.Jiang Y.H., Beaudet A.L. Human disorders of ubiquitination and proteasomal degradation. Curr. Opin. Pediatr. 2004;16:419–426. doi: 10.1097/01.mop.0000133634.79661.cd. [DOI] [PubMed] [Google Scholar]
- 7.Attaix D., Ventadour S., Codran A., Bechet D., Taillandier D., Combaret L. The ubiquitin-proteasome system and skeletal muscle wasting. Essays Biochem. 2005;41:173–186. doi: 10.1042/EB0410173. [DOI] [PubMed] [Google Scholar]
- 8.Fridell R.A., Harding L.S., Bogerd H.P., Cullen B.R. Identification of a novel human zinc finger protein that specifically interacts with the activation domain of lentiviral Tat proteins. Virology. 1995;209:347–357. doi: 10.1006/viro.1995.1266. [DOI] [PubMed] [Google Scholar]
- 9.Frosk P., Weiler T., Nylen E., Sudha T., Greenberg C.R., Morgan K., Fujiwara T.M., Wrogemann K. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am. J. Hum. Genet. 2002;70:663–672. doi: 10.1086/339083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoser B.G., Frosk P., Engel A.G., Klutzny U., Lochmuller H., Wrogemann K. Commonality of TRIM32 mutation in causing sarcotubular myopathy and LGMD2H. Ann. Neurol. 2005;57:591–595. doi: 10.1002/ana.20441. [DOI] [PubMed] [Google Scholar]
- 11.Saccone V., Palmieri M., Passamano L., Piluso G., Meroni G., Politano L., Nigro V. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2H. Hum. Mutat. 2008;29:240–247. doi: 10.1002/humu.20633. [DOI] [PubMed] [Google Scholar]
- 12.Chiang A.P., Beck J.S., Yen H.J., Tayeh M.K., Scheetz T.E., Swiderski R.E., Nishimura D.Y., Braun T.A., Kim K.Y., Huang J., et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet–Biedl syndrome gene (BBS11) Proc. Natl Acad. Sci. USA. 2006;103:6287–6292. doi: 10.1073/pnas.0600158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yokota T., Mishra M., Akatsu H., Tani Y., Miyauchi T., Yamamoto T., Kosaka K., Nagai Y., Sawada T., Heese K. Brain site-specific gene expression analysis in Alzheimer's disease patients. Eur. J. Clin. Invest. 2006;36:820–830. doi: 10.1111/j.1365-2362.2006.01722.x. [DOI] [PubMed] [Google Scholar]
- 14.Jerusalem F., Engel A.G., Gomez M.R. Sarcotubular myopathy. A newly recognized, benign, congenital, familial muscle disease. Neurology. 1973;23:897–906. doi: 10.1212/wnl.23.9.897. [DOI] [PubMed] [Google Scholar]
- 15.Muller-Felber W., Schlotter B., Topfer M., Ketelsen U.P., Muller-Hocker J., Pongratz D. Phenotypic variability in two brothers with sarcotubular myopathy. J. Neurol. 1999;246:408–411. doi: 10.1007/s004150050374. [DOI] [PubMed] [Google Scholar]
- 16.Shokeir M.H., Kobrinsky N.L. Autosomal recessive muscular dystrophy in Manitoba Hutterites. Clin. Genet. 1976;9:197–202. doi: 10.1111/j.1399-0004.1976.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 17.Shokeir M.H., Rozdilsky B. Muscular dystrophy in Saskatchewan Hutterites. Am. J. Med. Genet. 1985;22:487–493. doi: 10.1002/ajmg.1320220306. [DOI] [PubMed] [Google Scholar]
- 18.Tobin J.L., Beales P.L. Bardet–Biedl syndrome: beyond the cilium. Pediatr. Nephrol. 2007;22:926–936. doi: 10.1007/s00467-007-0435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blacque O.E., Leroux M.R. Bardet–Biedl syndrome: an emerging pathomechanism of intracellular transport. Cell. Mol. Life Sci. 2006;63:2145–2161. doi: 10.1007/s00018-006-6180-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Runeberg-Roos P., Saarma M. Neurotrophic factor receptor RET: structure, cell biology, and inherited diseases. Ann. Med. 2007;39:572–580. doi: 10.1080/07853890701646256. [DOI] [PubMed] [Google Scholar]
- 21.Parnaik V.K., Manju K. Laminopathies: multiple disorders arising from defects in nuclear architecture. J. Biosci. 2006;31:405–421. doi: 10.1007/BF02704113. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz M.S., Sargeant M., Swash M. Longitudinal fibre splitting in neurogenic muscular disorders—its relation to the pathogenesis of ‘myopathic’ change. Brain. 1976;99:617–636. doi: 10.1093/brain/99.4.617. [DOI] [PubMed] [Google Scholar]
- 23.Banker B.Q., Engel A.G. Basic reactions in muscle. In: Engel A.G., Franzini-Armstrong C., editors. Myology. Vol. 1. McGraw-Hill, Inc.; 1994. pp. 832–888. [Google Scholar]
- 24.Zhu Q., Couillard-Despres S., Julien J.P. Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp. Neurol. 1997;148:299–316. doi: 10.1006/exnr.1997.6654. [DOI] [PubMed] [Google Scholar]
- 25.Elder G.A., Friedrich V.L., Jr., Bosco P., Kang C., Gourov A., Tu P.H., Lee V.M., Lazzarini R.A. Absence of the mid-sized neurofilament subunit decreases axonal calibers, levels of light neurofilament (NF-L), and neurofilament content. J. Cell. Biol. 1998;141:727–739. doi: 10.1083/jcb.141.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lariviere R.C., Julien J.P. Functions of intermediate filaments in neuronal development and disease. J. Neurobiol. 2004;58:131–148. doi: 10.1002/neu.10270. [DOI] [PubMed] [Google Scholar]
- 27.Liu Q., Xie F., Siedlak S.L., Nunomura A., Honda K., Moreira P.I., Zhua X., Smith M.A., Perry G. Neurofilament proteins in neurodegenerative diseases. Cell. Mol. Life Sci. 2004;61:3057–3075. doi: 10.1007/s00018-004-4268-8. [DOI] [PubMed] [Google Scholar]
- 28.Mersiyanova I.V., Perepelov A.V., Polyakov A.V., Sitnikov V.F., Dadali E.L., Oparin R.B., Petrin A.N., Evgrafov O.V. A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am. J. Hum. Genet. 2000;67:37–46. doi: 10.1086/302962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Georgiou D.M., Zidar J., Korosec M., Middleton L.T., Kyriakides T., Christodoulou K. A novel NF-L mutation Pro22Ser is associated with CMT2 in a large Slovenian family. Neurogenetics. 2002;4:93–96. doi: 10.1007/s10048-002-0138-4. [DOI] [PubMed] [Google Scholar]
- 30.Fabrizi G.M., Cavallaro T., Angiari C., Bertolasi L., Cabrini I., Ferrarini M., Rizzuto N. Giant axon and neurofilament accumulation in Charcot-Marie-Tooth disease type 2E. Neurology. 2004;62:1429–1431. doi: 10.1212/01.wnl.0000120664.07186.3c. [DOI] [PubMed] [Google Scholar]
- 31.Jordanova A., De Jonghe P., Boerkoel C.F., Takashima H., De Vriendt E., Ceuterick C., Martin J.J., Butler I.J., Mancias P., Papasozomenos S., et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain. 2003;126:590–597. doi: 10.1093/brain/awg059. [DOI] [PubMed] [Google Scholar]
- 32.Canete-Soler R., Wu J., Zhai J., Shamim M., Schlaepfer W.W. p190RhoGEF binds to a destabilizing element in the 3′ untranslated region of light neurofilament subunit mRNA and alters the stability of the transcript. J. Biol. Chem. 2001;276:32046–32050. doi: 10.1074/jbc.M104104200. [DOI] [PubMed] [Google Scholar]
- 33.Canete-Soler R., Reddy K.S., Tolan D.R., Zhai J. Aldolases a and C are ribonucleolytic components of a neuronal complex that regulates the stability of the light-neurofilament mRNA. J. Neurosci. 2005;25:4353–4364. doi: 10.1523/JNEUROSCI.0885-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cullheim S., Ulfhake B. Observations on the morphology of intracellularly stained gamma-motoneurons in relation to their axon conduction velocity. Neurosci. Lett. 1979;13:47–50. doi: 10.1016/0304-3940(79)90073-9. [DOI] [PubMed] [Google Scholar]
- 35.Cullheim S., Ulfhake B. Relations between cell body size, axon diameter and axon conduction velocity of triceps surae alpha montoneurons during the postnatal development in the cat. J. Comp. Neurol. 1979;188:679–686. doi: 10.1002/cne.901880410. [DOI] [PubMed] [Google Scholar]
- 36.Cullheim S., Kellerth J.O. A morphological study of the axons and recurrent axon collaterals of cat alpha-motoneurones supplying different functional types of muscle unit. J. Physiol. 1978;281:301–313. doi: 10.1113/jphysiol.1978.sp012423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wigston D.J., English A.W. Fiber-type proportions in mammalian soleus muscle during postnatal development. J. Neurobiol. 1992;23:61–70. doi: 10.1002/neu.480230107. [DOI] [PubMed] [Google Scholar]
- 38.Balastik M., Ferraguti F., Pires-da Silva A., Lee T.H., Alvarez-Bolado G., Lu K.P., Gruss P. Deficiency in ubiquitin ligase TRIM2 causes accumulation of neurofilament light chain and neurodegeneration. Proc. Natl Acad. Sci. USA. 2008;105:12016–12021. doi: 10.1073/pnas.0802261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El-Husseini A.E., Vincent S.R. Cloning and characterization of a novel RING finger protein that interacts with class V myosins. J. Biol. Chem. 1999;274:19771–19777. doi: 10.1074/jbc.274.28.19771. [DOI] [PubMed] [Google Scholar]
- 40.Ohkawa N., Kokura K., Matsu-Ura T., Obinata T., Konishi Y., Tamura T.A. Molecular cloning and characterization of neural activity-related RING finger protein (NARF): a new member of the RBCC family is a candidate for the partner of myosin V. J. Neurochem. 2001;78:75–87. doi: 10.1046/j.1471-4159.2001.00373.x. [DOI] [PubMed] [Google Scholar]
- 41.Fiori M.G., Salvi F., Plasmati R., Tassinari C.A. Muscle fiber splitting, capillary internalization, and target-like fiber formation in familial amyloidotic polyneuropathy. Clin. Neuropathol. 1996;15:240–247. [PubMed] [Google Scholar]
- 42.Malandrini A., Bonuccelli U., Parrotta E., Ceravolo R., Berti G., Guazzi G.C. Myopathic involvement in two cases of Hallervorden-Spatz disease. Brain Dev. 1995;17:286–290. doi: 10.1016/0387-7604(95)00039-e. [DOI] [PubMed] [Google Scholar]
- 43.Limos L.C., Ohnishi A., Sakai T., Fujii N., Goto I., Kuroiwa Y. ‘Myopathic’ changes in chorea-acanthocytosis. Clinical and histopathological studies. J. Neurol. Sci. 1982;55:49–58. doi: 10.1016/0022-510x(82)90169-1. [DOI] [PubMed] [Google Scholar]
- 44.Trout J.J., Stauber W.T., Schottelius B.A. Fiber splitting in tonic and phasic skeletal muscles following denervation, chloroquine and triton WR-1339 treatments. Virchows Arch. B: Cell. Pathol. Incl. Mol. Pathol. 1982;39:111–123. doi: 10.1007/BF02892841. [DOI] [PubMed] [Google Scholar]
- 45.Borg K., Solders G., Borg J., Edstrom L., Kristensson K. Neurogenic involvement in distal myopathy (Welander). Histochemical and morphological observations on muscle and nerve biopsies. J. Neurol. Sci. 1989;91:53–70. doi: 10.1016/0022-510x(89)90075-0. [DOI] [PubMed] [Google Scholar]
- 46.Swash M., Schwartz M.S. Implications of longitudinal muscle fibre splitting in neurogenic and myopathic disorders. J. Neurol. Neurosurg. Psychiatry. 1977;40:1152–1159. doi: 10.1136/jnnp.40.12.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swash M., Schwartz M.S., Sargeant M.K. Pathogenesis of longitudinal splitting of muscle fibres in neurogenic disorders and in polymyositis. Neuropathol. Appl. Neurobiol. 1978;4:99–115. doi: 10.1111/j.1365-2990.1978.tb00551.x. [DOI] [PubMed] [Google Scholar]
- 48.Burke R.E., Levine D.N., Tsairis P., Zajac F.E., 3rd. Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J. Physiol. 1973;234:723–748. doi: 10.1113/jphysiol.1973.sp010369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burke R.E., Levine D.N., Salcman M., Tsairis P. Motor units in cat soleus muscle: physiological, histochemical and morphological characteristics. J. Physiol. 1974;238:503–514. doi: 10.1113/jphysiol.1974.sp010540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dum R.P., O'Donovan M.J., Toop J., Burke R.E. Cross-reinnervated motor units in cat muscle. I. Flexor digitorum longus muscle units reinnervated by soleus motoneurons. J. Neurophysiol. 1985;54:818–836. doi: 10.1152/jn.1985.54.4.818. [DOI] [PubMed] [Google Scholar]
- 51.Gordon T., Tyreman N., Rafuse V.F., Munson J.B. Fast-to-slow conversion following chronic low-frequency activation of medial gastrocnemius muscle in cats. I. Muscle and motor unit properties. J. Neurophysiol. 1997;77:2585–2604. doi: 10.1152/jn.1997.77.5.2585. [DOI] [PubMed] [Google Scholar]
- 52.Brown M.C., Holland R.L., Hopkins W.G. Motor nerve sprouting. Annu. Rev. Neurosci. 1981;4:17–42. doi: 10.1146/annurev.ne.04.030181.000313. [DOI] [PubMed] [Google Scholar]
- 53.Edds M.V., Jr. Collateral nerve regeneration. Q. Rev. Biol. 1953;28:260–276. doi: 10.1086/399699. [DOI] [PubMed] [Google Scholar]
- 54.Havton L.A., Hotson J.R., Kellerth J.O. Partial peripheral motor nerve lesions induce changes in the conduction properties of remaining intact motoneurons. Muscle Nerve. 2001;24:662–666. doi: 10.1002/mus.1052. [DOI] [PubMed] [Google Scholar]
- 55.Kramerova I., Kudryashova E., Tidball J.G., Spencer M.J. Null mutation of calpain 3 (p94) in mice causes abnormal sarcomere formation in vivo and in vitro. Hum. Mol. Genet. 2004;13:1373–1388. doi: 10.1093/hmg/ddh153. [DOI] [PubMed] [Google Scholar]
- 56.Talmadge R.J., Roy R.R. Electrophoretic separation of rat skeletal muscle myosin heavy-chain isoforms. J. Appl. Physiol. 1993;75:2337–2340. doi: 10.1152/jappl.1993.75.5.2337. [DOI] [PubMed] [Google Scholar]
- 57.Kohn T.A., Myburgh K.H. Electrophoretic separation of human skeletal muscle myosin heavy chain isoforms: the importance of reducing agents. J. Physiol. Sci. 2006;56:355–360. doi: 10.2170/physiolsci.RP007706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.