

Abstract
This review describes the use of low-energy collisionally activated dissociation (CAD) with both tandem quadrupole and ion-trap mass spectrometry toward to structural characterization of glycerophospholipids (GPLs), including classes of glycerophosphocholine, glycerophosphoethanolamine, glycerophosphoserine, glycerophosphoglycerol glycerophosphoinositol and glycerophosphatidic acid, as well as their lyso-, plasmanyl-, and plasmenylphospholipid subclasses.. The mechanisms underlying the fragmentation processes leading to structural characterization of GPLs in various ion forms desorbed by electrospray ionization in the positive-ion and negative-ion modes are also discussed. The tandem mass spectrometric approaches afford the identification of the polar head group, the fatty acid substituents and the location of the radyl groups on the glycerol backbone of all the GPLs.
Keywords: glycerophospholipids, tandem quadrupole mass spectrometry, ion-trap mass spectrometry, electrospray ionization, charge-remote fragmentation, charge-driven fragmentation
1. Introduction
The recent development of electrospray ionization (ESI) [1,2] and matrix-assisted laser desorption/ionization (MALDI) [3,4] mass spectrometry (MS) permits direct analysis of intact biological substances. ESI/MS analyses of phospholipids are 2 to 3 orders of magnitude more sensitive than that achieved by FAB/MS [5–8], one traditional soft ionization method that is suitable for analysis of biomolecules. The superb sensitivity and ease of continuous sample introduction with ESI, coupled with tandem mass spectrometry provide opportunities to explore the structure and fragmentation process of complex lipids in greater detail [8–10]. The ions generated by ESI have also minimized some of the problems, such as the complication of spectra with matrix ions associated with FAB/MS.
In contrast, structural and mechanistic studies on lipids using MALDI/MS were limited by the fact that the ionization technique for lipid analysis is generally hampered by abundance of the matrix-derived noise in the mass range of interest, by lack of reproducibility, by complication stemmed from various adduct ion formation and from dissociation of the molecular ion species during ionization, and by lack of universal matrices for sensitive detection. However, the research on lipids with this ionization method appears to be rising [11], and new matrices have been introduced to improve the sensitivity [12].
In this review, we will describe the current state of knowledge about the mechanisms involved in the formation of various product ions following collisional activation of molecular ion species generated by ESI in the positive-ion and/or negative-ion modes of common glycerophospholipids (GPLs), including glycerophosphocholine (GPCho), glycerophosphoethanolamine (GPEtn), glycerophosphoserine (GPSer), glycerophosphatidic acid (GPA), glycerophosphoinositol (GPIno), and glycerophosphoglycerol (GPGro). Phospholipid classes that undergo similar fragmentation processes will be grouped together, starting with positive-ion in the order of [M + H]+, [M + Alk]+, and [M − H + 2Alk]+ (Alk = Na, Li) if applicable, and followed by negative-ion proceeding with [M −H]−, and [M − 2H + Alk]− if applicable.
2. Techniques and Methods
2.1. Tandem mass spectrometry
The ESI source is compatible with nearly all tandem mass spectrometers, including tandem sector, tandem quadrupole, ion-trap (IT), ion-cyclotron resonance (ICR), TOF/TOF, and hybrid instruments such as the quadrupole-time-of-flight (Q-oTOF) and QIT-TOF. However, most structural characterizations and studies on the fragmentation processes of complex lipids have been achieved mainly with triple stage quadrupole (TSQ) mass spectrometer [5–10], because of its early development with ESI, its high efficiency in collection of fragment ions in the RF-only collision cell, its ease of operation, and its relatively low cost. Recently, studies on lipids also have been carried out by IT [13–17], and more complicated instruments such as Q-oTOF [18], and FTICR [19] coupled to ESI.
Structural characterization achieved by magnetic sector instruments, which involve high-energy collisionally activated dissociation (CAD) using keV translational energy [20, 21], may yield additional structural information [22], but the sensitivity is poor, as compared to that obtained by TSQ, which is usually operated with collision energies below 100 eV [23]. In addition, with the relative ease in applying skimmer CAD, MS/MS/MS experiment can be carried out by TSQ to reveal the fragmentation processes in greater detail. In this respect, ITMS is particularly attractive, because MSn spectra (n = 2,3, --) can be obtained in series by time [24,25]. However, its sensitivity has been limited by the number of the ions that can be trapped, and the low-mass cut-off nature of the instrument [25] also restrains its application in the structural characterization that requires information of low-mass ions. With the recent development in ion-trap techniques, such as linear ion-trap (LIT) that permits signifcantly more ions to be trapped, it is anticipated that studies on complex lipids and structural characterization with IT mass spectrometry will increase.
Fragment ions generated by high-energy CAD with sector and by low-energy CAD with TSQ or ITMS instruments are similar. The gas-phase ion chemistry and the mechanisms underlying the fragmentation processes are generally interchangeable among the various methods applied, although differences may exist. The mechanisms of fragmentation of lipids described in this article are based primarily on the studies in the author’s laboratory, where the experiments were carried out mainly with a Finnigan TSQ-7000 triple quadrupole mass spectrometer operated at unit resolution, and a Finnigan LCQ-DECA ITMS, with which MSn product-ion spectra (n = 2, 3) were obtained. Samples were infused continuously at a flow rate of 1 to 3μl/min into the mass spectrometers. Tandem mass spectra are acquired in profile mode at 3 s/scan, and accumulated for a period of time that was determined by the quality of the spectra, typically 3–10 min.
2.2. Nomenclature
The designations and abbreviations that are recommended by IUPAC (http://www.chem.qmul.ac.uk/iupac/lipid/) are used. Examples for glycerophospholipid nomenclatures are shown below: diacyl-, 1-O-alkyl-2-acyl- (plasmanyl-), and 1-O-alk-1′-enyl-2-acyl (plasmalogen) phospholipids, e.g., the dipalmitoyl-, 1-O-hexadecyl-2-palmitoyl-, and 1-O-hexadec-1′-enyl-2-palmitoyl-sn-glycero-3-phosphocholines are designated as 16:0/16:0-PC, a16:0/16:0-PC, and p16:0/16:0-PC, respectively. The product-ion spectra obtained from the MSn (n = 2,3) experiments were designated as the MSn-spectra (n = 2,3).
3. Molecular species formed by electrospray ionization
Nearly all phospholipids are readily ionizable, when subjected to ESI. However, the formation of the molecular species is governed by the polar head groups that distinguish the various classes. Early studies with ESI analyses of the phospholipid platelet activating factor (PAF) demonstrated that choline glycerophospholipids could be successfully analyzed as protonated ([M + H]+) and alkali metal ([M + Alk]+) adducts in the positive-ion mode or as chloride adducts ([M + Cl]−) in the negative-ion mode [5]. Adduct ions of GPCho in the forms of [M + HCO2]−, [M + CH3CO2]−, [M −15]− [26,27], [M + Alk]+ (Alk = Li, Na, K) (6,26,28) and of [M + CF3CO2H + K]+ [14], are later reported. However, the sensitivity of GPCho in positive ion mode is 102-order of magnitude greater than that in the negative ion mode, attributable to the fact that GPCho contains a quaternary nitrogen with a fixed positive charge and more readily forms positive than negative ions by ESI.
In the negative-ion mode, GPEtn, GPSer, GPGro, GPIno, and GPA all yield prominent [M − H]− ions. GPSer also forms [M − 2H + Alk]− adduct ion in the presence of Alk+[29]. In the positive-ion mode, GPEtn, GPSer, GPGro, GPIno, and GPA all form [M + H]+ ions or alkali metal adduct ions in the form of [M + Alk]+, [M + 2Alk − H]+ or [M + 3Alk − 2H]+, dependent on the concentration of alkali metal or H+ ion present in the samples. The sensitivity in the positive-ion mode, in general, is at least 1-order of magnitude less than that observed in the negative-ion mode.
Ions in the forms of [M − 2H + Na + Co]+ and [M − 2H + Na + Ni]+ were also reported for phospholipid classes of GPEtn, GPGro, and of GPSer [30], but the sensitivity is not expected to be as great as that observed for [M −H]− ions. Other phospholipid species, such as cardiolipin, and phosphatidylinositol polyphosphates, which bear multiple phosphoric acid charge sites, form [M − H]−, [M + Na − 2H]− and [M − 2H]2− ions in the negative-ion mode [31–33]. The formation of these ion species is strictly affected by the concentration of Na+ and the pH of the solution, with which the ESI events take place. Dimeric molecular species are often formed, if high concentration of GPL solution is ionized.
Structural characterization of GPLs relies on the rich fragment ions generated by CAD of the precursor ions, and the molecular ions formed by ESI under different conditions for various classes of GPLs vary greatly. The mechanisms described in this review are written in the premise that the molecular species subjected to CAD tandem mass spectrometry for structural characterization are readily formed by ESI, in the positive-ion and/or negative-ion mode(s). Mechanistic studies and structural characterization of the molecular species, such as the [M − 2H + Na + Ni]+ and [M + CF3CO2H + K]+ adduct ions will not be described here.
4. Structural characterization by tandem mass spectrometry and fragmentation processes
4.1. Fragmentation processes of the [M + H]+ ions
4.1.1. Glycerophosphocholine (GPCho)
The protonated molecular species ([M + H]+) are readily formed by GPCho, which possess a positive charge site on the quaternary nitrogen. The product-ion spectra arising from the [M + H]+ ions are often simple and therefore, are less applicable although achievable, for structural characterization [6,8,28,34]. For example, the product-ion spectrum of the [M + H]+ ions of 16:0/18:1-PC at m/z 760.6 (Fig. 1A) is dominated by the m/z 184 ion, representing a phosphocholine ion, and ions regarding to the structural information are of low abundance. Studies on the product-ion spectra of various GPCho including deuterium-labeled analogs indicate formation of the m/z 184 ion involves the participation of mainly the α-hydrogen of the fatty acyl at sn-2 (Scheme 1) [34]. The studies also revealed that the α-hydrogen of the fatty acyl at sn-2 is more labile than that at sn-1, resulting in the more favorable formation of the [M + H – R′ 2CH=CO]+ ion than the [M + H - R1CH=CO]+ ion, arising from losses of the fatty acyl substituents at sn-2 and at sn-1 as ketenes, respectively. Therefore, the position of the fatty acyl moieties on the glycerol backbone can be assigned.
Fig. 1.

The product-ion spectra of the [M + H]+ ions of 16:0/18:1-PC at m/z 760 (A), 16:0/18:2-PE at m/z 716 (B), 18:0/18:1-PS at m/z 790 (C), 16:0/18:1-PG at m/z 749 (D), and of 16:0/18:1-PA at m/z 675 (E).
Scheme 1.

The mechanism proposed for formation of the phosphocholine ion from GPCho.
4.1.2. Glycerophosphethanolamine (GPEtn)
In contrast, the product-ion spectra of GPEtn are dominated by [M + H – 141]+ (a ion, Scheme 2) arising from elimination of the phosphoethanolamine moiety via the same bond cleavage as that of GPCho as described above. The product-ion spectrum of the [M + H]+ ion of 16:0/18:2-PE at m/z 716 (Fig. 1B) is dominated by the m/z 575 ([M + H – 141]+) ion, which gives rise to the 16:0-, and 18:2-acylium ions at m/z 239 (R1CO+) and 263 (R2CO+), respectively (Scheme 2) [35]. The phosphoethanolamine and [M − 141] moieties arising from cleavage of the CH2-OP compete proton for ion formation. The observation of [M + H – 141]+ rather than a protonated phosphoethanolamine at m/z 142 indicates that the phosphoethanolamine (having a zwitterion ion form) is less competitive for proton. In contrast, the phosphocholine moiety arising from the same cleavage from GPCho is a zwitterion containing a N(CH3)3 group that is more basic and formation of the protonated phosphocholine ion of m/z 184 is a more facile process and the spectrum is dominated by the m/z 184 ion. The fragmentation process leading to the [M + H – 141]+ ion also involves the participation of the α-hydrogen of the fatty acyl substituent, mainly at sn-2 [35].
Scheme 2.

The fragmentation processes and the fragment ions observed for the [M + H]+ ions of the phospholipids other than GPCho.
4.1.3. Glycerophosphoserine (GPSer), GPGro and GPA
The striking similarities in the product-ion spectra of the [M + H]+ ions arising from GPSer and GPEtn indicate that the fragmentation processes and the gas phase basicity of GPSer and GPEtn are similar. The product-ion spectrum of the [M + H]+ ion of 16:0/18:1-GPSer at m/z 762 (Fig. 1C) is dominated by the m/z 577 ([M + H − 185]+) ion, arising from elimination of the phosphoserine moiety via the same pathway as that of GPEtn (Scheme 2), and a protonated phosphoserine ion expected at m/z 186 is not present. The spectrum also contains ions at m/z 339 and 313, arising from further dissociation of m/z 577, by losses of the fatty acyl substituents at sn-1 and sn-2 as ketenes, respectively. The acylium ions are observed at m/z 265 (R2CO+) and 239 (R1CO+).
Similarly, the product-ion spectra of the [M + H]+ ions of 16:0/18:1-PG at m/z 749 (Fig. 1D) and of 16:0/18:1-PA (Fig. 1E) at m/z 675 are also dominated by the m/z 577 ([M + H – (HO)2P(O)OX]+) ions arising from losses of phosphoglycerol and phosphate moieties, respectively (a ion, Scheme 2), consistent with the notion that the gas-phase phosphoglycerol and phosphatic acid are less competitive for proton for formation of the protonated (HO)2P(O)OX ion. The fatty acyl substituents are also reflected by the acylium ions at m/z 265 (18:1) and 239 (16:0), and by the ions at m/z 339 and 313 arising from further dissociation of m/z 577. However, these ions are of low abundance and to assign the position of fatty acyl substituents on the glycerol backbone based on the differences in the abundances of these ion pairs is not reliable. In contrast, the R2CO+ ion at m/z 265 is substantially more abundant than the R1CO+ ion at m/z 239 in the MS3-spectrum of the m/z 577 ion (749 → 577), for example, arising from 16:0/18:1-PS. Thus, the position of the fatty acid substituents on the glycerol backbone can be determined [29].
The product-ion spectra arising from the [M + H]+ ions as described above are simple and contain only a few prominent ions that are unique to the individual lipid classes. Therefore, precursor-ion scan of m/z 184 has been used to achieve simultaneous qualification and semi-quantification of the GPCho molecular species in biological specimen [36, 37]. Neutral loss scans of 141 and 185 also have been previously used for tandem mass spectrometric analysis of GPEtn and GPSer, respectively [36]. However, the fragment ions leading to structural characterization arising from CAD of the [M + H]+ ions are often of low abundance, and thus are less useful for structural identification. In contrast, fragment ions arising from alkali adduct ions, in particular, the [M + Li]+ ions (see section 4.2.), as well as from the [M − H]− ions (see section 4.3.) are abundant and are suitable for unambiguous structural identification.
4.2. The fragmentation processes of the [M + Li]+ and [M − H + 2Li]+ (if applicable) ions
Formation of Li+ and other metal adduct ions as means of tailoring fragmentation of biomolecules was dated to the 1980s [38–40]. Among the earlier observations in the study of phospholipids by tandem mass spectrometry with ESI are that fragment ions arising from the [M + Li]+ adduct ions of GPCho lipids are abundant, and structural identification and distinction among isomers can be achieved [28]. In contrast, product-ion spectra from the [M + Na]+, [M + K]+, or [M + H]+ ions yield little information about the fatty-acid substituents. Following CAD on their [M + Li]+ adduct ions generated by ESI, structural characterization of GPEtn [35], GPSer [29], GPGro, GPA [41], and lysophosphatidylcholines [42] by tandem mass spectrometry has been sucessfully employed.
GPEtn, GPSer, GPGro, GPIno and GPA also form dilithiated adduct ions ([M + 2Li -H]+) upon ESI in the presence of more concentrated Li+ [17,29,35]. Tandem mass spectra of the [M + 2Li - H]+ ions are also applicable for structural charcterization.
4.2.1. GPCho
a. Diacylphosphatidylcholines
Charge-remote fragmentation is thought to be the major fragmentation process leading to the ion formation for the [M + Li]+ ions of GPCho under low-energy CAD [34]. The α-hydrogen at the fatty acyl chain involves in the elimination of the adjacent fatty acyl substituent as an acid. Because the α-hydrogens of the fatty acyl at sn-2 are more labile than those at sn-1, preferential loss of R1CO2H to yield the [M + Li - R1CO2H]+ ion over the loss of R2CO2H that yields [M + Li - R2CO2H]+ (route a, Scheme 3) was observed [34]. Studies with source CAD tandem mass spectrometry and IT with MS3 of the [M + Li - 59]+ ion demonstrated that loss of trimethylamine ([M + Li - 59]+) is the primary step leading to further fragmentations [34]. The preferential loss of R1CO2H using the α-hydrogens of the fatty acyl at sn-2 also gives rise to more prominent [M + Li – 59 - R1CO2H]+ ion than the [M + Li – 59 - R2CO2H]+ ion, following primary loss of the trimethylamine moiety (route b). The disparity in the formation of the above ion pairs results in the characterization of GPCho and positional isomers such as 16:0/18:1-PC (Fig. 2A) and 18:1/16:0-PC (Fig. 2B) can be differentiated [28,34].
Scheme 3.

The proposed mechanisms of fragmentation of lithiated GPCho.
Fig. 2.

The product-ion spectra of the [M + Li]+ ions of 16:0/18:1-PC (A), of 18:1/16:0-PC at m/z 766 (B), and of 16:0/20:4-PC at m/z 788 (C).
The m/z 707 ion ([M + Li - 59]+) (Fig. 2A and Fig. 2B) further dissociates to the [M + Li - 183]+ ion at m/z 583 and the [M + Li - 189]+ ion at m/z 577, via losses of ethylene phosphate and lithium ethylene phosphate residues, respectively (route b′). The m/z 577 ion is more abundant than m/z 583, but the abundances of the analogous ion pairs at m/z 605 ([M + Li - 183]+) and m/z 599 ([M + Li - 189]+) are reversed in the product-ion spectrum of the lithiated 16:0/20:4-PC (Fig. 2C), of which the fatty acyl chain at sn-2 is highly unsaturated.
The m/z 583 ([M + Li - 183]+) ion arising from 16:0/18:1-PC (Fig. 2A) is a lithiated ion and gives rise to m/z 303 by further loss of the 18:1-fatty acyl substituent at sn-2 as an α,β-unsaturated fatty acid (loss as 18:2-fatty acid) (route b”, Scheme 3). This fragmentation also results in the ion at m/z 329 (583 – 254), reflecting loss of the 16:0-fatty acid substituent at sn-2 as an α,β-unsaturated fatty acid (loss as 16:1-fatty acid), in the product-ion spectrum of 18:1/16:0-PC (Fig. 2B). In contrast, ions arising from loss of the fatty acid at sn-1 as an α,β-unsaturated fatty acid were not observed. These unique fragmentation processes are also seen for the [M + Li]+ ion of triacylglycerol [43] and provide additional information to confirm the position of the fatty acid substituents at the glycerol backbone. Similarly, the ion at m/z 577 ([M + Li-189]+) is a protonated ion and gives rise to acylium ions at m/z 239 and 265, as described earlier (Scheme 2).
The product-ion spectrum of 16:0/20:4-PC (Fig. 2C) contains a unique ion at m/z 313, arising from m/z 599 ([M + Li - 189]+) by further loss of the 20:4- fatty acid as a ketene. This fragmentation process is only observed for phospholipid classes consisting of a highly unsaturated fatty acyl moiety at sn-2, including diacyl GPCho and GPEtn.
b. 1-O-alkyl-2-acyl-sn-glycero-3-phosphocholines (plasmanylphosphocholines) and 1-O-alk-1′-enyl-2-acyl-sn-glycero-3-phosphocholines (plasmalogens)
Both the product-ion spectra of the [M + Li]+ ions of 1-O-alkyl-2-acyl-sn-glycero-3-phosphocholines and 1-O-alk-1′-enyl-2-acyl-sn-glycero-3-phosphocholines are dominated by the [M + Li - 59]+ ion; while the [M + Li - 59 - RxCO2H]+ ion (x = 1 or 2) is of low abundance and the [M + Li - RxCO2H]+ ion (x = 1 or 2) is not present. The findings are consistent with the concept that the α-hydrogen at the fatty acyl chain involves in the elimination of the adjacent fatty acyl substituent as an acid. Neither the 1-O-alkyl nor the 1-O-alk-1′-enyl moiety contains the labile α-hydrogens that are required for the R2CO2H loss, and thus, the pathways leading to the formation of the [M + Li - R2CO2H]+ and [M + Li – 59-R2CO2H]+ ions are not operative. This is illustrated by the product-ion spectrum of the [M + Li]+ ion of a16:0/18:1-PC at m/z 752.6 (Fig. 3A), in which the ions expected at m/z 470 and 411 are absent. The ions expected at m/z 510 ([M + Li - R1OH]+) and 451 ([M + Li + 59 -R1OH]+) that arise from loss of the 1-O-alkyl substituent at sn-1 involving the participation of the α-hydrogen at sn-2 are also absent, possibly due to the fact that elimination of the 1-O-alkyl substituent as an alcohol is less facile than the acid loss as seen for diacyl-GPCho. The lack of α-hydrogen at sn-1 also accounts for the rise of the ([M + Li - 59]+) ions, which would have been significantly reduced by the further dissociation process involving the α-hydrogen.
Fig. 3.

The product-ion spectra of the [M + Li]+ ions of a16:0/18:1-PC at m/z 752 (A), p16:0/18:1-PC at m/z 750 (B), lyso1/18:1-PC at m/z 528 (C), and of 18:1/lyso2-PC at m/z 528 (D), a16:0/lyso2-PC at m/z 488 (E), and of p16:0/lyso2-PC at m/z 486 (F).
The product-ion spectrum of the [M + Li]+ ion of p16:0/18:1-PC at m/z 750.6 (Fig. 3B), is similar to that of a16:0/18:1-PC. However, the spectrum contains a prominent ion at m/z 279 (561 - C17H33CO2H), arising from further loss of the 18:1-fatty acyl moiety at sn-2 from the m/z 561 ([M + Li - 189]+) ion, which is less abundant than the analogous ion at m/z 563 seen in Fig. 3B. The decline of the [M + Li - 189]+ ion at m/z 561 caused by its subsequent dissociation is consistent with the prominence of the m/z 279 ion ([M + Li – 189 - R2CO2H]+) from the secondary dissociation process. This spectrum feature is unique to plasmenylcholine and is readily applicable in its distinction from a plasmanyl GPCho [42]. The distinction between the two subclasses can also be seen by the findings that the product-ion spectrum of plasmanyl GPCho also contains a R1+ ion, arising from cleavage of the C-O ether bond of the 1-O-alkyl moiety at sn-1. This alkyl ion gives rise to a series of (CH2)nH+ (where n = 4, 5, --, 13) ions at m/z 57, 71, --, and 183 (Fig. 3B, inset) [42]. In contrast, the ion series is not present in the product-ion spectrum from plasmalogen GPCho (Fig. 3A). Similar results are seen for plasmanylethanolamine (see section 4.2.2.).
c. Lysophosphatidylcholines
The ions reflecting losses of fatty acid substituents (i.e., [M + Li - RxCO2H]+ and [M + Li – 59 - RxCO2H]+ ions) (x = 1 or 2) are not present in the product-ion spectra of the [M + Li]+ ions of 2- or 1-lysophophatidylcholine, because of its lack of the fatty acid substituent at sn-1 or at sn-2. The profile of the product-ion spectrum of lyso1/18:1-PC at m/z 528 (Fig. 3C) is similar to that of a16:0/18:1-PC (Fig. 3A). The abundances of the ions at m/z 345 ([M + Li - 183]+) and 339 ([M + Li - 189]+) are close to the analogous ions seen in the spectrum of a diacyl-PC. However, the [M + Li - 59]+ ion at m/z 469 is prominent, and the ions expected at m/z 246 ([M + Li - R2CO2H]+) and 187 ([M + Li – 59 - R2CO2H]+), arising from the fragmentations involving loss of the fatty acid substituent at sn-2 are absent. Both the lyso1/18:1-PC and a16:0/18:1-PC share a structure of which the fatty acyl substituent at sn-2 possesses a pair of α-hydrogens and the α-hydrogens at sn-1 are not present. Therefore, the resemblance of the two spectra is partially attributed to the fact that the major pathways leading to ion formations are initiated by the labile α-hydrogen of the fatty acyl at sn-2. In contrast, the similar fragmentations observed for 18:1/lyso2-PC are initiated by the α-hydrogens of the fatty acyl at sn-1, which are less labile. Thus, a distinct product-ion spectrum featured by a prominent ion at m/z 469 ([M + Li - 59]+), along with the less prominent ions at m/z 345 ([M + Li - 183]+) and 339 ([M + Li - 189]+) was seen (Fig. 3D). The consecutive dissociation processes differentiated by the origins of α-hydrogen lead to the prominence of m/z 339 and 345 and to the decline of m/z 469 for lyso1/18:1-PC (Fig. 3C), which can be easily distinguished from the 18:1/lyso2-PC isomer that yields a prominent ion at m/z 469, and the less prominent ions at m/z 339 and 345 [42]. The distinction between the product-ion spectra of the [M + Na]+ ions of 1- and of 2-lysophosphatidyl isomers of GPEtn or of GPCho is also substantial and readily applicable for distinction among lysophospholipid isomers [44].
d. Lysoplasmanyl- and lysoplasmenylphosphocholines
The product-ion spectra of the [M + Li]+ ions of a16:0/lyso2-PC at m/z 488 (Fig. 3E) and of p16:0/lyso2-PC at m/z 486.4 (Fig. 3F) are similar to that of 18:1/lyso2-PC (Fig. 3C). However, the [M + Li - 189]+ ions expected at m/z 299 ([488 - 189]+) (Fig. 3E) and m/z 297 ([486 - 189]+) (Fig. 3F), are absent, and the [M + Li - 183]+ ions at m/z 305 ([488 - 183]+) (Fig. 3E), and at m/z 303 ([486 - 183]+) (Fig. 3F) are of low abundance. The lack of α-hydrogen in both the sn-1 and sn-2 moieties, as seen for the two compounds, hampers the dissociation process leading to [M + Li - 189]+ from [M + Li - 59]+, and results in the drastic decline in the production of the [M + Li - 189]+ ions. As a result, the [M + Li - 59]+ ion becomes prominent, consistent with the concept that the α-hydrogens of the fatty acyl substituent participate in the formation of these ions.
4.2.2. Glycerophosphethanolamine (GPEtn)
When ionized in the presence of Li+, GPE forms both monolithiated ([M + Li]+) and dilithiated ([M − H + 2Li]+) adduct ions [35]. The intensities of the [M + Li]+ and the [M −H + 2Li]+ ions are dependent on the concentration of the Li+ ion.
a. The [M + Li]+ ions of diacyl GPEtn
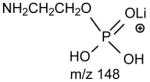
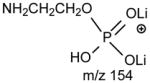
The profiles of the product-ion spectra and the fragmentation processes of the [M + Li]+ ions of GPEtn are similar to those of GPC. As shown in Fig. 4A, the product-ion spectrum of the [M + Li]+ ion of 16:0/18:2-GPE at m/z 722 contains an abundant ion at m/z 679 ([M + Li - 43]+) (a), arising from loss of aziridine to a monolithiated 16:0/18:2-GPA ion (see section 4.2.3.), which further dissociates to m/z 575 ([M + Li - 147]+) (c) and m/z 581 ([M + Li - 141]+) (d) by elimination of LiH2PO4 and H3PO4, respectively (Scheme 4). These ions along with the ions at m/z 148 (b) and 105, corresponding to a lithiated ethanolaminephosphate ([(HO)2PO2(CH2)2NH2Li]+) and a lithiated phosphoric acid ([(HO)3POLi]+) ions, respectively, are indicative of the phosphoethanolamine head group and are characteristic of GPEtn.
Fig. 4.

The product-ion spectra of the [M + Li]+ ions of 16:0/18:2-PE at m/z 722 (A), of 16:0/20:4-PE at m/z 746 (B), of p18:0/18:1-PE at m/z 736 (C), and of a18:0/18:0-PE at m/z 740 (D).
Scheme 4.

The fragmentation mechanisms proposed for the [M + Li]+ ions of glycerophospholipids (other than GPCho).
The notion that loss of the fatty acyl substituent at sn-1 as an acid is more favorable than the analogous loss at sn-2 is also accountable for the observation of a greater abundance of the ion at m/z 466 ([M + Li - R1CO2H]+) than the ion at m/z 442 ([M + Li – R2CO2H]+), and a greater abundance of the m/z 423 ([M + Li - 43 - R1CO2H]+) ion than the m/z 399 ([M + Li - 43 – R2CO2H]+) ion. The [M + Li –147]+ ion at m/z 575 is equivalent to an [M + Li –189]+ ion as described for GPCho, and further dissociates to the 18:2- and 16:0-acylium ions at m/z 263 and 239. The product-ion spectrum of the [M + Li]+ ion of 16:0/20:4-GPE at m/z 746 (Fig. 4B) also contains a unique ion at m/z 313, arising from m/z 599 ([M + Li –147]+) by further loss of the 20:4-fatty acid at sn-2 as a ketene, similar to that previously observed for GPCho (section 4.2.1.). This further fragmentation also results in the decline of the [M + Li - 147]+ ion at m/z 599 (relative to the [M + Li - 141]+ ion at m/z 605).
b. The [M + Li]+ ions of plasmalogen GPEtn and plasmanyl GPEtn
The ions at m/z 693 ([M + Li - 43]+), 595 ([M + Li - 141]+), 589 ([M + Li - 147]+), and at m/z 148 commonly seen for GPEtn were also observed in the product-ion spectrum of the [M + Li]+ ion of p18:0/18:1-PE at m/z 736 (Fig. 4C). The spectrum contains the ion at m/z 425, arising from the combined losses of aziridine and the alk-1′-enyl residue at sn-1 as an alcohol (C16H33CH=CHOH), and the ion at m/z 307, arising from further loss of the 18:1-fatty acid substituent at sn-2 as a free acid from m/z 589. These two ions identify the alkenyl ether at sn-1 and the fatty acid at sn-2, respectively. In contrast, the analogous ion pairs are absent in the product-ion spectrum of a18:0/18:0-PE at m/z 740 (Fig. 4D), a plasmanyl-GPEtn.
The observation of the ion pairs that reflect both the sn-1 and sn-2 substituents affords the complex structures of, for example, a plasmalogen GPEtn from bovine brain consisting of p18:0/20:4-PE and p16:0/22:4-PE structures to be unveiled [35]. In contrast, the product-ion spectra obtained from the [M − H]− ions only give one carboxylate anion from sn-2 (section 4.3.8.). Therefore, confirmation of the structural assignments requires a comparison of the two mass spectra of the GPEtn obtained before and after destructive removal of plasmalogen GPEtn by acid treatment, resulting in severe sample losses [45,46]. The structural assignment of the [M + Li]+ ion of a18:0/18:0-PE at m/z 740 (Fig. 4D) is based on the observation of the ions at m/z 431 ([M + Li – 43 – R2′CH=CO]+), arising from the combined losses of aziridine and the 18:0-fatty acid at sn-2 as a ketene, along with the series of the (CH2)nH+ ions starting from m/z 253 (R1+) that represents the 18:0-alkyl moiety at sn-1. The ion series is similar to those observed for plasmanyl-GPCho (section 4.2.1.) and is signature of the plasmanyl subclasses of GPEtn and GPCho.
c. The [M − H + 2Li]+ ions of GPEtn
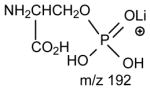
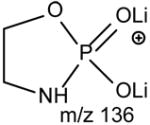
The ions corresponding to losses of the phosphoethanolamine residues (loss of 141 and 147, respectively), as seen for the [M + Li]+ ion are absent in the product-ion spectrum from its [M − H + 2Li]+ ion. For example, the product-ion spectrum of 16:0/18:2-PE at m/z 728 (Fig. 5A) is dominated by the m/z 599 ([M − H + 2Li - 129]+) (f ion, Scheme 5) and m/z 136 ([LiO)2P+(OCH2CH2NH]) (e) ions, arising from cleavage of the C3O-P bond, and the ions at m/z 587 (loss of 141) and 581 (loss of 147) are not present. Again, the more labile α-hydrogen at the fatty acyl at sn-2 participates in the R1CO2H loss and leads to that the ion at m/z 472 ([M − H + 2Li – R1CO2H]+) is more abundant than m/z 448 ([M − H + 2Li –R2CO2H]+); the ion at m/z 429 ([M − H + 2Li – aziridine - R1CO2H]+) is more abundant than the ion at m/z 405 ([M − H + 2Li – aziridine – R2CO2H]+); the ion at m/z 343 (599 –R1CO2H) is more abundant than the ion at m/z 319 (599 – R2CO2H). The similar losses of the fatty acid substituents as the lithium salt give rise to m/z 423 ([M − H + 2Li - aziridine -R1CO2Li]+), 399 ([M − H + 2Li - aziridine - R2CO2Li]+), m/z 337 (599 - R1CO2Li) and 313 (599 – R2CO2Li), respectively. An ion corresponding to a lithiated 18:2 fatty acid from sn-2 was observed at m/z 287, but an analogous ion deriving from the sn-1 fatty acid was not observed. This provides direct assignment of the sn-2 fatty acid substituent.
Fig. 5.

The product-ion spectra of the [M + 2Li - H]+ ions of 16:0/18:1-PE at m/z 728 (A), and of p16:0/20:4-PE at m/z 736 (B).
Scheme 5.

The proposed fragmentation mechanisms of the [M − H + 2Li]+ ions of glycerophopholipids.
The product-ion spectrum of the dilithiated p16:0/20:4-PE at m/z 736 (Fig. 5B), is also dominated by the m/z 607 ([M − H + 2Li - 129]+) and m/z 136 ([LiO)2P+(OCH2CH2NH]) ions, arising from cleavage of the CO-P bond, and the ions that are indicative of the alkenyl ether at sn-1 and the fatty acid at sn-2 are not present [35].
4.2.3. GPSer, GPGro, Glycerophosphoinositol (GPIno), and GPA
The similarities among the GPEtn, GPSer and GPGro classes observed by ESI mass spectrometry, not only lies on the formation of the molecular species, but also on their tandem mass spectra, and therefore, the fragmentation pathways underlying the ion formation under low-energy CAD are nearly identical. Table 1 lists the common ions arising from the [M + Li]+ ions of GPEtn, GPSer, GPGro, and of GPA. The common fragment ions arising from the [M − H + 2Li]+ ions of GPEtn, GPSer, GPGro, GPIno and of GPA are listed in Table 2. When ionized in the presence of Li+, the sensitivities of GPSer, GPGro, and GPA observed as the [M + Li]+ and [M − H + 2Li]+ adduct ions and of GPIno observed as the [M − H + 2Li]+ ion are significantly poorer than those observed as the [M − H]− ions in negative-ion mode.
Table 1.
Head group-related ions observed in the product-ion spectra of the [M+Li]+ ions
| * Phospholipid | Head group (YH) | a | 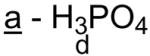 |
b | |
|---|---|---|---|---|---|
| GPEtn |
|
[M + Li - 43]+ | [M + Li - 147]+ | [M + Li - 141]+ |
|
| GPSer |
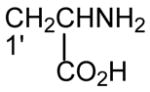
|
[M + Li - 87]+ | [M + Li - 191]+ | [M + Li - 185]+ |
|
| GPGro |
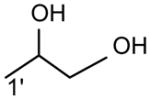
|
[M + Li - 74]+ | [M + Li - 178]+ | [M + Li - 172]+ |

|
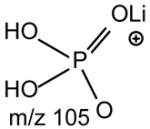
| GPA | H | [M + Li ]+ | [M + Li- 104]+ | [M + Li- 98]+ |
|
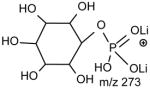
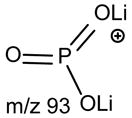
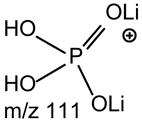
No study was conducted on the [M+Li]+ ions of GPIns.
Table 2.
Head group-related ions observed in the product-ion spectra of the [M − H + 2Li]+ ions
| Phospholipid | Head group (YH) | g | f | e | h |
|---|---|---|---|---|---|
| GPEtn |
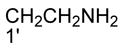
|
[M + 2Li -H - 43]+ | [M + 2Li -H - 129]+ |
|
|
| GPSer |
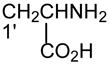
|
[M + 2Li -H - 87]+ | - |
|

|
| GPGro |
|
[M + 2Li -H - 74]+ | [M + 2Li -H - 160]+ |
|
|
| GPIns |
|
[M + 2Li -H - 162]+ | [M + 2Li -H - 248]+ |
|
|
| GPA | H | [M + 2Li -H]+ | [M + 2Li -H - 86]+ |
|
|
a. The [M + Li]+ ions of GPSer, GPGro and GPA
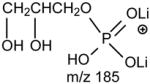
One of the most interesting findings in the product-ion spectrum of the [M + Li]+ ion from GPSer is that an ion corresponding to loss of a phosphoric acid is also observed. The ion is seen at m/z 670, in the product-ion spectra of 16:0/18:1-PS at m/z 768 obtained with a TSQ (Fig. 6A) and with an ITMS (Fig. 6B), however, the ion is the most prominent in the latter spectrum. The results are consistent with the notion that the ion may arise from internal loss of the phosphoric acid (98 Da) via rearrangement processes. The loss is also consistent with the observation of the ion at m/z 595 from further loss of the serine residue in the IT MS3-spectrum of the ion at m/z 670 ([M + Li – 98]+) [29]. The product-ion spectrum of the [M + Li]+ ion of GPGro, for example, of 16:0/18:1-PG at m/z 755 (Fig. 6C), is similar to that arising from GPSer (Fig. 6A), and the analogous ions at m/z 599 and 179 arising from cleavage of the O-P bond of the phosphoglycerol group (Table 1) are prominent. However, the product-ion spectrum of the [M + Li]+ ion of 16:0/18:1-PA at m/z 681 (Fig. 6D) is dominated by the ions at m/z 577 and 583, reflecting losses of H3PO4 and LiH2PO4, respectively, and the lithiated phosphoric ion expected at m/z 105 is not present. The similarities in the product-ion spectra of the [M + Li]+ ions arising from GPA and from GPEtn are consistent with the proposed mechanism, in which the aziridine residue was first cleaved from GPEtn to give a GPA-like ion and undergoes secondary dissociation.
Fig. 6.

The product-ion spectra of the [M + Li]+ ions of 16:0/18:1-PS at m/z 768 obtained with TSQ (A), and ITMS (B) instruments. The TSQ product-ion spectra of the [M + Li]+ ions of 16:0/18:1-PG at m/z 755, and of 16:0/18:1-PA at m/z 681 are shown in Panel C and Panel D, respectively.
b. The [M + 2Li –H]+ ions of GPSer, GPGro, GPIno, and GPA
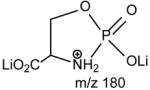
Upon low-energy CAD in a TSQ instrument, the dilithiated adduct ion of 16:0/18:1-PS at m/z 774 (Fig. 7A) yields a prominent dilithiated GPA-like ion at m/z 687 by loss of the serine residue ([(M + 2Li – H) - 87]+). This is followed by further losses of the fatty acyl substituents at sn-1 and sn-2, respectively, to give ions at m/z 431 (687 – R1CO2H) and 405 (687 – R2CO2H). The charge-remote fragmentation processes lead to preferential formation of the m/z 431 ion over the m/z 405 ion, attributable to the fact that the α-hydrogens of the sn-2 fatty acyl substituent that involves in the loss of 16:0-fatty acid (R1CO2H) is more labile. The cleavage of C3-OP bond to eliminate a dilithium salt of phosphoserine ([(M + 2Li – H) -197]+) gives rise to m/z 577 ion, along with m/z 198 (Scheme 5, route c) (Table 2) [29].
Fig. 7.

The product-ion spectra of the [M + 2Li - H]+ ions of 16:0/18:1-PS at m/z 774 (A), of 16:0/18:1-PG at m/z 761 (B), of 16:0/18:2-PI at m/z 847 (C), and of 16:0/18:1-PA at m/z 687 (D).
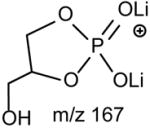
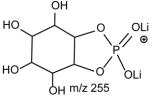
The preferential loss of the fatty acid substituent at sn-1 over that at sn-2 are seen for all of the [M + 2Li – H]+ precursors, including GPGro, GPIno, and GPA. These are shown by the product-ion spectra of 16:0/18:1-PG at m/z 761 (Fig. 7B), of 16:0/18:2-PI at m/z 847 (Fig. 7C), and of 16:0/18:1-PA at m/z 687 (Fig. 7D). The cleavage of the C3O-P bond gives rise to the prominent ion at m/z 167, along with the ions at m/z 601 and 607 that are of low abundance (Scheme 5, route a), and the ion from cleavage of the C3-OP bond as observed for GPSer is absent in the product-ion spectrum of 16:0/18:1-PG at m/z 761 (Fig. 7B). This preferential formation of m/z 167 over the m/z 601 ion via cleavage of the C3O-P bond indicates that the gas-phase (LiO)(HO)P=O(OX) moiety (where X=glycerol –H2O) may be more competitive than the diacylglycerol moiety for Li+ for ion formation. Similar results are also seen for the [M + 2Li – H]+ ion of 16:0/18:2-PI at m/z 847 (Fig. 7C), which gives rise to a prominent [(LiO)(HO)P=O(OX) + Li]+ (where X=inositol –H2O) (e ion) ion at m/z 255, and the lithiated diacylglycerol ion (f ion) at m/z 599 is of low abundance.
With employment of multiple-stage tandem mass spectrometry with a linear ion-trap instrument the characterization of GPLs as lithiated adduct ions affords not only the identification of the fatty acid substituents and their location on the glycerol backbone as described above, but also permits the location of the double bond(s) of the unsaturated fatty acid substituents to be unveiled in detail. This mass spectrometric approach leading to a nearly complete structural characterization of GPLs using LIT MSn has been recently demonstrated by Hsu and Turk [17].
4.3. The Fragmentation processes observed in negative-ion mode
4.3.1. Glycerophosphocholine (GPCho)
One of the most interesting findings in the earlier studies on GPCho by FAB/MS is that a set of three ions corresponding to [M − 15]−, [M − 60]−, and [M − 86]− were observed, when subjected to FAB ionization in the negative-ion mode [47]. The high-energy CAD product-ion spectra of the [M − 15]−, [M − 60]−, and [M − 86]− ions are readily distinguishable [40,47]. In the presence of X (where X = Cl−, Br−, CH3CO2−, HCO2−, CF3CO2−, etc), GPCho forms various [M + X]− adduct ions, together with the [M − 15]− ion ([M + X – CH3X]−) by ESI [5,6,26,27,48]. The [M − 60]− and [M − 86]− ions were also seen together with, for example, the [M + CF3CO2]− adduct ion [48]. The tandem quadrupole product-ion spectra of the ions at m/z 744 ([M − 15]−) (Fig. 8A), 699 ([M − 60]−) (Fig. 8B), and at m/z 673 ([M − 86]−) (Fig. 8C) from 16:0/18:1-PC are similar to those obtained by FAB/MS/MS [47], indicating that the fragmentation processes under high- and low-energy CAD are similar. The product-ion spectra obtained with a TSQ are dominated by the carboxylate anions at m/z 255 (R1CO2−) and 281 (R2CO2−) (Fig.s 8A-C). The fact that the m/z 281 ion is more abundant than the m/z 255 ion in the product-ion spectrum arising from the [M − 15]− at m/z 744 (Fig. 8A) generated by FAB and ESI has been previously applied for locating the fatty acid substituents on the glycerol backbone [5,6,25,26,47]. A FAB tandem mass spectrometric approach for locating the fatty acid substituents based on the findings that the ion at m/z 391 arising from loss of the 18:1-fatty acid at sn-2 is more abundant than the ion at m/z 417, arising from loss of the 16:0-fatty acid substituent at sn-1 in the product-ion spectrum of the m/z 673 ([M − 86]−) ion was also reported [49].
Fig. 8.

The product-ion spectra of the [M − 15]− ion at m/z 744 (A), the [M − 60]− ion at m/z 699 (B), and of the [M − 86]− ion at m/z 673 (C). These precursor ions were generated from 16:0/18:1-PC by ESI in the presence of CF3CO2− in negative-ion mode.
The product-ion spectrum of the [M − 86]− ion of 16:0/18:1-PC generated by FAB [49] and ESI (Fig. 8C) is identical to that of the [M − H]− ion of 16:0/18:1-PA at m/z 673 (section 4.3.2., Fig. 9A); while the fragmentation mechanisms for the [M − 15]− at m/z 744 and for the [M − 60]− at m/z 699 under low-energy CAD are similar to those observed for the [M − H]− ion of GPEtn (section 4.3.3).
Fig. 9.

The product-ion spectra of the [M − H]− ion of 16:0/18:1-PA at m/z 673 obtained with TSQ (A), ITMS (B), and its MS3-spectra of the ions at m/z 391 ([M − H – R2CO2H]−) (673 →391) (C), m/z 417 ([M − H – R1CO2H]−) (673 → 417) (D), m/z 409 ([M − H – R′2CHC=O]−) (673 → 409) (E), and at m/z 435 ([M − H – R′1CHC=O]−) (673 → 435) (F) o riginated from m/z 673.
Although the approaches are useful for identification of the fatty acid substituents and location of their position on the glycerol backbone, the adduct ion formation with anionic counter ions (X) is unpredictable and sensitivity is often poor. In addition, molecules of interest are seen in various forms, resulting in confusion in identification of molecular species and decline in sensitivity, in particular, from the GPL species in a mixture.
4.3.2. GPA
The major pathways leading to formation of the [M − H – RxCO2H]− and [M − H –Rx′CH=C=O]− (Rx=Rx′CH2, x= 1, 2) ions via losses of the fatty acyl substituent as an acid or as a ketene, respectively, for the [M − H]− ion of GPA are thought to be charge-driven fragmentation (CDF) processes that involve the participation of the exchangeable hydrogen of the phosphate head group [50]. The observations of a higher abundance of the [M − H –R2CO2H]− ion at m/z 391 than the [M − H – R1CO2H]− ion at m/z 417 and of a higher abundance of the [M − H – R2′CH=C=O]− ion at m/z 409 than the [M − H – R1′CH=C=O]− ion at m/z 435 in the product-ion spectrum of 16:0/18:1-GPA ( Fig. 9A) are possibly attributable to the fact that loss of the fatty acyl substituent at sn-2 as an acid or as a ketene is sterically more favorable than the analogous loss at sn-1. This speculation is based on the notion that the phosphate charge site that initiates the fragmentation processes by CDF is in the closer proximity to the fatty acyl moiety at sn-2 than that at sn-1 [51]. The ions at m/z 391 and 417 from losses of the 18:1- and 16:0-fatty acid substituents, respectively are also more abundant than the ions at m/z 409 and 435, respectively, arising from the analogous losses of the fatty acid substituents as ketenes, possibly attributable to the fact that the gaseous [M − H]− of GPA is an acidic ion, which undergoes more facile acid loss to the [M − H – RxCO2H]− ion than the ketene loss to the [M − H – Rx′CH=C=O]− ion (x = 1 or 2) (Scheme 6) [50].
Scheme 6.

The proposed fragmentation mechanisms of the [M − H]− ions of phosphatidic acid.
The fragment ions arising from losses of the fatty acid substituents as acids and as ketenes are prominent in the IT MS2-spectra of the [M − H]− ions of phospholipids [13]. As shown in Fig. 9B, the IT MS2-spectrum of 16:0/18:1-GPA, is dominated by the [M − H –RxCO2H]− ions (x = 1 or 2) at m/z 391 and 417; while the [M − H – Rx′CH=C=O]− ions at m/z 409 and 435, and the RxCO2− ions at m/z 255 and 281 are of low abundance. The decline in the [M − H – RxCO2H]− ions at m/z 391 and 417 and the rise of the RxCO2− ions of m/z 255 and 281 in the product-ion spectrum obtained with TSQ (Fig. 9A) as compared to those observed by ITMS in Fig. 9B, are consistent with the notion that the collisional activation occurred in a TSQ instrument involves multiple collisions that result in consecutive fragmentations, while resonance excitation is the major fragmentation process occurred in an ion-trap, and fragment ions derived from secondary fragmentations are minimal. The consecutive processes as seen by the IT MS3-spectra of the ions at m/z 391 ([M − H –R2CO2H]−) (673 → 391) (Fig. 9C) and 417 ([M − H – R1CO2H]−) (673 → 417) (Fig. 9D) gives rise to ions at m/z 255 and 281, respectively, by neutral loss of 136 (Scheme 6A, route c1a′ and c2a′). The consecutive fragmentation processes also involve the nucleophilic attack of the anionic charge site of the [M − H – RxCO2H]− precursor ions on the C2 or C3 of the glycerol to expel RxCO2−. The fragmentation processes are consistent with the notion that the [M − H – RxCO2H]− ions (x = 1 or 2) become basic ions; while the [M − H – Rx′CH=C=O]− ions at m/z 409 and 435 become more acidic than their precursors of [M − H]− due to gaining protons from the ketene loss, and undergo further acid loss to m/z 153 ([M − H – R1(or 2) ′ CH=C=O – R2 (or 1)CO2H]−) (Fig. 9E and Fig. 9F) (Scheme 6C, route a′1b). Because the m/z 391 ([M − H – R2CO2H]−) ion is sterically more favorably formed than the m/z 417 ([M − H –R1CO2H]−) ion from the primary fragmentation step, the m/z 255 ion (R1CO2−) resulting from m/z 391 becomes more abundant than the m/z 281 (R2CO2−) ion, resulting from secondary dissociation of m/z 417.
4.3.3. Glycerophosphoserine (GPSer)
Upon ESI in the negative-ion mode, GPSer forms the [M − H]− ion along with the [M − H - 87]− ion arising from loss of the serine residue, indicating that GPSer is labile. The product-ion spectrum of the [M − H]− ion of 16:0/18:1-PS at m/z 760 obtained with a TSQ ( Fig. 10A) is very similar to that from the [M − H]− ion of 16:0/18:1-PA (Fig. 9A), because loss of serine to [M − H - 87]− is the primary fragmentation process leading to further fragmentation for PS. These fragmentation processes have been supported by the previous study with IT MS2 on the [M − H]− ion of 16:0/18:1-PS at m/z 760, which is dominated by the ion at m/z 673, an ion equivalent to a [M − H]− of 16:0/18:1-PA, and by the IT MS3 production spectrum of m/z 673 (760 → 673), which is identical to that arising from 16:0/18:1-PA [29].
Fig. 10.

The product-ion spectra of the [M − H]− ion of m/z 760 (A), and of the [M − 2H + Li]− ion of m/z 766 (B) from16:0/18:1-PS.
GPSer also forms [M − 2H + Alk]− (Alk = Na, Li) ions when subjected to ESI in the presence of Alk+, due to that it possesses two anionic charge sites of which one can attach an Alk+. The product-ion spectrum of the [M − 2H + Li]− ion of 16:0/18:1-PS at m/z 766 (Fig. 10B) contains the major ions at m/z 502 and 528 arising from loss of the 18:1- and 16:0-fatty acyl substituents as ketenes, respectively. The spectrum also contains the ions at m/z 484 and 510 arising from the corresponding losses of the fatty acyl substituents as acids. These latter two ions are respectively less prominent than the former two ions, suggesting that the gaseous ion of [M − 2H + Li]− is more basic than that of [M − H]− because the substitution of a proton by a Li+ in the former ion. The m/z 502 ([M − 2H + Li – R′2CH=CO]−) ion reflecting loss of the fatty acyl ketene at sn-2 is more abundant than the m/z 528 ([M − 2H + Li –R′1CH=CO]−) ion arising from the similar loss at sn-1. This is consistent with the notion that loss of fatty acid substituents as ketenes is the favored pathway for basic precursor ions and the process is sterically more favorable at sn-2 than that at sn-1.
4.3.4. Glycerophosphoethanolamine (GPEtn)
The product-ion spectra of the 16:0/18:1-PE (Fig. 11A) and the 18:1/16:0-PE (Fig. 11B) at m/z 716 are readily distinguishable. Both charge-remote and charge-driven are the major fragmentation pathways that lead to the ion formation for GPEtn observed with TSQ instrument. The gas-phase [M − H]− ion of GPE is basic, and undergoes preferential losses of fatty acyl substituents as ketenes ([M − H – Rx′CH=C=O]−) over as acids ([M − H –RxCO2H]−) [52]. The anionic charge site renders nucleophilic attack on either the α-hydrogen of the fatty acyl substituent, or on the carbon of the glycerol to which the fatty acid substituent attached. The attack on the α-hydrogen of the fatty acyl results in the [M − H –Rx′CH=C=O]− ion series at m/z 478 and 452 (Scheme 7A), while the latter process leads to formation of RxCO2− ions at m/z 281 and 255 by charge-transfer (Scheme 7B) (Fig. 11A and 11B). Both the fragmentation processes are sterically more favorable at sn-2 than at sn-1 and result in that the m/z 452 ion ([M − H – R2′CH=C=O]-) is more abundant than the m/z 478 ([M − H – R1′CH=C=O]−) ion, as well as the m/z 281 ion (R2CO2−) is more abundant than the m/z 255 ion (R1CO2−) for the 16:0/18:1-PE isomer (Fig. 11A); while the abundances of the above ion pairs are reversed in the product-ion spectrum of 18:1/16:0-PE (Fig. 11B).
Fig. 11.

The TSQ product-ion spectra of the [M − H]− ions of 16:0/18:1-PE (A), and of 18:1/16:0-PE at m/z 716 (B). The ITMS MS2-spectrum of 16:0/18:2-PE at m/z 714 is shown in Panel C.
Scheme 7.

The fragmentation mechanisms proposed for the [M − H]− ions of GPEtn.
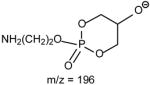
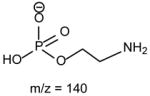
The fragmentation processes leading to formation of the [M − H – RxCO2H]− ions may arise from CRF. The methine hydrogen at C-2 of the glycerol backbone may participate in the loss of R1CO2H (Scheme 8A), while the methylene hydrogens at C-1 involve in R2CO2H loss (Scheme 8B). The ion at m/z 434 ([M − H – R2CO2H]−) is slightly more abundant than the ion at m/z 460 ([M − H – R1CO2H]−)(Fig. 11A), probably attributable to the fact that the hydrogen participates in the R1CO2H loss is one, the methine hydrogen; while the R2CO2H loss involves the methylene hydrogens at C-1, is two [36]. The ions reflecting the polar head group are observed at m/z 140 and 196 (Table 3) which are 43 Da (ethyleneamine) higher than the analogous ions at m/z 97 and 153 observed for GPA, indicating that these ions arise from similar fragmentation processes.
Scheme 8.

The fragmentation mechanisms proposed for the losses of RxCO2H for the [M − H]− ion of GPEtn.
Table 3.
The proposed configurations of the polar head group-related ions observed for the [M − H]− ions of major phospholipid classes
| Phospholipid | Head group | [M − H - R2(1) CO2H - R1(2)CH=CO]− | [M − H - R2CO2H - R1CO2H]− | 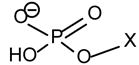 |
|---|---|---|---|---|
| GPEtn |
|
|

|
|
| * GPCho(M − 15)− |
|
|
|
|
| GPGro |
|
|
|
|
| GPIns |
|
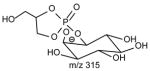
|
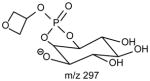
|
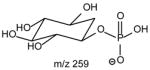
|
| GPA | H |
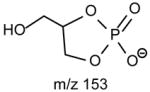
|
||
| PS |
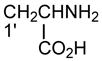
|
Same as PA |
The [M − 15]- ion from GPCho was formed from an adduct anion (such as Cl−) by loss of a methyl group (loss of methyl chloride).
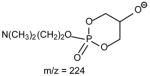
The profile of the IT product-ion spectrum from MS2 of 16:0/18:2-PE at m/z 714 (Fig. 11C) is similar to that obtained with TSQ [52]. However, the ions at m/z 452 ([M − H –R′2CH=CO]−) and 476 ([M − H – R′1CH=CO]−), and the ions at m/z 279 (R2CO2−) and 255 (R1CO2−) are prominent; while the ions at m/z 434 and 458 arising from losses of the fatty acyl as free fatty acids are of low abundance. This is consistent with the concept that losses of fatty acyl substituents as ketene and formation of carboxylate anions are the major fragmentation processes for GPEtn, which is a basic phospholipid; while losses of the fatty acyl substituents as acids are the major pathways for acidic precursors such as GPA, as described earlier. The profiles of both the product-ion spectra of the [M − 15]− ion at m/z 744 (Fig. 8A) and of the [M − 60]− ion at m/z 699 (Fig. 8B) as observed for 16:0/18:1-PC are nearly identical to that arising from the [M − H]− ion of 16:0/18:1-PE, indicating that the gas phase basicities of the former two ions are close to the [M − H]− ion of GPEtn. The results are consistent with the suggestion that the [M − 15]− and [M − 60]− ions represent a deprotonated phosphatidyl-ethanol-N,N′-dimethylamine and a deprotoned phosphatidylethylene ions, respectively [36,47]. The presence of the phosphatidyl-ethanol-N,N′-dimethylamine polar head group is also consistent with the observation of the ions at m/z 224, 206 and 168 as observed in Fig. 8A (Table 3).
4.3.5. Glycerophosphoglycerol (GPGro)
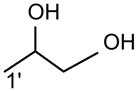
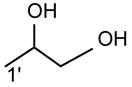
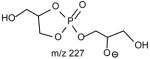
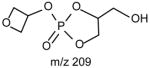
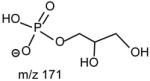
The m/z 465 ([M − H- R2CO2H]−) and the m/z 483 ([M −H – R′2CH=C=O]−) ions are respectively more abundant than the analogous ions at m/z 491 ([M − H - R1CO2H]−) and m/z 509 ([M − H –R′1CH=C=O]−) in the product-ion spectrum of 16:0/18:1-PG at m/z 747 (Fig. 12A) obtained by TSQ (48), because the preferential losses of the fatty acid substituent at sn-2, similar to that described for GPA and GPEtn. However, the ion at m/z 483 ([M − H –R′2CH=C=O]−) is more abundant than the ion at m/z 465 ([M −H - R2CO2H]−), arising from loss of the fatty acid substituent at sn-2 as a ketene and as a free acid, respectively; while the ion at m/z 491 ([M − H - R1CO2H]−) is more abundant than ion at m/z 509 ([M −H –R′1CH=C=O]−), arising from the analogous losses of the fatty acid substituent at sn-1, respectively. This is consistent with the notion that GPGro is a weakly acidic phospholipid [53], and the gas-phase basicity of the [M − H]− ion is between that of GPEtn and GPA. The results are also consistent with the notion that the α-hydrogen of the fatty acid substituent at sn-2 is more labile and undergoes more facile loss of ketene. The m/z 391 ion arising from a combined losses of the sn-2 fatty acid substituent and the polar head group ([M − H -R2CO2H - 74]−) is also more abundant than the m/z 417 ion ([M − H - R1CO2H - 74]−) arising from the analogous losses at sn-1. These features along with that the R2CO2− at m/z 281 is more abundant than the R1CO2- ion at m/z 255 are readily applicable for determination of 16:0/18:1-PG structure and permit its differentiation from the 18:1/16:0-PG isomer (Fig. 12B). The ions that are indicative of the polar head groups are observed at m/z 227, 209, 171 and 153 (53) (Table 3), arising from the similar processes as described for GPA and GPEtn. Both CRF and CDF are the fragmentation processes underlying the formation of the [M − H - RxCO2H]− ions for GPGro [53]. The evidence lies on the studies on the deuterated d2-16:0/18:1-PG analog (Fig. 12C), which show two separate processes by which the CDF process leads to the formation of the m/z 466 ([M − H – R2CO2D]−) and 492 ([M − H –R1CO2D]−) ions involving the participation of the exchangeable hydrogen of the glycerol head group; while CRF process which requires the participation of a non-exchangeable hydrogen at glycerol backbone lead to form the m/z 467 ([M − H – R2CO2H]−) and 493 ([M −H – R1CO2H]−) ions [53]. The occurrence of CRF and CDF to GPGro further supports the concept that acid loss for acidic precursor such as GPA, is a CDF process, which involves the exchangeable hydrogen, while the similar loss for basic phospholipid class, such as GPEtn, is a CRF process that involves the hydrogen at the glycerol backbone. In contrast, CDF initiated by resonance excitation is the fragmentation processes for GPGro observed by ITMS. This is reflected by the IT MS2-spectrum of 16:0/18:1-PG (Fig. 12D), of which the [M − H – RxCO2H]− ions at m/z 491 and 465 are respectively more abundant than the [M − H – R′xCH=CO]− ions at m/z 509 and 483. The results also indicate that the gas-phase ion of the [M − H]− ion of GRGro is slightly acidic. The influences of the gas-phase basicities of the precursor ions on the fragmentation processes are also observed for other phospholipid classes, including GPIno (section 4.3.6.)
Fig. 12.

The TSQ product-ion spectra of the [M − H]− ions of 16:0/18:1-PG (A), and of 18:1/16:0-PG (B) at m/z 747, and of d2-16:0/18:1-PG at m/z 749 (C). Panel (D) is the ITMS MS2-spectrum of 16:0/18:2-PG at m/z 747.
4.3.6. Glycerophosphoinositol (GPIno)
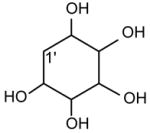
The fact that GPIno is a strong acid [54] is in accord with what has been observed in the product-ion spectra of the [M − H]− ion of 16:0/18:2-PI at m/z 833 obtained by low-energy CAD with TSQ (Fig. 13A) and with ITMS (Fig. 13B) instruments. Both the spectra are featured by that the ions at m/z 553 ([M − H – R2CO2H]−) and 577 ([M − H – R1CO2H]−) arising from losses of the fatty acid substituents as acids are respectively more abundant than the ions at m/z 571 ([M − H – R′2CH=C=O]−) and 595 ([M − H – R′2CH=C=O]−), arising from the corresponding losses of the fatty acids as ketenes. The m/z 391 ([M − H – R2CO2H –(inositol –H2O)]− (or [M − H – R′2CH=C=O - inositol]−)) ion from consecutive loss of the inositol head group is also more abundant than the m/z 417 ([M − H – R1CO2H – (inositol –H2O)]− (or [M − H – R′1CH=C=O - inositol]−)) ion (Scheme 9), due to that the fragment ions reflecting those losses arise from charge-driven processes, which are preferentially occurred at sn-2 position [33].
Fig. 13.

The product-ion spectrum of the [M − H]− ion of 16:0/18:2-PI at m/z 833 obtained with a TSQ (A), and an ITMS (B) instruments.
Scheme 9.

The fragmentation mechanisms proposed for the [M − H]− ion of GPIns.
The relative intensity of the R2CO2− ion at m/z 279 is close to that of the R1CO2− ion at m/z 255 in Fig. 13A. However, an appreciable difference in the abundances of the R1CO2− and R2CO2− ions was seen for the other phospholipid classes that yield two carboxylate anions. Three pathways lead to formation of the RxCO2− ions for GPIno. The predominant pathway arises from nucleophilic attack of the phosphate anionic charge site to expel the sn-1 or sn-2 fatty acyl substituents to R1CO2− or R2CO2−. This pathway is a CDF process, which is sterically more favorable at sn-2 than at sn-1, and results in that the abundance of m/z 279 (R2CO2−) > m/z 255 (R1CO2−). The R1CO2− ion at m/z 255 can also arise from further decomposition of the ions at m/z 391 ([M − H – R′2CH=C=O - inositol]−) (same m/z value as [M − H – R2CO2H – (inositol – H2O)]−), 553 ([M − H – R2CO2H]−), and at m/z 571 ([M − H –R′2CH=C=O]−). Similarly, the m/z 279 (R2CO2−) can also arise from the ions at m/z 415 ([M −H – R′1CH=C=O - inositol]−) (same m/z value as [M − H – R1CO2H – (inositol – H2O)]−), 577 ([M − H – R1CO2H]−) and at m/z 595 ([M − H – R′1CH=C=O]−). These secondary fragmentation processes yielding the RxCO2− ions via multiple collisions within a TSQ instrument prevail when a higher collision energy is applied. This consecutive dissociation results in the abundance of m/z 255 (R1CO2−) > 279 (R2CO2−), because the precursor ions of m/z 255 at m/z 391, 553 and 571 are preferentially formed from the primary dissociation involving elimination of the fatty acyl moiety at sn-2. In contrast, the ions at m/z 415, 577 and 595 that give rise to m/z 279 are less abundant, because they arise from losses of the analogous moieties at sn-1. Therefore, under a optimal collision energy for GPIno ( 45eV), the m/z 255 ion is slightly more abundant than the m/z 279 ion, while the abundances of the two ions are reversed, when [M − H]− ion of 16:0/18:2-PI is obtained at a lower collision energy (≤ 35 eV) [33].
The multiple collisions under the collision energy optimized for acquiring the product-ion spectrum with TSQ instrument for GPIno also cause further degradation of the carboxylate anions, dependent on the degree of unsaturation (see section 4.3.8). Thus, the relative intensities of the carboxylate anions from GPIno are varied with the collision energy. Consequentially, locating the fatty acid substituents on the glycerol backbone based on the abundances of the carboxylate anions in the product-ion spectra obtained with a TSQ instrument is not reliable. In contrast, the consecutive fragmentation processes occurred in an ITMS is minimal and the fragment ions are mainly derived from primary fragmentation. Thus, the RxCO2− ions at m/z 255 and 279 are of low abundance in the product-ion spectrum of 16:0/18:2-PI obtained with an ITMS (Fig. 13B).
The ions reflecting the inositol head group are observed at m/z 315, 297, 279, 259, 241 and 223. The m/z 297 ion ([M − H - R1CO2H – R2CO2H]−) arises from consecutive losses of the fatty acyl substituents as acids. The first acid loss involves an exchangeable hydrogen of the inositol head group, while the subsequent loss of the fatty acid substituent can involve an exchangeable hydrogen on the inositol head group, or the non-exchangeable hydrogen on the glycerol backbone, suggesting that both CDF and CRF processes contribute to formation of m/z 297.
4.3.7. Lysophospholipid, plasmenylphospholipid (plasmalogen), and plasmanylphospholipid
The product-ion spectra of both lyso1/18:1-PE (Fig. 14A) and 18:1/lyso2-PE (Fig. 14B) are dominated by the sole carboxylate anion at m/z 281 that identifies the 18:1-fatty acyl moiety of the molecules. The m/z 214 ion, reflecting loss of the 18:1-fatty acyl ketene ([M − H – R1CH=C=O]−) is more abundant than m/z 196, corresponding to loss of the 18:1-fatty acid ([M − H – R1CO2H]−) in the spectrum from 1-lysophosphatidylethanolamine; while the abundances of the two ions are reversed in the spectrum from 2-lysophosphatidylethanolamine. This is consistent with the notion that the gas-phase [M − H]− ions of 1- and of 2-lysophosphatidylethanolamine are weakly basic ions and undergo more facile loss of the fatty acyl substituent as a ketene at sn-2 than at sn-1. Thus, positional isomers of 1- and 2-lyso-PE can be differentiated. However, acidic lysophospholipids such as the [M − H]− ions of 1- and 2-lysophosphatic acids undergo mainly acid loss via CDF processes. This is seen by the MS3-spectrum of the m/z 409 ion (673 → 409) (Fig. 9E) (the ion of m/z 409 is equivalent to the [M − H]− ion of 16:0/lyso2-PA, a 2-lyso-PA), which is dominated by the ion at m/z 153 arising from loss of 16:0-fatty acid substituent (see Scheme 6C). The spectrum is nearly identical to that arising from m/z 435 (673 → 435) (Fig. 9F), which is equivalent to a lyso1/18:1-PA ion, a 1-lyso-PA. These results indicate that differentiation between 1- and 2-lysophosphatic acid isomers by their product-ion spectra may not be feasible. Similarly, distinction between acidic isomers of 1- and 2-lysophosphatidylinositol is likely not possible.
Fig. 14.

The product-ion spectra of the [M − H]− ions of lyso1/18:1-PE at m/z 478 (A), of 18:1/lyso2-PE at m/z 478 (B), of p16:0/18:0-PE at m/z 698 (C), and of a16:0/18:0-PE at m/z 704 (D).
Both the product-ion spectra of plasmenyl (p16:0/18:2-PE, Fig. 14C), and plasmanyl (a16:0/18:0-PE, Fig. 14D) GPEtn are dominated by a single set of ions of RCO2−, [M − H –RCO2H]− and [M − H – R′CH=CO]−, that identify the fatty acid subtituent but not its location in the glycerol backbone. Ions reflecting the 1-O-alkyl or the 1-O-1-alkenyl moiety are absent, and structural characterization, in particular, differentiation between plasmanyl- and plasmenyl phospholipid subclasses are not applicable. However, multiple-stage LIT mass spectrometry with application of MS3 or MS4 on the [M − H – R2CO2H – polar head group]− ion arising from the [M − H]− ions of plasmenyl- and plasmanyl GPLs readily yield spectra applicable for distinction between a plasmenyl- and a plasmanyl GPLs [15]. The product-ion spectra arising from the [M + Li]+ ions of these two subclasses as described earlier (section 4.2.3.) also give distinction between these two ether phospholipid subclasses.
4.3.8. Phospholipid consisting of polyunsaturated fatty acid substituents
GPL molecules consisting of polyunsaturated fatty acyl substituents are unique intheir class, regarding to their CAD tandem mass spectrometry. The uniqueness in the product-ion spectra of the [M + Li]+ ions leads to their structural assignment (section 4.2.2. and section 4.2.3.). In negative-ion mode, the product-ion spectra of the [M − H]− ions of GPLs consisting of polyunsaturated fatty acyl substituents are also readily distinguishable from those consisting of saturated fatty acid substituents. The distinction between them by tandem mass spectrometry is mainly attributable to the fact that the polyunsaturated carboxylate anions formed by CAD undergo vigorous secondary dissociation, while the saturated carboxylate anions undergo minimal secondary dissociation after formation in a TSQ instrument [33,55].
This consecutive dissociation process results in a drastic change in the abundances of the R2CO2− and R1CO2− anions observed in the spectra [33]. For example, the tandem quadrupole product-ion spectrum of the 16:0/20:4-PE (Fig. 15A), contains ions at m/z 303 and 255, representing a 20:4- and a 16:0-carboxylate anions, respectively, but the spectrum also contains the m/z 259 ion generated by loss of CO2 from m/z 303. The product-ion spectrum of 16:0/22:6-PE (Fig. 15B) contains the 16:0- and 22:6-carboxylate anions at m/z 255 and 327, but an abundant ion at m/z 283 (327 – CO2) is also present. This further elimination of CO2 from polyunsaturated carboxylate anion is also observed for 18:0/22:6-PE (Fig. 15C), resulting in drastic decline in the abundance of m/z 327, corresponding to the 22:6 carboxylate anion at sn-2, and the rise of m/z 283 (327 - CO2) (C21H31−, F.W. = 283.242), which overlaps with the 18:0-carboxylate anion (C17H35CO2−, F.W. = 283.264) arising from sn-1 because they are not resolvable by a quadrupole mass spectrometer. Similar result is also seen in the product-ion spectrum of 18:0/22:6-PG (Fig. 15D), in which the ion at m/z 327 arising from 22:6-fatty acid substituent at sn-2 is less abundant than the ion at m/z 283, corresponding to the 18:0-fatty acid anion at sn-1, a reversal to that observed for 16:0/18:2-PE (Fig. 4A) and 16:0/18:1-PG (Fig. 12A), in which secondary dissociation of the carboxylate anions is minimal.
Fig. 15.

The product-ion spectra of the [M − H]− ions of 16:0/20:4-PE at m/z 738 (A), of 16:0/22:6-PE at m/z 762 (B), of 18:0/22:6-PE at m/z 790 (C), and of 22:6/22:6-PG at m/z 865 (D).
The product-ion spectra obtained with a TSQ instrument are often dominated by the RxCO2− ions, which are informative for identification of the fatty acid substituents. The differential formation of the R1CO2− and R1CO2− ions also has been previously suggested for determination of the position of the fatty acid substituents on the glycerol backbone [36,47]. However, the abundances of the R2CO2− and R1CO2− ions are governed not only by the stability of carboxylate anions after they were generated by CAD as described above, but also by the polar head group that distinguishes the various phospholipid classes. An unusual abundance ratio of R2CO2−/R1CO2− has also been reported for GPLs that contain very short chain fatty acyl (< C10) (49) and the GPGro species consisting of Δ316:1-fatty acid substituent [16]. In contrast, the fragment ions arising from losses of the fatty acyl substituent as an acid or as a ketene at sn-2 are more abundant than those arising from the corresponding losses at sn-1 in the product ion spectra of the [M − H]− ions from all of GPL classes, regardless of the chain length, the number and the position of the unsaturated bond(s) of the fatty acid substituents. Thus, the position of the fatty acyl substituents on the glycerol backbone can be unambiguously determined. In this respect, structural characterization using ITMS is attractive, because the ions reflecting those losses in the MS2-spectra are often prominent and rarely undergo secondary degradation.
5. Conclusions
The awareness of the mechanisms underlying the fragmentation processes under low-energy CAD with tandem quadrupole mass spectrometry and with multiple-stage ion-trap mass spectrometry as seen for the various molecular species generated by ESI in negative-and positive-ion modes has facilitated structural identification of the more complicated phospholipid species such as acyl-PG [56], cardiolipins [31,32,57], phosphatidylinositol mannosides [58,59], and GPGro containing unique fatty acid substituent [16],. The structural characterization of these complex lipids using the traditional methods would involve laborious analytical techniques including chromatographic separation, chemical reaction and NMR spectroscopy. The tandem mass spectrometric approaches as described here, in contrast, are relatively simple and the structures of complex lipids, in particular, from biological specimen that often contain various isomers can be revealed in detail [32,58,59]. However, understanding the mechanisms underlying the fragmentation processes for various phopholipid classes is required to avoid misinterpretation.
Acknowledgments
The author acknowledges the support of US Public Health Service Grants P41-RR-00954, R37-DK-34388, P60-DK-20579, P30-DK56341 and P01-HL-57278.
Abbreviations
- GPLs
glycerophospholipids
- GPCho
glycerophosphocholine
- GPEtn
glycerophosphoethanolamine
- GPSer
glycerophosphoserine
- GPGro
glycerophosphoglycerol
- GPIno
glycerophosphoinositol
- GPA
glycerophosphatidic acid
- CAD
collisionally activated dissociation
- IT
ion-trap
- LIT
linear ion-trap
- TSQ
triple-stage quadrupole
- ESI
electrospray ionization
- MALDI
matrix-assisted laser desorption/ionization
- MS
mass spectrometry
- FAB
fast-atom bombardment
- FT-ICR
Fourier-transform ion-cyclotron-resonance
- TOF
time-of-flight
- Q-oTOF
quadrupole-time-of-flight
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1989;246:64. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 2.Yamashita M, Fenn JB. J Phys Chem. 1984;88:4451. [Google Scholar]
- 3.Karas M, Hillenkamp F. Anal Chem. 1988;60:2299. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 4.Karas M, Bachmann D, Bahr U, Hillenkamp F. Int J Mass Spectrom Ion Proc. 1987;78:53. [Google Scholar]
- 5.Weintraub ST, Pinckard RN, Hail M. Rapid Commun Mass Spectrom. 1991;5:309. doi: 10.1002/rcm.1290050702. [DOI] [PubMed] [Google Scholar]
- 6.Han X, Gross RW. J Am Soc Mass Spectrom. 1995;6:1202. doi: 10.1016/1044-0305(95)00568-4. [DOI] [PubMed] [Google Scholar]
- 7.Kim HY, Wang TC, Ma YC. Anal Chem. 1994;66:3977. doi: 10.1021/ac00094a020. [DOI] [PubMed] [Google Scholar]
- 8.Pulfer M, Murphy RC. Mass Spectrom Rev. 2003;22:332. doi: 10.1002/mas.10061. [DOI] [PubMed] [Google Scholar]
- 9.Griffiths WJ. Mass Spectrom Rev. 2003;22:81. doi: 10.1002/mas.10046. [DOI] [PubMed] [Google Scholar]
- 10.Han X, Gross RW. Mass Spectrom Rev. 2005;24:367. doi: 10.1002/mas.20023. [DOI] [PubMed] [Google Scholar]
- 11.Schiller J, Suss R, Fuchs B, Muller M, Zschornig O, Arnold K. 2007;12:2568. doi: 10.2741/2255. [DOI] [PubMed] [Google Scholar]
- 12.Li YL, Gross ML, Hsu FF. J Am Soc Mass Spectrom. 2005;16:679. doi: 10.1016/j.jasms.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Larsen A, Uran S, Jacobsen PB, Skotland T. Rapid Commun Mass Spectrom. 2001;15:2393. doi: 10.1002/rcm.520. [DOI] [PubMed] [Google Scholar]
- 14.Ho YP, Huang PC. Rapid Commun Mass Spectrom. 2002;16:1582. doi: 10.1002/rcm.751. [DOI] [PubMed] [Google Scholar]
- 15.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2007 ;18:2065. doi: 10.1016/j.jasms.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu FF, Turk JJ, Williams TD, Welti R. J Am Soc Mass Spectrom. 2007;18:783. doi: 10.1016/j.jasms.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2008;19:1681. doi: 10.1016/j.jasms.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beckedorf AI, Schaffer C, Messner P, Peter-Katalinic J. J Mass Spectrom. 2002;37:1086. doi: 10.1002/jms.369. [DOI] [PubMed] [Google Scholar]
- 19.Fridriksson EK, Shipkova PA, Sheets ED, Holowka D, Baird B, McLafferty FW. Biochemistry. 1999;38:8056. doi: 10.1021/bi9828324. [DOI] [PubMed] [Google Scholar]
- 20.Jennings KR. Int J Mass Spectrom Ion Phys. 1968;1:227. [Google Scholar]
- 21.McLafferty FW. Tandem mass spectrometry. New York: John Wiley & Sons; 1983. [Google Scholar]
- 22.Gross ML. Mass Spectrom Rev. 1989;8:165. [Google Scholar]
- 23.Yost RA, Enke CG. J Am Chem Soc. 1978;100:2274. [Google Scholar]
- 24.Stafford GC, Kelley PE, Jr, Syka JEP, Reynolds WE, Todd JFJ. Int J Mass Spectrom Ion Processes. 1984;60:85. [Google Scholar]
- 25.Louris JN, Cooks RG, Syka JEP, Kelley GCPE, jr, Stafford JF, Todd J. Anal Chem. 1987;59:1677. [Google Scholar]
- 26.Kerwin JL, Tuininga AR, Ericsson LH. J Lipid Res. 1994;35:1102. [PubMed] [Google Scholar]
- 27.Harrison KA, Murphy RC. J Mass Spectrom. 1995;30:1772. doi: 10.1002/(SICI)1096-9888(199904)34:4<330::AID-JMS798>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 28.Hsu FF, Bohrer A, Turk J. J Am Soc Mass Spectrom. 1998;9:516. doi: 10.1016/S1044-0305(98)00012-9. [DOI] [PubMed] [Google Scholar]
- 29.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2005;16:1510. doi: 10.1016/j.jasms.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 30.Ho YP, Huang PC, Deng KH. Rapid Commun Mass Spectrom. 2003;17:114. doi: 10.1002/rcm.880. [DOI] [PubMed] [Google Scholar]
- 31.Hsu FF, Turk J, Rhoades ER, Russell DG, Shi Y, Groisman EA. J Am Soc Mass Spectrom. 2005;16:491. doi: 10.1016/j.jasms.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 32.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2006;17:420. doi: 10.1016/j.jasms.2005.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2000;11:986. doi: 10.1016/S1044-0305(00)00172-0. [DOI] [PubMed] [Google Scholar]
- 34.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2003;14:352. doi: 10.1016/S1044-0305(03)00064-3. [DOI] [PubMed] [Google Scholar]
- 35.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2000;11:595. doi: 10.1016/S1044-0305(00)00172-0. [DOI] [PubMed] [Google Scholar]
- 36.Murphy RC, Harrison KA. Mass Spectrom Rev. 1994;13:57. [Google Scholar]
- 37.Brugger B, Erben G, Sandhoff R, Wieland FT, Lehmann WD. Proc Natl Acad Sci U S A. 1997;94:2339. doi: 10.1073/pnas.94.6.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adams J, Gross ML. Org Mass Spectrom. 1988;23:307. [Google Scholar]
- 39.Jensen NJ, Tomer KB, Gross ML. J Am Chem Soc. 1985;107:1863. [Google Scholar]
- 40.Adams J. Mass Spectrom Rev. 1990;9:141. [Google Scholar]
- 41.F.F. Hsu, J. Turk, Unpublished results.
- 42.Hsu FF, Turk J, Thukkani AK, Messner MC, Wildsmith KR, Ford DA. J Mass Spectrom. 2003;38:752. doi: 10.1002/jms.491. [DOI] [PubMed] [Google Scholar]
- 43.Hsu FF, Turk J. J Am Soc Mass Spectrom. 1999;10:587. doi: 10.1016/S1044-0305(99)00035-5. [DOI] [PubMed] [Google Scholar]
- 44.Han X, Gross RW. J Am Chem Soc. 1996;118:451. [Google Scholar]
- 45.Kayganich KA, Murphy RC. Anal Chem. 1992;64:2965. doi: 10.1021/ac00047a015. [DOI] [PubMed] [Google Scholar]
- 46.Ramansdham S, Hsu FF, Bohrer A, Nowatzke W, Ma WZ, Turk J. Biochemistry. 1998;37:4553. doi: 10.1021/bi9722507. [DOI] [PubMed] [Google Scholar]
- 47.Jensen NJ, Tomer KB, Gross ML. Lipids. 1986;21:580. doi: 10.1007/BF02534056. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, Reid GE. Int J Mass Spectrom. 2006;252:242. doi: 10.1016/j.ijms.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang ZH, Gage DA, Sweeley CC. J Am Soc Mass Spectrom. 1992;3:71. doi: 10.1016/1044-0305(92)85020-K. [DOI] [PubMed] [Google Scholar]
- 50.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2000;11:797. doi: 10.1016/S1044-0305(00)00151-3. [DOI] [PubMed] [Google Scholar]
- 51.Hitchcock PB, Mason R, Thomas KM, Shipley GG. Proc Natl Acad Sci USA. 1974;71:3036. doi: 10.1073/pnas.71.8.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2000;11:892. doi: 10.1016/S1044-0305(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 53.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2001;12:1036. doi: 10.1016/S1044-0305(00)00194-X. [DOI] [PubMed] [Google Scholar]
- 54.Christie WW. Lipid analysis (2nd ed): isolation, separation, identification, and structural analysis of lipids: Chapter 1: The structure, chemistry and occurrence of lipids. Pergamon Press; New York: 1992. p. 1. [Google Scholar]
- 55.Kerwin JL, Wiens AM, Ericsson LH. J Mass Spectrom. 1996;31:184. doi: 10.1002/(SICI)1096-9888(199602)31:2<184::AID-JMS283>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 56.Hsu FF, Turk J, Shi Y, Groisman EA. J Am Soc Mass Spectrom. 2004;15:1. doi: 10.1016/j.jasms.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 57.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2006;17:1146. doi: 10.1016/j.jasms.2006.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsu FF, Turk J, Owens RM, Rhoades ER, Russell DG. J Am Soc Mass Spectrom. 2007;18:479. doi: 10.1016/j.jasms.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsu FF, Turk J, Owens RM, Rhoades ER, Russell DG. J Am Soc Mass Spectrom. 2007;18:466. doi: 10.1016/j.jasms.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]