

Abstract
Haploinsufficiency of the transcription co-activator EYA1 causes branchio–oto–renal syndrome, congenital birth defects that account for as many as 2% of profoundly deaf children; however, the underlying cause for its dosage requirement and its specific role in sensory cell development of the inner ear are unknown. Here, an allelic series of Eya1 were generated to study the basis of Eya1 dosage requirements for sensory organ development. Our results show different threshold requirements for the level of Eya1 in different regions of the inner ear. Short and disorganized hair cell sterocilia was observed in wild-type/null heterozygous or hypomorphic/hypomorphic homozygous cochleae. Patterning and gene-marker analyses indicate that in Eya1 hypomorphic/null heterozygous mice, a reduction of Eya1 expression to 21% of normal level causes an absence of cochlear and vestibular sensory formation. Eya1 is initially expressed in the progenitors throughout the epithelium of all six sensory regions, and later on during sensory cell differentiation, its expression becomes restricted to the differentiating hair cells. We provide genetic evidence that Eya1 activity, in a concentration-dependent manner, plays a key role in the regulation of genes known to be important for sensory development. Furthermore, we show that Eya1 co-localizes with Sox2 in the sensory progenitors and both proteins physically interact. Together, our results indicate that Eya1 appears to be upstream of very early events during the sensory organ development, hair cell differentiation and inner-ear patterning. These results also provide a molecular mechanism for understanding how hypomorphic levels of EYA1 cause inner-ear defects in humans.
INTRODUCTION
Haploinsufficiency for human EYA1 causes branchio–oto–renal (BOR) or branchio–oto (BO) syndrome, congenital birth defects that account for as many as 2% of profoundly deaf children (1–3). These syndromes are transmitted as autosomal-dominant disorders with high but incomplete penetrance and variable expressivity (4,5). The otic anomalies in BOR/BO syndrome involve malformation of the outer, middle and inner ears, and hearing loss, which ranges from mild to profound, is conductive, sensorineural, or both (6). The inner-ear defects include either an absence of cochlea or an undercoiled cochlea, and lacking or abnormal semicircular canals of the vestibular apparatus (5). Although functional levels of EYA1 are clearly critical for normal development, the molecular basis of EYA1 haploinsufficiency and the effects of varying EYA1 concentration on the development of inner ear sensory regions are unclear.
We previously reported that the Eya1 heterozygotes show a conductive hearing loss similar to BOR syndrome, whereas Eya1 homozygotes show an arrest of inner-ear development at the otocyst stage (7,8). The Eya1 gene encodes a transcription co-activator containing a divergent N-terminal activation domain and a conserved C-terminal Eya domain that mediates protein–protein interactions with Sine oculis and Dachshund proteins (9–11). During mammalian inner-ear morphogenesis, Eya1 functions upstream of and genetically interacts with Six1, a member of the homeodomain transcription factor Six family that is homologous to the Drosophila Sine oculis (12). Consistent with this interaction, Six1-deficient mice show defects in the ear similar to that observed in the Eya1 mutants (12), and haploinsufficiency for the human SIX1 gene also causes BOR syndrome (13). We previously demonstrated that the mutations of EYA1 or SIX1 identified from BOR patients affect the Eya1–Six1 interaction (13,14), thus providing a potential mechanistic understanding of how the disruption of combinatorial interactions of these transcription factors can lead to specific defects in BOR syndrome. However, the downstream molecular effects of altered Eya1 or Six1 dosage in the inner ear are poorly understood.
In the inner ear, there are six distinct sensory organs: five vestibular organs fundamental for balance and one auditory organ—the organ of Corti, necessary for hearing. Each of these sensory regions is composed of sensory hair cells and their associated nonsensory supporting cells; both arise from a common progenitor (15). At present, little is known about the molecular mechanisms involved in the specification of this sensory cell lineage. Recent genetic studies have shown that the SOXB1-HMG box transcription factor Sox2 is required for establishing the prosensory domain in the inner ear and may function upstream of Atoh1, which belongs to the basic helix–loop–helix transcription factor family and plays an essential role for hair cell differentiation but not for their initial specification (16–19). However, it is unclear whether Eya1 or Six1 plays a role in controlling the specification of sensory progenitors and their differentiation, and the early arrest of inner-ear development in their null mutants precluded genetic evaluation of the specific roles of these genes in the sensory patches (20).
To dissect out the potential roles of Eya1 in the development of ear sensory epithelia and to determine the effect of Eya1 dosage on these roles, here we report the examination of systematic dosage reduction of Eya1 on the development of sensory organs by generating an allelic series combing the wild-type, hypomorphic (Eya1bor) (21) and null (knockout allele, Eya1−) (7) alleles of Eya1. Eya1 mRNA and protein expression levels in Eya1bor/bor homozygous and Eya1bor/− compound heterozygous ears were decreased to 40 and 21% of wild-type levels, respectively. Patterning and gene-marker analyses indicate that some sensory formation occurred in Eya1bor/bor mutant inner ears but the prosensory domain was not well defined. In contrast, Eya1bor/− compound heterozygous inner ears exhibited a complete absence of cochlear and some vestibular sensory formation. These results together with a complete absence of inner-ear sensory formation in Eya1-null mice indicate that Eya1 is essential for establishing sensory epithelia in the inner ear. To determine whether Eya1 plays a specific role during sensory cell development, we have compared the expression of Eya1 with Sox2 domain in the developing sensory epithelia of the inner ear. Co-localization of Eya1 with Sox2 expression in the future organ of Corti suggests that Eya1 plays a role not only in the early development of sensory progenitors but also in the hair cell differentiation. Furthermore, our results show that Eya1 physically interacts with Sox2 protein, and five specific Eya domain mutations identified in affected BOR patients disturbed the Eya1–Sox2 interaction. Finally, we provide genetic evidence that Eya1 induces differences in downstream responses in a dose-dependent manner. These results provide a molecular and developmental mechanism for understanding how hypomorphic levels of EYA1 cause inner-ear defects in humans.
RESULTS
A dosage-dependent role for Eya1 in the sensory epithelium
To characterize the mechanisms underlying EYA1 haploinsufficiency and explore potential involvement of Eya1 dosage in sensory organ development and patterning of the inner ear, we generated an allelic series of Eya1 dose by combing the wild-type, hypomorphic (Eya1bor) and null (Eya1−) alleles of Eya1. Quantitative reverse transcription (RT)–PCR and western blot with tissue extracts prepared from E13.5 inner ears revealed that Eya1 mRNA and protein expression levels were decreased to 72.5 ± 3, 48.5 ± 2, 39.5 ± 4, 21.3 ± 1 and 0% of normal levels in Eya1bor/+,Eya1+/−,Eya1bor/bor,Eya1bor/− and Eya1−/− mice, respectively (the values are the mean and the variation from the mean that were determined from four independent experiments) (Fig. 1). To reveal the dosage effects of Eya1 protein level on the expressivity of inner-ear phenotype, we first analyzed the inner ears from these mice at E18.5 by paintfilling (Fig. 2A–G; Table 1). All Eya1bor/+ mice (n = 15) appeared structurally normal (data not shown). However, a few Eya1+/− heterozygotes showed mild or severe inner-ear defects including malformation of the cochlea, and truncation of the anterior or posterior ampullae and the endolymphatic duct (Fig. 2B; Table 1). In contrast, inner ears of Eya1bor/bor homozygotes were more severely affected. The posterior ampullae were absent, and their associated semicircular canals were either absent or truncated in all cases examined (n = 20) (Fig. 2C and D; Table 1), whereas 13 of 20 Eya1bor/bor mice also exhibited an absence of the anterior ampullae and truncation of their associated canals. In contrast, the lateral cristae and canals were present but also malformed (n = 20). Furthermore, the saccule and utricle were also severely affected (Fig. 2C and D), and cochleae were undercoiled, varying from a three-quarter turn (Fig. 2C) to the most basal one-quarter turn (Fig. 2D; Table 1). Further reduction of Eya1 in Eya1bor/− compound heterozygotes resulted in cochlear agenesis with 100% penetrance (Fig. 2E–G; Table 1). Only one semicircular canal that appeared to be the anterior canal was present (n = 10), and five of 10 mice lacked recognizable saccule (Fig. 2E and F) and endolymphatic duct/sac (Fig. 2G). No inner-ear structure forms in Eya1−/− embryos (7,8).
Figure 1.

Analyses of Eya1 expression levels in different Eya1 mutants. (A) Semi-quantitative RT–PCR of E13.5 inner-ear extracts. β-Actin was used as an internal control for RT–PCR. Eya1 expression in wild-type was taken as baseline (100%) and its expression in the null was designated as zero. The relative amount of Eya1 expression in the mutants was calculated as percentage relative to the amount of wild-type and null mice. Each sample was normalized by the expression of β-actin (n= 4 for each genotype). An average result of four independent experiments is shown. Error bar represents standard deviation. The lower panel shows one representative result of RT–PCR products analyzed on 1% agarose gel. (B) Western blot of E13.5 inner ears with a purified rabbit anti-Eya1 peptide antibody showing Eya1 protein expression levels in different genotypes. Two aberrant bands were observed in Eya1−/− mice, but the normal Eya1 product was undetectable. Quantification of western blot results coincided with the RT–PCR results.
Figure 2.

Inner-ear malformation and sensory defects in Eya1 hypomorphic mutant mice. (A–G) Paintfilled inner ears at E18.5 from wild-type, Eya1+/− heterozygotes, Eya1bor/bor homozygotes and Eya1bor/− compound heterozygotes. Asterisks indicate truncated semicircular canals and missing ampullae in mutants. Arrow in (B) points to truncated endolymphatic duct (ed). Arrow in (D) points to the shortened cochlea (co). (H–M) E18.5 inner-ear sections stained with an anti-MYO7A antibody showing sensory epithelium of the semicircular canal, utricle and saccule in Eya1bor/+ heterozygotes and malformation of these structures in Eya1bor/bor and Eya1bor/− mutants. aa, anterior ampulla; ac, anterior crista; asc, anterior semicircular canal; la, lateral ampulla; lc, lateral crista; lsc, lateral semicircular canal; pa, posterior ampulla; psc, posterior semicircular canal; s, saccule; u, utricle. Scale bars: 50 µm.
Table 1.
Inner-ear defects in Eya1bor/+, Eya1+/−, Eya1bor/bor and Eya1bor/− hypomorphic mutant embryos at E18.5
| Abnormalities | bor/+, n = 30 (15) | +/−, n = 44 (22) | bor/bor, n = 40 (20) | bor/−, n = 20 (10) | |
|---|---|---|---|---|---|
| Endolymphatic duct/sac | 0 | 3a (4) | 20a (16) | 7b (5), 7c (7) | |
| Semicircular canals | |||||
| Anterior | 0 | 2a (2) | 22a (13) | Formed one canal | |
| Posterior | 0 | 2a (2) | 40b (20) | ||
| Lateral | 0 | 0 | 0 | ||
| Ampulla | |||||
| Anterior | 0 | 1a (1) | 22b (13), 18c (11) | 20b (10) | |
| Posterior | 0 | 1a (1) | 40b (20) | 20b (10) | |
| Lateral | 0 | 40c (20) | 20c (10) | ||
| Saccule | 0 | 5c (5) | 40c (20) | 16b (9), 4c (3) | |
| Utricle | 0 | 0 | 40c (20) | 20c (10) | |
| Cochlea | 0 | 15d (9) | 40d (20) | 20b (10) |
n, number of ears (number of embryos).
Fifteen ears of 44 Eya1+/− ears (nine of 22 mice) in C3HeB/FeJ strain showed shortened or/and malformed cochlea at E18.5. Among them, two ears (from two embryos) had most severe phenotype: one ear retained only about one-half turn and another ear retained about three-quarter turn. Majority of the affected ears lost around one-half turn of the cochlea and exhibited variable malformations of inner-ear structures. In contrast, all Eya1bor/bor and Eya1bor/− embryos in the same strain showed more severe inner-ear defects.
aTruncated.
bAbsent.
cMalformed.
dShortened.
Next, we examined the sensory regions in the mutant animals at E18.5 by staining with early hair cell differentiation marker MYO7A (22) and scanning electron microscopy (SEM). The vestibular organs that developed in Eya1bor/bor animals demonstrated a clear reduction of sensory epithelium and hair cells of the utricular and saccular maculae and the cristae when compared with controls (Fig. 2H, I, K and L; Table 2). In Eya1bor/− animals, the epithelium at the sites where one would normally see the lateral crista and utricular macula was identifiable by its thickened appearance but was largely reduced in size when compared with normal animals (Fig. 2J). Three of five mice (four of 10 ears) showed an extremely small sensory epithelium in the saccule with very few hair cells (Fig. 2M; Table 2). All other sensory regions were absent in the compound heterozygous animals.
Table 2.
Eya1 gene dosage effect on hair cell numbers
| Genotype | Number of animals | Counts per macula |
Counts per 100 μm |
||||
|---|---|---|---|---|---|---|---|
| HCs-utricle | HCs-saccule | IHCs | OHCs | HCs | OHC/IHC | ||
| +/+ | 2 | 1679 ± 33 | 1705 ± 56 | 13.5 ± 0.8 | 44.1 ± 1.2 | 56.6 ± 1.6 | 3.27 |
| bor/+ | 2 | 1670 ± 88* | 1686 ± 53* | 13.0 ± 0.9# | 43.2 ± 1.0# | 56.2 ± 1.7# | 3.32 |
| +/− | 2 | 1575 ± 80** | 1508 ± 36*** | 14.5 ± 10** | 43.3 ± 1.8* | 57.8 ± 1.2### | 2.99 |
| +/− apex | 2 | 20.5 ± 2.0** | 42.2 ± 2.2* | 63.7 ± 3.8### | 2.06 | ||
| bob/bor | 2 | 1169 ± 67*** | 875 ± 65*** | 15.2 ± 1.0*** | 51.3 ± 16*** | 66.5 ± 1.5*** | 3.38 |
| bor/bor-apex | 2 | 24.3 ± 2.0*** | 51.6 ± 2.3*** | 78.0 ± 4.0*** | 2.12 | ||
| bor/− | 2 | 713 ± 88*** | 37 (1)a | – | – | – | – |
Hair cells were determined for two animals (four ears) at E18.5. The indicated values are the mean and the variation from the mean.
aOnly one Eya1bor/− inner ear had a total of 37 hair cells in the saccule and the others had no hair cells. For the cochlea, inner hair cells (IHCs) and outer hair cells (OHCs) were counted separately for each indicated genotype and were then combined to get total numbers of hair cells (HCs). Counts were expressed in numbers of hair cells per 100 μm from the middle turn of the cochleae to take into account the slightly different lengths that were counted for each cochlea. Counts were also expressed as outer cell versus inner cell ratios (OHC/IHC). For Eya1+/− and Eya1bor/bor cochleae, hair cells in the middle and apical regions of the cochleae were counted separately. Significant increases in hair cell numbers were observed in the apical regions of Eya1+/− cochleae and in Eya1bor/bor cochleae. The numbers of the inner hair cells were more increased in the apical region of the cochleae in Eya1+/− animals. Significance was determined by comparing each of the mutant groups with the wild-type animals, using StatView’s t- test.
*P = 0.7814.
**P = 0.0534.
***P < 0.0001.
#P = 0.4475.
##P = 0.0023.
###P = 0.1086.
In the cochlea, differentiated hair cells with hair bundles were clearly visible in Eya1bor/+ mice at E18.5 and morphologically indistinguishable from wild-type embryos by staining with an anti-MYO7A antibody (Fig. 3A) and lectin that binds to hair cell stereocilia (Fig. 3D). Eya1+/− cochleae demonstrated no significant changes in hair cell numbers in each 100 µm in the basal and middle cochlea, respectively, when compared with controls (Table 2; data not shown), although occasional extra inner hair cells were observed in the middle region (Fig. 3B and E). In contrast, in the apical region, Eya1+/− cochleae had many extra hair cells (nearly two rows of inner hair cells) (Table 2; data not shown). This observation was further confirmed with hair cell counts (Table 2) and by SEM (arrows, Fig. 3H). To examine whether hair cells in Eya1+/− cochleae differentiated normally, we analyzed Eya1+/− cochleae at P7 and adult stages (∼6 weeks old) by SEM. Interestingly, the hair cells developed in Eya1+/− cochleae exhibited short and disorganized stereocilia bundles, with outer hair cell bundles missing in some in the middle and apical regions at these stages in all animals examined (n = 6, Fig. 3K; data not shown). In contrast, Eya1bor/bor cochleae showed many extra inner and outer hair cells, and the rows of hair cells were very disorganized (Fig. 3C and F). Hair cell counts from the middle and apical regions of the cochleae demonstrated that Eya1bor/bor cochleae have significantly more hair cells than controls (Table 2). SEM confirmed the disorganization of cochlea both in the patterning of the sensory cell rows and, at a single cell level, in the loss of polarity and disorganization of many hair cell stereocilia bundles (Fig. 3I and L). In the less severe cases in which the cochlea almost reached three-quarter turn, the patterning of the sensory cell rows was less affected in the very basal regions (data not shown) than in the middle and apical regions (Fig. 3I and L), which showed continuous but irregular rows of hair cells. No evidence of cochlea formation was observed in Eya1bor/− mice between E11.5 and P0 by SEM and histological analyses (data not shown). Thus, progressive depletion of Eya1 protein resulted in increasing severity of inner-ear malformations, with a ∼79% reduction being sufficient to block cochlea formation. A complete aplasia of the cochlear and some vestibular sensory areas in the compound heterozygous together with the observation of no sensory formation in the null inner ears indicate that Eya1 is essential for establishing sensory epithelia in the inner ear. Our results also indicate that Eya1 may have a role in the development or maintenance of the stereocilia bundles.
Figure 3.

Altered hair cell patterning in Eya1+/− and Eya1bor/bor cochleae. (A–C) E18.5 cochlear sections stained with an anti-MYO7A antibody and (D–F) lectin-stained cochleae show one row of inner (arrow) and three rows (arrowheads) of outer hair cells in Eya1bor/+. In Eya1+/− cochleae, hair cell number and patterning in the basal regions are relatively normal, whereas extra inner hair cells are observed from the middle regions towards the apical regions (arrows). The hair cells in Eya1bor/bor cochleae are extremely disorganized with many extra inner and outer hair cells. (G–L) SEM of cochleae of the indicated genotypes at E18.5 (G–I, J and L) and 6 weeks old of age (H and K). The extra inner hair cells and short and disorganized stereocilia with missing patches of outer hair cells are observed in the apex of Eya1+/− cochleae at 6 weeks old of age (K). Many rows hair cells are observed in the apex of Eya1bor/bor mice. (G–I) Low-power views, and (J–L) higher power views. Scale bars: 50 µm for panels (A–I) and 25 µm for panels (J–L).
Eya1 is expressed in sensory precursors
Since it is unclear from these data whether Eya1 plays a direct role in patterning of the hair cells, we examined the expression of Eya1 in the sensory patches and compared its expression with the expression domain of Sox2, an early marker for sensory progenitors in the inner ear and a gene required for sensory cell development. We have recently generated an Eya1lacZ knockin allele (see Materials and Methods), and X-gal staining of Eya1lacZ/+ embryos recaptured endogenous Eya1 expression detected by in situ hybridization (data not shown). Thus, these mice provide a sensitive model for assessing Eya1 expression.
X-gal staining for the expression of Eya1lacZ allele in Eya1lacZ/+ heterozygotes revealed that Eya1 is expressed in all six sensory regions in the inner ear (Fig. 4). In the vestibular sensory epithelia, Eya1 is expressed throughout the thickness of the epithelia at E13.5 (Fig. 4A and B) and showed overlapping pattern with Sox2 (Fig. 4C; data not shown; 19). From E15.5, Eya1 expression became progressively restricted to the differentiating hair cells (Fig. 4D and E), distinct from that of Sox2 expression, which was strongly detected in the supporting cells (data not shown; 19).
Figure 4.
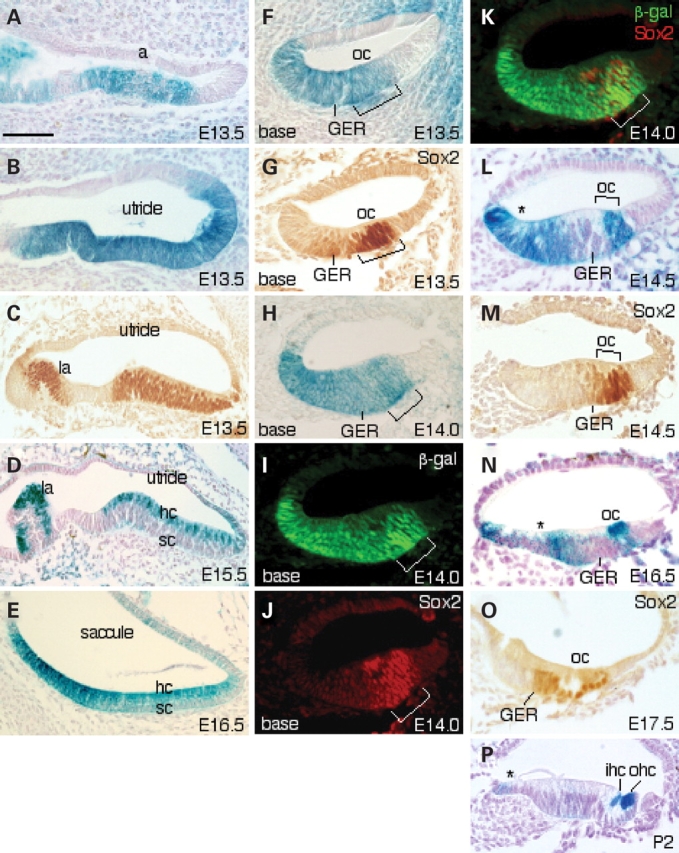
Eya1 expression and its relation with Sox2 in the sensory patches of the inner ears. (A, B, D–F, H, L, N) Sections of X-gal-stained Eya1lacZ/+ inner ears showing Eya1 expression in all six sensory regions. (C, G, M and O) Inner-ear sections stained with an anti-Sox2 antibody. (I–K) Cochlear sections stained with both an anti-Sox2 antibody and an anti-β-gal antibody. (A–E) In the vestibular sensory epithelia, Eya1 is expressed throughout the thickness of the epithelium in all sensory regions at E13.5 and becomes progressively restricted to differentiating hair cells (hc) at E15.5 and 16.5. (C) Sox2 is expressed in all sensory epithelial cells in the vestibular regions at E13.5. (F) Cochlear sections showing Eya1 expression in the future organ of Corti (oc) and in the great epithelial ridge (GER) at E13.5. (G) Cochlear section showing Sox2 expression in the organ of Corti and in the border cells in the GER adjacent to the future organ of Corti at 13.5. (H) At E14.0, Eya1 expression becomes weaker in the border cells but remains in other regions. (I–K) Studies showing that Eya1 and Sox2 co-localize within the progenitors at the site of future organ of Corti (brackets). (L) Eya1 expression disappears in the border cells in the GER at E14.5. (M) Sox2 is expressed in the site of organ of Corti and in the border cells in the GER at E14.5. (N) At E16.5, Eya1 becomes abundantly expressed in the differentiating hair but disappears in supporting cells. Its expression remains in the region that will give rise to inner sulcus (asterisk). (O) Sox2 expression is restricted to the supporting cells (sc) and border cells. (P) At P2, Eya1 is strongly distributed in the differentiating hair cells and weakly in the epithelial cells outside of the GER (asterisk). a, ampulla; la, lateral ampulla. Scale bars: 75 µm for panels (A–D) and 50 µm for all other panels.
In the developing sensory epithelium of the cochlea, cellular commitment and differentiation in the organ of Corti begin in the mid-basal region at about E13.5 and proceed in a wave that progresses towards both the base and the apex. At E13.5, Eya1 expression was observed in a broad group of cells in the less mature apical region throughout the thickness of the epithelium including the site of the future organ of Corti, and overlaps with Sox2 domain (data not shown). In the more mature basal regions, Eya1 is expressed in the future organ of Corti, but its expression extends into the entire epithelium in the great epithelial ridge (GER) (Fig. 4F). In contrast, Sox2 is expressed strongly in the future organ of Corti and extends weakly into adjacent GER region (Fig. 4G), which shows overlapping with Eya1 domain.
From E14.0, we have observed developmental changes in the pattern of Eya1 expression in the cochlea. Eya1 expression gradually disappeared in the border cells in the GER adjacent to the future organ of Corti (Fig. 4H, L and N), where the level of Sox2 distribution was gradually increased (Fig. 4M and O). In contrast, Eya1 expression remained in the epithelial cells of the GER that will give rise to the inner sulcus cells where Sox2 is not expressed (asterisk, Fig. 4H, L and N). In the developing organ of Corti, double-labeling with both anti-Sox2 and anti-β-gal antibodies confirmed co-localization of Eya1 with Sox2 in the sensory progenitors at the site of future organ of Corti at E14.0 (Fig. 4I–K; data not shown). From E16.5, the sensory progenitor cells are differentiating into hair and supporting cells, and Eya1 became abundantly distributed in the differentiating hair cells but almost completely disappeared in the supporting cells (Fig. 4N). In contrast, Sox2 expression became restricted to the supporting cells at E17.5 (Fig. 4O). At postnatal stages, shorter X-gal staining of Eya1lacZ/+ inner ears revealed that Eya1 expression was most abundant in the hair cells (Fig. 4P). Eya1 expression in the epithelial cells outside of the GER was also visible on sections (asterisk, Fig. 4O), whereas its expression in the GER next to the border cells was very faint and became undetectable on sections. Nonetheless, the overlapping and distinct pattern of Eya1 and Sox2 expression suggests that Eya1 may play not only an early role in inner-ear sensory cell specification but also a specific role in sensory hair cell differentiation.
Impaired patterning of precursor cells in the organ of Corti in Eya1bor/bor mice
To seek an explanation for the morphological defects observed in Eya1bor/bor mutant mice, we investigated whether Eya1 has a role in specifying or maintaining the sensory progenitors in the cochlea by analyzing the expression of several genes that are known to be important for sensory organ formation. At E14.0, the prosensory domain was well established in Eya1bor/+ and Eya1+/− cochlea as demonstrated by immunostaining for the expression of the cyclin-dependent kinase inhibitor p27Kip1 (Fig. 5A and B), an established marker of the prosensory domain in the cochlea (23). In contrast, the P27Kip1 expression domain in Eya1bor/bor cochleae appeared to be diffused and not as sharply defined as in controls (Fig. 5C). This observation was further confirmed with the expression of Sox2 (Fig. 5D–F).
Figure 5.
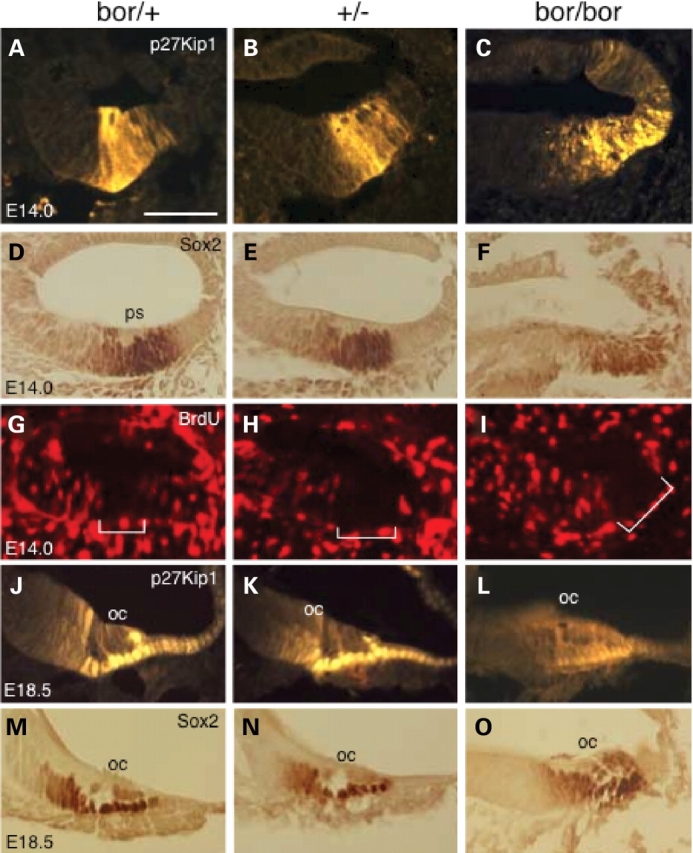
Altered expression of p27Kip1 and Sox2 in Eya1bor/bor cochleae. (A–F) Immunostaining of E14.0 (A–I) and E18.5 (G–L) cochlear sections with an anti-p27KIP1 (A–C, J–L), anti-BrdU (G–I) or anti-Sox2 (D–F, M–P). Brackets indicate the prosensory domain (ps) marked by p27KIP1 and Sox2 expression and lack of BrdU-positive cells. The total length of the cochlea duct (between two small arrows) is thinner in the Eya1 mutants. Scale bars: 50 µm.
Next, we analyzed whether Eya1 dosage affects cell cycle of the sensory progenitors by performing BrdU-labeling at E13.5, 14.0 and 14.5, a time when most hair cells have exited the cell cycle (24–26). After the administration of an 8 h pulse of BrdU (three injections), BrdU-positive cells were present throughout the cochlear epithelium (Fig. 5G), indicating that they are still in the cell cycle. The sensory progenitor cells at the site of the future organ of Corti already became postmitotic at E13.5–14.5, as seen by the lack of BrdU incorporation in this region (bracket, Fig. 5G). In Eya1+/− or Eya1bor/bor cochlear epithelium, the region where p27Kip1 is expressed also lacked BrdU-positive cells (Fig. 5H and I), indicating that the sensory progenitors do exit cell cycle and become postmitotic in the mutant cochleae.
At E18.5, p27Kip1 expression becomes restricted to the differentiating supporting cells as well as in the border cell in the GER (Fig. 5J). Although the cochlea epithelium appears to be thinner in Eya1+/− animals (Fig. 5K), the pattern of p27Kip1 expression was not altered. In contrast, Eya1bor/bor cochleae showed weak and disorganized p27Kip1 expression (Fig. 5L). Similarly, the expression of Sox2, detected in supporting and border cells in the GER, showed disorganized pattern (Fig. 5M–P). These results suggest that Eya1 gene dosage may be critical for defining the prosensory domain in the cochleae by mediating the creation or maintenance of sensory/nonsensory boundaries.
Eya1 gene dosage critically affects Sox2, Otx, Bmp4 and Lfng expressions in the otocyst
Since Sox2 is expressed in the otic ectoderm from very early stages, we sought to examine whether Eya1 gene dosage affects Sox2 expression in the otocyst from early stages. Interestingly, Sox2 expression in the otocysts was reduced in Eya1bor/bor, more reduced in Eya1bor/− and very faint in Eya1−/− embryos at E9.5 by both whole-mount (Fig. 6A–D) and section (Fig. 6E–H) in situ hybridization, whereas its expression in the neural tube appeared to be near normal in all mutants.
Figure 6.
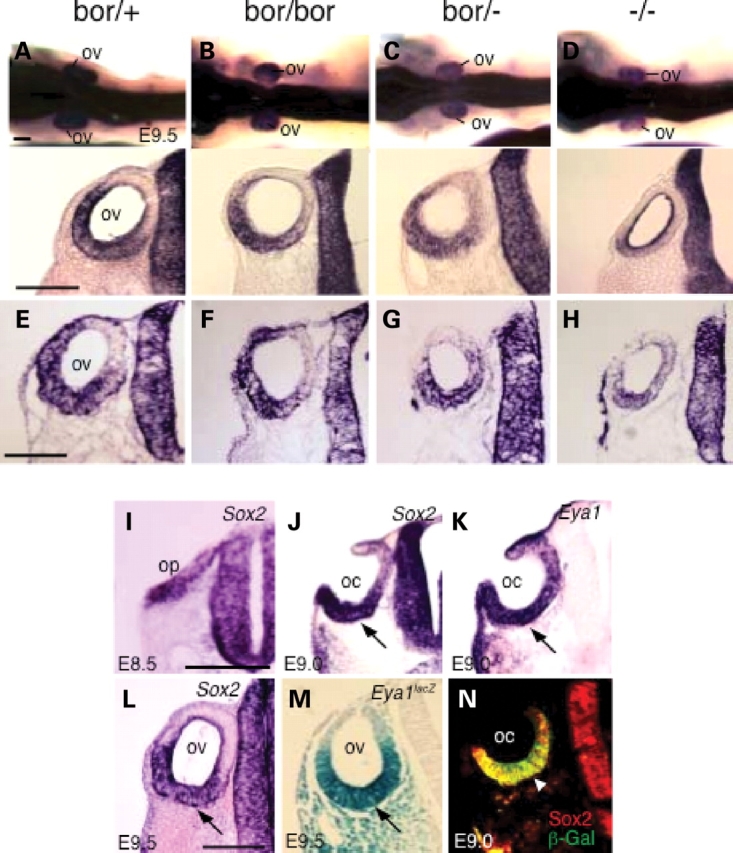
Eya1 gene dosage critically affects the expression of Sox2 in the otocysts. (A–D) Whole-mount in situ hybridization at E9.5 showing reduction of Sox2 expression in the otic vesicle (ov). Lower panels are sections through the otic region of whole-mount in situ embryos, which confirm the reduction of Sox2 expression specifically in the otic vesicle. (E–H) Section in situ hybridization showing reduction of Sox2 expression in the otic vesicle in the mutants. (I, J and L) Sections stained with Sox2 probe at E8.5 (I), E9.0 (J) and E9.5 (L). (K) Section stained with Eya1 probe at E9.0. (M) Section stained with X-gal for the Eya1lacZ allele at E9.5. (N) Section stained with anti-β-gal and Sox2 antibodies at E9.0. Scale bars: 100 µm.
In chick, Sox2 is expressed in the otic placode (27,28); however, the onset of Sox2 expression during mouse inner-ear development has not been examined. To gain better understanding of the relation between Sox2 and Eya1 during early inner-ear development, we analyzed the expression of both genes on adjacent sections from E8.5 to E10.5. Similar to Eya1 (8), Sox2 is expressed stronger in the ventral rim and weaker in the dorsal rim of the otic placode and cup at E8.5–E9.0 (Fig. 6I–K). The expression of both genes became restricted to the ventral wall of the ototcyst at E9.5 (Fig. 6L and M). Co-localization studies confirmed that both Eya1 and Sox2 show overlapping patterns in the otic ectoderm between E8.5 and E10.5 (Fig. 6N; data not shown). Together, although we cannot exclude the possibility that the reduction of Sox2 expression is caused by reduced number of cells present in the mutants, our results indicate that Eya1 gene dosage critically affects Sox2 expression from otocyst stage and the different levels of reduction in Sox2 transcripts are presumably in response to a 60 (in Eya1bor/bor) to 100% (in Eya1−/−) reduction in Eya1 protein levels.
To further explore the effects of Eya1 activity on sensory organ patterning, we analyzed several other genes that are known to be important for sensory organ development. The homeobox genes Otx1 and Otx2 are regionally expressed in the otocyst and their inactivation in mice causes selective loss or malformation of inner-ear sensory organs (29,30). Otx1 is expressed in a region destined to form the lateral canal and ampulla, utricle and saccule and cochlea, whereas Otx2 is expressed mostly in the region destined to form the cochlea. Both genes are essential for the morphogenesis of the lateral semicircular canal and ampulla, the saccule and the cochlea. The expression of both genes was detected in Eya1bor/bor embryos at reduced levels, but was undetectable in Eya1bor/− or Eya1−/− embryos at around E10.25 and 10.5 by both section and whole-mount in situ hybridization (Fig. 7A–H). The bone morphogenetic protein 4 (Bmp4), a member of the Tgfβ superfamily, has also been shown to play a role in the early sensory organ development, in interaction with Fgf10 (31,32). At E10.5, Bmp4 expression is normally restricted to two areas, which mark the sensory anlagen of the cristae, but was largely diminished in both domains in Eya1bor/bor otocysts (Fig. 7I and J). Eya1bor/− otocysts showed further reduction of Bmp4 expression, with the dorsal expression domain absent and the lateral domain weakened greatly (Fig. 7K). In contrast, Bmp4 expression became undetectable in Eya1−/− otocysts (Fig. 7L; 8). The Notch signaling modulator lunatic fringe (Lfng) is also implicated in the early sensory organ development (33) and is expressed in a broad domain at E10.25 but stronger in the anteroventral domain which corresponds to the neurogenic region (arrowhead, Fig. 7M) and weaker in the medial region (arrow, Fig. 7M). Its expression was largely weakened in Eya1bor/bor embryos (Fig. 7N) and was only weakly retained in the anteroventral domain of Eya1bor/− otocysts (arrowhead, Fig. 7O). No expression was observed in Eya1−/− embryos (Fig. 7P). Since we have previously found that in the absence of Eya1, the inner-ear neuorgensis is initiated normally, and the neuronal precursors are able to delaminate from otocyst but fail to form a morphologically detectable ganglion due to abnormal apoptosis from as early as E9.5 (20), it is possible that the Lfng-expressing cells in Eya1bor/− ototcyst are neuronal precursors. To further clarify this, we analyzed the expression of Neuord, an early neuronal differentiation marker (34). Similar to Lfng, a very faint expression of Neurod was observed in the anteroventral domain corresponding to the neurogenic region in Eya1bor/− otocyst (data not shown; D. Zou et al., manuscript in preparation), further suggesting that the Lfng-labeling cells in the neurogenic region of Eya1bor/− otocysts are likely to be neuronal precursors. Together, our results indicate that Eya1 gene dosage critically affects the expression of otic genes that are known to be important for sensory organ development from early stages, either directly or indirectly. The dramatic responses of these genes to a 60 (in Eya1bor/bor) to 79% (in Eya1bor/−) reduction in a transcription factor imply that the spatiotemporal regulation of Eya1 expression in the developing inner ear is critical, tightly regulated and without compensatory mechanisms such as feedback or genetic redundancy.
Figure 7.

Eya1 gene dose-dependent responses of sensory gene expression. (A–D) Whole-mount in situ showing reduced expression of Otx1 in Eya1bor/bor and no expression in Eya1bor/− or Eya1−/− embryos. (E–H) Section in situ showing Otx2 expression in the otocyst at E10.5, reduced expression in Eya1bor/bor and no expression in Eya1bor/− or Eya1−/− embryos. (I–L) Section in situ showing reduced Bmp4 expression in Eya1bor/bor or Eya1bor/− embryos and no expression in Eya1−/− embryos. (M–P) Whole-mount in situ showing Lfng expression in Eya1bor/+, reduced expression in Eya1bor/bor or Eya1bor/−, no expression in Eya1−/− embryos, respectively. Arrowhead points to the expression in the neurogenic domain, and arrow points to the expression in the medial region. There is no endolymphatic duct outgrowth in Eya1−/− embryos (8). Scale bars: 100 µm.
Eya1 physically interacts with Sox2 protein
Since Eya1 co-localizes with Sox2 in the sensory progenitor cells in the inner ear, we next investigated whether these two proteins physically interact. To test whether Eya1 forms a complex with Sox2, we performed an immunoprecipitation analysis. Cell extracts from the 293 cells transfected with mouse Flag–Eya1 and Sox2 were incubated with Flag-antibody-coupled agarose beads or with anti-Sox2 antibody, recovered by protein A beads, and analyzed on SDS–PAGE followed by western blotting with anti-Sox2 or anti-Flag antibody. As shown in Figure 8A, the Flag–Eya1 was co-immunoprecipitated with Sox2. We then examined whether the point mutations that cause single-amino acid substitutions identified in the conserved Eya1 domain in BOR patients affect Eya1–Sox2 complex formation (1,14) (Kumar et al., 1998; Azuma et al., 2000). Interestingly, all five single amino acid substitutions of E330K, G393S, S454P, L472R or R514G identified in the conserved domain region resulted in decreased complex formation of Eya1–Sox2 (Fig. 8). Similar results were obtained in p19 cells, which endogenously express Sox2 (Fig. 8A). Thus, our data demonstrated that Eya1 can interact with Sox2 in cultured cells, and five missense mutations identified in patients affected the Eya1–Sox2 complex formation, providing new insights into the molecular mechanisms of inner-ear defects that occurred in human BOR patients.
Figure 8.

Eya1 and Sox2 physically interact. (A) Western blot and co-immunoprecipitation in 293 cells or p19 cells. Five micrograms of the cell extracts isolated from 293 cells transfected with Flag–Eya1F/pcDNA3 alone, both Flag–Eya1F/pcDNA3 and Sox2/pcDNA3 together or Sox2/pcDNA3 alone were loaded on SDS–PAGE and analyzed by western blotting with anti-Sox2 or anti-Flag antibody for detecting Sox2 or Flag–Eya1. For co-immunoprecipitation experiments, 40 µg of the same extracts was incubated with anti-Sox2 antibody and precipitated by protein A beads or precipitated by Flag-antibody-coupled agarose beads. The precipitates were dissolved in SDS sample buffer followed by western blot analysis with anti-Flag or anti-Sox2 antibody, respectively. For p19 cells, only Flag–Eya1F was transfected and cell extracts were prepared. Anti-Flag or anti-Sox2 was used for immunoprecipitation, respectively, and both antibodies were used for western blot. (B) Western blot and co-immunoprecipitation. Cell extracts from 293 cells co-transfected with pcDNA3 vector alone, Flag–Eya1F/pcDNA3 or each of its missense mutants and Sox2/pcDNA3 were loaded on SDS–PAGE and analyzed by western blotting with anti-Sox2 or anti-Flag antibody for detecting Sox2 or Flag–Eya1. Two amino acid substitutions, E330K and G393S, resulted in smaller products. For co-immunoprecipitation experiments, 40 µg of the same nuclear extracts was incubated with and precipitated by Flag-antibody-coupled agarose beads. The precipitates were dissolved in SDS sample buffer followed by western blot analysis with anti-Sox2 antibody. (C) Effects of the conserved Eya domain missense mutations on the formation of Eya1–Sox2 complexes. The relative amount of wild-type Eya1–Sox2 complex detected by anti-Sox2 antibody after co-immunoprecipitation was 100%. The relative amount of the mutant Eya1–Sox2 complex was calculated as percentage relative to the amount of wild-type complex. Each sample was normalized by the input amount of Eya1 and Sox2 as detected by western blot shown in (B). An average result of two similar independent experiments is shown. Error bar represents standard deviation.
DISCUSSION
The Eya1 gene is required for normal development of the inner ear, but the early arrest of inner-ear development at the otocyst stage of Eya1 null embryos has thus far precluded genetic evaluation of the specific role of Eya1 in sensory epithelial histogenesis. Here, we demonstrate that Eya1 acts in a gene dose-sensitive manner to regulate the development of sensory regions and expression of specific sets of sensory genes during inner-ear development. Our studies provide the first direct evidence for the requirement of Eya1 for normal hair cell development.
The role of Eya1 in the generation and specification of sensory patches in the otocyst
The sensory organs of the inner ear are all derived from the otocyst, which forms from the otic placode next to the hindbrain neural tube. It has been proposed that the different sensory epithelia are all derived from a common prosensory region in the ventromedial wall of the otocyst and may be specified by their proximity to the boundaries between different compartments within the otocyst defined by the asymmetric gene expression patterns (29,35). Recent studies have implicated several transcription factors, growth factors and Notch signaling in specifying sensory patches in the otocyst (35–38). However, the molecular controls for the determination of a common prosensory region and the specification of six different sensory patches are largely unknown. Our studies have shown that Eya1 is expressed in the otic placode before invagination and its expression becomes progressively restricted to the ventral half of the otocyst within which the sensory organs form (Fig. 7) (7). Thus, Eya1 may play a critical role in a very early event specifying the prosensory area in the ventral wall of the otocyst. In the absence of Eya1, the prosensory progenitor cells may not be specified, thus leading to a complete absence of sensory formation in Eya1−/− mutant (Fig. 9). Consistent with this view, we failed to detect the expression of Lfng and Jag1 in Eya1−/− mutant (Fig. 7; and data not shown), whose expression defines the prosensory area in the otocyst. We also failed to detect the expression of other early sensory markers, including Bmp4, Fgf3 and Fgf10 in the sensory patches in Eya1−/− otocyst (Fig. 7;) (7,8), further suggesting that the sensory patches are not specified in the absence of Eya1.
Figure 9.

Schematic drawing of Eya1 expression domains in relation to Sox2 and Atoh1 expression in the otocyst and developing cochlea, cristae and maculae and its possible roles in sensory cell development. In the otocyst, Eya1 (red) and Sox2 (green) are co-expressed in the ventromedial wall of the otocyst where the prosensory epithelia are formed. These two genes may act together to specify a common prosensory area in the otocyst at this early stage by turning on other sensory genes including Jag1, Lfng, Bmp4 and Fgfs, and in the absence of Eya1, the other sensory markers are not expressed. In the developing cochlea, at E12.5, the floor of cochlear duct will give rise to the organ of Corti (oc), the inner (is) and outer (os) sulci. Eya1 and Sox2 are co-expressed in the prosenory progenitors of the organ of Corti, and this co-localization suggests that both genes may act together to regulate the cell cycle of the prosensory progenitors. Once the sensory progenitor cells become postmitotic, they express p27Kip1, and at around E13.5–14.5, Atoh1 is activated in the postmitotic progenitors that become committed to hair cell fate and is co-expressed with Eya1 in the differentiating hair cells. However, Sox2 expression becomes restricted to supporting cells and border cells in the GER. Therefore, Eya1 and Sox2 may act together to regulate hair cell commitment by activating Atoh1; however, Eya1 but not Sox2 acts to maintain or upregulate Atoh1 expression and to function together with Atoh1 for hair cell differentiation. Eya1 may act as a competence factor but Atoh1 may act as a differentiation factor. Therefore, Eya1-positive nonsensory cells are competent for hair cell fate, and they will differentiate into hair cells when Atoh1 is ectopically expressed in those cells. This explains why there is a region unique to Eya1 expression, and cells within that region can be converted to hair cells by overexpression of Atoh1 at postnatal stages (47). Similar function of these genes may be involved in the sensory cell development of the cristae and maculae.
The expression pattern of Eya1 between E8.5 and E9.5 is nearly identical to that of Sox2, and Sox2 expression was reduced in Eya1 mutant otic ectoderm (Fig. 6). Sox2 has been shown to be required for sensory cell development in the inner ear, and it may act after the early sensory specification of prosensory patches, as its expression was downregulated in Jag1 conditional mutants (19,39). However, the specific role of Sox2 during inner-ear development remains unclear, as its expression is broader and precedes Jag1. If prosensory progenitor cells are not specified in Eya1−/− embryos, why do we still detect Sox2 expression in the ventral region? One likely explanation is that both Eya1 and Sox2 are required to specify a subset of epithelial cells in the ventral wall of otocyst to acquire a prosensory cell fate, and the Sox2-expressing cells present in Eya1−/− ototcysts are uncommitted epithelial cells.
Sox2 is expressed in at least three types of stem cells, neural stem cells, embryonic stem cells and trophoblast stem cells, and mounting evidence has shown that it is a key regulator in the maintenance of progenitor cell identity but not in their differentiated derivatives (40–43). However, it is unknown whether Sox2 interacts with other genetic pathways implicated in this process, and if so, how. In addition, it is unclear whether Sox2 is a key regulator for specification and maintenance of this stem cell type and function in sensory patch regeneration in mammalian inner ear. Recently, it has been shown that ablation of Sox2 expression from neural progenitors inhibits their proliferation and differentiation (42). We have previously shown that Eya1 regulates otic epithelial cell proliferation and survival (7,8). We also have shown that in the absence of Eya1, the generic neural differentiation program in the cranial neurogenic placodes is not initiated (20). Eya1 and Six4 are expressed very early in the preplacodal domain and are involved in otic placode specification, whereas Six1 is expressed in the invaginating otic placode at around E8.75 (8,12). It is possible that Eya1 functions in parallel to Sox2 in the generation of progenitor cells in the otocyst. Consistent with this view, we found that Eya1 and Sox2 form a complex in cultured 293 cells, which do not endogenously express Eya1 or Sox2. Nonetheless, altered expression of Sox2 in Eya1−/− embryos suggests that Eya1 is required either directly or indirectly for normal expression of Sox2 during inner-ear development.
The role of Eya1 in hair cell development
The cochlea develops from the ventral otocyst as the cochlear duct from around E11 and at around E12.5; the floor of the cochlear duct will give rise to three distinct regions—the organ of Corti and the inner and outer sulci (Fig. 9). Hair cell development begins in the vestibular system around E12.5 and in the cochlea around E13.5 (24,25). After the prosensory patches are specified, the progenitor cells become postmitotic and are comprised of uncommitted hair and supporting cells (15,19,24–26,44). Subsequently during development, a subpopulation of the progenitor cells in each patch is selected to develop as hair cells. To date, however, how exactly the sensory cell lineage is specified and how the hair cell fate is determined are poorly understood. Sox2 is required for the specification of sensory cell lineage, whereas Atoh1 has been shown to be a key regulator for hair cell development. Deletion of Atoh1 leads to a complete loss of hair cells, whereas overexpression of Atoh1 is sufficient to induce hair cell formation in the nonsensory cells in the GER in embryonic, postnatal and adult inner ears (45–47). However, the onset of Atoh1 expression during sensory cell development and its specific role remain dispute. In addition, how Atoh1 expression is activated and the molecular genetic pathways regulating hair cell development are essentially unknown. In this study, we carefully assessed the spatial expression of Eya1, using the sensitive X-gal staining for the newly generated Eya1lacZ allele during sensory cell development, and its relation with Sox2 expression. The differential changes in its distribution associated with hair cell differentiation suggest that Eya1 may have multiple roles: an early role in which Eya1 specifies or maintains the sensory organ; a second role in which Eya1 regulates hair cell differentiation; a later one in which it plays a role in hair cell patterning by mediating the creation or maintenance of sensory/nonsensory boundaries in the inner ear. In addition, possible together with Sox2, Eya1 may regulate cell-cycle exit of future hair cells before the initiation of Atoh1 expression in these cells (Fig. 9).
In the cochlea, Eya1 is initially expressed throughout the thickness of sensory epithelia and overlaps with Sox2 domain in the prosensory progenitors (Fig. 9). However, Eya1 expression domain is much broader than Sox2 and extends into the epithelial cells in the GER and the cells outside of the GER, which normally give rise to inner sulcus cells. During sensory cell differentiation, Eya1 expression becomes progressively restricted to the hair cells but gradually disappears in the border cells in the GER and in the supporting cells where Sox2 is expressed. The overlap and distinct pattern of Eya1 and Sox2 distributions during sensory cell development suggests several possibilities of their actions in regulating hair cell development. First, Eya1 probably acts together with Sox2 in specifying the sensory cell lineage. Once the sensory progenitors are specified, both Eya1 and Sox2 are involved in the initial selection of hair cell fate by activating Atoh1 expression. Indeed, we found that Eya1 and Sox2 physically interact.
After postmitotic cells initiated hair cell differentiation by expressing Atoh1, Eya1 but not Sox2 is required to both maintain Atoh1 expression and act together with Atoh1 to regulate hair cell differentiation. Consistent with this view, we found that abundant Eya1 expression becomes progressively restricted to differentiating hair cells (Fig. 4). Interestingly, in addition to differentiating hair cells, Eya1 is expressed in epithelial cells that normally give rise to inner sulcus in the cochlea, which coincides precisely with the region where overexpression of Atoh1 converted those cells to hair cells in postnatal rat inner ear (47). It will be important to gain a molecular insight into the regulation of Atoh1 by Eya1 and Sox2.
Differential sensitivity to Eya1 gene dosage
One intriguing observation from our studies was that the six sensory regions were differentially affected by altered Eya1 gene dosage. The most obvious change was cochlear agenesis in Eya1bor/− animals as the Eya1 dosage decreased from 40–21%, whereas a very small number of hair cells differentiated in the maculae and the cristae. In Eya1bor/bor animals, the numbers of hair cells and the sensory epithelia were largely reduced in the maculae and cristae, whereas hair cell differentiation patterns varied on the basis of their apical or basal location of the cochlea. Therefore, different thresholds for Eya1 action could be part of the mechanism by which Eya1 controls specific sensory cell fate decisions by modulating the expression of distinct downstream genes in different sensory areas. Eya1 may be required at a specific level at the right time to specify a cochlear fate, which requires the activation of a set of genes that are particularly sensitive to the concentration of Eya1. Biophysical interaction between Eya1 and Six1 provides a mechanism for enhancing transcriptional activity, which may amplify gene activity at particular times or locations during inner-ear development. The expression pattern of Otx1, Otx2, Bmp4, Fgf3, Fgf10 and Lfng in Eya1bor/− mutants is almost identical to that seen in the Six1 null embryos (7,8,12). Therefore, these genes may represent common downstream targets of Eya1–Six. It is possible that Eya1 may integrate diverse transcription factors into multiple pathways to regulate distinct downstream targets in different regions of the inner ear, and Sox2 may be the common factor in the sensory region that cooperatively interacts with Eya1. Further expression studies of these genes in their complementary mutant backgrounds should help define the relationship between Sox2 and Eya–Six.
How are we to explain the differential effect of the mutation on the basal and apical portions of the cochlea? Why do extra and irregular rows often occur in the apical regions? For example, although the cochlea is largely shortened in Eya1bor/bor animals, the very basal regions exhibited normal patterning in hair cell formation. However, in the middle and apical regions of the cochlea, the hair cells were often arranged in multiple rows. Similar observation was obtained in Eya1+/− cochleae. Interestingly, SEM revealed that Eya1+/− hair cell stereocilia appeared shorter and disorganized at P7 and adult stages (Fig. 3; data not shown). In addition, patches of outer hair cells were frequently missing in the middle towards apical regions of Eya1+/− cochleae at these stages. One possible interpretation for the multiple rows of inner hair cells observed in the apical regions of Eya1+/− or Eya1bor/bor mutants is that it is caused by defects in their arrangement due to the shorter cochlea. Recent studies of mouse mutants with defects in planar cell polarity and polarized extension indicated that the organ of Corti is formed from a defined number of postmitotic progenitors through cellular intercalation (known as convergent extension) from the base to the apex in parallel to the basal-to-apical differentiation of hair cells (48–51). This explains why multiple rows of hair cells frequently occurred in the apical but not in the basal regions of the cochlea in Eya1+/− or Eya1bor/bor animals. Interestingly, similar differential effect between basal and apical regions of the cochlea was also observed in Sox2, Jag1, Fgfr1, Neurog1 and Foxg1 mouse mutants (19,39,52–55).
Why were the inner and outer hair cells differentially affected by altered Eya1 gene dosage? This may be a result of differential regulation of the development and maintenance between inner and outer hair cells, as suggested by previous evidence. For example, a selective loss of inner or outer hair cells and an age-dependent selective loss of outer hair cells have been previously observed in several mouse mutants (19,56–58). Alternatively, this also could be a result of a late function of Eya1, in which it regulates hair cell patterning by modulating the Notch signaling through lateral inhibition (reviewed in 37,38,59,60). The abundant Eya1 expression in the differentiating hair cells may be required for the maintenance of the differentiated state by preventing their immediate neighbors in the developing sensory epithelium from differentiating into hair cells. In support of this, we found that the expression of Notch signaling is affected by altered Eya1 gene dosage from early stages. Mutations in the modulators involved in the Notch signaling, including Jag1, Notch1 and Hes1, often led to two rows of inner hair cells (Zine et al., 2000; 39,47,52,61). It will be important to examine whether Eya1 may modulate the Notch signaling during sensory hair cell development in the inner ear directly and/or through its effect on hair cells.
It should be noted that Eya1 hypomorphic mutant mice display additional and more severe defects in nonsensory regions than the Sox2 mutants (19) and there is no inner-ear formation in Eya1-null mice (7,8). Previous studies suggested that all sensory regions in the inner ear have some influence on nonsensory development through a shared lineage (39,61–63). Both Bmp4 and Fgf10, two early genes important for canal crista development, are known to regulate canal development (31,32,64,65). In contrast, the morphogenesis of the lateral canal depends on the expression of Otx1 (30,66) and develops even in the absence of the lateral canal crista (55). Numerous upstream patterning genes influence ear morphogenesis (64,67), but it remains unclear how they are linked to the earlier-outlined downstream effectors. Since Fgf10, Bmp4 and Otx genes are downregulated in the Eya1 mutant mice, the morphogenetic defects observed in the Eya1 mutants may directly relate to an epistatic regulation of those genes by Eya1. Nonetheless, our results suggest that it is likely that other unknown downstream targets contribute to the inner-ear defects observed in the Eya1 mutant animals.
Eya1 and haploinsufficiency
There is considerable variation in the type and severity of inner-ear malformations in BOR syndrome, even among individuals with identical EYA1 mutations. The basis for such clinical diversity is unknown. Specific hypotheses to explain the defects associated with altered dosage of transcription factors have been proposed on the basis of the fact that most act as part of protein complexes (68,69). It was hypothesized that a narrow window exists, outside of which slight changes in the level of expression of the transcription factors cause an imbalance that leads to a significant impact on target gene transcription levels. As Eya proteins have been shown to act with Six proteins to regulate transcription activation, we believe that transcriptional synergy may be responsible for EYA1 haploinsufficiency in BOR/BO patients. Biophysical interaction between Eya1 and Six1 provides a mechanism for enhancing transcriptional activity, which may amplify gene activity at particular times or locations during inner-ear development. The observation of physical interaction between Eya1 and Sox2 suggests that Sox2 may also be involved in Eya1 transcriptional synergy, and the missense mutations identified in patients are likely to influence the Eya1–Sox2 complex formation, which in turn affects downstream gene expression and causes disease. The highly variable expressivity in BOR/BO patients can be explained by slightly altered levels of EYA1 expression among different individuals carrying same mutations that may be caused by random environmental events or sequence variations of genetic modifier such as PAX3 (70). Detailed analyses of the molecular defects and molecular pathways associated with human EYA1-related inner-ear defects have been difficult to assess in patients. Our analyses of an allelic series of Eya1 mutations in the mouse have confirmed a critical relationship between gene dosage of Eya1 and the severity of inner-ear malformation. The severity of hair cell malformation observed in Eya1+/− or Eya1bor/bor mutants provide the first evidence for the basis of hearing loss occurring in human patients at the level of hair cell function.
MATERIALS AND METHODS
Mice and genotyping
Eya1+/− and Eya1bor/+ mutant mice in C3HeB/FeJ strain were used for this study. Genotyping for these mice was performed as described previously (7,21).
The Eya1lacZ mutant allele was recently created by replacement of the endogenous start codon as well as the exon 1 with a promoterless Escherichia coli ATG-lacZ-poly(A) cassette and the PGK-neo gene. Mutant mice carrying Eya1lacZ allele were obtained using gene-targeting technology. A set of lacZ primers were used to detect the mutant allele and a set of primers within the exon 1 of Eya1 gene were used to detect Eya1 wild-type allele.
Semi-quantitative RT–PCR and western blot
Total RNAs were isolated from mouse inner ears at E13.5 of +/+, Eya1bor/+, Eya1+/−, Eya1bor/bor, Eya1bor/− and Eya1−/− embryos using RNAzol B (Biotecx Laboratories). Absence of residual genomic DNA was checked by PCR control reactions using RNA as template. Generation of cDNAs was carried out by RT using 1 µg of DNA-free total RNA as template with Superscript™ reverse transcriptase (Promega). Eya1 mRNA expression was amplified and quantitated by RT–PCR using 5 µl of the RT products and a primer pair specific for and internal to Eya1 (1.3 kb; forward primer 5′-GGAAAGTGGATTGTCACAGT-3′ and reverse primer 5′-CAGGTACTCTAATTCCAAGGC-3′). A primer pair specific for mouse β-actin (448 bp; forward primer 5′-GCTGTGTTCCCATCCATCGTGG-3′ and reverse primer 5′-GACGCATGATGGCGGTG-TGGCA-3′) was used to amplify the β-actin expression as an internal control for quantitation. Thirty microliter sample of each PCR reaction product was analyzed by 1% agarose gels and visualized and quantified with the VerserDoc system (Fisher). The PCR quantification reactions were performed in duplicate for each of the cDNA preparations. The RT–PCRs were repeated four times and the results were reproducible.
For western blot, extracts from E13.5 mouse inner ears of +/+, Eya1bor/+, Eya1+/−, Eya1bor/bor, Eya1bor/− and Eya1−/− embryos were prepared according to standard procedures. The protein concentrations of the extracts were determined using a protein assay kit (Bio-Rad), analyzed on 12% SDS–PAGE and transferred to a nitrocellulose membrane. A rabbit anti-Eya1 peptide antibody was used as the primary antibody, and HRP-coupled goat anti-mouse or goat anti-rabbit antiserum was used as the secondary antibody. An ECL kit (Pierce) was used for detection. Signals were quantified with the VerserDoc system (Fisher).
Paintfilling and SEM
The paintfilling of the ears at E18.5 was performed as described (65). For SEM, ears from E18.5, P7 and 6-week-old mice were dissected, fixed in 4% PFA, followed by 0.5% OsO4, decalcified, dehydrated, critical point dried, mounted on stubs, sputter coated and viewed with a Hitachi SEM (53).
Immunohistochemistry, X-gal staining and in situ hybridization
Embryos were fixed and processed using standard procedures. For immunohistochemistry, primary antibodies were specific for MYO7A (Proteus Biosciences INC), p27KIP1 (BD Biosciences pharmingen), SOX2 (Chemicon), β-galactosidase (Sigma), Jag1 (Santa Cruz) and mouse monoclonal anti-BrdU (Sigma). For whole-mount lectin staining, the ears were dissected out and incubated with a biotinylated lectin (Griffonia simplifonia I, Vector Lab) diluted 1:100. An FITC-labeled avidin secondary reagent was used to visualize the hair cells. X-gal staining was as described previously (12).
Whole-mount and section in situ hybridization were carried out according to standard procedures with digoxigenin-labeled riboprobes specific for Sox2, Eya1, Otx1 and Lfng and with radioisotope-labeled riboprobes for Otx2 and Bmp4. We used six embryos for each genotype at each stage for each probe, and the staining was consistent in each embryo.
Hair cell counts
Hair cell counts from lectin-stained whole-mounts for cochlea
Hair cell counts were performed on mid-basal regions (300 µm in length) of the lectin-stained cochleae of wild-type, Eya1bor/+ and Eya1+/− mice. For Eya1bor/bor mice, hair cell counts were performed on mid-basal (200 µm in length) and mid-apical (100 µm in length) regions, respectively, and cochleae that reached at least three-quarter turn were used for counts. Each genotype contained counts from three or four different embryos. After the capture of high-resolution images of the cochlear, counts and measurements were performed using Nikon elements research imaging software.
Hair cell counts from MYO7A-immunostained sections for utricle and saccule
To quantify the number of hair cells in the utricle and saccule, we cut serial sections at 7 µm through complete inner ears and stained the sections with the hair-cell marker Myosin VIIa. We quantitatively evaluated the entire utricle and saccule for two animals (four ears) at E18. We counted cells in every second section in the microscope and analyzed optical sections through deconvolution microscopy. The numbers were multiplied by two to obtain the final values for each ear.
Cell proliferation
To examine cell proliferation, timed pregnant female mice were injected i.p. three times at 2 h intervals with 5-bromodeoxyuridine (BrdU; Sigma) in PBS at 100 mg/kg and processed as described (12).
Cell culture, transfection assays, co-immunoprecipitation and western blot
Two hundred and ninety-three cells derived from human embryonic kidney fibroblast were cultured in Dulbecco’s modified medium supplemented with 10% fetal bovine serum, 100 U penicillin/ml and 100 µg streptomycin/ml at 37°C under 6% CO2. Transfection was performed using FuGENE™ 6 transfection reagent (Roche) according to the manufacturer’s instructions. Forty-eight hours after the transfection, the cells were used for extract preparation.
Cell lysates were prepared from cells co-transfected with a mouse Sox2/pcDNA3 expression plasmid, plus an empty vector, mouse Flag–Eya1 wild-type or mutant plasmid according to Buller et al. (14). The protein concentrations of the extracts were determined using a protein assay kit (Bio-Rad). Proteins were incubated with Flag-antibody-coupled agarose beads for co-immunoprecipitation analysis as described (14). Proteins were analyzed on 12% SDS–PAGE and transferred to a nitrocellulose membrane. Anti-Flag or anti-Sox2 was used as the primary antibody and HRP-coupled goat anti-mouse or goat anti-rabbit antiserum was used as secondary antibody. An ECL kit (Pierce) was used for detection.
FUNDING
This work was supported by NIH RO1 DC005824 (P.-X.X.) and RO1 DC005590 (B.F.).
ACKNOWLEDGEMENTS
We thank L. Schwandt, K. Shields, D. Silvius, B. Chen, H. Sun, J. Sun and L. Huang for technical assistance.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Abdelhak S., Kalatzis V., Heilig R., Compain S., Samson D., Vincent C., Weil D., Cruaud C., Sahly I., Leibovici M., et al. A human homologue of the Drosophila eyes absent gene underlies branchio–oto–renal (BOR) syndrome and identifies a novel gene family. Nat. Genet. 1997;15:157–164. doi: 10.1038/ng0297-157. [DOI] [PubMed] [Google Scholar]
- 2.Fraser F.C., Sproule J.R., Halal F. Frequency of the branchio–oto–renal (BOR) syndrome in children with profound hearing loss. Am. J. Med. Genet. 1980;7:341–349. doi: 10.1002/ajmg.1320070316. [DOI] [PubMed] [Google Scholar]
- 3.Vincent C., Kalatzis V., Abdelhak S., Chaib H., Compain S., Helias J., Vaneecloo F.M., Petit C. BOR and BO syndromes are allelic defects of EYA1. Eur. J. Hum. Genet. 1997;5:242–246. [PubMed] [Google Scholar]
- 4.Heimler A., Lieber E. Branchio–oto–renal syndrome: reduced penetrance and variable expressivity in four generations of a large kindred. Am. J. Med. Genet. 1986;25:15–27. doi: 10.1002/ajmg.1320250104. [DOI] [PubMed] [Google Scholar]
- 5.Konig R., Fuchs S., Dukiet C. Branchio–oto–renal (BOR) syndrome: variable expressivity in a five-generation pedigree. Eur. J. Pediatr. 1994;153:446–450. doi: 10.1007/BF01983410. [DOI] [PubMed] [Google Scholar]
- 6.Chen A., Francis M., Ni L., Cremers C.W., Kimberling W.J., Sato Y., Phelps P.D., Bellman S.C., Wagner M.J., Pembrey M., et al. Phenotypic manifestations of branchio–oto–renal syndrome. Am. J. Med. Genet. 1995;58:365–370. doi: 10.1002/ajmg.1320580413. [DOI] [PubMed] [Google Scholar]
- 7.Xu P.-X., Adams J., Peters H., Brown M.C., Heaney S., Maas R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat. Genet. 1999;23:113–117. doi: 10.1038/12722. [DOI] [PubMed] [Google Scholar]
- 8.Zou D., Silvius D., Rodrigo-Blomqvist S., Enerback S., Xu X. Eya1 regulates the growth of otic epithelium and interacts with Pax2 during the development of all sensory areas in the inner ear. Dev. Biol. 2006;298:430–441. doi: 10.1016/j.ydbio.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen R., Amoui M., Zhang Z., Mardon G. Dachshund and eyes absent proteins form a complex and function synergistically to induce ectopic eye development in Drosophila. Cell. 1997;91:893–903. doi: 10.1016/s0092-8674(00)80481-x. [DOI] [PubMed] [Google Scholar]
- 10.Pignoni F., Hu B., Zavitz K.H., Xiao J., Garrity P.A., Zipursky S.L. The eye-specification proteins So and Eya form a complex and regulate multiple steps in Drosophila eye development. Cell. 1997;91:881–891. doi: 10.1016/s0092-8674(00)80480-8. [DOI] [PubMed] [Google Scholar]
- 11.Xu P-X., Cheng J., Epstein J., Maas R.L. Activation function of the Eya gene products and their possible roles in connective tissue patterning. Proc. Natl Acad. Sci. USA. 1997;94:11974–11979. doi: 10.1073/pnas.94.22.11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng W., Huang L., Wei Z.-B., Silvius D., Tang H., Xu X. The role of Six1 in mammalian auditory system development. Development. 2003;130:3989–4000. doi: 10.1242/dev.00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruf R.G., Xu P.-X., Silvius D., Otto E.A., Beekmann F., Muerb U.T., Kumar S., Neuhaus T.J., Kemper M.J., Berkman J., et al. SIX1 mutations cause branchio–oto–renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc. Natl Acad. Sci. USA. 2004;101:8090–8095. doi: 10.1073/pnas.0308475101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buller C., Xu X., Marquis V., Schwanke R., Xu P.-X. Molecular effects of Eya1 domain mutations causing organ defects in BOR syndrome. Hum. Mol. Genet. 2001;10:2775–2781. doi: 10.1093/hmg/10.24.2775. [DOI] [PubMed] [Google Scholar]
- 15.Fekete D.M., Muthukumar S., Karagogeos D. Hair cells and supporting cells share a common progenitor in the avian inner ear. J. Neurosci. 1998;18:7811–7821. doi: 10.1523/JNEUROSCI.18-19-07811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bermingham N.A., Hassan B.A., Price S.D., Vollrath M.A., Ben-Arie N., Eatock R.A., Bellen H.J., Lysakowski A., Zoghbi H.Y. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–1841. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- 17.Chen P., Johnson J.E., Zoghbi H.Y., Segil N. The role of Math1 in inner ear development: uncoupling the establishment of the sensory primordium from hair cell fate determination. Development. 2002;129:2495–2505. doi: 10.1242/dev.129.10.2495. [DOI] [PubMed] [Google Scholar]
- 18.Fritzsch B., Pauley S., Matei V., Katz D.M., Xiang M., Tessarollo L. Mutant mice reveal the molecular and cellular basis for specific sensory connections to inner ear epithelia and primary nuclei of the brain. Hear. Res. 2005;206:52–63. doi: 10.1016/j.heares.2004.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiernan A.E., Pelling A.L., Leung K.K., Tang A.S., Bell D.M., Tease C., Lovell-Badge R., Steel K.P., Cheah K.S. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434:1031–1035. doi: 10.1038/nature03487. [DOI] [PubMed] [Google Scholar]
- 20.Zou D., Silvius D., Fritzsch B., Xu X. Eya1 and Six1 are essential for early steps of sensory neurogenesis in mammalian cranial placodes. Development. 2004;131:5561–5572. doi: 10.1242/dev.01437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson K.R., Cook S.A., Erway L.C., Matthews A.N., Sanford L.P., Paradies N.E., Friedman R.A. Inner ear and kidney anomalies caused by IAP insertion in an intron of the Eya1 gene in a mouse model of BOR syndrome. Hum. Mol. Genet. 1999;8:645–653. doi: 10.1093/hmg/8.4.645. [DOI] [PubMed] [Google Scholar]
- 22.Mburu P., Liu X.Z., Walsh J., Saw D, Jr, Cope M.J., Gibson F., Kendrick-Jones J., Steel K.P., Brown S.D. Mutation analysis of the mouse myosin VIIA deafness gene. Genes Funct. 1997;1:191–203. doi: 10.1046/j.1365-4624.1997.00020.x. [DOI] [PubMed] [Google Scholar]
- 23.Lowenheim H., Furness D.N., Kil J., Zinn C., Gultig K., Fero M.L., Frost D., Gummer A.W., Roberts J.M., Rubel E.W., Hackney C.M., Zenner H.P. Gene disruption of p27(Kip1) allows cell proliferation in the postnatal and adult organ of Corti. Proc. Natl Acad. Sci. USA. 1999;96:4084–4088. doi: 10.1073/pnas.96.7.4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee Y.S., Liu F., Segil N. A morphogenetic wave of p27Kip1 transcription directs cell cycle exit during organ of Corti development. Development. 2006;133:2817–2826. doi: 10.1242/dev.02453. [DOI] [PubMed] [Google Scholar]
- 25.Matei V., Pauley S., Kaing S., Rowitch D., Beisel K.W., Morris K., Feng F., Jones K., Lee J., Fritzsch B. Smaller inner ear sensory epithelia in Neurog1 null mice are related to earlier hair cell cycle exit. Dev. Dyn. 2005;234:633–650. doi: 10.1002/dvdy.20551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruben R.J. (1967) Development of the inner ear of the mouse: a radioautographic study of terminal mitoses. Acta Otolaryngol. 220(suppl):1–44. [PubMed] [Google Scholar]
- 27.Neves J., Kamaid A., Alsina B., Giraldez F. Differential expression of Sox2 and Sox3 in neuronal and sensory progenitors of the developing inner ear of the chick. J. Comp. Neurol. 2007;503:487–500. doi: 10.1002/cne.21299. [DOI] [PubMed] [Google Scholar]
- 28.Wood H.B., Episkopou V. Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre-gastrulation to early somite stages. Mech. Dev. 1999;86:197–201. doi: 10.1016/s0925-4773(99)00116-1. [DOI] [PubMed] [Google Scholar]
- 29.Fritzsch B., Beisel K.W., Jones K., Farinas I., Maklad A., Lee J., Reichardt L.F. Development and evolution of inner ear sensory epithelia and their innervation. J. Neurobiol. 2002;53:143–156. doi: 10.1002/neu.10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morsli H., Tuorto F., Choo D., Postiglione M.P., Simeone A., Wu D.K. Otx1 and Otx2 activities are required for the normal development of the mouse inner ear. Development. 1999;126:2335–2343. doi: 10.1242/dev.126.11.2335. [DOI] [PubMed] [Google Scholar]
- 31.Chang W., Brigande J.V., Fekete D.M., Wu D.K. The development of semicircular canals in the inner ear: role of FGFs in sensory cristae. Development. 2004;131:4201–4211. doi: 10.1242/dev.01292. [DOI] [PubMed] [Google Scholar]
- 32.Pauley S., Wright T.J., Pirvola U., Ornitz D., Beisel K., Fritzsch B. Expression and function of FGF10 in mammalian inner ear development. Dev. Dyn. 2003;227:203–215. doi: 10.1002/dvdy.10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang N., Martin G.V., Kelley M.W., Gridley T. A mutation in the lunatic fringe gene suppresses the effects of a Jagged2 mutation on inner hair cell development in the cochlea. Curr. Biol. 2000;10:659–662. doi: 10.1016/s0960-9822(00)00522-4. [DOI] [PubMed] [Google Scholar]
- 34.Kim W.Y., Fritzsch B., Serls A., Bakel L.A., Huang E.J., Reichardt L.F., Barth D.S., Lee J.E. NeuroD-null mice are deaf due to a severe loss of the inner ear sensory neurons during development. Development. 2001;128:417–426. doi: 10.1242/dev.128.3.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fekete D.M. Cell fate specification in the inner ear. Curr. Opin. Neurobiol. 1996;6:533–541. doi: 10.1016/s0959-4388(96)80061-4. [DOI] [PubMed] [Google Scholar]
- 36.Barald K.F., Kelley M.W. From placode to polarization: new tunes in inner ear development. Development. 2004;131:4119–4130. doi: 10.1242/dev.01339. [DOI] [PubMed] [Google Scholar]
- 37.Bryant J., Goodyear R.J., Richardson G.P. Sensory organ development in the inner ear: molecular and cellular mechanisms. Br. Med. Bull. 2002;63:39–57. doi: 10.1093/bmb/63.1.39. [DOI] [PubMed] [Google Scholar]
- 38.Fritzsch B., Beisel K.W., Hansen L.A. The molecular basis of neurosensory cell formation in ear development: a blueprint for hair cell and sensory neuron regeneration? Bioessays. 2006;28:1181–1193. doi: 10.1002/bies.20502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiernan A.E., Xu J., Gridley T. The Notch ligand JAG1 is required for sensory progenitor development in the mammalian inner ear. PLoS Genet. 2006;2:e4. doi: 10.1371/journal.pgen.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G., et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bylund M., Anderson E., Novitch B.G., Muhr J. Vertebrate neurogenesis is counteracted by Sox1-3 activity. Nat. Neurosci. 2003;6:1162–1168. doi: 10.1038/nn1131. [DOI] [PubMed] [Google Scholar]
- 42.Ferri A.L., Cavallaro M., Braida D., Di Cristofano A., Canta A., Vezzani A., Ottolenghi S., Pandolfi P.P., Sala M., DeBiasi S., Nicolis S.K. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development. 2004;131:3805–3819. doi: 10.1242/dev.01204. [DOI] [PubMed] [Google Scholar]
- 43.Graham V., Khudyakov J., Ellis P., Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39:749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- 44.Lang H., Fekete D.M. Lineage analysis in the chicken inner ear shows differences in clonal dispersion for epithelial, neuronal, and mesenchymal cells. Dev. Biol. 2001;234:120–137. doi: 10.1006/dbio.2001.0248. [DOI] [PubMed] [Google Scholar]
- 45.Kawamoto K., Ishimoto S., Minoda R., Brough D.E., Raphael Y. Math1 gene transfer generates new cochlear hair cells in mature guinea pigs in vivo. J. Neurosci. 2003;23:4395–4400. doi: 10.1523/JNEUROSCI.23-11-04395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woods C., Montcouquiol M., Kelley M.W. Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat. Neurosci. 2004;7:1310–8131. doi: 10.1038/nn1349. [DOI] [PubMed] [Google Scholar]
- 47.Zheng J.L., Gao W.Q. Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat. Neurosci. 2000;3:580–386. doi: 10.1038/75753. [DOI] [PubMed] [Google Scholar]
- 48.Curtin J.A., Quint E., Tsipouri V., Arkell R.M., Cattanach B., Copp A.J., Henderson D.J., Spurr N., Stanier P., Fisher E.M., et al. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr. Biol. 2003;13:1129–1133. doi: 10.1016/s0960-9822(03)00374-9. [DOI] [PubMed] [Google Scholar]
- 49.Lu X., Borchers A.G., Jolicoeur C., Rayburn H., Baker J.C., Tessier-Lavigne M. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature. 2004;430:93–98. doi: 10.1038/nature02677. [DOI] [PubMed] [Google Scholar]
- 50.Montcouquiol M., Rachel R.A., Lanford P.J., Copeland N.G., Jenkins N.A., Kelley M.W. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature. 2003;423:173–177. doi: 10.1038/nature01618. [DOI] [PubMed] [Google Scholar]
- 51.Wang J., Mark S., Zhang X., Qian D., Yoo S.J., Radde-Gallwitz K., Zhang Y., Lin X., Collazo A., Wynshaw-Boris A., Chen P. Regulation of polarized extension and planar cell polarity in the cochlea by the vertebrate PCP pathway. Nat. Genet. 2005;37:980–985. doi: 10.1038/ng1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kiernan A.E., Ahituv N., Fuchs H., Balling R., Avraham K.B., Steel K.P., Hrabe de Angelis M. The Notch ligand Jagged1 is required for inner ear sensory development. Proc. Natl Acad. Sci. USA. 2001;98:3873–3878. doi: 10.1073/pnas.071496998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma Q., Anderson D.J., Fritzsch B. Neurogenin 1 null mutant ears develop fewer, morphologically normal hair cells in smaller sensory epithelia devoid of innervation. J. Assoc. Res. Otolaryngol. 2000;1:129–143. doi: 10.1007/s101620010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pirvola U., Ylikoski J., Trokovic R., Hébert J.M., McConnell S.K., Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- 55.Pauley S., Lai E., Fritzsch B. Foxg1 is required for morphogenesis and histogenesis of the mammalian inner ear. Dev. Dyn. 2006;235:2470–2482. doi: 10.1002/dvdy.20839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McFadden S.L., Ding D., Reaume A.G., Flood D.G., Salvi R.J. Age-related cochlear hair cell loss is enhanced in mice lacking copper/zinc superoxide dismutase. Neurobiol. Aging. 1999;20:1–8. doi: 10.1016/s0197-4580(99)00018-4. [DOI] [PubMed] [Google Scholar]
- 57.Schrott A., Stephan K., Spoendlin H. Auditory brainstem response thresholds in a mouse mutant with selective outer hair cell loss. Eur. Arch. Otorhinolaryngol. 1990;247:8–11. doi: 10.1007/BF00240940. [DOI] [PubMed] [Google Scholar]
- 58.Whitlon D.S., Gabel C., Zhang X.J. Cochlear inner hair cells exist transiently in the fetal Bronx Waltzer (bv/bv) mouse. Comp. Neurol. 1996;364:515–522. doi: 10.1002/(SICI)1096-9861(19960115)364:3<515::AID-CNE9>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 59.Kelley M.W. Hair cell development: commitment through differentiation. Brain Res. 2006;1091:172–185. doi: 10.1016/j.brainres.2006.02.062. [DOI] [PubMed] [Google Scholar]
- 60.Kelley M.W. Regulation of cell fate in the sensory epithelia of the inner ear. Nat. Rev. Neurosci. 2006;7:837–849. doi: 10.1038/nrn1987. [DOI] [PubMed] [Google Scholar]
- 61.Brooker R., Hozumi K., Lewis J. Notch ligands with contrasting functions: Jagged1 and Delta1 in the mouse inner ear. Development. 2006;133:1277–1286. doi: 10.1242/dev.02284. [DOI] [PubMed] [Google Scholar]
- 62.Daudet N., Lewis J. Two contrasting roles for Notch activity in chick inner ear development: specification of prosensory patches and lateral inhibition of hair-cell differentiation. Development. 2005;132:541–551. doi: 10.1242/dev.01589. [DOI] [PubMed] [Google Scholar]
- 63.Fritzsch B., Matei V.A., Nichols D.H., Bermingham N., Jones K., Beisel K.W., Wang V.Y. Atoh1 null mice show directed afferent fiber growth to undifferentiated ear sensory epithelia followed by incomplete fiber retention. Dev. Dyn. 2005;233:570–583. doi: 10.1002/dvdy.20370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang W., Cole L.K., Cantos R., Wu D.K. Molecular genetics of vestibular organ development. In: Highstein S.M., Fay R.R., Popper A.N., editors. The Vestibular System. New York: Springer Verlag; 2004. pp. 11–56. [Google Scholar]
- 65.Morsli H., Choo D., Ryan A., Johnson R., Wu D.K. Development of the mouse inner ear and origin of its sensory organs. J. Neurosci. 1998;18:3327–3335. doi: 10.1523/JNEUROSCI.18-09-03327.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fritzsch B., Signore M., Simeone A. Otx1 null mutant mice show partial segregation of sensory epithelia comparable to lamprey ears. Dev. Genes Evol. 2001;211:388–396. doi: 10.1007/s004270100166. [DOI] [PubMed] [Google Scholar]
- 67.Fritzsch B., Pauley S., Beisel K.W. Cells, molecules and morphogenesis: the making of the vertebrate ear. Brain Res. 2006;1091:151–171. doi: 10.1016/j.brainres.2006.02.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Papp B., Pal C., Hurst L.D. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424:194–197. doi: 10.1038/nature01771. [DOI] [PubMed] [Google Scholar]
- 69.Veitia R.A. Exploring the etiology of haploinsufficiency. Bioessays. 2002;24:175–184. doi: 10.1002/bies.10023. [DOI] [PubMed] [Google Scholar]
- 70.Epstein J.A. Pax3 and vertebrate development. Methods Mol. Biol. 2000;137:459–470. doi: 10.1385/1-59259-066-7:459. [DOI] [PubMed] [Google Scholar]