

Abstract
Dystonia is a neurological disorder characterized by excessive involuntary muscle contractions that lead to twisting movements or abnormal posturing. Traditional views place responsibility for dystonia with dysfunction of basal ganglia circuits, yet recent evidence has pointed towards cerebellar circuits as well. In the current studies we used two strategies to explore the hypothesis that the expression of dystonic movements depends on influences from a motor network that includes both the basal ganglia and cerebellum. The first strategy was to evaluate the consequences of subthreshold lesions of the striatum in two different animal models where dystonic movements are thought to originate from abnormal cerebellar function. The second strategy employed microdialysis to search for changes in striatal dopamine release in these two animal models where the cerebellum has been already implicated. One of the animal models involved tottering mice, which exhibit paroxysmal dystonia due to an inherited defect affecting calcium channels. In keeping with prior results implicating the cerebellum in this model, surgical removal of the cerebellum eliminated their dystonic attacks. In contrast, subclinical lesions of the striatum with either 6-hydroxydopamine (6OHDA) or quinolinic acid (QA) exaggerated their dystonic attacks. Microdialysis of the striatum revealed dystonic attacks in tottering mice to be associated with a significant reduction in extracellular striatal dopamine. The other animal model involved the induction of dystonia via pharmacological excitation of the cerebellar cortex by local application of kainic acid in normal mice. In this model the site of stimulation determines the origin of dystonia in the cerebellum. However, subclinical striatal lesions with either 6OHDA or QA again exaggerated their generalized dystonia. When dystonic movements were triggered by pharmacological stimulation of the cerebellum, microdialysis revealed significant reductions in striatal dopamine release. These results demonstrate important functional relationships between cerebellar and basal ganglia circuits in two different animal models of dystonia. They suggest that expression of dystonic movements depends on influences from both basal ganglia and cerebellum in both models. These results support the hypothesis that dystonia may result from disruption of a motor network involving both the basal ganglia and cerebellum, rather than isolated dysfunction of only one motor system.
Keywords: dystonia, cerebellum, basal ganglia, animal model
Introduction
Dystonia is a motor disorder described by excessive involuntary muscle contractions (Tarsy and Simon, 2006). Muscle contraction is disproportionate for intended movements, and the contractions spread to nearby muscles not normally involved. Sometimes overflow contractions include muscles that antagonize the action of the primary muscles. The patterns of muscles involved determine the final outcome, which is expressed as twisting movements that are often sustained or repetitive. Dystonia may affect nearly any part of the body. Most often an isolated region is affected, such as the neck in cervical dystonia or the hand in writer's cramp. However, generalized forms of dystonia exist with broad involvement of the body.
The cause of most dystonias is unknown, though some cases can be linked with inherited or acquired defects of the nervous system. Clinical studies historically implicate the basal ganglia, but recent evidence has pointed to cerebellar circuits too (Jinnah and Hess, 2006). Experimental studies in animals also have linked dystonia with defects in the basal ganglia (Gernert et al., 2002; Tabbal et al., 2006; Cuny et al., 2008) or cerebellum (LeDoux et al., 1993; Campbell et al., 1999; Pizoli et al., 2002). Two plausible and interrelated hypotheses have been advanced to explain these differing results regarding the neural substrates for dystonia (Jinnah and Hess, 2006). One is that the superficial similarities among different forms of dystonia belie a heterogeneous disorder with varied neural substrates. Some dystonias may arise from isolated dysfunction of the basal ganglia, while others may involve only the cerebellum. Another hypothesis is that dystonia arises from dysfunction of a motor network involving both, the basal ganglia and cerebellum, rather than independent dysfunction of either motor system. The current studies address the anatomical basis for dystonic motor behaviour in light of the motor network hypothesis.
Methods
Genetic dystonia model
Tottering mutant mice were used as the genetic dystonia model. They were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and bred at Johns Hopkins University along with congenic normal C57BL/6J mice. Both male and female mice were used at 2–4 months of age. Animals were housed in groups of four to five and maintained on a 12 h light/dark cycle with free access to food and water. All procedures were conducted in accordance with the ‘Guide for Care and Use of Laboratory Animals’ of the National Institutes of Health and approved by the Institutional Animal Care and Use Committee.
Tottering mutants exhibit paroxysmal dystonia due to a point mutation in the Cacna1a gene (Fletcher et al., 1996; Wakamori et al., 1998). The mutation impairs activity of CaV2.1 (P/Q-type) calcium channels, but dystonic motor behaviour arises from a form of maladaptive molecular plasticity involving secondary up-regulation of CaV1.2 (L-type) calcium channels in the cerebellum (Campbell and Hess, 1998; Campbell et al., 1999). These mutants provide an especially convenient tool for the current studies for several reasons. Defects in the same gene cause paroxysmal or focal dystonia in humans (Giffin et al., 2002; Spacey et al., 2005), increasing the likelihood of providing results relating to humans. Further, unlike many genetic animal models that lack motor abnormalities resembling dystonia, tottering mice exhibit a motor disorder that closely resembles the clinical and electrophysiological characteristics of human dystonia, and videos of their disorder have been published (Hess and Jinnah, 2005; Jinnah et al., 2005; Shirley et al., 2007). Their dystonia occurs as discrete spontaneous attacks superimposed on a baseline of mild ataxia, and attacks can be triggered by caffeine (Fureman et al., 2002). The influence of experimental manipulations therefore can be compared at baseline and when dystonic movements occur. The discrete attacks further allow quantification of their frequency, duration and severity.
Behaviour assessments were conducted by an observer blinded to experimental group at least 2 weeks after recovery following any experimental procedure. Baseline motor behaviour was evaluated with a motor disability rating scale (Jinnah et al., 2000). Briefly, mice were placed singly in Plexiglas cages and observed undisturbed for 1 min every 10 min for 2 h. At each interval a score from 0 to 4 was assigned based on motor disability (Table 1). A score of 3 or higher defined a dystonic attack in tottering mice. The frequency of spontaneous attacks was estimated by monitoring the animals undisturbed for 14 h split into two separate 7 h epochs on different days.
Table 1.
Motor disability rating scale
| Score | Behaviour |
|---|---|
| D0 | Normal motor behavior |
| D1 | No functional impairment; slightly slowed or abnormal motor behaviour |
| D2 | Mild impairment; limited ambulation unless disturbed, transient abnormal postures, infrequent falls |
| D3 | Moderate impairment; limited ambulation even when disturbed, frequent abnormal postures, frequent falls but upright for majority of time |
| D4 | Severe impairment; almost no ambulation, sustained abnormal postures, not upright for majority of time |
The motor disability scores provided estimates of functional impairment, but could not describe the nature of the disability. Additionally, the duration of spontaneous dystonic attacks in tottering mice was not readily determined because they occurred at random. To characterize the duration of attacks and nature of abnormal movements, attacks were triggered with 5 mg/kg subcutaneous caffeine and monitored every 1 min at 10 min intervals for 2 h with a dyskinesia rating scale as previously described (Devanagondi et al., 2007; Shirley et al., 2007). At each interval multiple abnormal behaviours were recorded according to body part. These included tremor (face, neck, trunk and limbs); tonic flexion or extension (face, neck, trunk and limbs); twisting (neck and trunk); clonus (face and limbs); head bobbing (vertical displacement) and head wagging (horizontal displacement). Several other problems not readily ascribed to an isolated body part also were recorded including falling (with all four limbs off the ground), listing (tilting to one side with two limbs off the ground), stumbling, widened stance, circling (360° turn with forward ambulation), spinning (360° turn without forward ambulation) and reverse locomotion. The total dyskinesia score reflected an arithmetic sum of all abnormal behaviours in any body part, with a maximum total score of 30. Several subscores for clinically defined motor disorders also were calculated by combining similar behaviours across body parts (Devanagondi et al., 2007; Shirley et al., 2007). The dystonia score reflected a combination of scores for all twisting movements and sustained abnormal postures. Total scores for tremor and clonus were similarly derived.
Cerebellar lesions
Cerebellectomies were performed on five tottering mice to verify a role for the cerebellum in their dystonic movements. The procedure was performed as previously described (Devanagondi et al., 2007). After tribromoethanol anaesthesia, the scalp over the occipital region was shaved and cleaned with Betadine. A 1 cm vertical incision was made and a 25 mm2 area of bone overlying the posterior cerebellum was removed. Suction was used to eliminate the entire cerebellum and deep cerebellar nuclei via their attachments to the rest of the brain at the peduncles. The cavity was filled with a collagen-based haemostat (Avitene, Davol, Cranston RI, USA). A control group of five tottering mice was subject to a sham procedure in which the cerebellum was exposed but not removed. Wounds were sutured and animals placed on a 30°C heating pad for recovery. They received 50 mg/kg chloramphenicol, 0.02 mg/kg buprenorphine and 10% glucose in 0.45% saline twice daily for 3 days. Operated animals resumed normal ingestive behaviours in 1–2 days. They lost ∼10% of their body weight in the immediate postoperative period but recovered their preoperative weights by 2 weeks after surgery. Spontaneous and caffeine-induced attacks were monitored as described above.
Pharmacologic dystonia model
To verify that experimental findings were not peculiar to tottering mutants, we also examined a second, unrelated pharmacological dystonia model. In this model, microinjection of kainic acid into the cerebellar cortex of normal C57BL/6J mice induces generalized dystonia as a result of local excitation of glutamate receptors (Pizoli et al., 2002). The dystonic movements match clinical and electrophysiological criteria for human dystonia, and video demonstrations of their disorder have been published (Hess and Jinnah, 2005). This model also is particularly convenient for the current studies, because the origin of dystonic movements is determined by the microinjection site in the cerebellum, and the animals have an otherwise intact nervous system. Additionally, movements emerge within minutes of injection, permitting quantitative studies of onset, duration and severity.
Briefly, kainic acid was dissolved in 0.9% saline at a concentration of 50 μg/ml. A midline incision was made over the posterior skull, a small hole drilled at the midline and 0.5 μl of kainic acid was injected into the anterior cerebellum at a depth of 2 mm. The short-acting anaesthetic isoflurane was used for the procedure to permit rapid recovery and immediate behavioural assessments.
Baseline motor behaviour was evaluated with the motor disability scale as described above for the genetic dystonia model, and via automated photocell activity chambers (Jinnah and Hess, 2005). For the activity chambers, mice were tested individually during the light phase for a total of 2 h without a habituation period in 20 × 40 cm Plexiglas cages with four infrared beams across the short axis and eight infrared beams across the long axis (San Diego Instruments, San Diego, CA, USA). The numbers of beam breaks were measured in 10 min bins for 1 h.
The severity and duration of dystonia triggered by kainic acid application in the cerebellum were estimated via the disability rating scale (Table 1). Unlike the genetic model, spontaneous attacks of dystonia do not occur in the pharmacological model, so spontaneous dystonia was not scored. Also unlike the genetic model, dystonia in the pharmacological model is not accompanied by other abnormal movements. Therefore, the nature of the motor disorder was not profiled with the dyskinesia rating scale.
Striatal lesions
To address contributions of the basal ganglia, dystonic motor behaviour was evaluated in both of the dystonia animal models following subthreshold lesions of this region. Lesions were conducted with two different toxins, each in separate groups of animals with a parallel control group. The goal was to produce a subclinical lesion that would modify basal ganglia function without confounding the assessment of dystonia.
The consequences of damage to nigrostriatal projections by intrastriatal microinjection of 6-hydroxydopamine (6OHDA) were explored first, because many studies have implicated dopaminergic dysfunction in dystonia (Perlmutter and Mink, 2004). Bilateral lesions were made in view of evidence for contralateral compensation after unilateral lesions (Roedter et al., 2001). Bilateral 6OHDA causes a severe hypokinetic syndrome with significant mortality that precludes proper assessment of motor behaviour, so preliminary dose-finding experiments were conducted to find the maximum dose of 6OHDA that would cause minimal persisting motor impairment. The final amount was 10 nmol/μl, delivered in 1 μl over 5 min in 0.9% saline with 0.01% ascorbic acid. A parallel control group received only vehicle. For these injections, mice were anaesthetized with 20 ml/kg of 2% intraperitoneal tribromoethanol in saline and mounted in a stereotactic frame (Kopf Instruments, Tujunga, CA, USA). Injections were made at the following coordinates from bregma: AP = +1.0, ML = ± 2.1, DV = −3.5. Animals receiving 6OHDA exhibited moderate hypoactivity with aphagia and adipsia for 2–4 days before recovering normal motor and ingestive behaviour. During this period all mice received 0.02 mg/kg buprenorphine and 10% glucose in 0.45% saline twice daily.
To verify that experimental findings were not peculiar to 6OHDA lesions, we explored an unrelated method for striatal injury. Specifically, we explored the consequences of lesions of striatal neurons, because prior studies implicate damage to these neurons as another cause for dystonia (Berardelli et al., 1998). These lesions were made by intrastriatal microinjection of quinolinic acid (QA), which causes selective lesions of these neurons (Schwarcz et al., 1983). Lesions were made at the same coordinates used for 6OHDA. Preliminary dose-finding experiments revealed that bilateral injection of 15 nmol QA in 1 μl produced minimal mortality with 1–2 days of transient hyperactive motor behaviour.
Histology
Lesions were verified in all animals following behavioural testing. Mice were anaesthetized with methoxyflurane (Mallinckrodt Veterinary, Mundelein, IL, USA) and perfused transcardially with 4% paraformaldehyde in 0.1 M phosphate buffer at pH 7.4. Brains were removed and placed in fixative overnight at 4°C. They then were equilibrated in 30% sucrose in 0.1 M phosphate buffer. Sections were cut at 35 μm on a Microm HM440E sliding microtome (Dolbey-Jamison, Limerick, PA, USA) and stored in cryopreservative consisting of 30% glycerol and 30% ethylene glycol in 0.1 M phosphate buffer at −20°C. Every sixth section was stained with cresyl violet.
Parallel sections through the lesion were evaluated via immunohistochemistry. An antibody to tyrosine hydroxylase (EMD Biosciences Inc, San Diego, CA, USA) was used to identify 6OHDA-induced damage to nigrostriatal fibres. An antibody to calbindin (Chemicon, Temecula, CA, USA) was used to define loss of striatal neurons. Both antibodies were used at 1 : 1000 dilution with staining conducted as previously described (Egami et al., 2007).
Microdialysis
Microdialysis was performed in alert, freely moving mice as previously described (Fan and Hess, 2007). Briefly, mice were anaesthetized with tribromoethanol and a microdialysis probe was implanted in the striatum at coordinates AP = +0.6 AP, ML = 1.7, DV = −4.5. The probe was then perfused continuously with artificial cerebrospinal fluid at a flow rate of 0.6 μl/min via a liquid swivel connected to a microinjection pump. Sample collection began 14–16 h after surgery with intervals of 20 min. It was not feasible to use caffeine to induce dystonic attacks, since caffeine itself alters striatal dopamine overflow. Instead, attacks were induced by individually transferring tottering mice from their small home cage to a larger cage, a method that induces dystonia in ∼75% of cases (Fureman et al., 2002). Six samples were collected from tottering mice both before and during a dystonic attack. For kainic acid-induced dystonia, six baseline samples were collected and then mice were anaesthetized and microinjected with 0.5 μl of vehicle, 50 or 100 μg/ml of kainic acid into the cerebellum. Mice were allowed to recover for 10 min after microinjection, and six more samples were collected. Samples were supplemented with 250 μM ascorbic acid and frozen immediately at −80°C for HPLC analysis of monoamines. After microdialysis, brains were removed and probe location was confirmed histologically.
Microdialysis samples were analysed by HPLC (MD-150 column, 150 mm length; 3 mm I.D.; ESA, Chelmsford, MA, USA) equipped with a 5014B coulometric microdialysis cell at a flow rate of 0.6 ml/min as previously described (Fan and Hess, 2007). Values were expressed as nanogram per millilitre after adjustment for recovery rate of the microdialysis probe. For each mouse, average dopamine concentrations were calculated for baseline and dystonic states.
Data analysis
Mean results for individual behavioural analyses were compared via Student's t-test. Microdialysis results were evaluated by analysis of variance (ANOVA) with a post hoc Fisher's t-test to identify group effects. Samples collected before and during dystonia in the same animals were compared with a paired t-test. For behavioural studies involving multiple measures of the same animals over time, comparisons between experimental groups were made via a two-way ANOVA with time as a repeated measure. For all analyses, a P-value <0.05 was considered statistically significant.
Results
Genetic dystonia model
The involvement of the cerebellum in the expression of dystonic attacks in tottering mice was confirmed by surgical removal of the cerebellum. Tottering mice subject to cerebellectomy (n = 5) recovered well, with some expected worsening of their baseline ataxia following surgery. However, spontaneous and caffeine-induced dystonic attacks were absent (Fig. 1). Tottering mutants subject to sham surgery (n = 5) also recovered well. All sham-operated tottering controls exhibited their typical spontaneous and caffeine-induced dystonic attacks (Fig. 1). Post-mortem evaluation of the brains confirmed near-complete elimination of the cerebellum in all lesioned mice, with no damage to nearby structures (data not shown). These results confirm the cerebellum is required for the expression of dystonic attacks in tottering mutants (Campbell et al., 1999).
Fig. 1.

Caffeine-induced dystonic attacks in tottering mice. Results are average values ± SEM. Mice subject to cerebellectomy are shown in black (n = 5), and those with sham lesions are shown in grey (n = 5). A two-way ANOVA, with time as a repeated measure revealed significant effects for group [F(1,11) = 219, P < 0.001] and time [F(11,110) = 4.0, P < 0.001]. The interaction between group and time was also significant [F(11,88) = 4.5, P < 0.001].
In the tottering mutants, bilateral 6OHDA striatal lesions resulted in 2–4 days of severe but transient hypokinesis. Two weeks following lesions, mice recovered baseline motor function and were indistinguishable from controls. The motor disability scale revealed no differences in the baseline motor function of the 6OHDA-lesioned (n = 10) and control animals (n = 10) after recovery (P = 0.62, Fig. 2A).
Fig. 2.

Spontaneous dystonic attacks in tottering mice. Results are average values ± SEM. Mice with 6OHDA lesions are shown in black on (n = 10), and parallel sham-operated controls are grey (n = 10). Baseline motor disability is shown in (A), and attack frequency is shown in (B). The proportion of scores reaching the maximum score of 4 is shown as an estimate for severity in (C). Results were compared via Student's t-test, and P < 0.05 was noted with an asterisk as statistically significant.
Though baseline behaviour was not overtly affected, spontaneous dystonic attacks in 6OHDA-lesioned totterings became more frequent than those in the sham-lesioned control group. The average frequency of spontaneous dystonic attacks nearly doubled in the 6OHDA-lesioned group (P < 0.001, Fig. 2B). The severity of dystonic attacks was estimated by comparing the proportion of motor disability scores reaching the maximum level of 4. There was a trend for increased severity in the 6OHDA lesioned animals that was not statistically significant (P = 0.34, Fig. 2C).
To obtain a more complete picture of the nature and duration of abnormal movements, dystonic attacks were triggered by caffeine and animals monitored using the dyskinesia scale until they returned to baseline. Only those animals exhibiting an attack within 30 min of drug administration were scored. Attacks in the 6OHDA-lesioned mice (n = 6) were significantly longer than those of sham-lesioned controls (n = 6); attacks averaged 91.7 ± 11.8 min in lesioned mice compared with 53.3 ± 5.4 min in the sham-lesioned controls (P < 0.01, Fig. 3A and B). Despite the increase in frequency and duration, the nature of the abnormal movements was unaffected by the lesions. The majority of abnormal movements in both 6OHDA-lesioned and control mice were dystonic, with less frequent clonic movements and tremor (Fig. 3C).
Fig. 3.

Dystonic attacks induced by 5 mg/kg caffeine in tottering mice given intrastriatal saline (n = 6, grey) or 6OHDA (n = 6, black). Results are average values ± SEM. The temporal profile of total dyskinesia scores is shown in (A). Two-way ANOVA with time as a repeated measure revealed significant effects for group [F(1,11) = 18.7, P < 0.01] and time [F(11,110) = 12.5, P < 0.001], but the interaction between group and time was not significant [F(11,110) = 10, P = 0.4]. (B) Shows the average attack duration for all animals. (C) Shows the distribution of abnormal movements contributing to the total dyskinesia score. Results in (B) and (C) were compared via Student's t-test, and P < 0.05 was noted with an asterisk as statistically significant.
Post-mortem histological studies confirmed 6OHDA produced consistent lesions in all animals. In lesioned animals, tyrosine hydroxylase immunostaining was lost in a discrete patch from each side of the striatum (Fig. 4A–C). Nissl stains showed minimal disruption of the striatal neuropil, and calbindin immunostains revealed no loss of striatal neurons (data not shown). These results demonstrate that partial 6OHDA lesions of striatal dopamine fibres increase the frequency and duration of dystonic attacks in tottering mutant mice, without changing the quality or severity of abnormal movements.
Fig. 4.
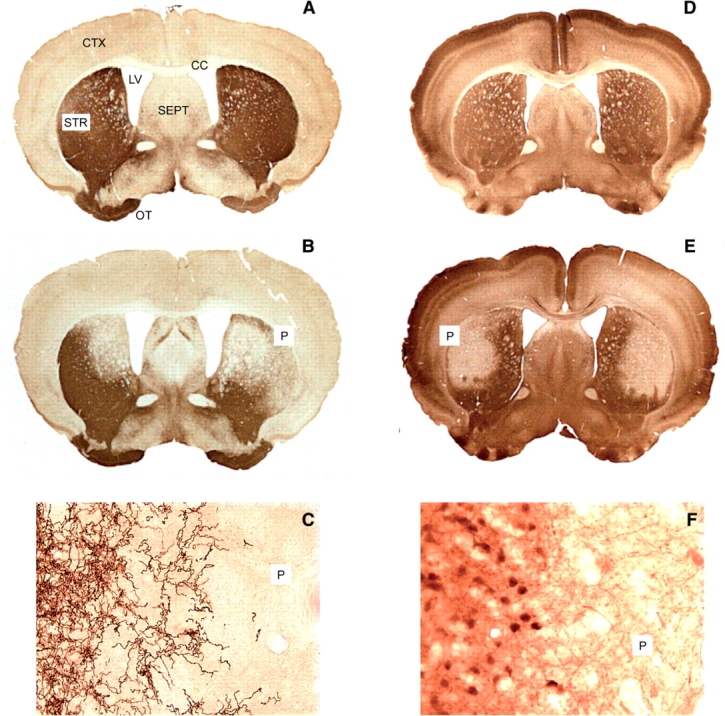
Histology of the striatum after staining with tyrosine hydroxylase (A–C) or calbindin (D–F). TH immunostaining in controls (A) revealed a dense network of fibres in the striatum (STR) and olfactory tubercle (OT), sparse fibres in cerebral cortex (CTX) and septum (SEPT) and no staining in corpus callosum (CC). In animals with bilateral 6OHDA injections (B), there were circumscribed patches bilaterally (P) that stained poorly for tyrosine hydroxylase. Higher power (40×) magnification at the edge of the patch showed a transition from dense fibre staining to a zone with few or no stained fibres (C). Nissl stains revealed no cell loss (data not shown). Calbindin immunostaining in controls (D) revealed labelled neurons and fibres in many regions. In animals treated with QA (E), there were circumscribed patches bilaterally (P) that stained poorly. Higher power (40×) magnification at the edge of the patch showed a transition from dense fibre staining to a zone with few stained cells or fibres (F). The loss of cells could also be seen with a Nissl stain (data not shown).
In the tottering mutants, bilateral QA lesions resulted in 2–4 days of mild hyperkinesis. Two weeks following the lesions, motor behaviour returned to normal and was indistinguishable from sham-lesioned controls. Motor disability scores showed no differences between QA-lesioned and the control animals (P = 0.42, Fig. 5A).
Fig. 5.

Spontaneous dystonic attacks in tottering mice with or without QA lesions. Results are average values ± SEM. Mice with QA lesions are shown in black (n = 10), and parallel sham-operated controls are grey (n = 8). Baseline motor disability is shown in (A), and attack frequency is shown in (B). The proportion of scores reaching the maximum score of 4 is shown as an estimate for severity in (C). Results were compared via Student's t-test, and P < 0.05 was noted with an asterisk as statistically significant.
Like the 6OHDA-lesioned mice, however, QA lesioned mice (n = 10) exhibited a significant increase in the frequency and duration of dystonic attacks compared with sham-lesioned controls (n = 8). The average frequency of spontaneous dystonic attacks doubled in the QA-lesioned group (P < 0.001, Fig. 5B), with an increase in severity too (P = 0.03, Fig. 5C). The duration of dystonic attacks triggered by caffeine in QA-lesioned mice increased significantly to an average of 88.9 ± 7.2 min from attacks in sham-lesioned controls that were 60 ± 5.8 min (P < 0.01, Fig. 6A and B). There was no change in the quality of abnormal movements in lesioned animals (Fig. 6C).
Fig. 6.

Dystonic attacks induced by 5 mg/kg caffeine in tottering mice given intrastriatal saline (n = 7, grey) or QA (n = 9, black). Results are average values ± SEM. The temporal profile of total dyskinesia scores is shown in (A). Two-way ANOVA with time as a repeated measure revealed significant effects for group [F(1,14) = 10.6, P < 0.01] and time [F(11,154) = 14.8, P < 0.001], as well as a significant interaction [F(11,154) = 6.5, P < 0.001]. (B) Shows the average attack duration for all animals. (C) Shows the distribution of abnormal movements contributing to the total dyskinesia score. Results for (B) and (C) were compared via Student's t-test, and P < 0.05 was noted with an asterisk as statistically significant.
Post-mortem histology confirmed lesions in all QA-treated mice. Calbindin immunostaining was lost in discrete patches from each side of the striatum (Fig. 4D–F). There was a loss of striatal neurons in both calbindin- and Nissl-stained sections. There was no associated loss of fibres immunostained for tyrosine hydroxylase (data not shown). These results demonstrate that subclinical lesions of striatal neurons increase the frequency and duration of dystonic attacks in tottering mice, without changing severity or quality.
Pharmacological dystonia model
Like tottering mutant mice, 6OHDA lesions in normal C57BL/6J mice resulted in 2–4 days of severe but transient hypokinesis. Two weeks following lesions, mice recovered baseline motor function and appeared indistinguishable from controls. Baseline motor disability scores revealed no measurable differences between 6OHDA-lesioned (n = 14) and sham-lesioned control (n = 13) mice (P = 0.40, Fig. 7A). Further assessments of baseline motor behaviour using automated activity chambers also revealed no differences between the lesioned and control mice (P = 0.44, Fig. 7B). Though baseline behaviour was normal, dystonic movements triggered by microinjection of kainic acid into the cerebellum were significantly more severe and prolonged in 6OHDA-lesioned mice in comparison with sham-lesioned controls (P < 0.02, Fig. 7C).
Fig. 7.

Microinjection of kainic acid into the cerebellum following 6OHDA lesions in normal mice. Mice with 6OHDA lesions (n = 14) are shown in black and saline controls (n = 13) are shown in grey. Results are average values ±SEM. Baseline motor function is shown as motor disability scores (A) or gross motor activity in automated photocell chambers (B). (C) Shows the temporal profile of motor disability due to dystonic motor behaviour after intra-cerebellar microinjection of 50 μg/ml kainic acid in 0.5 μl at time 0. Two-way ANOVA with time as a repeated measure revealed significant effects for time [F(11,275) = 91.4, P < 0.001] and group [F(11,25) = 6.3, P = 0.019]. The interaction between time and group was not significant [F(11,275) = 0.8, P = 0.65].
Also like tottering mutants, bilateral QA lesions in normal mice resulted in 2–4 days of transient hyperkinesis. Two weeks later, motor behaviour returned to normal and was indistinguishable from sham-lesioned controls (P = 0.43, Fig. 8A and B). Despite normal baseline behaviour, dystonic movements triggered by kainic acid in the cerebellum again were significantly worse in the lesioned mice compared with controls (P < 0.01, Fig. 8C).
Fig. 8.

Microinjection of kainic acid into the cerebellum following QA lesions in normal mice. Mice with QA lesions (n = 14) are shown in black and saline controls (n = 13) are shown in grey. Results are average values ± SEM. Baseline motor function is shown as motor disability scores (A) or gross motor activity in automated photocell chambers (B). (C) Shows the temporal profile of motor disability due to dystonic motor behaviour after intra-cerebellar microinjection of 50 μg/ml kainic acid in 0.5 μl at time 0. Two-way ANOVA with time as a repeated measure revealed significant effects for time [F(11,275) = 56.9, P < 0.001] and group [F(1,25) = 10.3, P < 0.005]. The interaction between time and group was not significant [F(11,275) = 1.1, P = 0.33].
Histological studies in all mice revealed the consequences of 6OHDA and QA to be similar to those observed for tottering mice shown in Fig. 4. These results demonstrate that subclinical 6OHDA lesions of striatal dopamine fibres or QA lesions of striatal neurons both worsen dystonic movements provoked in normal mice by pharmacological excitation of glutamate receptors in the cerebellar cortex.
Microdialysis of striatum
To determine if dystonia arising from the cerebellum modifies function in the intact basal ganglia, dopamine overflow in the unlesioned striatum was monitored by microdialysis in both the genetic and pharmacological dystonia models. In tottering mutant mice, the effect of dystonia on extracellular striatal dopamine concentrations was determined by comparing overflow before and after the induction of dystonia. Dystonia in tottering mice was associated with a significant reduction in striatal dopamine measured by microdialysis, compared with baseline (P < 0.05, Table 2).
Table 2.
Striatal dopamine overflow before and after induction of dystonia
| N | Baseline (ng/ml) | Dystonia (ng/ml) | |
|---|---|---|---|
| Genetic model | |||
| Tottering mice | 9 | 3.68 ± 0.55 | 2.85 ± 0.44* |
| Pharmacological model | |||
| Vehicle | 8 | 2.55 ± 0.25 | 2.58 ± 0.36 |
| 50 μg/ml kainic acid | 10 | 2.37 ± 0.27 | 1.78 ± 0.19* |
| 100 μg/ml kainic acid | 7 | 2.77 ± 0.23 | 1.56 ± 0.13*** |
Data are expressed as means ± SEM. N = sample size; *P< 0.05, ***P< 0.001 as determined by paired t-test.
In the pharmacological model, induction of dystonia via microinjection of kainic acid into the cerebellum also decreased striatal dopamine overflow. Both 50 and 100 μg/ml microinjections of kainic acid into the cerebellum significantly reduced striatal dopamine overflow compared with baseline (Table 2). After determining that baseline extracellular dopamine was comparable among the vehicle and kainic acid treatment groups [one-factor ANOVA; F(2,22) = 0.60, P > 0.5], a between-subjects comparison was performed. One-factor ANOVA revealed a significant dose-dependent reduction in striatal dopamine overflow [F(2,22) = 4.4, P < 0.05] and a Fisher's post hoc test demonstrated significant differences between vehicle and both doses of kainic acid (P < 0.05). Compared with vehicle, 50 μg/ml kainic acid reduced dopamine overflow by ∼30% and 100 μg/ml kainic acid reduced dopamine overflow by ∼40%. In contrast, dopamine overflow after microinjection of vehicle was not different from baseline.
Discussion
These studies provide support for the motor network model for dystonia. In mutant tottering mice, prior studies showed that abnormal cerebellar output is essential for the generation of dystonic movements (Campbell and Hess, 1998; Campbell et al., 1999). The current studies support a role for the cerebellum in these mutants by showing that dystonia is eliminated following surgical removal of the cerebellum. In a second pharmacological model, abnormal cerebellar output caused by direct application of the glutamate receptor agonist kainic acid in the cerebellar cortex is the source of dystonia (Pizoli et al., 2002). Although abnormal cerebellar function may be the source of dystonic movements in both the genetic and pharmacological models, the current studies reveal that the basal ganglia also play a role, which becomes apparent after they are injured. Subclinical lesions of the striatum increased the frequency and duration of dystonic movements without changing their quality in the genetic model, and they increased the duration and severity of dystonia in the pharmacological model. The exaggeration of dystonic movements by such lesions suggests the basal ganglia may normally afford a previously unrecognized protective effect in the models evaluated. These results are best accommodated by a motor network model in which both cerebellum and basal ganglia play a role in the expression of dystonia.
Dystonia and the basal ganglia
The basal ganglia traditionally have been implicated in human dystonia by studies showing correlations between dystonia and imaging or pathological evidence for basal ganglia dysfunction. Among patients with dystonia and overt lesions in structural imaging studies such as CT or MRI, the basal ganglia and especially the putamen are most frequently implicated (Marsden et al., 1985; Obeso and Gimenez-Roldan, 1988; Bhatia and Marsden, 1994). Even when overt lesions cannot be identified, functional imaging studies such as fMRI or PET often reveal areas of abnormal activity in the basal ganglia for many types of dystonia (Meunier et al., 2003; Asanuma et al., 2005). The basal ganglia are implicated further by neurosurgical treatments showing that dystonia improves in patients following pallidotomy or deep brain stimulation of the internal segment of the globus pallidus (Vidailhet et al., 2005; Kupsch et al., 2006).
The basal ganglia also are implicated in experimental studies of animals. Selective lesions of nigrostriatal dopamine pathways with 6OHDA or MPTP in both rodents and primates results in dystonic movements either spontaneously or after treatment with dopamine-related drugs (Perlmutter et al., 1997; Winkler et al., 2002; Tabbal et al., 2006). The neurotoxin 3-nitropropionic acid causes dystonia in both rodents and primates in association with selective striatal pathology (Palfi et al., 2000; Fernagut et al., 2002; Ghorayeb et al., 2002; Cuny et al., 2008). Focal destructive lesions of the posterior putamen also cause dystonia in primates (Burns et al., 1995). Together, the clinical and experimental studies provide strong converging support that the basal ganglia play an important role in dystonia.
Dystonia and the cerebellum
On the other hand, several structural imaging studies also link defects of the cerebellum or its connections with dystonia (Krauss et al., 1997; LeDoux and Brady, 2002; Draganski et al., 2003; Le Ber et al., 2006; O’Rourke et al., 2006; Delmaire et al., 2007; Hagenah et al., 2007; Obermann et al., 2007). Multiple functional imaging studies also note abnormal activity of the cerebellum in dystonia. In some cases, abnormal activity is limited to the cerebellum or quantitatively greater than abnormalities elsewhere (Kluge et al., 1998; Odergren et al., 1998; Lehericy et al., 2004). The cerebellum is an essential contributor to network models of regional abnormalities in functional imaging studies of dystonia (Meunier et al., 2003; Asanuma et al., 2005), and recent studies suggest that its role is not adequately explained as a secondary phenomenon (Carbon et al., 2007). Finally, surgical studies have shown some patients with dystonia respond to deep brain stimulation of the ventral posterior thalamus or targeted lesions of this area (Krauss et al., 2004). This region of the thalamus is the major recipient of cerebellar afferents. It is not an integral part of the basal ganglia.
Additional evidence implicating the cerebellum in dystonia comes from experimental animal studies. The dt rat model exhibits abnormal movements with both clinical and electrophysiological hallmarks of dystonia (LeDoux and Lorden, 1998). Metabolic mapping and electrophysiological studies demonstrate abnormal activity of the cerebellum. Surgical removal of the cerebellum eliminates the dystonic movements and replaces them with ataxic movements. Cerebellectomy also eliminates dystonic movements in mutant tottering (as mentioned above) or lethargic mice (Devanagondi et al., 2007). The current studies confirm that pharmacological excitation of the cerebellum by local application of kainic acid acutely evokes dystonic movements even in normal mice (Pizoli et al., 2002). Together, the clinical and experimental studies provide strong converging support that the cerebellum plays an important role in dystonia.
The motor network model
The motor network model for dystonia provides a broad conceptual paradigm that accommodates results from the current studies, as well as results from prior clinical and animal studies that have pointed either to the basal ganglia or cerebellum. In principle, dystonia may result from dysfunction in any node of the network, or abnormalities in their interactions. Though the current studies and many prior ones have focused on the basal ganglia and cerebellum, the complete motor network may involve other nodes. For example, there is evidence that localized disruption of specific regions within the thalamus can lead to dystonia (Lee and Marsden, 1995; Kim, 2001; Lehericy et al., 2001). Dystonia also may arise from abnormal interactions among the regions. An example is the two-hit rat model for blepharospasm, where combining partial injury to nigral neurons with abnormal proprioceptive feedback from weakened orbicularis oculi muscles results in blepharospasm, when neither abnormality alone is sufficient (Schicatano et al., 1997). Additional studies are needed to define the whole network and how various insults in different nodes may result in dystonia.
How might the nodes interact in the expression of dystonia? The mechanisms for communication between the basal ganglia and cerebellum are only partly understood. The traditional conceptualization is that communication occurs predominantly via separate thalamic relays to shared regions of cerebral cortex. In fact, a common phenomenon among many different forms of human dystonia is enhanced cortical excitability, or impaired cortical inhibition (Hallett, 2006). Cortical excitability is regulated by both basal ganglia and cerebellum, with some evidence that they exert opposing influences (Liepert et al., 2004; Tamburin et al., 2004; Hallett, 2006). Thus, the influences of the basal ganglia and cerebellum may converge upon cortical excitability as a common final pathway for dystonic movement.
In addition to shared targets in cerebral cortex, there are more direct subcortical connections between the basal ganglia and cerebellum. In primates and rodents, the deep nuclei of the cerebellum project via disynaptic or trisynaptic pathways involving the thalamus to the putamen or globus pallidus (Ichinohe et al., 2000; McFarland and Haber, 2000; Hoshi et al., 2005). In cats a pathway from the deep cerebellar nuclei to nigrostriatal dopamine neurons has been described (Snider et al., 1976). More recently, there is growing appreciation for connections between basal ganglia and cerebellum through the zona incerta, fields of Forel and red nucleus (Pong et al., 2007).
There is also neurochemical evidence for communication between the basal ganglia and cerebellum via these subcortical connections. In our studies, dystonia arising from abnormal cerebellar function was associated with reduced dopamine in microdialysis samples from the striatum (Table 2). Others have shown that unilateral electrical stimulation of the cat deep cerebellar nuclei increased dopamine turnover in the contralateral striatum (Nieoullon et al., 1978). Conversely, unilateral electrolytic lesions of the deep cerebellar nuclei in rats reduced contralateral striatal dopamine turnover (Tellerman et al., 1979). Together, these studies show that cerebellar activity can influence the dynamics of striatal dopamine, a central neuromodulator of basal ganglia function that has frequently been implicated in dystonia (Perlmutter and Mink, 2004).
At the functional level, another potential mechanism for interactions between basal ganglia and cerebellum in dystonia involves the somewhat overlapping functions of the two motor systems, and the possibility that damage to one may lead to compensatory reactions of another. For example, rats with hemicerebellectomy display changes in the strength of corticostriatal signalling that parallel functional recovery of ataxic motor behaviour (Centonze et al., 2008). These findings suggest the basal ganglia may provide functional compensation for damage to the cerebellum. In this example, the compensatory changes reflect an adaptive response to restore normal motor function. Conversely, damage to the basal ganglia may lead to compensatory reactions in the cerebellum. These compensatory changes may not always produce an adaptive outcome, if the cerebellum is not well suited for the task and cannot operate with the same fidelity as the basal ganglia. In fact, a recent imaging study of regional metabolic patterns suggested a mechanism involving a primary defect in the basal ganglia that leads to secondary changes in the cerebellum as a cause for dystonia (Carbon et al., 2007). In this situation, dystonia results from a maladaptive cerebellar response rather than the primary pathology in the basal ganglia. Further studies in both animals and humans will be valuable for distinguishing primary defects from compensatory changes, and whether the compensations are adaptive or maladaptive.
Limitations of the current studies
Although the current studies support the motor network model for dystonia, some limitations must be acknowledged. It is well recognized that results from any single animal model may not generalize to other animal models or to the human disease (Jinnah et al., 2005; Jinnah and Hess, 2008). The current studies address this limitation by demonstrating the same outcome in two distinctly different animal models, each with two very different lesion methods. Obtaining similar results across four different experimental settings increases the likelihood of revealing more general principles regarding the neural substrates for dystonia.
Another limitation of the current studies involves the use of the lesion method to infer localization of function or dysfunction (Brazis et al., 2006). While examining the consequences of focal lesions in the nervous system traditionally has been one of the most extensively used strategies for localization in the dystonia literature, findings ultimately must be supported by converging sources of information from other methods. The current studies address some of the limitations of the classical lesion method by evaluating the effects of subclinical lesions that do not directly interfere with the motor disorder being evaluated. Additionally, microdialysis studies revealed a functional alteration in striatal dopamine in two different animal models where cerebellar dysfunction is thought to cause dystonia, even in the non-lesioned state.
Despite many years of research, the exact functions of the cerebellum and basal ganglia in normal motor control remain uncertain. The functions of these regions often are addressed separately for heuristic reasons, but normal motor behaviour obviously requires seamless integration of their overlapping functions. Different types of motor dysfunction also are assigned traditionally to one or another region. Cerebellar defects are most commonly associated with ataxia, while basal ganglia defects are associated with hypokinetic or hyperkinetic movement disorders. However, it is feasible that some disorders, such as dystonia, result more specifically from a defect in their interactions.
Acknowledgements
This work was supported by the Dystonia Medical Research Foundation and by NIH grants NS40470, NS33592 and NS48458.
Glossary
Abbreviations:
- ANOVA
analysis of variance
- QA
quinolinic acid
- 6OHDA
6-hydroxydopamine
References
- Asanuma K, Carbon-Correll M, Eidelberg D. Neuroimaging in human dystonia. J Med Invest. 2005;52:272–79. doi: 10.2152/jmi.52.272. [DOI] [PubMed] [Google Scholar]
- Berardelli A, Rothwell JC, Hallett M, Thompson PD, Manfredi M, Marsden CD. The pathophysiology of primary dystonia. Brain. 1998;121:1195–212. doi: 10.1093/brain/121.7.1195. [DOI] [PubMed] [Google Scholar]
- Bhatia KP, Marsden CD. The behavioral and motor consequences of focal lesions of the basal ganglia in man. Brain. 1994;117:859–76. doi: 10.1093/brain/117.4.859. [DOI] [PubMed] [Google Scholar]
- Brazis PW, Masdeu JC, Biller J. New York: Lippincott Williams & Wilkins; 2006. Localization in clinical neurology. [Google Scholar]
- Burns LH, Pakzaban P, Deacon TW, Brownell AL, Tatter SB, Jenkins BG. Selective putaminal excitotoxic lesions in non-human primates model the movement disorder of Huntington disease. Neuroscience. 1995;64:1007–17. doi: 10.1016/0306-4522(94)00431-4. [DOI] [PubMed] [Google Scholar]
- Campbell DB, Hess EJ. Cerebellar circuitry is activated during convulsive episodes in the tottering (tg/tg) mutant mouse. Neuroscience. 1998;85:773–83. doi: 10.1016/s0306-4522(97)00672-6. [DOI] [PubMed] [Google Scholar]
- Campbell DB, North JB, Hess EJ. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268–78. doi: 10.1006/exnr.1999.7171. [DOI] [PubMed] [Google Scholar]
- Carbon M, Ghilardi MF, Argyelan M, Dhawan V, Bressman SB, Eidelberg D. Increased cerebellar activation during sequence learning in DYT1 carriers: an equiperformance study. Brain. 2007;131:146–54. doi: 10.1093/brain/awm243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Rossi S, De Bartolo P, De Chiara V, Foti F, Musella A, et al. Adaptations of glutamatergic synapses in the striatum contribute to recovery from cerebellar damage. Eur J Neurosci. 2008;27:2188–96. doi: 10.1111/j.1460-9568.2008.06182.x. [DOI] [PubMed] [Google Scholar]
- Cuny E, Ghorayeb I, Guehl D, Escola L, Bioulac B, Burbaud P. Sensory motor mismatch within the supplementary motor area in the dystonic monkey. Neurobiol Dis. 2008;30:151–61. doi: 10.1016/j.nbd.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Delmaire C, Vidailhet M, Elbaz A, Bourdain F, Bleton JP, Sangla S, et al. Structural abnormalities in the cerebellum and sensorimotor circuit in writer's cramp. Neurology. 2007;69:376–80. doi: 10.1212/01.wnl.0000266591.49624.1a. [DOI] [PubMed] [Google Scholar]
- Devanagondi R, Egami K, LeDoux MS, Hess EJ, Jinnah HA. Neuroanatomical substrates for paroxysmal dyskinesias in lethargic mice. Neurobiol Dis. 2007;27:249–57. doi: 10.1016/j.nbd.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draganski B, Thun-Hohnstein C, Bogdahn U, Windler J, May A. Motor circuit gray matter changes in idiopathic cervical dystonia. Neurology. 2003;61:1228–31. doi: 10.1212/01.wnl.0000094240.93745.83. [DOI] [PubMed] [Google Scholar]
- Egami K, Yitta S, Kasim S, Lewers JC, Roberts RC, Lehar M, et al. Basal ganglia dopamine loss due to defect in purine recycling. Neurobiol Dis. 2007;26:396–407. doi: 10.1016/j.nbd.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Hess EJ. D2-like dopamine receptors mediate the response to amphetamine in a mouse model of ADHD. Neurobiol Dis. 2007;26:201–11. doi: 10.1016/j.nbd.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernagut PO, Diguet E, Stefanova N, Biran M, Wenning GK, Canioni P, et al. Subacute systemic 3-nitropropionic acid intoxication induces a distinct motor disorder in adult C57BL/6 mice: behavioural and histopathological characterisation. Neuroscience. 2002;114:1005–17. doi: 10.1016/s0306-4522(02)00205-1. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Lutz CM, O'Sullivan TN, Shaughness JD, Hawkes R, Frankel WN, et al. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–17. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- Fureman BE, Jinnah HA, Hess EJ. Triggers of paroxysmal dyskinesias in the calcium channel mouse mutant tottering. Pharmacol Biochem Behav. 2002;73:631–37. doi: 10.1016/s0091-3057(02)00854-7. [DOI] [PubMed] [Google Scholar]
- Gernert M, Bennay M, Fedrowitz M, Fehders JH, Richter A. Altered discharge pattern of basal ganglia output neurons in an animal model of idiopathic dystonia. J Neurosci. 2002;22:7244–53. doi: 10.1523/JNEUROSCI.22-16-07244.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorayeb I, Fernagut PO, Stefanova N, Wenning GK, Bioulac B, Tison F. Dystonia is predictive of subsequent altered dopaminergic responsiveness in a chronic 1-methyl4-phenyl-1,2,3,6-tetrahydropyridine +3-nitropropionic acid model of striatonigral degeneration in monkeys. Neurosci Lett. 2002;335:34–38. doi: 10.1016/s0304-3940(02)01137-0. [DOI] [PubMed] [Google Scholar]
- Giffin NJ, Benton S, Goadsby PJ. Benign paroxysmal torticollis of infancy: four new cases and linkage to CACNA1A mutation. Dev Med Child Neurol. 2002;44:490–93. doi: 10.1017/s0012162201002407. [DOI] [PubMed] [Google Scholar]
- Hagenah J, Reetz K, Zuhlke C, Rolfs A, Binkofski F, Klein C. Predominant dystonia with marked cerebellar atrophy: a rare phenotype in famililal dystonia. Neurology. 2007;68:2157. doi: 10.1212/01.wnl.0000269478.69285.7e. [DOI] [PubMed] [Google Scholar]
- Hallett M. Pathophysiology of dystonia. J Neural Transm Suppl. 2006;70:485–88. doi: 10.1007/978-3-211-45295-0_72. [DOI] [PubMed] [Google Scholar]
- Hess EJ, Jinnah HA. Mouse models of dystonia. In: Le Doux MS, editor. Animal models of movement disorders. San Diego: Elsevier Academic Press; 2005. pp. 265–77. [Google Scholar]
- Hoshi E, Tremblay L, Feger J, Carras PL, Strick PL. The cerebellum communicates with the basal ganglia. Nat Neurosci. 2005;8:1491–93. doi: 10.1038/nn1544. [DOI] [PubMed] [Google Scholar]
- Ichinohe N, Mori F, Shoumura K. A di-synaptic projection from the lateral cerebellar nucleus to the laterdorsal part of the striatum via the central lateral nucleus of the thalamus in the rat. Brain Res. 2000;880:191–97. doi: 10.1016/s0006-8993(00)02744-x. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Hess EJ. Animal models of movement disorders. San Diego: Elsevier Academic Press; 2005. The assessment of movement disorders in mice. In: Le Doux MS, editor; pp. 55–71. [Google Scholar]
- Jinnah HA, Hess EJ. A new twist on the anatomy of dystonia: the basal ganglia and the cerebellum. Neurology. 2006;67:1740–41. doi: 10.1212/01.wnl.0000246112.19504.61. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Hess EJ. Experimental therapeutics for dystonia. Neurotherapeutics. 2008;5:198–209. doi: 10.1016/j.nurt.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnah HA, Hess EJ, LeDoux MS, Sharma N, Baxter MG, DeLong MR. Rodent models for dystonia research: characteristics, evaluation, and utility. Mov Disord. 2005;20:283–92. doi: 10.1002/mds.20364. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, Sepkuty JP, Ho T, Yitta S, Drew T, Rothstein JD, et al. Calcium channel agonists and dystonia in the mouse. Mov Disord. 2000;15:542–51. doi: 10.1002/1531-8257(200005)15:3<542::AID-MDS1019>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Kim JS. Delayed onset mixed involuntary movements after thalamic stroke. Brain. 2001;124:299–309. doi: 10.1093/brain/124.2.299. [DOI] [PubMed] [Google Scholar]
- Kluge A, Kettner B, Zschenderlein R, Sandrock D, Munz DL, Hesse S, et al. Changes in perfusion pattern using EDC-SPECT indicated frontal lobe and cerebellar involvement in exercise-induced paroxysmal dystonia. Mov Disord. 1998;13:125–34. doi: 10.1002/mds.870130124. [DOI] [PubMed] [Google Scholar]
- Krauss JK, Seeger W, Jankovic J. Cervical dystonia associated with tumors of the posterior fossa. Mov Disord. 1997;12:443–47. doi: 10.1002/mds.870120329. [DOI] [PubMed] [Google Scholar]
- Krauss JK, Yianni J, Loher TJ, Aziz TZ. Deep brain stimulation for dystonia. J Clin Neurophysiol. 2004;21:18–30. doi: 10.1097/00004691-200401000-00004. [DOI] [PubMed] [Google Scholar]
- Kupsch A, Benecke R, Muller J, Trottenberg T, Schneider GH, Poewe W, et al. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med. 2006;355:1978–90. doi: 10.1056/NEJMoa063618. [DOI] [PubMed] [Google Scholar]
- Le Ber I, Clot F, Vercueil L, Camzuat A, Viemont M, Benamar N, et al. Predominant dystonia with marked cerebellar atrophy: a rare phenotype in familial dystonia. Neurology. 2006;67:1769–73. doi: 10.1212/01.wnl.0000244484.60489.50. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, Brady KA. Secondary cervical dystonia associated with structural lesions of the central nervous system. Mov Disord. 2002;18:60–69. doi: 10.1002/mds.10301. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, Lorden JF. Abnormal cerebellar output in the genetically dystonic rat. In: Fahn S, Marsden CD, DeLong M, editors. Dystonia 3. Vol. 78. Philadelphia: Lippincott-Raven Publishers; 1998. pp. 63–78. [PubMed] [Google Scholar]
- LeDoux MS, Lorden JF, Ervin JM. Cerebellectomy eliminates the motor syndrome of the genetically dystonic rat. Exp Neurol. 1993;120:302–10. doi: 10.1006/exnr.1993.1064. [DOI] [PubMed] [Google Scholar]
- Lee MS, Marsden CD. Movement disorders following lesions of the thalamus or subthalamic region. Mov Disord. 1995;9:493–507. doi: 10.1002/mds.870090502. [DOI] [PubMed] [Google Scholar]
- Lehericy S, Gerardin E, Poline JB, Meunier S, Van de Moortele PF, Le Bihan D, et al. Motor execution and imagination networks in post-stroke dystonia. NeuroReport. 2004;15:1887–90. doi: 10.1097/00001756-200408260-00010. [DOI] [PubMed] [Google Scholar]
- Lehericy S, Grand S, Pollak P, Poupon F, LeBas JF, Limousin P, et al. Clinical characteristics and topography of lesion in movement disorders due to thalamic lesions. Neurology. 2001;57:1055–66. doi: 10.1212/wnl.57.6.1055. [DOI] [PubMed] [Google Scholar]
- Liepert J, Kucinski T, Tuscher O, Pawlas F, Baumer T, Weiller C. Motor cortex excitability after cerebellar infarction. Stroke. 2004;35:2484–88. doi: 10.1161/01.STR.0000143152.45801.ca. [DOI] [PubMed] [Google Scholar]
- Marsden CD, Obeso JA, Zarranz JJ. The anatomical basis of symptomatic dystonia. Brain. 1985;108:463–83. doi: 10.1093/brain/108.2.463. [DOI] [PubMed] [Google Scholar]
- McFarland NR, Haber SN. Convergent inputs from thalamic motor nuclei and frontal cortical areas to the dorsal striatum in the primate. J Neurosci. 2000;20:3798–813. doi: 10.1523/JNEUROSCI.20-10-03798.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier S, Lehericy S, Garnero L, Vidailhet M. Dystonia: lessons from brain mapping. Neuroscientist. 2003;9:76–81. doi: 10.1177/1073858402239593. [DOI] [PubMed] [Google Scholar]
- Nieoullon A, Cheramy A, Glowisnki J. Release of dopamine in both caudate nuclei and both substantia nigrae in response to unilateral stimulation of cerebellar nuclei in the cat. Brain Res. 1978;148:143–52. doi: 10.1016/0006-8993(78)90384-0. [DOI] [PubMed] [Google Scholar]
- O’Rourke K, O’Riordan S, Gallagher J, Hutchinson M. Paroxysmal torticollis and blepharospasm following bilateral cerebellar infarction. J Neurol. 2006;253:1644–45. doi: 10.1007/s00415-006-0202-3. [DOI] [PubMed] [Google Scholar]
- Obermann M, Yaldizli Z, De Greiff A, Lachenmayer ML, Buhl AR, Tumczak F, et al. Morphometric changes of sensorimotor structures in focal dystonia. Mov Disord. 2007;22:1117–23. doi: 10.1002/mds.21495. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Gimenez-Roldan S. Clinicopathologic correlation in symptomatic dystonia. Adv Neurol. 1988;50:113–22. [PubMed] [Google Scholar]
- Odergren T, Stone-Elander S, Ingvar M. Cerebral and cerebellar activation in correlation to the action-induced dystonia in writer's cramp. Mov Disord. 1998;13:497–508. doi: 10.1002/mds.870130321. [DOI] [PubMed] [Google Scholar]
- Palfi S, Leventhal L, Goetz CG, Hantraye T, Roitberg BZ, Sramek J, et al. Delayed onset of progressive dystonia following subacute 3-nitropropionic acid treatment in Cebus apella monkeys. Mov Disord. 2000;15:524–30. [PubMed] [Google Scholar]
- Perlmutter JS, Mink JW. Dysfunction of dopaminergic pathways in dystonia. Adv Neurol. 2004;94:163–70. [PubMed] [Google Scholar]
- Perlmutter JS, Tempel LW, Black KJ, Parkinson D, Todd RD. MPTP induces dystonia and parkinsonism. Clues to the pathophysiology of dystonia. Neurology. 1997;49:1432–38. doi: 10.1212/wnl.49.5.1432. [DOI] [PubMed] [Google Scholar]
- Pizoli CE, Jinnah HA, Billingsley ML, Hess EJ. Abnormal cerebellar signaling induces dystonia in mice. J Neurosci. 2002;22:7825–33. doi: 10.1523/JNEUROSCI.22-17-07825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pong M, Horn KM, Gibson AR. Pathways for control of face and neck musculature by the basal ganglia and cerebellum. Brain Res Rev. 2008 doi: 10.1016/j.brainresrev.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Roedter A, Winkler C, Samii M, Walter GF, Brandis A, Nikkhah G. Comparison of unilateral and bilateral intrastriatal 6-hydroxydopamine induced axon termanl lesions: evidence for interhemispheric functional coupling of the two nigrostriatal pathways. J Comp Neurol. 2001;432:217–29. doi: 10.1002/cne.1098. [DOI] [PubMed] [Google Scholar]
- Schicatano EJ, Basso MA, Evinger C. Animal model explains the origins of the cranial dystonia benign essential blepharospasm. J Neurophysiol. 1997;77:2842–46. doi: 10.1152/jn.1997.77.5.2842. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Whetsell WO, Mangano RM. Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science. 1983;219:316–18. doi: 10.1126/science.6849138. [DOI] [PubMed] [Google Scholar]
- Shirley TL, Rao LM, Hess EJ, Jinnah HA. Paroxysmal dyskinesias in mice. Mov Disord. 2008;23:259–264. doi: 10.1002/mds.21829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider RS, Maiti A, Snider SR. Cerebellar pathways to ventral midbrain and nigra. Exp Neurol. 1976;53:714–28. doi: 10.1016/0014-4886(76)90150-3. [DOI] [PubMed] [Google Scholar]
- Spacey SD, Materek LA, Szczygielski BI, Bird TD. Two novel CACNA1A gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch Neurol. 2005;62:314–16. doi: 10.1001/archneur.62.2.314. [DOI] [PubMed] [Google Scholar]
- Tabbal SD, Mink JW, Antenor JAV, Carl JL, Moerlein SM, Perlmutter JS. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced acute transient dystonia in monkeys with low striatal dopamine. Neuroscience. 2006;141:1281–87. doi: 10.1016/j.neuroscience.2006.04.072. [DOI] [PubMed] [Google Scholar]
- Tamburin S, Fiaschi A, Marani S, Andreoli A, Manganotti P, Zanette G. Enhanced intracortical inhibition in cerebellar patients. J Neurol Sci. 2004;217:205–10. doi: 10.1016/j.jns.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Tarsy D, Simon DK. Dystonia. N Engl J Med. 2006;355:818–29. doi: 10.1056/NEJMra055549. [DOI] [PubMed] [Google Scholar]
- Tellerman K, Astrow A, Fahn S, Snider SR, Snider RS, Glassgold JM. Cerebellar control of catecholaminergic activities: implications for drug therapy of movement disorders. Int J Neurol. 1979;12:135–55. [PubMed] [Google Scholar]
- Vidailhet M, Vercueil L, Houeto JL, Krystkowiak P, Benabid AL, Cornu P, et al. Bilateral deep-brain stimulation of the globus pallidus in primary generalized dystonia. N Engl J Med. 2005;352:459–67. doi: 10.1056/NEJMoa042187. [DOI] [PubMed] [Google Scholar]
- Wakamori M, Yamazaki K, Matsunodaira H. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem. 1998;52:34857–67. doi: 10.1074/jbc.273.52.34857. [DOI] [PubMed] [Google Scholar]
- Winkler C, Kirik D, Bjorklund A, Cenci MA. L-DOPA-induced dyskinesias in the intrastriatal 6-hydroxydopamine model of Parkinson's disease: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis. 2002;10:165–86. doi: 10.1006/nbdi.2002.0499. [DOI] [PubMed] [Google Scholar]