

Summary
Cell proliferation requires close coordination of cell growth and division to ensure constant cell size through the division cycles. IQGAP1, an effector of CDC42 GTPase has been implicated in the modulation of cell architecture, regulation of exocytosis and in human cancers. The precise mechanism underlying these activities is unclear. Here, we show that IQGAP1 regulates cell proliferation, which requires phosphorylation of IQGAP1 and binding to CDC42. Expression of the C-terminal region of IQGAP1 enhanced cellular transformation and migration, but reduced the cell size, whereas expression of the N-terminus increased the cell size, but inhibited cell transformation and migration. The N-terminus of IQGAP1 interacts with mTOR, which is required for IQGAP1-mediated cell proliferation. These findings are consistent with a model where IQGAP1 serves as a phosphorylation-sensitive conformation switch to regulate the coupling of cell growth and division through a novel CDC42-mTOR pathway, dysregulation of which generates cellular transformation.
Keywords: Cell growth, IQGAP1, Protein synthesis
Introduction
Cell proliferation, which is generally controlled by the rate of protein synthesis, requires tight coordination of cell growth and division and a diverse number of proteins have been shown to affect both cell growth and cell cycle progression (Yang et al., 2008; Tapon et al., 2001). The concept of the coupling of the cell cycle progression to cell growth was pioneered in the early studies of yeast cell cycle in terms of cyclins and cyclin-dependent kinases (reviewed by Cross, 1995; Bean et al., 2006). Treatment with the TOR-specific growth inhibitor rapamycin or nutrient withdrawal delays passage through the cell cycle, indicating that cell growth and the cell cycle are tightly coupled processes (reviewed by Cross, 1995). However, in multicellular organisms, the action of different growth factors fine-tunes the level of control, uncoupling cell growth from cell division during the development of different tissues (Tapon et al., 2001), indicating a highly regulated process. The evolutionarily conserved serine/threonine kinase, mTOR has been suggested to mediate this link by integrating nutrient and growth factor signals through the PI3K-mTOR pathway, where PI3K controls the cell cycle and mTOR regulates the cell growth (Tapon et al., 2001; Fingar and Blenis, 2004; Sabatini, 2006; Wullschleger et al., 2006). Consistent with this view, tumors arising from perturbation of the TOR wing of the pathway are benign (tuberous sclerosis), whereas those arising in combination with alterations in the PI3K wing are malignant (Sabatini, 2006). However, the precise mechanism by which this coupling occurs is still unraveling. Although the GTPase CDC42 has been observed to affect cell growth and proliferation in response to growth factor signaling (Cerione, 2004), a direct link with mTOR is unknown.
IQGAP1, an effector of CDC42, is a conserved multifunctional protein implicated in tumorigenesis and invasive growth. Several human cancers such as colon (Nabeshima et al., 2002), metastatic melanoma (Clark et al., 2000), glioblastoma (Balenci et al., 2006) and gastric carcinomas (Takemoto et al., 2001) display changes in IQGAP1 expression or localization. Moreover, knockout of Iqgap1 in mice produced gastric hyperplasia and lung adenoma (Li et al., 2000), and its expression was implicated in tumorigenesis (Jadeski et al., 2007). The mechanisms underlying the presumptive transforming effect of IQGAP1 remain largely unknown.
However, several aspects of IQGAP1 functions offered important clues. The yeast IQGAP1 ortholog (Iqg1p) has been implicated in cytokinesis and polarized growth, acting both upstream and downstream of CDC42 (Osman and Cerione, 1998; Osman et al., 2002; Osman and Cerione, 2005). Mammalian IQGAP1 was shown to assemble actin cytoskeleton through binding to CDC42, the myosin-essential light chain, Arp2/3 and N-WASP (Brown and Sacks, 2006; Le Clainche et al., 2007; Bensenor et al., 2007), to capture microtubule plus-ends via CLIP-170 (Fukata et al., 2002) and to integrate signaling networks (Mateer et al., 2003; Roy et al., 2005). Furthermore, a new role for IQGAP1 in secretion is emerging: it regulates cell-cell junctions (Fukata et al., 1999; Fukata et al., 2001) considered to be sites for polarized exocytosis (Grindstaff et al., 1998; Kreitzer et al., 2003), and influences membrane trafficking in gastric parietal cells (Zhou et al., 2003).
Our laboratory has demonstrated a regulatory role of IQGAP1 in polarized growth that might provide a platform to understanding its mechanism(s) in cell proliferation. We have shown that IQGAP1 binds the exocyst-septin complex and the ER-translocon subunit Sec61β through its N-terminus and promotes protein synthesis and secretion. CDC42, which binds the C-terminal region of IQGAP1, disrupts this interaction and inhibits IQGAP1-mediated secretion (Rittmeyer et al., 2008).
Here, we investigate a role for IQGAP1 in integrating cell growth with cell division. We tested the hypothesis that CDC42-bound IQGAP1 promotes cell division whereas exocyst-bound IQGAP1 promotes cell growth and migration. Our results demonstrate that IQGAP1 promotes cell growth via interactions of its N-terminal domain with mTOR and accelerates cell division via interactions of its C-terminal domain with CDC42. Our data suggest that IQGAP1 switches between a pro-growth and pro-cell-division/migration conformation, in part through phosphorylation of Ser1443 and that failure to switch between them leads to uncontrolled cell proliferation and transformation. Our findings demonstrate a regulatory role of IQGAP1 in cell proliferation and position it as a novel anti-cancer target for rapamycin.
Results
Differential effects of IQGAP1 domains on cytokinesis
To begin investigating whether IQGAP1 affects cytokinesis, HeLa cells stably expressing low levels (∼10% of wild type) of V5-IQGAP1 constructs (Fig. 1A,B) were examined by fluorescence microscopy. The V5-tagged constructs appeared to be localized correctly (Fig. 1D), as previously observed, to the ER, the cytoplasm and plasma membrane (Rittmeyer et al., 2008). When plated at low density, mammalian cells can potentially become bi-nucleated (Kanda et al., 2005), therefore, we scored cells that contained three or more nuclei. Fig. 1C,D suggest that, in contrast to the expression of C-terminus IQGAP1 (IQGAP1-C) or full-length IQGAP1 (IQGAP1-F), expression of IQGAP1-IR-WW [or N-terminus IQGAP1 (IQGAP1-N), not shown] produces highly multinucleated cells, suggesting that it impairs cytokinesis. This suggested that IQGAP1 influences cytokinesis and that its deregulation can either arrest (Fig. 1) or accelerate cytokinesis. To test the latter idea, we assayed proliferation in fibroblast NIH3T3 cells, a classic system for transformation studies, because HeLa are highly transformed cells.
Fig. 1.

IQGAP1 regulates cytokinesis. (A) Schematics of V5-IQGAP1 or HA-IQGAP1 stably expressed in HeLa or in NIH3T3 cells, respectively. F, IQGAP1-F; N, IQGAP1-N; C, IQGAP1-C; IR-WW, IQGAP1-IR-WW. (B) Immunoblot demonstrating similar stable expression levels of IQGAP1 constructs in HeLa cells. Equal amounts of the cell lysate from each cell line was loaded on 10-20% gradient gels and blotted with V5 antibody. One-tenth of the lysate loaded for the constructs was resolved on the same gel and blotted with antibody against IQGAP1 to detect the endogenous protein as a control. (C) Quantification of stable HeLa cells containing three or more nuclei was scored for n=50 cells each from three independent experiments and shown as the means ± s.e.m. and average significance value of P<0.005. Cells expressing the vector (V) were included as a negative control. (D) Differential effects of IQGAP1 on cytokinesis. Cells were seeded at low density, grown for 6 days, and double stained with V5 antibodies to visualize the IQGAP1 constructs and DAPI for nuclei. Scale bar: 10 μm.
IQGAP1 influences cell-cycle progression rate and controls proliferation
To determine how IQGAP1 influences cell division, we measured the cell cycle progression rate of the stable NIH3T3 cells expressing equal levels of the domains (Fig. 2A), using the BrdU (thymidine analog, bromodeoxyuridine) incorporation assay. Fig. 2B,C demonstrates that stable expression of IQGAP1-C (second column) enhanced DNA synthesis in cells to levels almost twice that observed with IQGAP1-N (third column) and IQGAP1-F (first column), suggesting that IQGAP1 differentially regulates cell division via different domains. Further, IQGAP1-C acceleration of cell division indicated that it might induce cellular transformation.
Fig. 2.

IQGAP1 regulates cell cycle progression. (A) Immunoblot demonstrating equal expression of HA-IQGAP1 constructs in NIH3T3 stable cell lines. Equal amounts of cell lysates from each cell line was loaded and blotted with anti-HA antibody. One-fifth of the lysate loaded for detecting the construct was loaded on the same gel and blotted with IQGAP1 antibody to detect the endogenous protein as a control. (B) Photomicrographs of immunofluorescence in asynchronous NIH3T3 cells stably expressing the indicated IQGAP1 constructs and triple stained with antibodies against HA (left panels), BrdU (middle panels) and DAPI for nuclei (right panels). Cells expressing the vector (V) were included as negative controls. Scale bar: 50 μm. (C) Quantification of the percentage of cells incorporating BrdU after 16 hours is shown as mean ± s.e.m. for four clones measured in triplicate. Average statistical significance between highest and lowest values, *P<0.001.
To test this idea definitively, we used transformation assays as described in the Materials and Methods to measure the proliferation rates of the stable cell lines in high serum, low serum and in soft agar. Fig. 3A-C shows that IQGAP1-C cells grew three times faster in high serum and that they efficiently proliferated in low-serum and formed numerous foci ≥50 μm in diameter in soft agar compared with control (V) and with IQGAP1-N cells. IQGAP1-F cells exhibited a similar phenotype, but they grew at a slower rate than IQGAP1-C cells (Fig. 3A-C). Thus, by four independent measurement criteria: cell cycle progression rate, saturation density, growth in low serum and soft agar, stable expression of IQGAP1-C consistently enhanced proliferation rate producing transformed phenotypes, whereas IQGAP1-N induced none. Thus, relative to IQGAP1-F, and taken together with the data in Fig. 1, IQGAP1-C appears to serve as promoter of cell division whereas IQGAP1-N might serve as inhibitor (see Fig. 1C,D).
Fig. 3.

IQGAP1 regulates cell proliferation in fibroblast NIH3T3 cells. (A) Lack of contact inhibition was measured over 6 days as the saturation density of stable cell lines growing in high-serum (10%) and presented as the means ± s.e.m. for n=4 (*P≤0.001). (B) Upper panel, proliferation rate in low serum (1%) was determined as saturation density over 6 days and presented as mean ± s.e.m. for n=4 experiments. Lower panel, photomicrographs of the cells on day 6. (C) Upper panel, growth in soft agar to measure anchorage-independent growth was determined as the number of foci ≥50 μm in diameter. Means ± s.e.m. for n=3 experiments are shown for four clones done in duplicate (*P<0.001). Lower panel, photomicrographs of the cells on day 10. (D) Endogenous IQGAP1 is required for transformation. Growth in low-serum medium for control (V) and IQGAP1-C cells untreated as positive control, or treated with scrambled (S) or two IQGAP1 siRNAs (R) that reduced the endogenous level by ∼90% as shown on the blot. Actin was detected as a loading control. P values indicate significant difference between highest and lowest values.
To test whether expression of IQGAP1-C alone is sufficient for transformation, we depleted the endogenous IQGAP1 with an N-terminus-targeted siRNA (Fig. 3D). IQGAP1-C cells that were treated with two IQGAP1 siRNAs that reduced endogenous protein by 90% (Fig. 3D, blot), and did not affect IQGAP1-C or other IQGAP-family members (Rittmeyer et al., 2008), were unable to proliferate in low serum compared with cells untreated or treated with control RNAi (Fig. 3D), indicating that IQGAP1-C requires endogenous IQGAP1 for transformation (see Discussion). These results suggest that IQGAP1 differentially regulates cell proliferation by different domains, thus its cellular activity must be regulated.
Requirement of IQGAP1 phosphorylation and CDC42 binding for proliferation
Phosphorylation is commonly used by cells for regulating their proteins. We observed doublets of IQGAP1 in asynchronously proliferating cells, specifically those treated with EGF (Fig. 4A, lower panels) or expressing IQGAP1-F or IQGAP-C (Fig. 4A, first panel). Their expression increases the cellular level of active GTP-CDC42 (Grohmanova et al., 2004; Rittmeyer et al., 2008), which, in turn binds phosphorylated IQGAP1 (Grohmanova et al., 2004). Moreover, treatment of the cell lysate with calf intestine phosphatase eliminated the mobility shift (not shown). To identify potential phosphorylation sites on IQGAP1, we searched the ExPASy proteomic server (http://ca.expasy.org/). Among several potential sites of different kinases, two PKCε sites, S1443 and S1441 were identified in the C-terminus. S1443 was more attractive because in addition to residing at the C-terminal domain responsible for CDC42 binding and activation, mass spectrometry demonstrated its phosphorylation by PKCε during dissociation of cell-cell contacts (Grohmanova et al., 2004), which is a hallmark of cellular transformation. Indeed, the upper band immunoprecipitated and detected by IQGAP1 antibodies (Fig. 4A, first panel), was also detected by Ser-P PKC-substrate antibodies and demonstrated that cells expressing IQGAP1-F or IQGAP1-C contained more of this protein (Fig. 4A, second panel, pSer, compare lanes F and C with lanes V and N). Treatment of the cells with EGF for 5 minutes significantly enhanced the intensity of the upper band, suggesting an increase in IQGAP1-P (Fig. 4A, lower panel). Thus mitogenic signals or expression of IQGAP1, which activate CDC42, appears to trigger its phosphorylation, suggesting that cellular IQGAP1 might be regulated by phosphorylation perhaps in a cell-cycle-dependent manner to promote cell division in cooperation with CDC42.
Fig. 4.

IQGAP1 phosphorylation and binding to CDC42 are required for transformation. (A) Immunoblots of equal amount of extracts from cells untreated (upper panels) or treated with EGF for 5 minutes (lower panels) immunoprecipitated and blotted with antibodies for IQGAP1 or phosphoserine (pSer). Actin was detected as a loading control. The blot represents three experiments with identical results. (B) Top panel, blot demonstrating equal expression levels of IQGAP1 mutants in total lysates of the NIH3T3 stable cell lines: C,IQGAP1-C; E, IQGAP1-C-S1443E; 9, IQGAP1-C-ΔMK24; 1, IQGAP1-C-S1443A. Actin and endogenous IQGAP1 were detected as loading controls. Lower panel, proliferation rate of mutant cells in low serum (1%) medium. Means ± s.e.m. for n=3 experiments are shown for three clones each done in duplicate (*P<0.001). (C) Lack of contact inhibition was measured as saturation density in high serum (10%) and expressed as mean ± s.e.m. for n=3 experiments for three clones each done in duplicate (*P<0.001). (D) Anchorage-independent growth for the mutants in soft agar was scored after 10 days as the percentage of colonies ≥50 μm in diameter. Error bars are means ± s.e.m. for n=3 experiments (*P<0.001). P values indicate significant difference between highest and lowest values.
This hypothesis was tested genetically and by RNAi, focusing on S1443, because mass spectrometry showed S1441 to be a minor phosphorylation site (Li et al., 2005). We assayed proliferation of cells stably expressing phosphomimetic IQGAP1-CS1443E, phosphodefective IQGAP1-CS1443A or IQGAP1-CΔMK24 containing a deletion of the CDC42-binding and activation region. The transformation assays shown in Fig. 4B-D demonstrated that unlike the phosphodefective (1), the phosphomimetic mutant (E) caused transformed phenotypes. These data, although indicating that S1443 is crucial for regulation of IQGAP1 function in cell division, do not eliminate contribution by other phosphorylation sites.
Binding to CDC42 was also required for transformation, because IQGAP1-CΔMK24 inhibited proliferation as measured by the three transformation assays (Fig. 4B-D), which we then verified by RNAi. Two shRNAs that reduced the level of endogenous CDC42, but not IQGAP1, by more than 50% (Fig. 5A), impaired proliferation of IQGAP1-C cells in low-serum medium and their capacity to form colonies in soft agar by ∼fourfold (Fig. 5B-C). Moreover, we found that the constitutively active mutant CDC42F28L demonstrated previously to induce tumorigenesis in mice (Lin et al., 1997), requires IQGAP1 for transformation because depletion of IQGAP1 impaired their proliferation in low serum more than twofold (Fig. 5D). Thus these data indicate that IQGAP1 and CDC42 mutually control proliferation via IQGAP1-C and CDC42 raising a question about the role of IQGAP1-N in proliferation.
Fig. 5.

Mutual requirement of IQGAP1 and CDC42 for cellular transformation. (A) Upper panel, western blot of transformed IQGAP1-C cells that were treated with two shRNAs targeting CDC42 and evaluated by blotting with antibodies for CDC42 and for IQGAP1 as a control. Lower panel, densitometry quantification of the CDC42 band in control (first column) and shRNA-treated (second and third columns) bands demonstrating ∼33% average knockdown of endogenous CDC42 by shRNA treatment. (B) Growth in low-serum medium of IQGAP1-C and control (V) NIH3T3 stable cells untreated or treated with CDC42 shRNA or control shRNA. (C) Growth in soft agar of the same cell lines. Upper panel, quantification of colonies greater than 50 μm in diameter. Means ± s.e.m. for n=3 experiments are shown for four clones done in duplicate. Growth of IQGAP1-C cells with and without control siRNA was significantly greater than for control cells (*P<0.001). Lower panels, representative photomicrographs of colonies formed by the different cell lines on day 10. (D) Growth in low-serum medium of transformed CDC42F28L and vector-control NIH3T3 cells untreated or treated with specific IQGAP1-siRNA.
IQGAP1 modulates cell size via mTOR
Our results suggest that IQGAP1-N serves as inhibitor of cell proliferation. However, our previous findings indicated that IQGAP1-N binds to the exocyst and the ER translocon, and enhances protein synthesis and exocytosis (Rittmeyer et al., 2008) required for cell growth. Moreover, a subunit of the conserved mTOR complex 1 (TORC1), which regulates protein synthesis and cell growth (Harrington et al., 2005; Wullschleger et al., 2006; Sabatini, 2006), was identified as a binding partner in a yeast two-hybrid screen with Iqg1p (M.A.O., unpublished results). Cells must increase their mass before division to ensure constant size upon proliferation, therefore we examined whether IQGAP1-N contributes to proliferation by promoting cell growth via mTOR.
The data in Fig. 6A (left) suggest that endogenous IQGAP1 and mTOR reciprocally co-immunoprecipitate from untransfected HeLa cell lysates. To examine the domain responsible for the interaction, exogenous V5-tagged IQGAP1 domains stably expressed in HeLa cells (Fig. 1B) were used for immunoprecipitation. V5-IQGAP1-F or V5-IR-WW, but not V5-IQGAP1-C or the vector control V, also co-immunoprecipitated with endogenous mTOR (Fig. 6A, right), suggesting that IQGAP1-IR-WW directly or indirectly mediates the interaction. Direct interaction studies with purified proteins produced faint bands, indicating the requirement of larger complexes (not shown). These results implicate IQGAP1 as a component of the mTOR-signaling pathway that regulates cell growth and size.
Fig. 6.

IQGAP1 binds mTOR, regulates the cell size and requires mTOR for proliferation. (A) Left, endogenous IQGAP1 and mTOR reciprocally co-immunoprecipitate from untransfected HeLa cells. Mock denotes control immunoprecipitation with V5 antibodies. Right, exogenous V5-IQGAP1 and endogenous mTOR were co-immunoprecipitated with V5 antibodies from stable HeLa cell lysate and blotted with mTOR and V5 antibodies. (B) HeLa cells stably expressing IQGAP1 domains were seeded at 70% confluency, grown overnight to 80% and evaluated by FACS. The x-axis indicates relative cell size from equal numbers of cells in each cell line. The cell size distribution curve of the vector control, in solid grey, is included in each image for comparison. (C) IQGAP1 requires mTOR for proliferation. IQGAP1-C cells were untreated, treated with DMSO (the drug-vehicle as a control) or with 100 nM mTOR-inhibitor rapamycin, grown in low-serum medium and scored every other day for 6 days. Growth of vector-control cell (V) in low-serum was included for comparison. Means ± s.e.m. for n=3 experiments are shown (P≤0.001).
To determine the biological significance of the IQGAP1-mTOR interaction and test our hypothesis that IQGAP1-N contributes to proliferation by enhancing cell growth, we measured the cell size. Since IQGAP1-WW arrests cytokinesis, HeLa cells were plated at higher densities to eliminate any effects on cell size generated by multi-nuclei accumulation (Kanda et al., 2005). Cells were grown for 1 day then evaluated by FACS to determine DNA content and cell size. Cells that were in G1 and G2 phases of the cell cycle were scored for size. Fig. 6B shows that cells expressing IQGAP1-F and IQGAP1-IR-WW that interact with mTOR, became larger, whereas IQGAP1-C cells did not increase their size in G1 and became smaller in G2. The increase in size was not due to DNA content, because cells with similar DNA contents were compared, but such an increase is consistent with the finding that the IR-WW domain promotes protein synthesis and secretion (Rittmeyer et al., 2008).
Next, we examined any relevance to cell proliferation; we hypothesized that IQGAP1 proliferation probably requires mTOR, which we tested pharmacologically with the mTOR-specific-inhibitor rapamycin. By day six of treatment with rapamycin, proliferation of IQGAP1-C-transformed cells was inhibited more than threefold, compared with IQGAP1-C cells that were untreated or treated with vehicle DMSO as a control (Fig. 6C), and they grew similarly to negative vector-control cells (V). These data are a strong indication that promotion of cell growth by IQGAP1 is required for proliferation and that it occurs via the mTOR pathway. Thus, IQGAP1-C transformation ability not only requires endogenous IQGAP1 (Fig. 3D), but also apparently the interaction of endogenous IQGAP1 with mTOR.
The differential requirement of CDC42 for IQGAP1 transformation and IQGAP1-N for cell growth raised questions about the migratory capacity of the transformed cells, because we previously showed that CDC42 inhibited IQGAP1 exocytosis and displaced the exocyst (Rittmeyer et al., 2008). Therefore, we examined the migration rates of NIH3T3 cells stably expressing the IQGAP1 mutants by two complementary assays.
IQGAP1 phosphorylation and binding to CDC42 modulate migration
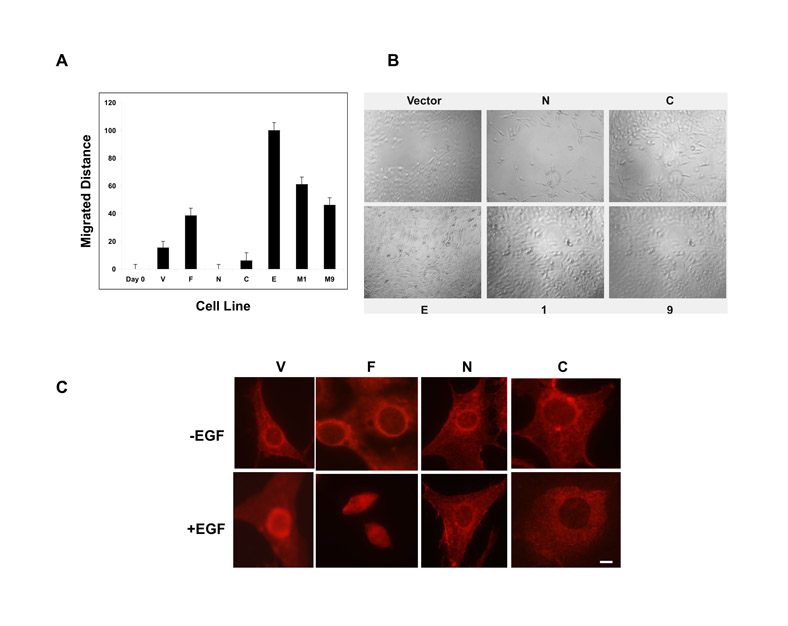
Cell migration in vitro has been reported to correlate with tumor invasion and metastasis in vivo (Brown and Bicknell. 2001) and IQGAP1 has been implicated in migration (Bensenor et al., 2007). The migration rates of stable cell lines were determined with a modified Boyden assay, which measures chemotaxis (Fig. 7A). Both IQGAP1-C and the phosphodefective mutant cells displayed migration rates that were four times higher (Fig. 7A, second and sixth columns respectively) than the control (V), whereas IQGAP1-F and the phosphomimetic mutant cells (first and fourth columns), migrated around three times faster than the control. By contrast, the CDC42-binding mutant IQGAP1-CΔMK24 cells migrated similarly to the control (Fig. 7A, fifth column). These results suggest that IQGAP1 phosphorylation is dispensable and might even attenuate migration (Fig. 7A, compare E and 1) whereas binding to CDC42 might be required. Unexpectedly, expression of IQGAP1-N, which enhances exocytosis (Rittmeyer et al., 2008), inhibited migration (Fig. 7A, third column).
Fig. 7.

IQGAP1 regulates cell migration. (A) Top, quantification of the number of migrated stable NIH3T3 cells with the Boyden assay. Means ± s.e.m. are shown for n=3 experiments (*P<0.001). Migration rates of IQGAP1-C and the phosphodefective mutant (I) cells were significantly greater than that of control cells. Lower, representative photomicrograph of migrated cells stained with Giemsa (dark spots). (B). Wound-healing assay of the indicated NIH3T3 stable cell lines. The scratched monolayer was photographed immediately (Day 0, is representative of all cells) and after 12 hours. Scale bars: 10 μm (upper left). For quantification of the migrated distances, see supplementary material Fig. S1A. The line in Day 0 was used as reference to calculate the migrated distances of the cell lines.
These results were tested with a wound-healing migration assay (Fig. 7B; supplementary material S1A,B), which also directly reflects cell-cell and cell-ECM (extracellular matrix) properties of cell migration (Niinaka et al., 2001) because IQGAP1 has been implicated in cadherin-mediated cell adhesion (Fukata et al., 2001; Fukata et al., 2002). Twelve hours after wounding, and as shown in Fig. 7B and supplementary material Fig. S1A-B, the phosphorylation mutants (1 and E) appear to have migrated best (100% closure for S1443E and 61% for S1443A) followed, in order, by IQGAP1-C (53.8%), IQGAP1-CΔMK24 (46.1%) then IQGAP1-F (38.5%). Whereas IQGAP1-C cells migrated slowly, IQGAP1-N cells were unable to migrate, suggesting a requirement of both domains for efficient migration by this assay. Furthermore, after 24 hours, both IQGAP1-CΔMK24 and the phosphodefective cells confluently closed their wound, whereas IQGAP1-C and the phosphomimetic cells did so to a lesser extent (supplementary material Fig. S1B). Similarly, IQGAP1-N cells remained unable to heal wounds (supplementary material Fig. S1B). Overall, these results indicated that binding to CDC42 helps at early time points, but is eventually dispensable for ECM-mediated migration and that sustained IQGAP1-phosphorylation slows migration (compare E and 1 in Fig. 7A,B), as demonstrated at the 24 hour time point (compare E, 1 and 9 in supplementary material Fig. S1B). Inability of IQGAP1-C cells to seal wounds was intriguing, but could be due to IQGAP1-C inhibition of IQGAP1 in cell scattering, because it inhibits translocation of β-catenin to the nucleus (supplementary material Fig. S1C) (Fukata et al., 2001). Collectively, these data suggest that IQGAP1 dynamically utilizes CDC42 and phosphorylation to modulate migration, and that sustained phosphorylation of IQGAP1 and binding to CDC42, although it enhances proliferation, attenuates migration.
Collectively, the data presented indicated that IQGAP1-N, which interacts with mTOR, enhanced the cell size, but inhibited cytokinesis and migration, whereas IQGAP1-C, which binds and activates CDC42, reduced the cell size, and enhanced cell division and migration. They further suggest that IQGAP1 couples cell growth with cell cycle progression, and that its deregulation alters cell proliferation.
Discussion
Cell proliferation is the product both of cell growth (mass or size) and of cell division (cell number), which determines the size, shape and fate of individual cells and ultimately organism size. Dysfunction of this process has serious consequences on animal development and health. Therefore, cells must tightly regulate the coupling of their growth and division both in time and in space. Our data demonstrate, for the first time, that IQGAP1 directly links CDC42, implicated in growth-factor-induced modulation of the actin cytoskeleton (Cerione, 2004) with the mTOR pathway (Fig. 6), implicated in regulating metabolism, cell growth and cell cycle progression in response to nutrient and growth factor signals. Thus, unlike IQGAP3, which was recently reported to induce cell proliferation exclusively through Ras-ERK signaling (Nojima et al., 2008), IQGAP1 appears to regulate cell proliferation through a novel CDC42-mTOR pathway. This appears to reflect differential roles for the IQGAP family and a unique mechanism by which IQGAP1 couples cell growth or size to cell cycle progression. Dysfunction of this role may predispose to the observed IQGAP1-associated cancers, which can be explained based on two scenarios.
First, IQGAP1 accelerates cell cycle progression producing transformed phenotypes (Figs 2, 3, 4, 5), which requires binding to CDC42 and phosphorylation of IQGAP1 on S1443 (Fig. 4). Thus, mutations sustaining IQGAP1 phosphorylation and its binding to CDC42, deregulate cell proliferation, favoring cell transformation. The finding that PKCε, the activating kinase of IQGAP1 (Grohmanova et al., 2004), controls cytokinesis (Saurin et al., 2008), further supports this view. Perhaps phosphorylation sustains IQGAP1 association with protein partners that promote cell division or alters its subcellular distribution, as observed in certain cancers (Takemoto et al., 2001), thus sequestering IQGAP1 from its regulators. Nevertheless, persistent cell division without a concerted increase in cell growth produced a smaller cell size (Fig. 6B). Hence, for sustained proliferation, the role of IQGAP1 in cell growth must also be activated, explaining the requirement for endogenous IQGAP1 for transformation (Fig. 3D). The capacity of IQGAP1 to coordinately promote cell growth and division can be explained based on its ability to act as a dynamic conformation switch regulated by phosphorylation and protein-protein interactions (Fig. 8).
Fig. 8.

A model for the role of IQGAP1 in coordinating cell growth and division. IQGAP1 acts as conformational switch regulated by phosphorylation and protein-protein interaction and exists in an auto-inhibitory state in quiescent cells (Le Clainche et al., 2007). An intermediate form generated by binding of exocyst, CDC42 and N-WASP promotes migration in response to motility signals. During growth cycles, in response to nutrients, IQGAP1 operates in a closed form generated by folding of the C-terminus (Rittmeyer et al., 2008) and interaction with mTOR (and the exocyst) to promote cell growth. At a specific cell size, mitogenic signals lead to phosphorylation of IQGAP1, activating CDC42 and promoting cell division. Dynamic exchange between phosphorylated and dephosphorylated IQGAP1 coordinates growth and division to regulate cell proliferation.
Second, a plausible mechanism, not observed in cell culture, involves IQGAP1 deactivation in cytokinesis (Fig. 1). Failure of cytokinesis results in a polyploid genome, which is known to constitute a transient initiating step in cancer lesions associated with genome instability (Caldwell et al., 2007). This in turn leads to aneuploidy; a hallmark feature of cancers associated with dysfunction of the key partners of IQGAP1, such as CLIP170 and APC mutations that block initiation of cytokinetic furrow in Min mice (Caldwell et al., 2007). Similarly, mutations of IQGAP1 that arrest cytokinesis in vivo might predispose to cancer pathogenesis, which requires analyses in live animals. Thus, activating or deactivating mutants of IQGAP1 would induce cellular transformation.
The function of IQGAP1 in the mTOR pathway probably represents its primary mechanism in controlling cell proliferation. Our data suggest a model whereby IQGAP1 regulates the coupling of cell growth through an IQGAP1-mTOR growth module to cell cycle progression through an IQGAP1-CDC42 cell division complex (Fig. 8). IQGAP1 interfaces and requires mTOR for proliferation (Fig. 6), shares cellular locations (Drenan et al., 2004; Rittmeyer et al., 2008) and key functions previously attributed to mTOR, such as regulating cell proliferation (Figs 2, 3, 4, 5), protein synthesis (Rittmeyer et al., 2008) and cell size (Fig. 6B). Furthermore, both IQGAP1 and mTOR were implicated in regulating cyclin D1, which controls passage through the critical G1-S transition of the cell cycle (Ewen and Lamb, 2004). Effect of mTOR on cell proliferation was attributed primarily to activating cyclin D1 expression (Nelson et al., 2003). Similarly, IQGAP1 is implicated in regulating cyclin D1 through APC, a part of the Wnt-signaling inhibitory complex that regulates β-catenin, which might explain the enhancement of DNA synthesis by IQGAP1 expression (Fig. 2). Currently, the mTOR pathway is a target for cancer and angiogenesis therapies (Sabatini, 2006), and our findings add IQGAP1 as potential target.
IQGAP1 modulates migration
Cell migration is an essential feature of invasive cancer cells in which IQGAP1 has been implicated. Thus the finding that expression of IQGAP1-N, which binds the exocyst and mTOR, inhibited migration and wound healing was intriguing. Perhaps the primary role of IQGAP1-N is to promote cell growth (Fig. 6B), whereas migration requires dynamic switching of IQGAP1 between CDC42-bound and exocyst- and/or mTOR-bound states where expression of IQGAP1-N prevents this dynamic by binding to IQGAP1 C-terminus (Le Clainche et al., 2007) (Fig. 8), thus inhibiting migration. This would mimic a cellular mechanism where IQGAP1 regulates migration by acting as a conformational switch (Fig. 8).
Enhancement of migration by expression of IQGAP1-C, which inhibits exocytosis, can arguably be explained based on differential interaction with exocyst subunits (Sakurai-Yageta et al., 2008) or on activation of N-WASP (Le Clainche et al., 2007; Bensenor et al., 2007). However, the latter appears more plausible, because (1) association of IQGAP1-C with the exocyst could be observed only with overexpressed proteins, and (2) a construct lacking the presumed exocyst-binding region at the C-terminus of IQGAP1 can still co-immunoprecipitate with the exocyst (Sakurai-Yageta et al., 2008), supporting our finding that IQGAP1 associates with the exocyst thorough its N-terminus (Rittmeyer et al., 2008). Furthermore, our data indicate that efficient migration requires both domains (Fig. 7), where perhaps IQGAP1 might assume an intermediary conformation, permitting interactions with both N-WASP (Le Clainche et al., 2007) and the exocyst (Fig. 8). Thus, our findings argue that IQGAP1 has a regulatory role in cell migration.
Materials and Methods
Plasmid construction, mutagenesis and stable cell-line selection
HA-IQGAP1 constructs were generated by high-fidelity PCR, cloned into the pJ4H vector and verified both by sequencing and production of the correct size proteins. Full-length and the C-terminus constructs were cloned into the SmaI site, producing plasmids pJ4H-IQGAP1-F and pJ4H-IQGAP1-C, respectively. The N-terminus was cloned into the SmaI-ClaI sites producing pJ4H-IQGAP1-N. Site-directed mutagenesis of pJ4H-IQGAP1-C using a QuikChange kit (Stratagene) produced phosphodefective S1443A (denoted 1) and phosphomimetic S1443E (denoted E) mutations. Primer sets: S1443A-F, 5′-CTGACAAGATGAAAAAGTCAAAAGCTGTAAAGGAAGACAGCAACC-3′ and S1443A-R, 5′-GGTTGCCTTCCTTTACAGCTTTTGACTTTTTCATCTTGTCAG-3′; S1443E-F, 5′-CCTGACAAGATGAAAAAGTCAAAAGAGGTAAAGGAAGACAGCAACC-3′ and S1443E-R, 5′-GGTTGCTGTCTTCCTTTACCTCTTTTGACTTTTTCATCTTGTCAGG-3′ were used. The pJ4H-IQGAP1-C-ΔMK24 (denoted 9) containing a deletion of amino acids M1054-K1077 of IQGAP1 required for CDC42 binding was created as previously described (Mataraza et al., 2002; Rittmeyer et al., 2008).
The constructs were stably expressed in NIH3T3 cells used for transformation studies. Briefly, cells growing in 60 mm plates were cotransfected with 0.2 μg pcDNA3-Neo and 3 μg each of the pJ4H-HA-IQGAP1 constructs. After 48 hours, cells were split into two 100 mm plates at 80% and 20% and maintained in DMEM supplemented with 5% calf serum (CS) and 700 μg/ml G418 (Invitrogen). After 10-14 days, G418-resistant colonies were subcultured in the same medium and tested for protein expression by immunoblotting. Cells that incorporated the vector (neomycin-resistant cells) were used as a control. HeLa cells stably expressing V5-IQGAP1 constructs on pCDNA3-TOPO were generated following the Invitrogen instruction manual. In both cell types, we were unable to obtain clones with high expression levels and all positive clones expressed IQGAP constructs below endogenous level. To avoid clonal differences, four clones with equal expression from each construct were compared.
Cell culture, transfection and synchronization
NIH3T3 cells were cultured in Dulbecco's modified Eagle medium (DMEM) plus 10% CS and 100 U/ml penicillin, 100 μg/ml streptomycin (Invitrogen) and incubated in a humidified 5% CO2, 95% air incubator at 37°C. HeLa cells (CCL-2, ATCC) were cultured in MEM plus 10% FBS under same conditions.
Fluorescence microscopy
Cells were cultured on dual-chamber slides (Nalge, Nunc) or coverslips, washed with PBS, fixed in –20°C methanol-acetone for 10 minutes, blocked with 1 mg/ml BSA in PBS, incubated with primary or control antibodies followed by secondary (Texas Red and Alexa Fluor 488, Molecular Probes) for 1 hour each at room temperature and the nuclei were stained with DAPI (Sigma). Microtubules were stained with FITC α-tubulin antibody (Sigma). IQGAP1 was detected with a polyclonal (Santa Cruz), HA (Covance) or V5 (Invitrogen) antibodies. Cells were examined with a Leica confocal microscope or an Olympus fluorescence microscope fitted with a Hamamatsu ORCAER monochrome CCD camera and the images were processed in Adobe Photoshop.
Immunoprecipitation and immunoblotting
Immunoprecipitation, SDS-PAGE and immunoblotting were performed following standard procedures. Briefly, cells were washed in ice-cold phosphate-buffered saline (PBS), scraped into lysis buffer (25 mM HEPES, pH 7.4, 15 mM MgCl2, 150 mM NaCl, 1% NP40, 10 μg/ml each of leupeptin and aprotinin and 0.2 mg/ml phenymethylsulfonic chloride) and centrifuged at 13,000 × g for 30 minutes. For all experiments, protein concentrations were determined (BCA kit, Pierce) and equal amount precleared with 15 μl PBS-equilibrated protein G or A beads for 1 hour at 4°C. Antibody and 40 μl beads were added, incubated with rotation for 3 hours at 4°C. The beads were washed four times with 1 ml lysis buffer, boiled for 10 minutes in 40 μl 2× SDS sample buffer, resolved on SDS-PAGE and immunoblotted. For immunoprecipitation with mTOR, detergent was replaced by 3% CHAPS buffer (Cell Signaling Technology) to preserve the TOR complex integrity (Sarbassov et al., 2005). Antibodies for mTOR and phosphoserine (pSer) PKC-substrate (#2261) were from Cell Signaling Technology and were used in combination with Signal Enhancer Hikari (Nacalai).
Cell-cycle progression assay
The DNA synthesis rate was measured with BrdU incorporation by immunofluorescence. 1×104 cells were seeded on dual-chamber slides, at 60% confluency 200 μM BrdU were added and grown for ∼16 hours. Cells were rinsed in PBS, fixed with 3.7% formaldehyde for 15 minutes and permeabilized with 0.5% Triton X-100. After rinsing with PBS, DNA was digested with 0.5 U/μl DNase 1 (Sigma) in H2O for 30 minutes at 37°C, rinsed and blocked with 2% BSA in PBS for 1 hour. Cells were incubated with BrdU (1:200) and HA (1:1000) antibodies followed by Texas red (1:200) or FITC (1:200) labelled secondary antibodies for 1 hour at room temperature. Nuclei were stained with DAPI for 5 minutes, rinsed in PBS and water and visualized.
Transformation assays
Three independent classic transformation assays were used to compare stably transformed cells with neomycin-resistant control cells. (1) Low-serum assay: to measure serum-independent growth, the cells were seeded at 10×104 in 12-well plates in DMEM plus 10% CS and incubated for 5 hours to allow the cells to attach. Thereafter, the medium was replaced with DMEM containing 1% CS and changed every other day for 6 days. At the indicated time-points (2, 4 and 6 days) the cells were washed extensively with PBS, trypsinized and counted with a haemocytometer. (2) Saturation density: to measure proliferation rate in high serum, the cells were processed as above except that the medium contained 10% CS and the cells were counted once on day 6 as above. (3) Soft agar assay: to measure anchorage-independent growth, 104 cells were mixed with DMEM containing 10% CS and 0.3% agarose and plated on top of a solidified layer of DMEM plus 0.5% agarose and 10% CS. Cells were fed weekly with 1 ml DMEM plus 10% CS and 0.3% agarose. At ∼2 weeks, colonies ≥50 μm were scored as a percentage of cells or colonies under a bright field microscope. Each of the experiment was done at least three times from at least three independent clones.
Migration assays
Modified Boyden assay: following Brown and Bicknel (Brown and Bicknel, 2001), millicell chambers (12 mm diameter, 8 μm pores, Millipore) were precoated on the membrane underside with 10 μg/ml fibronectin (Sigma) at room-temperature. Cells were serum-starved overnight and collected by limited trypsinization followed by addition of soybean trypsin inhibitor (Sigma). 10×104 cells were added to the upper compartment containing 300 μl migration medium (DMEM with 0.5% BSA) and the chambers placed into 24-well dishes containing 10 μg/ml fibronectin. After 4 hours at 37°C, cells remaining in the upper chamber were removed by cotton swabs. Migrated cells on the lower membrane surface were fixed with methanol and stained with Giemsa (1:20 in H2O). Migration values were determined as means ± s.e.m. from three independent experiments by counting five high-power (×40) random fields/chamber. Wound-healing assay: 1×104 cells were seeded in dual-chamber slides and grown to confluency. Cell monolayers were scratched down the middle with a sterile 200 μl pipette tip, washed with warm medium and photographed at 0 hour, then at 12 hours and 24 hours after wounding. The migrated distances were quantified as a percentage of time 0 from three independent experiments.
RNA interference
Human IQGAP1 siRNA, scrambled and siCONTROL oligomers from Dharmacon directed against IQGAP1-N that do not affect IQGAP1-C or other IQGAP family members, were previously described (Rittmeyer et al., 2008). A set of four shRNA plasmids against CDC42 and controls were transfected following manufacturer's protocol (OriGene).
FACS analysis
To determine cell size, exponentially growing cells at ∼80% confluency, were trypsinized and fixed with 70% methanol for 30 minutes at –20°C. Cells were washed with FACS buffer (0.1% Triton X-100, 2 mM MgCl2, 0.1% NaCl, 10 mM HEPES, pH 6.8), suspended in the same buffer containing 1 μg/ml DAPI (Sigma), incubated on ice for 30 minutes. The DNA content to determine cell cycle stage as well as the cell size of equal number of cells from each cell line were measured with a flow cytometer (BD Biosciences LSRII) and analyzed with BD FACSDiVA software.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/12/2024/DC1
This work was supported by grants to M.A.O. from the National Institutes of Health-NCI and the American Cancer Society. We thank Richard Cerione for the CDC42F28L stable cells, Dina Alameida, Weill Cornell Medical College and Samuel Kim for technical help, David Wilson and Bill Brown for comments on a previous draft. Deposited in PMC for release after 12 months.
References
- Balenci, L., Clarke, I. D., Dirks, P. B., Assard, N., Ducray, F., Jouvet, A., Belin, M. F. and Honnor, J. (2006). IQGAP1 protein specifies amplifying cancer cells in glioblastoma multiforme. Cancer Res. 66, 9074-9082. [DOI] [PubMed] [Google Scholar]
- Bean, J. M., Siggia, E. D. and Cross, F. R. (2006). Coherence and timing of cell cycle start examined at single cell resolution. Mol. Cell 21, 3-4. [DOI] [PubMed] [Google Scholar]
- Bensenor, L., Kan, H. M., Wang, N., Wallrabe, H., Davidson, L. A., Cai, Y., Schafer, D. A. and Bloom, S. G. (2007). IQGAP1 regulates cell motility by linking growth factor signaling to actin assembly. J. Cell Sci. 120, 658-669. [DOI] [PubMed] [Google Scholar]
- Brown, D. and Sacks, D. B. (2006). IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol. 16, 242-249. [DOI] [PubMed] [Google Scholar]
- Brown, N. and Bicknell, S. R. (2001). Cell migration and the boyden chamber. In Metastasis Research Protocols, Vol. 2: Analysis of Cell Behavior In Vitro and In Vivo (ed. S. A. Brooks and U. Schumacher), pp. 47-54. Totowa, NJ: Humana Press.
- Caldwell, C. M., Green, R. A. and Kaplan, K. B. (2007). APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J. Cell Biol. 178, 1109-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerione, R. A. (2004). Cdc42: new roads to travel. Trends Cell Biol. 14, 127-132. [DOI] [PubMed] [Google Scholar]
- Clark, E. A., Golub, T. R., Lander, E. S. and Hynes, R. O. (2000). Genomic analysis of metastasis reveals an essential role for RhoC. Nature 406, 532-535. [DOI] [PubMed] [Google Scholar]
- Cross, F. R. (1995). Starting the cell cycle: what's the point? Curr. Opin. Cell Biol. 7, 790-797. [DOI] [PubMed] [Google Scholar]
- Drenan, R. M., Liu, X., Bertram, P. G. and Zheng, X. F. S. (2004). FRAP/mTOR localization in the ER and the Golgi apparatus. J. Biol. Chem. 279, 772-778. [DOI] [PubMed] [Google Scholar]
- Ewen, M. E. and Lamb, J. (2004). The activities of cyclin D1 that drive tumorigenesis. Trends Mol. Med. 10, 158-162. [DOI] [PubMed] [Google Scholar]
- Fingar, D. C. and Blenis, J. (2004). Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23, 3151-3171. [DOI] [PubMed] [Google Scholar]
- Fukata, M., Kuroda, S., Nakagawa, M., Kawajiri, A., Itoh, N., Shoji, I., Matsuura, Y., Yonehara, S., Kikuchi, A. and Kaibuchi, K. (1999). Cdc42 and Racl regulate the interaction of IQGAP1 with β-catenin. J. Biol. Chem. 274, 26044-26050. [DOI] [PubMed] [Google Scholar]
- Fukata, M., Nakagawa, M., Itoh, N., Kawajiri, A., Yamaga, I. M., Kuroda, S. and Kaibuchi, K. (2001). Involvement of IQGAP1, an effector of Rac1 and Cdc42 GTPase, in cell-cell dissociation during cell scattering. Mol. Cell. Biol. 21, 2165-2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata, M., Watanabe, T., Noritake, J., Nakagawa, M., Yamaga, M., Kuroda, S., Matsuura, Y., Iwamatsu, A., Perez, F. and Kaibuchi, K. (2002). Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 109, 873-885. [DOI] [PubMed] [Google Scholar]
- Grindstaff, K., Yeaman, K. C., Anandasabapathy, N., Hsu, S. C., Rodriguez-Boulan, E., Scheller, R. H. and Wilson, J. W. (1998). Sec6/8 complex is recruited to cell-cell contacts and specifies transport vesicle delivery to the basal-lateral membrane in epithelial cells. Cell 93, 731-740. [DOI] [PubMed] [Google Scholar]
- Grohmanova, K., Schlaepfer, D., Hess, D., Gutierrez, P., Beck, M. and Kroschewski, R. (2004). Phosphorylation of IQGAP1 modulates its binding to Cdc42, revealing a new type of Rho-GTPase regulator. J. Biol. Chem. 279, 48495-48504. [DOI] [PubMed] [Google Scholar]
- Harrington, L. S., Findlay, G. M. and Lamb, R. F. (2005). Restraining PI3K: mTOR signaling goes back to the membrane. Trends Biochem. Sci. 30, 35-42. [DOI] [PubMed] [Google Scholar]
- Jadeski, L., Mataraza, J. M., Jeong, H. W., Li, Z. and Sacks, D. B. (2007). IQGAP1 stimulates proliferation and enhances tumorigenesis of human breast epithelial Cells. J. Biol. Chem. 283, 1008-1017. [DOI] [PubMed] [Google Scholar]
- Kanda, M., Akira, N. and Uyeda, T. Q. P. (2005). Adhesion-dependent and contractile ring-independent equatorial furrowing during cytokinesis in mammalian cells. Mol. Biol. Cell 16, 3865-3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer, G., Schmoranzer, J., Low, S. H., Li, X., Gan, Y., Weimbs, T., Simon, S. M. and Rodriguez-Boulan, E. (2003). Three-dimensional analysis of post-Golgi carrier exocytosis in epithelial cells. Nat. Cell Biol. 5, 126-136. [DOI] [PubMed] [Google Scholar]
- Le Clainche, C., Schlaepfer, D., Ferrari, A., Klingauf, M., Grohmanova, K., Veligodskiy, A., Dirdy, D., Le, D., Egile, C., Carlier, M. F. et al. (2007). IQGAP1 stimulates actin assembly through the N-Wasp-Arp2/3 pathway. J. Biol. Chem. 282, 426-435. [DOI] [PubMed] [Google Scholar]
- Li, S., Wang, Q., Chakladar, A., Bronson, R. T. and Bernards, A. (2000). Gastric hyperplasia in mice lacking the putative Cdc42 effector IQGAP1. Mol. Biol. Cell 20, 697-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., McNulty, D. E., Marler, K. J., Lim, L., Hall, C., Annan, R. S. and Sacks, D. B. (2005). IQGAP1 promotes neurite outgrowth in a phosphorylation-dependent manner. J. Biol. Chem. 280, 13871-13878. [DOI] [PubMed] [Google Scholar]
- Lin, R., Bagrodia, S., Cerione, R. A. and Manor, D. (1997). A novel Cdc42Hs mutant induces cellular transformation. Curr. Biol. 7, 794-797. [DOI] [PubMed] [Google Scholar]
- Mataraza, J. M., Li, Z. and Sacks, D. B. (2002). IQGAP1 is a component of Cdc42 signaling to the cytoskeleton. J. Biol. Chem. 277, 24753-24763. [DOI] [PubMed] [Google Scholar]
- Mateer, S. C., Wang, N. and Bloom, G. S. (2003). IQGAPs: integrators of the cytoskeleton, cell adhesion machinery, and signaling networks. Cell Motil. Cytoskeleton 55, 147-155. [DOI] [PubMed] [Google Scholar]
- Nabeshima, K., Shimao, Y., Inoue, T. and Koono, M. (2002). Immunohistochemical analysis of IQGAP1 expression in human colorectal carcinomas: its overexpression in carcinomas and association with invasion fronts. Cancer Lett. 176, 101-109. [DOI] [PubMed] [Google Scholar]
- Nelson, C. J., Rickheim, D. G., Tucker, M. M., Hansen, L. K. and Albrecht, J. H. (2003). Evidence that cyclin D1 mediates both growth and proliferation downstream of TOR in hepatocytes. J. Biol. Chem. 278, 3656-3663. [DOI] [PubMed] [Google Scholar]
- Niinaka, Y., Haga, A. and Raz, V. (2001). Quantification of cell motility. In Metastasis Research Protocols, Vol. 2: Analysis of Cell Behavior In Vitro and In Vivo (ed. S. A. Brooks and U. Schumacher), pp. 47-54. Totowa, NJ: Humana Press.
- Nojima, H., Adachi, M., Matsui, T., Okawa, K., Shoichiro Tsukita, S. and Tsukita, S. (2008). IQGAP3 regulates cell proliferation through the Ras/ERK signalling cascade. Nat. Cell Biol. 10, 971-978. [DOI] [PubMed] [Google Scholar]
- Osman, M. A. and Cerione, R. A. (1998). Iqg1p, a yeast homologue of the mammalian IQGAPs, mediates Cdc42p effects on the actin cytoskeleton. J. Cell Biol. 142, 443-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman, M. A. and Cerione, R. A. (2005). Actin doesn't do the locomotion: secretion drives cell polarization. In Protein Trafficking: Mechanisms and Regulation (ed. N. Segev). Georgetown, TX: Landes Bioscience.
- Osman, M. A., Konopka, J. B. and Cerione, R. A. (2002). Iqg1p links spatial and secretion landmarks to polarity and cytokinesis. J. Cell Biol. 159, 601-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittmeyer, E. N., Daniel, S., Hsu, S. C. and Osman, M. A. (2008). A dual role for IQGAP1 in regulating exocytosis. J. Cell Sci. 121, 391-408. [DOI] [PubMed] [Google Scholar]
- Roy, M., Li, Z. and Sacks, D. B. (2005). IQGAP1 is a scaffold for mitogen-activated protein kinase signaling. Mol. Cell. Biol. 25, 7940-7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini, D. (2006). mTOR and cancer: insights into a complex relationship. Nat. Rev. Cancer 6, 729-734. [DOI] [PubMed] [Google Scholar]
- Sakurai-Yageta, M., Recchi, C., Le Dez, G., Sibarita, J. B., Daviet, L., Camonis, C., D'Souza-Schorey, C. and Chavrier, P. (2008). The interaction of IQGAP1 with the exocyst complex is required for tumor cell invasion downstream of Cdc42 and RhoA. J. Cell Biol. 181, 986-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov, D. D., Guertin, D. A., Siraj, M. A. and Sabatini, D. M. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098-1101. [DOI] [PubMed] [Google Scholar]
- Saurin, A. T., Durgan, J., Cameron, A. J., Faisal, A., Marber, M. S. and Parker, P. J. (2008). The regulated assembly of a PKC epsilon complex controls the completion of cytokinesis. Nat. Cell Biol. 10, 891-901. [DOI] [PubMed] [Google Scholar]
- Takemoto, H., Yuchiro, D., Shiozaki, H., Imamura, H., Utsunomya, T., Miyata, H., Yano, M., Inoue, M., Fujiwara, Y. and Monden, M. (2001). Localization of IQGAP1 is inversely correlated with intercellular adhesion mediated by E-cadherin in gastric cancers. Int. J. Cancer 91, 783-788. [DOI] [PubMed] [Google Scholar]
- Tapon, N., Moberg, K. H. and Hariharan, I. (2001). The coupling of cell growth to the cell cycle. Curr. Opin. Cell Biol. 13, 731-737. [DOI] [PubMed] [Google Scholar]
- Wullschleger, S., Loewith, R. and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell 124, 471-484. [DOI] [PubMed] [Google Scholar]
- Yang, J. Y., Zong, C. S., Xia, W., Yamaguchi, H., Ding, Q., Xie, X., Lang, J. Y., Lai, C. C., Chang, C. J., Huang, W. C. et al. (2008). ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 10, 138-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, R., Guo, Z., Watson, C., Chen, E., Kong, R., Wang, W. and Yao, X. (2003). Polarized distribution of IQGAP proteins in gastric parietal cells and their roles in regulated epithelial cell secretion. Mol. Biol. Cell 14, 1097-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}