

Abstract
Blood-brain barrier (BBB) Na transporters are essential for brain water and electrolyte homeostasis. However, they also contribute to edema formation during the early hours of ischemic stroke by increased transport of Na from blood into brain across an intact BBB. We previously showed that a luminal BBB Na-K-Cl cotransporter is stimulated by hypoxia, aglycemia, and AVP and that inhibition of the cotransporter by intravenous bumetanide significantly reduces edema and infarct in the rat middle cerebral artery occlusion (MCAO) model of stroke. More recently, we found evidence that intravenous cariporide (HOE-642), a highly potent Na/H exchange inhibitor, also reduces brain edema after MCAO. The present study was conducted to investigate which Na/H exchange protein isoforms are present in BBB endothelial cells and to evaluate the effects of ischemic factors on BBB Na/H exchange activity. Western blot analysis of bovine cerebral microvascular endothelial cells (CMEC) and immunoelectron microscopy of perfusion-fixed rat brain revealed that Na/H exchanger isoforms 1 and 2 (NHE1 and NHE2) are present in BBB endothelial cells. Using microspectrofluorometry and the pH-sensitive dye BCECF, we found that hypoxia (2% O2, 30 min), aglycemia (30 min), and AVP (1–200 nM, 5 min) significantly increased CMEC Na/H exchange activity, assessed as Na-dependent, HOE-642-sensitive H+ flux. We found that AVP stimulation of CMEC Na/H exchange activity is dependent on intracellular Ca concentration and is blocked by V1, but not V2, vasopressin receptor antagonists. Our findings support the hypothesis that a BBB Na/H exchanger, possibly NHE1 and/or NHE2, is stimulated during ischemia to participate in cerebral edema formation.
Keywords: blood-brain barrier, stroke, cerebral ischemia, brain edema, cariporide
cerebral edema is a serious, rapidly developing complication of ischemic stroke. Previous studies showed that, during the early hours of ischemia, edema forms by a process involving increased net flux of Na and water into the brain across an intact blood-brain barrier (BBB) (4, 43, 65) and that barrier breakdown does not occur until after ≥4 h of ischemia (31, 42, 65). Early in edema formation, perivascular astrocytic end feet rapidly swell as ions and water are transported across the BBB into the brain (11, 25, 30), and, in this regard, BBB Na transporters facilitate ischemia-induced swelling of astrocytes. Consistent with these findings, previous studies showed that luminal BBB Na transporters appear to be rate limiting in ischemia-induced cerebral edema formation (6, 27, 42, 43, 65). We and others hypothesized that ischemic factors stimulate activity of one or more luminal BBB membrane Na transporters, which, via functional coupling to the abluminal Na-K-ATPase, facilitates vectorial transport of Na across the BBB in the brain (14, 19, 20, 29, 50, 53, 65). Studies from our laboratory provide evidence that the BBB Na-K-Cl cotransporter is a major participant in ischemia-induced edema formation. Nevertheless, other BBB Na transporters may well also contribute to ischemia-induced edema formation.
Previous studies provide some evidence that a BBB Na/H exchanger may contribute to cerebral edema formation during ischemic stroke. The Na/H exchange inhibitors amiloride and EIPA have been found to reduce basal intracellular pH (pHi) in cultured cerebral microvascular endothelial cells (CMEC) (23, 67), suggesting that Na/H exchange occurs in these cells. In other studies, CMEC have been reported to exhibit an EIPA-sensitive 22Na influx that is stimulated by endothelin, another factor increased in the brain during ischemia (28, 74). Furthermore, the Na/H exchange inhibitor SM-20220 has been shown to reduce increases in brain water after ischemia-reperfusion via transient middle cerebral artery occlusion (MCAO), as well as after 3 days of permanent MCAO in the rat (33, 70). We also found evidence that intravenous administration of the highly selective Na/H exchange inhibitor HOE-642 reduces edema and infarct after MCAO in the rat (73). Na/H exchange is known to serve multiple functions, including regulation of pHi, cell volume, and cell proliferation in epithelial and nonepithelial cells (10, 37, 59), and it participates in vectorial transport of ions across a variety of epithelia (54). Nine Na/H exchanger isoforms have been identified: NHE1, NHE2, NHE3, NHE4, and NHE5 in the plasma membrane and NHE6, NHE7, NHE8, and NHE9 in organelle membranes (16, 44, 55, 71). NHE1, which is ubiquitously distributed in tissues and is best known for its role in pH and cell volume regulation, also functions in transport of ions across epithelia (16, 48, 75). Although NHE1 is most commonly found in the basolateral membrane of epithelial cells, it resides in the apical membrane of some Na-absorbing epithelia (48, 75). A variety of studies have shown that NHE2 and NHE3 can also reside in the apical membrane and function in Na absorption across some epithelial cells (7, 16, 22, 47, 69). Together, these findings suggest that the Na/H exchanger may be another luminal membrane BBB Na transporter that is stimulated by ischemic conditions to increase Na transport across the BBB from blood into brain, thereby contributing to edema formation during ischemic stroke.
The present study was conducted to determine which Na/H exchanger isoforms are present in the BBB and to evaluate the effects of hypoxia, aglycemia, and AVP on Na/H exchange activity in CMEC. We report here that NHE1 and NHE2 are present in the luminal membrane of BBB endothelial cells and that Na/H exchange activity of CMEC is stimulated by hypoxia, aglycemia, and AVP. We further report that AVP stimulation of CMEC Na/H exchange activity appears to occur via a V1 vasopressin receptor- and Ca-dependent pathway.
MATERIALS AND METHODS
CMEC culture.
Bovine CMEC were maintained in DMEM that contained 5 mM d-glucose and 1 mM Na-pyruvate, supplemented with 2 mM l-glutamine, 50 μg/ml gentamicin, 1 ng/ml basic fibroblast growth factor, 5% calf serum, and 5% horse serum in an atmosphere of 95% air-5% CO2, as described previously (20). For Na/H exchange activity experiments, CMEC were grown on coverslips coated with collagen and attachment factor (Cell Systems, Kirkland, WA). For Western blot experiments, CMEC were grown on collagen- and fibronectin-coated 24-well plates for 8–10 days until confluent. Growth medium was replaced with fresh medium every other day. At 24–48 h before each experiment, cells were refed with a 50:50 (vol/vol) mixture of fresh DMEM containing 5% horse serum and 5% calf serum and astrocyte-conditioned medium (ACM) containing 10% FBS. ACM was prepared as described previously (50, 52).
Isolation of rat cerebral microvessels.
This study was conducted in accordance with the animal use and care guidelines issued by the National Institutes of Health, and the protocol was approved by the Animal Use and Care Committee at the University of California, Davis. Rat brains were removed within 5 min after pentobarbital sodium-induced euthanasia and placed in ice-cold 1× PBS. Brain tissue was maintained at 4°C in buffered medium containing protease inhibitors throughout the microvessel isolation procedure, except when the tissue was exposed to the digestive enzymes at 37°C. Under a dissecting microscope, the meninges were carefully removed, and pieces of the cerebrum were gently dissected away from the cortical surface. Cortical tissue was diced with a sharp razor blade, homogenized, treated with digestive enzymes, and then separated from single cells and larger vessels by filtration through a series of different pore-size mesh and finally through a glass bead column, as previously described (3). Once microvessels were isolated, they were lysed immediately and kept frozen in a −80°C freezer until they were needed for protein analysis.
Gel electrophoresis and Western blot analysis.
Western blot analysis was performed following methods we have described previously (52, 77). Briefly, CMEC monolayers on 24-well plates were rinsed twice with ice-cold PBS containing 5 mM EDTA (PBS-EDTA) + protease inhibitors (Complete Protease Inhibitor Cocktail tablet, Roche Diagnostic) and then lysed in PBS-EDTA containing 1% SDS + protease inhibitor. For preparation of lysates of rat cortical cerebral microvessels, freshly isolated microvessels were disrupted by sonication in PBS-EDTA containing 1% SDS + protease inhibitor. The protein concentration of CMEC and cerebral microvessels was then determined using the bicinchoninic acid method to ensure equal loading of membrane protein into each gel lane. Lysate samples and prestained molecular weight markers (Bio-Rad, Hercules, CA) were denatured in SDS reducing buffer containing DTT (Invitrogen NuPage, Carlsbad, CA) and heated to 70°C for 10 min and then loaded into gel lanes. Protein samples were electrophoretically separated on 7.5% Tris-glycine gels (PAGEr Gold Precast, Cambrex Rockland, ME; Mini-Protean II, Bio-Rad), and the resolved proteins were transferred to nitrocellulose membranes using a Bio-Rad Trans-Blot apparatus. The blots were then incubated in 7.5% nonfat dry milk + PBS-Tween for 1 h at room temperature. Subsequently, blots were incubated with NHE1 antibody {mouse monoclonal antibody 4E9 (Millipore, Bedford, MA) or rabbit polyclonal antibody XB17 [a gift from Gene Chang (9)]} or NHE2, NHE3, or NHE4 antibody (rabbit polyclonal AB3038, mouse monoclonal AB3136, or rabbit polyclonal AB3087, respectively; Millipore), rinsed three times with PBS-Tween, and then exposed to secondary antibodies (horseradish peroxidase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG; Zymed Laboratories, San Francisco, CA). After three PBS-Tween washes, protein bands were visualized using enhanced chemiluminescence (ECL, Amersham Biosciences, Little Chalfont Buckinghamshire, UK) with a Fuji Film LAS-3000 Imaging System (Medford, UK). Image Quant software (Molecular Dynamics, Sunnyvale, CA) was used to quantitate band density.
Immunocytochemistry.
Cultured CMEC grown on collagen-coated glass slides were fixed with 4% paraformaldehyde in 0.4 M phosphate buffer for 1 h, incubated with blocking solution (10% goat serum, 0.4% Triton X-100, and 1% BSA in PBS) for 1 h, and then incubated with rabbit polyclonal NHE1 antibody (XB17) or rabbit polyclonal NHE2 antibody in blocking solution overnight at 4°C and finally with donkey anti-rabbit biotin-conjugated antibody (1:500 dilution; Jackson ImmunoResearch Laboratories) for 1 h. Cells were exposed to Vectastain Elite ABS reagent to amplify the signal and then incubated (5 min) with diaminobenzidine reagent (Vector Laboratories). Cells were then counterstained with hematoxylin, which dyes the nuclei blue. Control cells were treated as described above but without primary antibody. Cell images were captured using a Leitz Diaplan microscope with a ProgRes 3012 digital camera and Adobe Photoshop 6.0 software.
Immunoelectron microscopy.
Rat brains were subjected to cardiac perfusion fixation for 60 min using 4% paraformaldehyde + 0.05% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were excised, postfixed in 4% paraformaldehyde overnight, and then subjected to freeze substitution, as described previously (21). Tissues were embedded in lowiacryl resin and sectioned onto carbon-coated grids and then immunolabeled with monoclonal NHE1 antibody (4E9) or polyclonal NHE2 antibody (AB3038) with 15-nm gold particle-conjugated anti-mouse IgG or anti-rabbit IgG, respectively, using methods we have described previously (53). Sections were stained with uranyl acetate and lead citrate and then observed using a Philips 410 electron microscope. For quantitatation of gold particle distribution in the microvessel plasma membranes, a total of 187 immunoelectron micrographs were examined, and gold particles were counted using a double-blind method. Data are presented as the percentage of total plasma membrane gold particles in the luminal membrane.
Na/H exchange activity assays.
The well-established NH4 prepulse method was used to assess Na/H exchange activity in cultured CMEC (12, 62). Briefly, CMEC monolayers grown on collagen- and attachment factor-coated glass coverslips were washed twice with HEPES-buffered medium that contained (in mM) 144 Na, 147 Cl, 5.8 K, 1.2 Ca, 0.4 HPO4, 0.4 H2PO4, 0.4 Mg, 0.4 SO4, 5.6 d-glucose, and 20 HEPES (pH 7.4, 290–300 mosM). CMEC were initially incubated in HEPES-buffered medium containing 5 μM BCECF-AM (Molecular Probes) for 30 min at 37°C and then placed in a closed, temperature-controlled perfusion-imaging chamber mounted on the stage of a Zeiss Axiovert inverted microscope. Before Na/H exchange activity measurement, CMEC were superfused with HEPES-buffered medium without BCECF-AM at a constant rate of 3 ml/min at 37°C for ≥10 min to wash out extracellular BCECF. For experiments evaluating hypoxia effects on Na/H exchange activity, CMEC were superfused with treatment medium containing varying levels of O2 for varying times. Treatment media were first equilibrated with the desired O2 level in a hypoxia chamber, and O2 levels in the media were verified by an O2 electrode (Corning). Equilibrated treatment medium was transferred to an airtight syringe that was then connected to the perfusion assembly while still in the hypoxia chamber, and the syringe and assembly were moved to the microscope stage. For experiments assessing the effects of aglycemia or AVP on Na/H exchange activity, CMEC monolayers were pretreated for 30 min with normoxic medium lacking glucose or containing AVP (0–200 nM), as described in previous studies (20). In some experiments, CMEC were also exposed to V1 and V2 vasopressin receptor agonists {[Phe2,Orn8]-vasotocin (Orn VP) or [deamino-Cys1,d-Arg8]-VP (DDAVP), respectively} or V1 or V2 vasopressin receptor antagonists {des-Gly9[phenylacetyl1,d-Tyr(Et)2,Lys6,Arg8]-VP (PhaaEt VP) and [d(CH2)51,d-Ile2,Ile4,Arg8,Ala-NH29]-VP (d-Ile VP), respectively} for 30 min. Also, in some experiments, CMEC were exposed to 17β-estradiol (0–100 nM) 24 h before use. For the Na/H exchange activity assay, pHi was calculated from the ratio of light intensities emitted at 535 nm after excitation at 440 and 490 nm. Images were collected every 10 s using a charge-coupled device camera and analyzed with OpenLab image-processing software. The high-K+-nigericin technique (13) was used to calibrate the ratio of fluorescence emissions (F490/F440) to pH. In each experiment, 20–30 cells were monitored per coverslip.
All experiments evaluating Na/H exchange activity were conducted using HEPES-buffered media that were nominally CO2 free (room air equilibrated) to minimize confounding activity of HCO3-dependent proton equivalent membrane transporters, as described previously (40, 41). After measurement of baseline pHi, cells were subjected to NH4 prepulse acidification by 5 min of exposure to 20 mM NH4Cl followed by NH4Cl washout in Na-free HEPES-buffered medium (HEPES) that contained (in mM) 147 Cl, 5.8 K, 1.2 Ca, 0.4 HPO4, 0.4 H2PO4, 0.4 Mg, 0.4 SO4, 5.6 d-glucose, 20 HEPES, and 147 choline (pH 7.4, 290–300 mosM). By this method, the mean baseline pHi and mean prerecovery pHi (observed after the prepulse at the start of the pH recovery period) did not differ significantly among experiments regardless of the experimental conditions (i.e., aglycemia, hypoxia, AVP, AVP agonists and antagonists, BAPTA, and estradiol). The initial rate of pH recovery from acidification was observed during the 60-s period after the medium was changed to one again containing Na (normal HEPES-buffered medium with 147 mM Na) and is expressed as ΔpHi/Δt. To determine H+ flux (J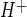 , in mM H+/min), this rate was multiplied by intracellular buffer capacity (β), assessed over the range of pHi values pertinent to these studies, as described previously (40, 41). Briefly, CMEC were perfused with progressively decreasing concentrations of NH4 in HEPES medium, and β was calculated as ΔNH4/ΔpHi. At the end of each experiment, cells were exposed to high-K medium (pH 7.0) containing 10 μM nigericin, and medium containing (in mM) 147 Cl, 113 K, 0.6 Ca, 1.02 Mg, 5.6 d-glucose, 20 HEPES, and 35.4 N-methyl-d-glucamine (a replacement for Na) was used for a calibration point.
, in mM H+/min), this rate was multiplied by intracellular buffer capacity (β), assessed over the range of pHi values pertinent to these studies, as described previously (40, 41). Briefly, CMEC were perfused with progressively decreasing concentrations of NH4 in HEPES medium, and β was calculated as ΔNH4/ΔpHi. At the end of each experiment, cells were exposed to high-K medium (pH 7.0) containing 10 μM nigericin, and medium containing (in mM) 147 Cl, 113 K, 0.6 Ca, 1.02 Mg, 5.6 d-glucose, 20 HEPES, and 35.4 N-methyl-d-glucamine (a replacement for Na) was used for a calibration point.
Materials.
DMEM and l-glutamine were purchased from GIBCO-BRL (Grand Island, NY); gentamicin from AG Scientific (San Diego, CA); FBS and calf serum from Hyclone (Logan, UT); horse serum, 17β-estradiol, and nigericin from Sigma (St. Louis, MO); BAPTA from EMD Biosciences/Calbiochem (San Diego, CA); BCECF-AM from Invitrogen/Molecular Probes (Carlsbad, CA); and AVP, as well as V1 and V2 vasopressin receptor agonists and antagonists, from Peninsula Laboratories (division of Bachem, San Carlos, CA). HOE-642 was a gift from Sanofi-Aventis Pharmaceuticals (Bridgewater, NJ).
Statistics.
Values are means ± SE. All experiments were done at least three times. Statistical analyses were done using StatView software (SAS Institute, Cary, NC). Comparisons of groups were made by ANOVA followed by Fisher's multiple-comparison test, unless otherwise indicated. P < 0.05 was considered significant.
RESULTS
Expression of NHE1 and NHE2 in cultured CMEC and freshly isolated rat microvessels.
If an Na/H exchanger contributes to Na transport across the BBB into the brain during cerebral ischemia, then the Na/H exchanger should be present in the luminal membrane of BBB endothelial cells. To test this notion, we first evaluated cultured bovine CMEC and freshly isolated rat microvessels for the presence of Na/H exchanger protein by Western blot analysis. Using specific antibodies that recognize NHE1, NHE2, NHE3, and NHE4, we evaluated CMEC and microvessels for the presence of these isoforms. As shown in Fig. 1, A and B, Western blots of lysates prepared from CMEC, as well as microvessels, revealed prominent bands for NHE1 and NHE2. Single ∼110- and ∼93-kDa bands were observed for blots probed with polyclonal NHE1 antibody (XB17; Fig. 1A) or polyclonal NHE2 antibody (AB3038; Fig. 1B, left and middle), respectively. A single ∼110-kDa band was also observed in Western blots using the monoclonal 4E9 NHE1 antibody. To validate the identity of the ∼93-kDa band as NHE2 protein, we conducted Western blot analysis after preincubation of the polyclonal NHE2 antibody with an NHE2 control peptide (AG784, Millipore). As shown in Fig. 1B (right), the ∼93-kDa band was no longer observed. In Western blots using NHE3 monoclonal antibodies, no immunoreactive bands were detected in CMEC or microvessel lysates, whereas a prominent band was observed for a kidney lysate-positive control (data not shown). In Western blots using a polyclonal NHE4 antibody, we did not find unequivocal evidence of NHE4 in CMEC, whereas a prominent band was present in the positive control kidney lysates (not shown).
Fig. 1.

Evaluation of Na/H exchange (NHE) protein isoforms in cerebral microvascular endothelial cells. A and B: lysates of bovine cerebral microvascular endothelial cells (CMEC, 5 μg) and freshly isolated rat cerebral microvessels (10 μg) were subjected to Western blot analysis (duplicate lanes are shown). A: blots were incubated with primary antibody to NHE1 protein (rabbit polyclonal antibody XB17 for CMEC and mouse monoclonal antibody 4E9 for microvessels) and then with secondary antibody, and bands were visualized by enhanced chemiluminescence. B: blots were incubated with primary antibody to NHE2 protein (rabbit polyclonal antibody AB3038 for CMEC and microvessels or NHE2 antibody in the presence of NHE control peptide) and then incubated with secondary antibody, and bands were visualized by enhanced chemiluminescence. Western blots are representative of 4 similar results for NHE1 and 3 similar results for NHE2. C and D: CMEC monolayers grown on collagen-coated glass slides were fixed and then incubated with NHE1 rabbit polyclonal primary antibody (XB17) or NHE2 polyclonal primary antibody (Millipore) and then with biotin-conjugated secondary antibody. Bound antibodies were visualized using diaminobenzidine (brown). Control slides were incubated with secondary antibody only. Slides were counterstained with hematoxylin for visualization of nuclei (blue). Images are representative of 3 similar results for NHE1 and 2 similar results for NHE2.
We also subjected CMEC to immunocytochemical analysis for NHE1 and NHE2. Confluent CMEC grown on collagen-coated glass slides were immunostained with NHE1 or NHE2 antibodies (see materials and methods). We observed NHE1 immunoreactivity (Fig. 1C, left), appearing as brown stain in the CMEC. When CMEC were exposed to secondary antibody only (Fig. 1C, right), no brown staining was observed. In these experiments, cells were counterstained with hematoxylin, which stains the nuclei blue. As shown in Fig. 1D, NHE2 protein was also observed in the CMEC (brown staining), whereas the negative control (secondary antibody only) shows no staining (only blue nuclei). Because the CMEC monolayers are very flat, it is not possible to reasonably discriminate between NHE1 and NHE2 staining that may be present in the cell membrane vs. the cytoplasm.
NHE1 and NHE2 are present in rat brain microvessel endothelial cells in situ.
The hypothesis that a BBB Na/H exchanger participates in Na transport from blood into the brain during ischemic stroke predicts that the Na/H exchanger will be present in the luminal membrane of BBB endothelial cells in situ. Thus we used immunoelectron microscopy to conduct experiments to evaluate the in situ distribution of Na/H exchanger proteins in cerebral microvessels of perfusion-fixed rat brains. Figure 2 shows representative immunoelectron micrographs generated using NHE1 (A and B) and NHE2 (C and D) antibodies (at 2 different dilutions each) and gold particle-conjugated secondary antibodies. We found that NHE1 protein is indeed present at the BBB luminal membrane, as indicated by the presence of gold particles in the images (Fig. 2, A and B). NHE2 is also present at the BBB luminal membrane (Fig. 2, C and D). Some electron micrographs showed evidence of NHE1 and NHE2 in the cytoplasm. However, the focus of these studies was to determine whether one or both Na/H exchanger isoforms reside at the luminal BBB membrane; thus the extent to which NHE1 and NHE2 are found in cytoplasmic compartments will need to be clarified in future studies. To quantitate the distribution of Na/H exchanger protein in BBB luminal and abluminal membranes, a total of 187 immunoelectron micrographs generated for both primary antibodies were examined. As shown in Fig. 3, we found that NHE1 and NHE2 are distributed predominantly in the luminal BBB membrane, with ∼67% of NHE1 in the luminal membrane (33% in the abluminal membrane) and ∼70% of NHE2 in the luminal membrane (30% in the abluminal membrane).
Fig. 2.

Immunoelectron microscopy localization of NHE proteins in rat brain microvascular endothelial membranes. Rat brains were perfusion fixed and then labeled with NHE1 primary antibody at dilutions of 1:1,000 and 1:2,000 (A and B, respectively) or NHE2 primary antibody at dilutions of 1:500 and 1:1,000 (C and D, respectively) and then with gold particle-conjugated secondary antibody. Images are representative micrographs. Vessel lumens are at the top of each image; astrocyte and neuronal elements are below the basal lamina underlying the endothelium. Arrowheads show locations of gold particles. EC, endothelial cell; N, nucleus. Scale bars, 0.2 μm.
Fig. 3.

NHE1 and NHE2 distribution between luminal and abluminal membranes of rat brain microvascular endothelial cells: quantitation of immunoelectron micrograph gold particles. Immunoelectron micrographs generated using NHE1 or NHE2 antibodies were evaluated for relative distribution of NHE isoforms in luminal (L) and abluminal (A) membranes of brain microvascular endothelial cells in perfusion-fixed rat brains. Values are means ± SE of 26 and 24 microvessels for NHE1 antibody dilutions of 1:1,000 and 1:2,000, respectively, and 56 and 81 microvessels for NHE2 antibody dilutions of 1:500 and 1:1,000, respectively.
Hypoxia, aglycemia, and vasopressin stimulate CMEC Na/H exchange activity.
The hypothesis addressed in these studies includes the prediction that Na/H exchange activity of BBB endothelial cells is stimulated by one or more factors during ischemia. Thus we determined whether hypoxia, aglycemia, and/or AVP, three prominent factors in cerebral ischemia, stimulate BBB Na/H exchange activity. We used the NH4 prepulse method with the pH-sensitive dye BCECF to evaluate Na/H exchange activity in CMEC (Fig. 4). A representative experiment demonstrating Na/H exchange activity in CMEC is shown in Fig. 4A. By this method, exposure of the cells to NH4 causes intracellular alkalinization, and subsequent exposure to NH4-free perfusion medium causes rapid acidification. The pH remains acidic and does not recover if the cells are maintained in Na-free medium (Na-free HEPES in Fig. 4A), nor does it recover if the cells are placed in medium containing Na with the Na/H exchange inhibitor HOE-642 (cariporide, 25 μM). When the cells are exposed to perfusion medium containing Na without HOE-642, pH rapidly recovers, indicating an Na-dependent, HOE-642-sensitive J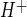 , or Na/H exchange activity, as described previously (40, 41). The trace shown in Fig. 4A represents the average values assessed for 30 cells in the field of view. For each experiment, J
, or Na/H exchange activity, as described previously (40, 41). The trace shown in Fig. 4A represents the average values assessed for 30 cells in the field of view. For each experiment, J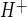 was calculated from the initial rate of pHi recovery after NH4 washout and return to normal Na+-, HOE-642-free perfusate. We found an HOE-642-sensitive and Na-dependent J
was calculated from the initial rate of pHi recovery after NH4 washout and return to normal Na+-, HOE-642-free perfusate. We found an HOE-642-sensitive and Na-dependent J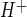 of 0.23 ± 0.01 mM H+/min (HEPES alone in Fig. 4A); in the absence of Na or in the presence of HOE-642, however, J
of 0.23 ± 0.01 mM H+/min (HEPES alone in Fig. 4A); in the absence of Na or in the presence of HOE-642, however, J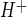 was not measurably different from zero (−0.003 ± 0.011 and 0.002 ± 0.002 mM H+/min for Na-free and HEPES + HOE-642, respectively). Because ACM has been shown to increase expression and activity of the CMEC Na-K-Cl cotransporter, we also tested whether ACM affected Na/H exchange activity in CMEC. Na/H exchange activities for bovine CMEC were 0.25 ± 0.01 and 0.26 ± 0.01 mM H+/min for cells treated without and with ACM, respectively (data not shown), suggesting that ACM does not have an effect on CMEC Na/H exchange activity. HOE-642 has been reported to be highly selective for NHE1, with an IC50 of 0.05 μM, but it also inhibits NHE2 at higher doses, with an IC50 of 3 μM (39). As an initial assessment of whether NHE1, NHE2, or both contribute to CMEC Na/H exchange activity, we evaluated the HOE-642 dose dependence for Na/H exchange inhibition. As shown in Fig. 4B, HOE-642 inhibited bovine CMEC Na/H exchange activity, with an IC50 of ∼7 μM, and it abolished Na/H exchange activity at 25 μM.
was not measurably different from zero (−0.003 ± 0.011 and 0.002 ± 0.002 mM H+/min for Na-free and HEPES + HOE-642, respectively). Because ACM has been shown to increase expression and activity of the CMEC Na-K-Cl cotransporter, we also tested whether ACM affected Na/H exchange activity in CMEC. Na/H exchange activities for bovine CMEC were 0.25 ± 0.01 and 0.26 ± 0.01 mM H+/min for cells treated without and with ACM, respectively (data not shown), suggesting that ACM does not have an effect on CMEC Na/H exchange activity. HOE-642 has been reported to be highly selective for NHE1, with an IC50 of 0.05 μM, but it also inhibits NHE2 at higher doses, with an IC50 of 3 μM (39). As an initial assessment of whether NHE1, NHE2, or both contribute to CMEC Na/H exchange activity, we evaluated the HOE-642 dose dependence for Na/H exchange inhibition. As shown in Fig. 4B, HOE-642 inhibited bovine CMEC Na/H exchange activity, with an IC50 of ∼7 μM, and it abolished Na/H exchange activity at 25 μM.
Fig. 4.

Evaluation of CMEC NHE activity as Na-dependent, HOE-642-sensitive H+ flux. NH4 prepulse technique was used to evaluate NHE activity in CMEC monolayers grown on collagen-coated glass coverslips. A: representative NH4 prepulse experiment showing Na-dependent, HOE-642-sensitive pH recovery from NH4 (20 mM) prepulse-induced intracellular acidification. Data are averages of 30 cells in the field of view. All HEPES-buffered media contain 147 mM Na, except Na-free HEPES. NHE inhibitor HOE-642 was present at 10 μM. At the end of each experiment, cells were exposed to pH 5.5–8.5 media containing high-K+-nigericin to calibrate pH. B: dependence of CMEC H+ efflux inhibition on dose of HOE-642. CMEC were assessed for NH4 prepulse-induced H+ flux in the presence of 0–80 μM HOE-642. HOE-642 inhibited Na-dependent H+ flux, with IC50 of ∼7 μM. Values are means ± SE of 3 separate dose-response experiments.
We next evaluated the effects of hypoxia, aglycemia, and AVP on CMEC Na/H exchange activity (Fig. 5). Exposure of cells to 30 min of glucose-free medium or hypoxia (∼2% O2) before and during assessment of Na/H exchange activity, as described for Fig. 4A, caused significant increases in CMEC Na/H exchange activity. For each set of experiments, the mean prerecovery pHi values did not differ significantly among experimental conditions. Thus, for the data shown in Fig. 5A, prerecovery pHi values were 6.588 ± 0.041 and 6.588 ± 0.041 for control and aglycemia, respectively (left), 6.670 ± 0.076 and 6.641 ± 0.055 for control and hypoxia, respectively (middle), and 6.430 ± 0.054 and 6.450 ± 0.021 for control and AVP, respectively (right). Exposure of CMEC to 5 min of AVP (100 nM) caused a robust increase in Na/H exchange activity from control levels to 0.61 ± 0.06 mM H+/min cells (Fig. 5A). In these experiments, exposure of cells to aglycemia or hypoxia before the NH4 prepulse did not cause a significant change in baseline pHi. Thus baseline pHi was 7.002 ± 0.042 and 6.986 ± 0.028 for control and aglycemia-exposed cells, respectively, and 7.018 ± 0.019 and 6.976 ± 0.017 for control and hypoxia-exposed cells, respectively. Baseline conditions were the same (cells were not treated with AVP until 5 min before Na/H exchange assay). Here, baseline pHi values were 6.958 ± 0.0.021 and 6.953 ± 0.025 for control and AVP experiments. These findings are valid for the remaining experiments reported in the present study (Figs. 6–8). None of the experimental conditions significantly altered baseline pHi before NH4 prepulse or the prerecovery pHi (data not shown).
Fig. 5.

Effects of AVP, hypoxia, and aglycemia on CMEC NHE activity measured as HOE-642-sensitive H+ flux. A: confluent CMEC monolayers grown on glass coverslips were exposed for 30 min to glucose or glucose-free medium (left), hypoxic (∼2% O2) or normoxic (middle) medium, or 0 or 100 nM AVP (right), and NHE activity was assessed. Values are means ± SE of 4 experiments each for control and aglycemia, 3 experiments each for control and hypoxia, and 6 and 3 experiments for control and 100 nM AVP, respectively. *Significantly different from control: P < 0.03, P < 0.05, and P < 0.0001 for aglycemia, hypoxia, and AVP, respectively (Student's t-test). B: CMEC monolayers were exposed to 0–200 nM AVP for 5 min, and NHE activity was assessed as HOE-642-sensitive H+ flux. AVP was also present during the assay. Values are means ± SE of 3 experiments for each condition. *Significantly different from control (i.e., 0 nM AVP): P < 0.013, P < 0.002, P < 0.0001, and P < 0.0001 for 10, 50, 100, and 200 nM AVP, respectively.
Fig. 6.

Effects V1 and V2 vasopressin receptor agonists and antagonists on NHE activity measured as HOE-642-sensitive H+ flux. A: CMEC monolayers grown on coverslips were exposed to 0 or 100 nM AVP, the V1 vasopressin receptor agonist [Phe2,Orn8]-vasotocin (Orn VP, 0 or 100 nM), or the V2 vasopressin receptor agonist [deamino-Cys1,d-Arg8]-VP (DDAVP, 0 or 100 nM) for 5 min, and NHE activity was assessed in the presence of AVP or the agonists. Values are means ± SE of 10, 5, 5, and 6 experiments for control, AVP, DDAVP, and Orn VP, respectively. *Significantly different from control: P < 0.0001 for 100 nM AVP and 100 nM Orn VP. B: bovine CMEC monolayers grown on glass coverslips were pretreated for 5 min in medium containing 0 or 100 nM AVP and the V1 vasopressin receptor antagonist des-Gly9[phenylacetyl1,d-Tyr(Et)2,Lys6,Arg8]-VP (PhaaEt VP, 0 or 100 nM) or the V2 vasopressin receptor antagonist [d(CH2)51,d-Ile2,Ile4,Arg8,Ala-NH29]-VP (d-Ile VP, 0 or 100 nM), and NHE activity was assessed in the presence of AVP ± antagonists. Values are means ± SE; n = 6 and 3 for controls without and with AVP, respectively; n = 12 and 10 for PhaaET VP without and with AVP, respectively; and n = 8 and 5 for d-Ile VP without and with AVP, respectively. *Significantly different from respective control (i.e., −AVP): P < 0.0001 for both with 100 nM AVP and 100 nM d-Ile VP with 100 nM AVP. C: CMEC were pretreated with 100 nM AVP + 0–100 nM PhaaEt VP or d-Ile VP, and NHE activity was assessed in the presence of the same concentrations of AVP and antagonists. Values are means ± SE for 38 individual experiments for d-Ile VP dose response and 46 separate experiments for PhaaET VP dose response. *Significantly different from NHE activity in the presence of AVP without antagonist: P < 0.0001 for all concentrations of antagonist tested. NHE activity in the presence of AVP with the V2 vasopressin receptor antagonist d-Ile VP was not significantly different from NHE activity in the presence of AVP alone at any concentration of d-Ile tested.
Fig. 8.

Estradiol inhibition of AVP-stimulated CMEC NHE activity. A: CMEC were pretreated with 0–100 nM 17β-estradiol (E2) 1 day before use, and NHE activity was measured as HOE-642-sensitive H+ flux. Values are means ± SE of 3, 3, 4, and 4 experiments for control, 1 nM E2, 10 nM E2, and 100 nM E2, respectively. *Significantly different from control without E2: P < 0.0026 for 1, 10, and 100 nM E2. B: CMEC monolayers on glass coverslips were treated with 0–100 nM E2 1 day before use. On the day of the experiment, cells were exposed to 100 nM AVP for 10 min, and NHE activity was measured as HOE-642-sensitive H+ flux in the presence of AVP and 1–100 nM E2. Values are means ± SE of 3 experiments for each condition. *Significantly different from control: P < 0.0001 for 100 nM AVP. #Significantly different from AVP without E2: P < 0.0001 for 1, 10, and 100 nM E2 with AVP, respectively.
Because of the magnitude of the AVP effect on Na/H exchange activity, we chose to focus the remainder of our investigation on the response of the CMEC Na/H exchange to AVP. We first evaluated the dose dependence of AVP effects on the Na/H exchanger. As shown in Fig. 5B, we found that 5 min of exposure of CMEC to ≥10 nM AVP significantly increased Na/H exchange activity.
AVP stimulates CMEC Na/H exchange activity via V1 vasopressin receptors.
Depending on the cell type, AVP can act via V1 or V2 vasopressin receptors, which are linked to activation of phospholipase C and adenylate cyclase, respectively (56). Furthermore, AVP has been reported to stimulate Na/H exchange activity by V1 vasopressin receptors in human platelets (1), but by V2 vasopressin receptors in renal collecting duct cells (61). To determine whether AVP stimulation of CMEC Na/H exchange activity occurs via V1 or V2 vasopressin receptors, we tested the effects of selective V1 and V2 vasopressin receptor agonists and antagonists on HOE-642-sensitive J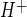 in CMEC. As shown in Fig. 6A, 5 min of treatment of CMEC with the V1 vasopressin receptor agonist Orn VP (100 nM) increased Na/H exchange activity to values not significantly different from those obtained with 100 nM AVP, whereas 30 min of treatment with the V2 vasopressin receptor agonist DDAVP (100 nM) did not affect Na/H exchange activity. Next, we tested the ability of V1 and V2 vasopressin antagonists to inhibit AVP-stimulated CMEC Na/H exchange activity (Fig. 6B). We found that stimulation of CMEC Na/H exchange activity induced by 5 min of treatment with AVP (100 nM) was abolished when the V1 vasopressin antagonist PhaaET VP (100 nM) was also present, whereas the V2 vasopressin receptor antagonist d-Ile VP (100 nM) did not significantly alter AVP-stimulated CMEC Na/H exchange activity. The dose response for the PhaaEt VP effect on AVP-induced Na/H exchange stimulation is shown in Fig. 6C: 5 nM PhaaEt VP significantly reduced Na/H exchange activity in the presence of 100 nM AVP, and 10 nM PhaaEt VP caused maximal inhibition. The V2 vasopressin receptor antagonist d-Ile VP had no significant effect on AVP-stimulated Na/H exchange activity in CMEC at any of the doses tested. Neither PhaaEt VP nor d-Ile VP in the absence of AVP altered Na/H exchange activity, i.e., HOE-642-sensitive J
in CMEC. As shown in Fig. 6A, 5 min of treatment of CMEC with the V1 vasopressin receptor agonist Orn VP (100 nM) increased Na/H exchange activity to values not significantly different from those obtained with 100 nM AVP, whereas 30 min of treatment with the V2 vasopressin receptor agonist DDAVP (100 nM) did not affect Na/H exchange activity. Next, we tested the ability of V1 and V2 vasopressin antagonists to inhibit AVP-stimulated CMEC Na/H exchange activity (Fig. 6B). We found that stimulation of CMEC Na/H exchange activity induced by 5 min of treatment with AVP (100 nM) was abolished when the V1 vasopressin antagonist PhaaET VP (100 nM) was also present, whereas the V2 vasopressin receptor antagonist d-Ile VP (100 nM) did not significantly alter AVP-stimulated CMEC Na/H exchange activity. The dose response for the PhaaEt VP effect on AVP-induced Na/H exchange stimulation is shown in Fig. 6C: 5 nM PhaaEt VP significantly reduced Na/H exchange activity in the presence of 100 nM AVP, and 10 nM PhaaEt VP caused maximal inhibition. The V2 vasopressin receptor antagonist d-Ile VP had no significant effect on AVP-stimulated Na/H exchange activity in CMEC at any of the doses tested. Neither PhaaEt VP nor d-Ile VP in the absence of AVP altered Na/H exchange activity, i.e., HOE-642-sensitive J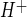 was 0.22 ± 0.00 and 0.23 ± 0.01 mM H+/min for cells treated with 100 nM PhaaEt VP alone and 100 nM d-Ile VP, respectively, compared with 0.22 ± 0.01 mM H+/min for control without AVP or antagonists.
was 0.22 ± 0.00 and 0.23 ± 0.01 mM H+/min for cells treated with 100 nM PhaaEt VP alone and 100 nM d-Ile VP, respectively, compared with 0.22 ± 0.01 mM H+/min for control without AVP or antagonists.
The binding of AVP to V1 vasopressin receptors has been shown to activate phospholipase C, generating inositol 1,4,5-triphosphate, which increases intracellular Ca concentration ([Ca]i), and diacylglycerol, which activates protein kinase C (64). If AVP stimulates CMEC Na/H exchange activity via V1 vasopressin receptors, it is predicted to be a [Ca]i-dependent event. To test this prediction, we examined the effects of the Ca chelator BAPTA-AM on AVP-induced stimulation of Na/H exchange activity in CMEC (Fig. 7). We found that stimulation of Na/H exchange activity by AVP (5 min, 100 nM) was abolished in cells pretreated with BAPTA-AM (30 min, 5 μM), whereas BAPTA-AM alone did not significantly alter Na/H exchange activity.
Fig. 7.

Ca dependence of AVP-stimulated CMEC NHE activity measured as HOE-642-sensitive H+ flux. CMEC monolayers were exposed to medium containing 0 or 5 μM BAPTA-AM at 37°C for 20 min and then to BAPTA-AM-free medium for 5 min. CMEC were then treated for 5 min with 0 or 100 nM AVP in BAPTA-AM-free medium and finally assayed for 5 min with AVP-containing, BAPTA-AM-free medium. Values are means ± SE of 5 and 6 experiments for control with and without AVP, respectively (both without BAPTA) and 5 and 3 experiments for cells treated with BAPTA with and without AVP, respectively. *Significantly different from control without AVP: P < 0.0001 for control with AVP.
Estradiol attenuates AVP-stimulated CMEC Na/H exchange activity.
Many studies have shown that estradiol is neuroprotective in stroke (18, 24, 72, 76). Our laboratory previously found that estradiol attenuates MCAO-induced cerebral edema and infarct in ovariectomized rats (51). We also found that acute and chronic exposures to estradiol reduce hypoxia- and AVP-induced stimulation of CMEC Na-K-Cl cotransporter activity, suggesting that estradiol's neuroprotective effects may be mediated in part by reducing ischemic factor stimulation of BBB cotransporter activity. If, as our studies suggest, ischemic factor stimulation of BBB Na/H exchange activity also contributes to ischemia-induced edema and infarct, then it is possible that estradiol has an inhibitory effect on the BBB Na/H exchanger. First, we evaluated the estradiol dose-dependent effects on the Na/H exchanger in CMEC. We found that exposure of CMEC to ≥1 nM estradiol for 1 day significantly reduced Na/H exchange activity of CMEC (Fig. 8A). We next evaluated the effect of acute estradiol exposure on AVP-stimulated Na/H exchange activity. We treated CMEC for 5 min with AVP (100 nM) alone or AVP + estradiol (1–100 nM). We found that 1 nM estradiol significantly reduced AVP-stimulated Na/H exchange activity in the cells and that 10 and 100 nM estradiol abolished AVP stimulation of Na/H exchange activity (Fig. 8B).
DISCUSSION
Previous studies provided evidence that a BBB Na/H exchanger may participate in ischemia-induced edema formation (33, 35, 70, 73). However, it has not been known what isoforms of the Na/H exchanger are present in BBB endothelial cells, nor is it known whether the Na/H exchanger is stimulated by factors present during cerebral ischemia. We demonstrate here that NHE1 and NHE2 proteins are expressed in brain microvascular endothelial cells and that both are present predominantly in the luminal BBB membrane. We also show that the ischemic factors hypoxia, aglycemia, and AVP are stimulators of Na/H exchange in brain microvascular endothelial cells. Together, these findings support the hypothesis that ischemia stimulates luminal BBB NHE1 and/or NHE2 to participate in edema formation during ischemic stroke. Our studies further reveal that AVP stimulation of BBB Na/H exchange activity occurs through a V1 vasopressin receptor- and Ca-dependent pathway. Finally, we provide evidence that estradiol, which reduces edema in ischemic stroke, may act in part by attenuating AVP-induced stimulation of BBB Na/H exchange activity.
Our studies are the first to demonstrate that brain microvascular endothelial cells express NHE1 and NHE2 proteins. This finding is consistent with previous reports that rat CMEC express mRNA for NHE1 (26) as well as NHE2 (67). However, these previous studies were limited to measurement of mRNA and did not evaluate the cells for expression of Na/H exchanger proteins. Rat brain endothelial cells have also been reported to express mRNA for NHE3 and NHE4 (67). However, we did not find compelling evidence for NHE3 or NHE4 in our Western blot studies. Whether this is because of species differences or because CMEC do not translate NHE3 and NHE4 mRNA to measureable levels of proteins remains to be clarified in future studies. NHE5 has also been reported in brain, but evidence suggests that it is neuron specific (2, 8, 55), and thus it was not included in the present investigation. We also did not include NHE6, NHE7, NHE8, and NHE9 in our investigation, largely because they appear to have an intracellular distribution (16, 44). However, it is possible that NHE5, NHE6, NHE7, NHE8, or NHE9 could be present in the BBB and, in some manner, participate in edema formation during stroke. Future studies are needed to address whether this is the case.
Although our Western blot and immunohistochemistry studies provide good evidence that cultured bovine CMEC and freshly isolated rat microvessels express NHE1 and NHE2 proteins, an important consideration is whether the proteins are present in BBB endothelial cells in situ and, if so, whether they are present in the luminal membrane as predicted for a role in transport of Na across the barrier into the brain. The immunoelectron microscopy experiments of the present study reveal that NHE1 and NHE2 are present in the luminal membrane of cerebral microvessels in situ, consistent with a role for both of these isoforms in BBB secretion of Na into the brain. These studies were conducted using perfusion-fixed brains of rats under normoxic conditions. Thus we do not know whether the in situ distribution of these Na/H exchanger isoforms is altered in ischemic brain.
Previous studies evaluating Na/H exchange activity in cultured brain microvascular endothelial cells include demonstrations of amiloride- and EIPA-sensitive J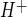 in piglet and rat brain endothelial cells, respectively (23, 46, 67). Rat brain endothelial cells have also been shown to exhibit Na/H exchange activity when assessed as EIPA-sensitive 22Na flux (74). In the present study, we chose to use the Na/H exchange inhibitor HOE-642 (cariporide), because it is highly selective for NHE1 while also inhibiting NHE2, with reported IC50 values of 0.05 and 3 μM, respectively (39). Our HOE-642 dose-response experiments reveal an IC50 of ∼7 μM for inhibition of CMEC J
in piglet and rat brain endothelial cells, respectively (23, 46, 67). Rat brain endothelial cells have also been shown to exhibit Na/H exchange activity when assessed as EIPA-sensitive 22Na flux (74). In the present study, we chose to use the Na/H exchange inhibitor HOE-642 (cariporide), because it is highly selective for NHE1 while also inhibiting NHE2, with reported IC50 values of 0.05 and 3 μM, respectively (39). Our HOE-642 dose-response experiments reveal an IC50 of ∼7 μM for inhibition of CMEC J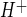 . This IC50 value is consistent with our observation that CMEC exhibit NHE1 and NHE2. Further study is needed to determine the extent to which the two isoforms contribute to CMEC Na/H exchange activity under control, as well as ischemic, conditions. Our observation that the Na/H exchange-mediated J
. This IC50 value is consistent with our observation that CMEC exhibit NHE1 and NHE2. Further study is needed to determine the extent to which the two isoforms contribute to CMEC Na/H exchange activity under control, as well as ischemic, conditions. Our observation that the Na/H exchange-mediated J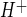 in CMEC is 0.23 ± 0.01 mM/min, which is equivalent to 0.003 pH unit/s in our studies, is comparable to Na/H exchange-mediated J
in CMEC is 0.23 ± 0.01 mM/min, which is equivalent to 0.003 pH unit/s in our studies, is comparable to Na/H exchange-mediated J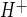 reported for other cells, including rat optic nerve astrocytes (0.003 pH unit/s) (38) and cortical astrocytes (0.005 pH unit/s) (32).
reported for other cells, including rat optic nerve astrocytes (0.003 pH unit/s) (38) and cortical astrocytes (0.005 pH unit/s) (32).
In the present study, we also demonstrate for the first time that Na/H exchange activity of BBB endothelial cells is stimulated by hypoxia, aglycemia, and AVP. CMEC Na/H exchange activity is significantly increased by 30 min of exposure to hypoxia (2% O2) or aglycemia or by 5 min of treatment with varying doses of AVP. In this regard, the CMEC Na/H exchanger responds to these ischemic factors in a manner similar to the CMEC Na-K-Cl cotransporter (20), supporting the hypothesis that both BBB Na transporters may participate in edema formation during ischemia. In these studies, which were conducted as an initial assessment of ischemic factor effects on the BBB Na/H exchanger, our finding that AVP caused a robust increase in CMEC Na/H exchange activity prompted us to focus the remainder of our investigations on AVP, leaving further experiments of hypoxia and aglycemia effects to future studies. Thus, just how the CMEC Na/H exchanger responds to aglycemia at different levels and durations of exposure remains to be determined.
There is much evidence that AVP appears to be an important contributor to ischemia-induced brain edema. During cerebral ischemia, AVP is released from extrahypothalamic neuronal processes terminating on brain microvessels (36, 68), which have been shown to possess AVP receptors (34, 58). Brattelboro rats, deficient in AVP, show less cerebral edema formation following MCAO-induced ischemia, and administration of AVP to these animals increases MCAO-induced edema (17). In another study, administration of a V1 vasopressin receptor antagonist reduced infarct volume in a rat focal cerebral ischemia model (66). Our finding that AVP stimulation of Na/H exchange activity is blocked by V1, but not V2, vasopressin receptor antagonists and mimicked by V1, but not V2, vasopressin receptor agonists, suggests that the AVP effect is mediated by a V1 vasopressin receptor. This is further supported by our finding that the effect of AVP on CMEC Na/H exchange activity is dependent on [Ca]i, as has been reported for V1 vasopressin receptor effects in other cells (45, 49, 56, 78), including our own previous studies showing that AVP stimulation of the CMEC Na-K-Cl cotransporter is also mediated by a V1 vasopressin receptor- and [Ca]i-dependent mechanism (50). Our findings are also consistent with previous reports that V1 vasopressin receptors are present in brain microvessels (56, 57) and that AVP-induced brain edema is mediated by a V1 vasopressin receptor (63). Collectively, these findings support the hypothesis that AVP-stimulated BBB Na/H exchange activity contributes to cerebral edema formation. Future studies are needed to address the mechanisms by which AVP, hypoxia, and aglycemia increase activity of cerebral microvessel Na/H exchange activity and to clarify whether ischemic conditions increase Na/H exchange activity in part by increasing the abundance of Na/H exchanger proteins in the cells. However, the increases in Na/H exchange activity observed in the present study are unlikely to be due to increased abundance of Na/H exchanger protein because of the short exposure times, i.e., 5 min for AVP and 30 min for hypoxia and aglycemia.
Increased activity of luminal BBB Na transporters during ischemia has been identified as a major contributing factor to ischemia-induced edema. Our previous studies demonstrated that intravenous administration of bumetanide to inhibit the BBB Na-K-Cl cotransporter reduces edema formation and infarct in the rat MCAO model of ischemic stroke (53). However, we also found evidence that intravenous administration of HOE-642 reduces edema and infarct in rats subjected to 3 h of permanent MCAO (73), suggesting that BBB Na/H exchanger also participates in formation of edema and infarct during ischemic stroke. Other studies provide some evidence that Na/H exchange inhibitors can reduce ischemic brain damage in rats after 3 days of permanent MCAO (33) or ischemia-reperfusion (70). In addition, studies using in situ perfusion and intracarotid bolus injection methods to analyze rates of Na transport across the BBB into brain demonstrated that the Na/H exchange inhibitors amiloride and dimethylamiloride significantly reduce Na uptake into the brain (5, 19), suggesting that a luminal BBB Na/H exchanger contributes to the low level of Na secretion into the brain during normoxia. The relative contributions of the BBB Na/H exchanger and Na-K-Cl cotransporter remain to be determined. However, we conducted initial studies to assess the effect of bumetanide + HOE-642 on edema formation in rats during MCAO and found evidence that the two inhibitors appear to have additive effects on reducing edema and infarct (data not shown) (73). Further study is needed to clarify the relative contributions of the cotransporter and exchanger to cerebral edema formation and also whether these contributions vary with time and degree of ischemia.
Although these studies collectively support a role for the BBB Na/H exchanger in vectorial transport of Na across the BBB into the brain, the Na/H exchanger might also participate in ischemia-induced swelling of BBB endothelial cells. In a recent study, we found significant increases in cell volume after CMEC were exposed to ≥3 h of hypoxia (1, 3, or 7.5% O2). We also found that CMEC swelling after 5 h of exposure to 7.5% O2 is significantly reduced by HOE-642 or bumetanide and abolished by HOE-642 + bumetanide (14). Thus Na/H exchange, as well as Na-K-Cl cotransport, may play a role in swelling of BBB endothelial cells as ischemia progresses over time.
The present study included an initial investigation of the effects of estradiol on BBB Na/H exchange activity, because previous studies from this and other laboratories showed that estradiol is neuroprotective in ischemic stroke, reducing edema and infarct (18, 24, 51, 60, 72). Furthermore, we previously showed that estradiol abolishes ischemic factor stimulation of BBB Na-K-Cl cotransporter activity (51). We demonstrate here that exposure of CMEC to estradiol (1–100 nM) for 24 h significantly reduces basal Na/H exchange activity and that a 5-min exposure to estradiol (10 and 100 nM) abolishes AVP stimulation of CMEC Na/H exchange activity. The finding that estradiol reduced Na/H exchange activity after only 5 min of treatment suggests a nongenomic mechanism of action, as we have reported for estradiol reduction of AVP effects on CMEC Na-K-Cl cotransporter activity (51). In those studies, we found that estradiol also diminished hypoxia or AVP-induced CMEC Na-K-Cl cotransporter abundance. Recently, estradiol has also been found to limit the shear stress-induced increase in NHE1 protein expression in bovine CMEC (15). Further studies are needed to clarify the mechanism by which estradiol inhibits the effect of AVP on the Na/H exchanger.
These findings indicate that the BBB Na/H exchanger and Na-K-Cl cotransporter respond similarly not only to ischemic factors that promote edema formation and infarct (i.e., hypoxia, aglycemia, and AVP), but also to estradiol, a factor known to reduce edema and infarct. This suggests that both BBB transport proteins are important participants in ischemia-induced cerebral edema formation, and both are thus potential targets for stroke therapy.
In summary, the results of the present study provide evidence that NHE1 and NHE2 are present in the luminal membrane of BBB endothelial cells and that CMEC Na/H exchange activity is stimulated by ischemic factors, including hypoxia, aglycemia, and AVP. Although we have yet to determine the full extent of hypoxia and aglycemia effects or the mechanism by which these factors stimulate the Na/H exchanger, we show here that AVP stimulation of the Na/H exchanger occurs via a V1 vasopressin receptor- and Ca-dependent mechanism. We further show that estradiol reduces AVP stimulation of the CMEC Na/H exchanger. Together with our previous studies, the present findings support the hypothesis that BBB NHE1 and/or NHE2, along with the BBB Na-K-Cl cotransporter, participate in ischemia-induced secretion of Na into the brain with consequent edema formation.
GRANTS
This work was supported by National Institutes of Health Grants NS-039953 (M. E. O'Donnell) and P01 AG-17164 (P. M. Wise) and American Heart Association Western States Affiliate Predoctoral Fellowship 0715071Y (T. I. Lam). This investigation was conducted in part in a facility constructed with support from National Center for Research Resources Research Facilities Improvement Program Grant C06 RR-17348-01.
REFERENCES
- 1.Aharonovitz O, Granot Y. Stimulation of mitogen-activated protein kinase and Na+/H+ exchanger in human platelets. Differential effect of phorbol ester and vasopressin. J Biol Chem 271: 16494–16499, 1996. [DOI] [PubMed] [Google Scholar]
- 2.Baird NR, Orlowski J, Szabó EZ, Zaun HC, Schultheis PJ, Menon AG, and Shull GE. Molecular cloning, genomic organization, and functional expression of Na+/H+ exchanger isoform 5 (NHE5) from human brain. J Biol Chem 274: 4377–4382, 1999. [DOI] [PubMed] [Google Scholar]
- 3.Bauer B, Hartz AM, Fricker G, Miller DS. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol Pharmacol 66: 413–419, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Betz AL Alterations in cerebral endothelial cell function in ischemia. Adv Neurol 71: 301–313, 1996. [PubMed] [Google Scholar]
- 5.Betz AL Sodium transport from blood to brain: inhibition by furosemide and amiloride. J Neurochem 41: 1158–1164, 1983. [DOI] [PubMed] [Google Scholar]
- 6.Betz AL, Keep RF, Beer ME, Ren X. Blood-brain barrier permeability and brain concentration of sodium, potassium, and chloride during focal ischemia. J Cereb Blood Flow Metab 14: 29–37, 1994. [DOI] [PubMed] [Google Scholar]
- 7.Biemesderfer D, Pizzonia J, Abu-Alfa A, Exner M, Reilly R, Igarshi P, Aronson PS. NHE3: a Na+/H+ exchanger isoform of renal brush border. Am J Physiol Renal Fluid Electrolyte Physiol 265: F736–F742, 1993. [DOI] [PubMed] [Google Scholar]
- 8.Bobulescu IA, Sole FD, Moe OW. Na+/H+ exchangers: physiology and link to hypertension and organ ischemia. Curr Opin Nephrol Hypertens 14: 485–494, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bookstein C, DePaoli AM, Xie Y, Niu P, Musch MW, Rao MC, Chang EB. Na+/H+ exchangers, NHE-1 and NHE-3, of rat intestine. Expression and localization. J Clin Invest 93: 106–113, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boron WF Regulation of intracellular pH. Adv Physiol Educ 28: 160–179, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Bourke RS, Kimelberg HK, Nelson LR, Barron KD, Auen EL, Popp AJ, Waldman JB. Biology of glial swelling in experimental brain edema. Adv Neurol Brain Edema 28: 99–109, 1980. [PubMed] [Google Scholar]
- 12.Boyarsky G, Ganz MB, Sterzel RB, Boron WF. pH regulation in single glomerular mesangial cells. I. Acid extrusion in absence and presence of HCO3. Am J Physiol Cell Physiol 255: C844–C856, 1988. [DOI] [PubMed] [Google Scholar]
- 13.Boyarsky G, Ransom B, Schlue WR, Davis MB, Boron WF. Intracellular pH regulation in single cultured astrocytes from rat forebrain. Glia 8: 241–248, 1993. [DOI] [PubMed] [Google Scholar]
- 14.Brillault J, Lam TI, Rutkowsky JM, Foroutan S, O'Donnell ME. Hypoxia effects on cell volume and ion uptake of cerebral microvascular endothelial cells. Am J Physiol Cell Physiol 294: C88–C96, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Chang E, O'Donnell ME, Barakat AI. Shear stress and 17β-estradiol modulate cerebral microvascular endothelial Na-K-Cl cotransporter and Na/H exchanger protein levels. Am J Physiol Cell Physiol 294: C363–C371, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Chesler M Regulation and modulation of pH in the brain. Physiol Rev 83: 1183–1221, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Dickinson LD, Betz AL. Attenuated development of ischemic brain edema in vasopressin-deficient rats. J Cereb Blood Flow Metab 12: 681–690, 1992. [DOI] [PubMed] [Google Scholar]
- 18.Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM. Estradiol protects against ischemic injury. J Cereb Blood Flow Metab 18: 1253–1258, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Ennis SR, Ren XD, Betz AL. Mechanisms of sodium transport at the blood-brain barrier studied with in situ perfusion of rat brain. J Neurochem 66: 756–763, 1996. [DOI] [PubMed] [Google Scholar]
- 20.Foroutan S, Brillault J, Forbush B, O'Donnell ME. Moderate to severe ischemic conditions increase activity and phosphorylation of the cerebral microvascular endothelial cell Na-K-Cl cotransporter. Am J Physiol Cell Physiol 289: C1492–C1501, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Golshani P, Liu XB, Jones EG. Differences in quantal amplitude reflect GluR4-subunit number at corticothalamic synapses on two populations of thalamic neurons. Proc Natl Acad Sci USA 98: 4172–4177, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoogerwerf WA, Tsao SC, Vevuyst O, Levine SA, Yun CHC, Yip JW, Cohen ME, Wilson PD, Lazenby AJ, Tse CM, Donowitz M. NHE2 and NHE3 are human and rabbit intestinal brush-border proteins. Am J Physiol Gastrointest Liver Physiol 270: G29–G41, 1996. [DOI] [PubMed] [Google Scholar]
- 23.Hsu P, Haffner J, Albuquerque ML, Leffler CW. pHi in piglet cerebral microvascular endothelial cells: recovery from an acid load. Proc Soc Exp Biol Med 212: 256–262, 1996. [DOI] [PubMed] [Google Scholar]
- 24.Hurn PD, Macrae IM. Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab 20: 631–652, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Iadecola C Mechanisms of cerebral ischemic damage. In: Cerebral Ischemia: Molecular and Cellular Pathophysiology, edited by W. Walz. Totowa, NJ: Humana, 1999, p. 3–34.
- 26.Kalaria RN, Premkumar DR, Lin CW, Kroon SN, Bae JY, Sayre LM, LaManna JC. Identification and expression of the Na+/H+ exchanger in mammalian cerebrovascular and choroidal tissues: characterization by amiloride-sensitive [3H]MIA binding and RT-PCR analysis. Brain Res Mol Brain Res 58: 178–187, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Kato H, Kogure K, Sakamoto N, Watanabe T. Greater disturbance of water and ion homeostasis in the periphery of experimental focal cerebral ischemia. Exp Neurol 96: 118–126, 1987. [DOI] [PubMed] [Google Scholar]
- 28.Kawai N, McCarron RM, Spatz M. The effect of endothelins on ion transport systems in cultured rat brain capillary endothelial cells. Acta Neurochir (Wien) 70: 138–140, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Keep RF (Editor). Potassium Transport at the Blood-Brain and Blood-CSF Barriers. New York: Plenum, 1993, p. 43–54. [DOI] [PubMed]
- 30.Kimelberg HK Cell swelling in cerebral ischemia. In: Cerebral Ischemia: Molecular and Cellular Pathophysiology, edited by W. Walz. Totowa, NJ: Humana, 1999, p. 45–68.
- 31.Kimelberg HK Current concepts of brain edema. Rev Lab Invest J Neurosurg 83: 1051–1059, 1995. [DOI] [PubMed] [Google Scholar]
- 32.Kintner DB, Su G, Lenart B, Ballard AJ, Meyer JW, Ng LL, Shull GE, Sun D. Increased tolerance to oxygen and glucose deprivation in astrocytes from Na+/H+ exchanger isoform 1 null mice. Am J Physiol Cell Physiol 287: C12–C21, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Kitayama J, Kitazono T, Yao H, Ooboshi H, Takaba H, Ago T, Fujishima M, Ibayashi S. Inhibition of Na+/H+ exchanger reduces infarct volume of focal cerebral ischemia in rats. Brain Res 922: 223–228, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Kretzschmar R, Ermisch A. Arginine-vasopressin binding to isolated hippocampal microvessels of rats with different endogenous concentrations of the neuropeptide. Exp Clin Endocrinol 94: 151–156, 1989. [DOI] [PubMed] [Google Scholar]
- 35.Kuribayashi Y, Horikawa N, Itoh N, Kitano M, Ohashi N. Delayed treatment of Na+/H+ exchange inhibitor SM-20220 reduces infarct size in both transient and permanent middle cerebral artery occlusion in rats. Int J Tissue React 21: 29–33, 1999. [PubMed] [Google Scholar]
- 36.Landgraf R Central release of vasopressin: stimuli, dynamics, consequences. Prog Brain Res 91: 29–39, 1992. [DOI] [PubMed] [Google Scholar]
- 37.Mahnensmith RL, Aronson PS. The plasma membrane sodium-hydrogen exchanger and its role in physiological and pathophysiological processes. Circ Res 56: 773–788, 1985. [DOI] [PubMed] [Google Scholar]
- 38.Mandal A, Delamere NA, Shahidullah M. Ouabain-induced stimulation of sodium-hydrogen exchange in rat optic nerve astrocytes. Am J Physiol Cell Physiol 295: C100–C110, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Masereel B, Pochet L, Laeckmann D. An overview of inhibitors of Na+/H+ exchanger. Eur J Med Chem 38: 547–554, 2003. [DOI] [PubMed] [Google Scholar]
- 40.McLean LA, Roscoe J, Jørgensen NK, Gorin FA, Cala PM. Malignant gliomas display altered pH regulation by NHE1 compared with nontransformed astrocytes. Am J Physiol Cell Physiol 278: C676–C688, 2000. [DOI] [PubMed] [Google Scholar]
- 41.McLean LA, Zia S, Gorin FA, Cala PM. Cloning and expression of the Na+/H+ exchanger from Amphiuma RBCs: resemblance to mammalian NHE1. Am J Physiol Cell Physiol 276: C1025–C1037, 1999. [DOI] [PubMed] [Google Scholar]
- 42.Menzies SA, Betz AL, Hoff JT. Contributions of ions and albumin to the formation and resolution of ischemic brain edema. J Neurosurg 78: 257–266, 1993. [DOI] [PubMed] [Google Scholar]
- 43.Menzies SA, Hoff JT, Betz AL. Extravasation of albumin in ischaemic brain oedema. Acta Neurochir (Wien) 51: 220–222, 1990. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura N, Tanaka S, Teko Y, Mitsui K, Kanazawa H. Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J Biol Chem 280: 1561–1572, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Nathanson MH, Moyer MS, Burgstahler AD, O'Carroll AM, Brownstein MJ, Lolait SJ. Mechanisms of subcellular cytosolic Ca2+ signaling evoked by stimulation of the vasopressin V1a receptor. J Biol Chem 267: 23282–23289, 1992. [PubMed] [Google Scholar]
- 46.Nicola PA, Taylor CJ, Wang S, Barrand MA, Hladky SB. Transport activities involved in intracellular pH recovery following acid and alkali challenges in rat brain microvascular endothelial cells. Pflügers Arch 456: 801–812, 2008. [DOI] [PubMed] [Google Scholar]
- 47.Noël J, Pouysségur J. Hormonal regulation, pharmacology, and membrane sorting of vertebrate Na+/H+ exchanger isoforms. Am J Physiol Cell Physiol 268: C283–C296, 1995. [DOI] [PubMed] [Google Scholar]
- 48.Noel J, Roux D, Pouyssegur J. Differential localization of Na+/H+ exchanger isoforms (NHE1 and NHE3) in polarized epithelial cell lines. J Cell Sci 109: 929–939, 1996. [DOI] [PubMed] [Google Scholar]
- 49.O'Donnell ME Endothelial cell sodium-potassium-chloride cotransport. Evidence of regulation by Ca2+ and protein kinase C. J Biol Chem 266: 11559–11566, 1991. [PubMed] [Google Scholar]
- 50.O'Donnell ME, Duong V, Suvatne S, Foroutan S, Johnson DM. Arginine vasopressin stimulation of cerebral microvascular endothelial cell Na-K-Cl cotransport activity is V1 receptor- and [Ca]-dependent. Am J Physiol Cell Physiol 289: C283–C292, 2005. [DOI] [PubMed] [Google Scholar]
- 51.O'Donnell ME, Lam TI, Tran LQ, Foroutan S, Anderson SE. Estradiol reduces activity of the blood-brain barrier Na-K-Cl cotransporter and decreases edema formation in permanent middle cerebral artery occlusion. J Cereb Blood Flow Metab 26: 1234–1249, 2006. [DOI] [PubMed] [Google Scholar]
- 52.O'Donnell ME, Martinez A, Sun D. Cerebral microvascular endothelial cell Na-K-Cl cotransport: regulation by astrocyte-conditioned medium. Am J Physiol Cell Physiol 268: C747–C754, 1995. [DOI] [PubMed] [Google Scholar]
- 53.O'Donnell ME, Tran L, Lam T, Liu XB, Anderson SE. Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab 24: 1046–1056, 2004. [DOI] [PubMed] [Google Scholar]
- 54.Orlowski J, Grinstein S. Na+/H+ exchangers of mammalian cells. J Biol Chem 272: 22373–22376, 1997. [DOI] [PubMed] [Google Scholar]
- 55.Orlowski J, Kandasamy RA, Shull GE. Molecular cloning of putative members of the Na/H exchanger gene family. J Biol Chem 267: 9331–9339, 1992. [PubMed] [Google Scholar]
- 56.Ostrowski NL, Lolait SJ, Bradley DJ, O'Carroll AM, Browstein MJ, Young WS. Distribution of V1a and V2 vasopressin receptor messenger ribonucleic acids in rat liver, kidney, pituitary and brain. Endocrinology 131: 533–535, 1992. [DOI] [PubMed] [Google Scholar]
- 57.Ostrowski NL, Lolait SJ, Young WS. Cellular localization of vasopressin V1a receptor messenger ribonucleic acid in adult male rat brain, pineal and brain vasculature. Endocrinology 135: 1511–1528, 1994. [DOI] [PubMed] [Google Scholar]
- 58.Pearlmutter AF, Szkrybalo M, Kim Y, Harik SI. Arginine vasopressin receptors in pig cerebral microvessels, cerebral cortex and hippocampus. Neurosci Lett 87: 121–126, 1988. [DOI] [PubMed] [Google Scholar]
- 59.Pedersen SF, O'Donnell ME, Anderson SE, Cala PM. Physiology and pathophysiology of Na+/H+ exchange and Na+-K+-2Cl− cotransporter in the heart, brain and blood. Am J Physiol Regul Integr Comp Physiol 291: R1–R25, 2006. [DOI] [PubMed] [Google Scholar]
- 60.Rau SW, Dubal DB, Bottner M, Gerhold LM, Wise PM. Estradiol attenuates programmed cell death after stroke-like injury. J Neurosci 23: 11420–11426, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rivarola V, Ford P, del Pilar Flamenco M, Galizia L, Capurro C. Arginine-vasopressin modulates intracelluar pH via V1 and V2 receptors in renal collecting duct cells. Cell Physiol Biochem 20: 549–558, 2007. [DOI] [PubMed] [Google Scholar]
- 62.Roos A, Boron WF. Intracellular pH. Physiol Rev 61: 296–434, 1981. [DOI] [PubMed] [Google Scholar]
- 63.Rosenberg GA, Estrada E, Kyner WT. Vasopressin-induced brain edema is mediated by the V1 receptor. Adv Neurol 52: 149–154, 1990. [PubMed] [Google Scholar]
- 64.Rozengurt E Early signals in the mitogenic response. Science 234: 161–166, 1986. [DOI] [PubMed] [Google Scholar]
- 65.Schielke GP, Moises HC, Betz AL. Blood to brain sodium transport and interstitial fluid potassium concentration during focal ischemia in the rat. J Cereb Blood Flow Metab 11: 466–471, 1991. [DOI] [PubMed] [Google Scholar]
- 66.Shuaib A, Xu Wang C, Yang T, Noor R. Effects of nonpeptide V1 vasopressin receptor antagonist SR-49059 on infarction volume and recovery of function in a focal embolic stroke model. Stroke 33: 3033–3037, 2002. [DOI] [PubMed] [Google Scholar]
- 67.Sipos H, Torocsik B, Tretter L, Adam-Vizi V. Impaired regulation of pH homeostasis by oxidative stress in rat brain capillary endothelial cells. Cell Mol Neurobiol 25: 141–151, 2005. [DOI] [PubMed] [Google Scholar]
- 68.Sorensen PS, Gjerris A, Hammer M. Cerebrospinal fluid vasopressin in neurological and psychiatric disorders. J Neurol Neurosurg Psychiatry 48: 50–57, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun AM, Liu Y, Dworkin LD, Tse CM, Donowitz M, Yip KP. Na+/H+ exchanger isoform 2 (NHE2) is expressed in the apical membrane of the medullary thick ascending limb. J Membr Biol 160: 85–90, 1997. [DOI] [PubMed] [Google Scholar]
- 70.Suzuki Y, Matsumoto Y, Ikeda Y, Kondo K, Ohashi N, Umemura K. SM-20220, a Na+/H+ exchanger inhibitor: effects on ischemic brain damage through edema and neutrophil accumulation in a rat middle cerebral artery occlusion model. Brain Res 945: 242–248, 2002. [DOI] [PubMed] [Google Scholar]
- 71.Szabo EZ, Numata M, Shull GE, Orlowski J. Kinetic and pharmacological properties of human brain Na+/H+ exchanger isoform 5 stably expressed in Chinese hamster ovary cells. J Biol Chem 275: 6302–6307, 2000. [DOI] [PubMed] [Google Scholar]
- 72.Toung TJ, Traystman RJ, Hurn PD. Estrogen-mediated neuroprotection after experimental stroke in male rats. Stroke 29: 1666–1670, 1998. [DOI] [PubMed] [Google Scholar]
- 73.Tran LQ, Anderson SE, O'Donnell ME. HOE-642 and bumetanide reduce edema formation and infarct following permanent rat middle cerebral artery occlusion (Abstract). FASEB J 18: A1069, 2004. [Google Scholar]
- 74.Vigne P, Ladoux A, Frelin C. Endothelins activate Na+/H+ exchange in brain capillary endothelial cells via a high affinity endothelin-3 receptor that is not coupled to phospholipase C. J Biol Chem 266: 5925–5928, 1991. [PubMed] [Google Scholar]
- 75.Wang XF, Yu MK, Lam SY, Leung KM, Jiang JL, Leung PS, Ko WH, Leung PY, Chew SBC, Liu CQ, Tse CM, Chan HC. Expression, immunolocalization, and functional activity of Na+/H+ exchanger isoforms in mouse endometrial epithelium. Biol Reprod 68: 302–308, 2003. [DOI] [PubMed] [Google Scholar]
- 76.Yang SH, Shi J, Day AL, Simpkins JW. Estradiol exerts neuroprotective effects when administered after ischemic insult. Stroke 31: 745–750, 2000. [DOI] [PubMed] [Google Scholar]
- 77.Yerby TR, Vibat CRT, Sun D, Payne JA, O'Donnell ME. Molecular characterization of the Na-K-Cl cotransporter of bovine aortic endothelial cells. Am J Physiol Cell Physiol 273: C188–C197, 1997. [DOI] [PubMed] [Google Scholar]
- 78.Zhao L, Brinton RD. Vasopressin-induced cytoplasmic and nuclear calcium signaling in cultured cortical astrocytes. Brain Res 943: 117–131, 2002. [DOI] [PubMed] [Google Scholar]