

Abstract
Charcot-Marie-Tooth disease type 1A (CMT1A) is caused by a 1.4 Mb duplication on chromosome 17p11.2, which contains the peripheral myelin protein-22 (PMP22) gene. Increased levels of PMP22 in compact myelin of peripheral nerves have been demonstrated and presumed to cause the phenotype of CMT1A. The objective of the present study was to determine whether an extra copy of the PMP22 gene in CMT1A disrupts the normally coordinated expression of PMP22 protein in peripheral nerve myelin and to evaluate PMP22 over-expression in patients with CMT1A and determine whether levels of PMP22 are molecular markers of disease severity. PMP22 expression was measured by taking skin biopsies from patients with CMT1A (n = 20) and both healthy controls (n = 7) and patients with Hereditary Neuropathy with liability to Pressure Palsies (HNPP) (n = 6), in which patients have only a single copy of PMP22. Immunological electron microscopy was performed on the skin biopsies to quantify PMP22 expression in compact myelin. Similar biopsies were analysed by real time PCR to measure PMP22 mRNA levels. Results were also correlated with impairment in CMT1A, as measured by the validated CMT Neuropathy Score. Most, but not all patients with CMT1A, had elevated PMP22 levels in myelin compared with the controls. The levels of PMP22 in CMT1A were highly variable, but not in HNPP or the controls. However, there was no correlation between neurological disabilities and the level of over-expression of PMP22 protein or mRNA in patients with CMT1A. The extra copy of PMP22 in CMT1A results in disruption of the tightly regulated expression of PMP22. Thus, variability of PMP22 levels, rather than absolute level of PMP22, may play an important role in the pathogenesis of CMT1A.
Keywords: PMP22, CMT1A, CMTNS, HNPP, Schwann cell, myelin, Charcot-Marie-Tooth disease
Introduction
Charcot-Marie-Tooth (CMT) disease refers to a group of inherited neuropathies with a prevalence of 1 in 2500 people (Skre, 1974). Dominantly inherited CMT can be separated into demyelinating (CMT1) and axonal (CMT2). At present, mutations in over 35 different genes have been identified and chromosomal localization of many other distinct inherited neuropathies have been mapped (Jani-Acsadi et al., 2008). Charcot-Marie-Tooth disease type 1A (CMT1A) is the most common form of CMT, affecting ∼50% of all CMT cases (Ionasescu et al., 1993; Wise et al., 1993) and 70% of patients with CMT1 (Nelis et al., 1996). CMT1A is caused by a 1.4 Mb duplication of chromosome 17p11.2-12 (Lupski et al., 1991; Raeymaekers et al., 1991). Deletion of this identical 1.4 Mb region causes a different inherited neuropathy, hereditary neuropathy with liability to pressure palsies (HNPP) (Chance et al., 1993). These two allelic neuropathies, CMT1A and HNPP, differ in many respects. Patients with CMT1A develop a slowly progressive, symmetrical, dymyelinating neuropathy with secondary axonal loss (Thomas et al., 1997; Krajewski et al., 2000). In contrast, patients with HNPP experience reversible multifocal weakness or sensory loss (Li et al., 2004). Pathological studies from CMT1A reveal diffuse ‘onion bulbs’ and length-dependent axonal degeneration (Lupski and Garcia, 1992; Gabreels-Festen et al., 1999). In contrast, nerves in HNPP are characterized by focal myelin thickenings known as ‘tomacula’ because of their sausage-like shape (Behse et al., 1972; Madrid and Bradley, 1975; Fewings et al., 1985; Yoshikawa and Dyck, 1991; Vallat et al., 1996; Gabreels-Festen et al., 1999). The peripheral myelin protein (PMP22) gene is contained within the region of chromosome 17p11.2-12 and its over- and under- expression is believed to be directly responsible for both diseases (Lupski et al., 1991; Raeymaekers et al., 1992; Chance et al., 1993; Inoue et al., 2001). A typical CMT1A phenotype is characterized by distal weakness, sensory loss, foot deformities and absent reflexes. However, the phenotypic presentation can be quite variable with severe cases presenting in infancy while occasional patients remain asymptomatic even in adulthood. The reasons for phenotypic variability in CMT1A are not known.
Morphological analyses of myelinated nerves from CMT1A patients have been primarily performed on sural nerve biopsies (Gabreels-Festen et al., 1995; Vallat et al., 1996). These biopsies require a surgical approach, may result in pain, and can only be performed twice for a given patient since humans have only two sural nerves. Since biopsies are not necessary to make the diagnosis, they are now rarely performed in CMT1A. We (Li et al., 2005; Sabet et al., 2006) and others (Nolano et al., 2003; Lombardi et al., 2005) have begun using 2–3 mm punch biopsies from glabrous skin to evaluate myelinated peripheral nerves. This minimally invasive procedure has shown that the morphological and molecular phenotypes of dermal myelinating Schwann cells behave similarly to myelinating Schwann cells ensheathing other peripheral nervous system axons (Li et al., 2005). In preliminary studies, using immuno-electron microscopy (immuno-EM), we observed that PMP22 was over-expressed in dermal Schwann cells from patients with CMT1A but we noticed that this over-expression seemed variable in the few patients we analysed (Li et al., 2005). Furthermore, the increased levels of PMP22 mRNA and PMP22 protein in CMT1A have been reported in a number of studies on sural nerve biopsies. The level of PMP22 appeared variable in some publications and was hypothesized to be secondary to demyelination (Hanemann et al., 1994; Yoshikawa et al., 1994; Vallat et al., 1996; Gabriel et al., 1997; Schenone et al., 1997).
The mutation causing CMT1A introduces an additional copy of PMP22 in tandem into one of the alleles of chromosome 17p11.2. In order to maintain PMP22 expression at normal level, this extra copy would have to be regulated in a coordinated fashion with the other two copies of PMP22, both temporally and spatially. However, the capacity to regulate an additional copy of PMP22 is not programmed or inherited in myelinating Schwann cells. Therefore, we hypothesized that PMP22 levels in CMT1A may be dysregulated and unpredictable between patients. Newly translated PMP22 in myelinated Schwann cells must undergo post-translational modifications. Levels of PMP22, like other myelin proteins, are tightly coordinated prior to reaching the compact myelin (Stahl et al., 1990; Pareek et al., 1997; Trapp et al., 2003). Normally, ∼90% of translated PMP22 is rapidly degraded and never inserted into myelin; less than 10% of the translated protein is therefore incorporated into myelin (Pareek et al., 1997). Over-expressed PMP22 in CMT1A would be subject to this same post-translational regulation. Whether abnormalities in post-transcriptional or post-translational regulation of PMP22 are involved in the pathogenesis of CMT1A or HNPP, in which there is only a single copy of the PMP22 gene, is unknown. With the establishment of our skin biopsy technique (Li et al., 2005), we had an opportunity to examine these issues in a cohort of patients with CMT1A by performing immuno-electron microscopy and real-time PCR to quantify PMP22 expression. These levels were then correlated with impairment of CMT1A patients, as defined by a previously validated CMT Neuropathy Score (CMTNS) (Shy et al., 2005). Our results show highly variable levels of both PMP22 protein and mRNA in CMT1A. In contrast, PMP22 levels appear uniform between different patients with HNPP. Variable levels of PMP22 protein or mRNA did not correlate with neurological disabilities in patients with CMT1A; thus they were not responsible for differences in the neurological impairment of this disease.
Methods
Patient evaluation
Subjects with CMT1A, HNPP and controls were prospectively evaluated at the CMT Neurology Clinic at Wayne State University. Evaluations consisted of a neurological history and examination and nerve conduction studies. Genetic testing at Athena Laboratories (Worcester, MA, USA) was performed in all patients or in first or second degree relatives to document the presence of a 1.4 Mb duplication on chromosome 17p11.2 (Lupski et al., 1991; Raeymaekers et al., 1991) indicating a diagnosis of CMT1A. If the subject had a first or second-degree relative with a documented duplication, a diagnosis was considered confirmed if the subject also had uniform motor conduction slowing of the median or ulnar nerve <35 and >10 m/s. Relatives with NCV outside this range required confirmation of CMT1A by genetic testing.
CMT Neuropathy Score and neuropathy impairment score
The severity of the peripheral neuropathy was evaluated in all patients by the CMT Neuropathy Score (CMTNS), a validated measurement of impairment in patients with CMT (Shy et al., 2005). The CMTNS is a composite score based on the history (total possible points = 12), the neurological examination (total possible points = 16) and clinical neurophysiology (total possible points = 8); the maximal possible score is 36. Patients with mild, intermediate and severe disability typically have a CMTNS between 1–10, 11–20 and 21 or greater.
Skin biopsy, real-time PCR and immuno-electron-microscopy
Glabrous skin biopsies were performed from the lateral aspect of the index finger for immuno-electron microscopy or distal forearm for real-time PCR. Total RNA was extracted from the forearm biopsies and subjected to the PCR analysis as described before. Immuno-electron microscopy was performed on biopsies to measure PMP22 or myelin basic protein (MBP) protein levels on the compact myelin (Fig. 1). The primary antibodies were polyclonal rabbit PMP22 (Lab Vision Corporation) and monoclonal mouse MBP (UltraClone Limited). The secondary antibodies were 12 nm gold-particle conjugated anti-rabbit and anti-mouse antibodies (Gold-AffiniPure, Jackson ImmunoResearch) (Li et al., 2005). Real-time PCR and immuno-electron microscopy levels were correlated with results from patients’ CMTNS.
Figure 1.
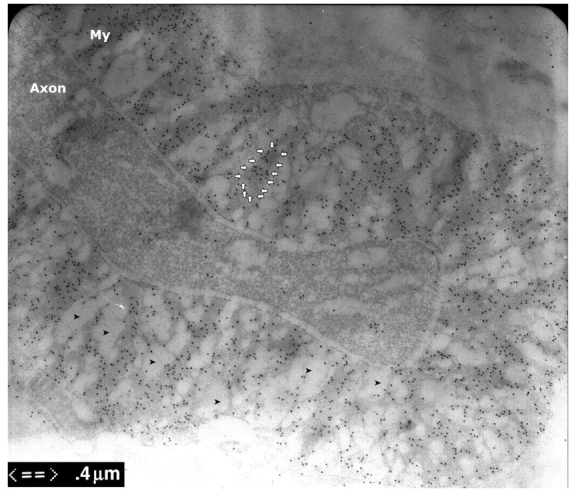
Immuno-electron microscopy of dermal myelinated nerve fibres. Skin biopsies were fixed for 3 h and embedded in LR white. The ultra-thin sections were stained with antibodies against PMP22 that were conjugated with gold particles. These particles were mainly found in compact myelin, but minimally in axons. Immuno-electron microscopy fixation techniques cannot completely preserve the compact myelin and usually results in vacuolated areas in the compact myelin (arrowheads). To exclude the effect of these vacuoles on our data, we only measured the grain density in the areas with intact myelin (arrow circle).
Statistical analysis
Comparisons of PMP22, MBP and S100 levels between patients with CMT1A, HNPP and control subjects were analysed using unpaired Student's t-test. Correlations between the CMTNS and immuno-electron microscopy and mRNA levels were performed using Pearson's correlation test. Prism 4 software (GraphPad Inc., 2003) was used for all the statistical analysis.
Results
PMP22 expression in Schwann cells is variable in CMT1A but not in HNPP
To compare PMP22 levels, we measured the density of PMP22 in compact myelin from skin biopsies of 20 patients with CMT1A, 6 patients with HNPP and 7 subjects with no history or evidence of neuropathy. We were able to identify an average of 6 myelinated nerve fibres in each of the 33 biopsies. An average of 980.2 μm2 myelin surface was found in each biopsy. Similar to our preliminary data (Li et al., 2005), values for normal controls were grouped at approximately 80 ± 19 grains/μm2. Densities for patients with HNPP were reduced, tightly clustered around 42 ± 13 grains/μm2. As predicted, values from CMT1A nerves were elevated with a mean value of 154 ± 68 grains/mm2. However, the distribution of grain densities from CMT1A nerves was highly variable, with a range between 57.7 and 301.8 grains/μm2 (Fig. 2).
Figure 2.

PMP22 and MBP grain densities in patients with CMT1A and normal controls. Skin biopsies were processed for immuno-electron microscopy studies. PMP22 grain densities were highly variable in CMT1A, but not in HNPP or normal controls. The densities were measured in grains/μm2.
We next evaluated clinical impairment in biopsied patients with CMT1A, using the CMTNS. The mean value for all patients was 20, at the high end of what is considered ‘moderate’ CMT (Shy et al., 2005). These results were also variable between patients with CMT1A; 5 had a CMTNS <10 (mild CMT), 10 had scores in the moderate range (11–20) and 5 patients had scores in the severe range (>20). We compared PMP22 grain densities with CMTNS results. There was no correlation between the two variables (r = −0.16; p = 0.5); thus variability in clinical impairment did not correlate with differing levels of PMP22 in dermal myelinated nerves (Fig. 3A).
Figure 3.

(A) Comparison of PMP22 expression and clinical impairment in CMT1A. There is no significant correlation between the PMP22 protein density in peripheral myelin and the CMTNS (n = 20, R = −0.16, Pearson's p = 0.5). (B) Although there is a trend between PMP22 mRNA level and CMTNS, the correlation does not reach statistical significance (n = 29, R = 0.33, Pearson's p = 0.08).
We were concerned that regional differences in demyelination of individual internodes may have influenced our findings, independent of PMP22 over-expression. We therefore measured MBP grain densities as a surrogate control of demyelination effect since MBP is myelin specific, not genetically altered in CMT1A and normally expressed in similar amounts to PMP22 in myelinating Schwann cells (Snipes and Suter, 1995). We compared grain densities for MBP in 11 patients with CMT1A (21 ± 13 grains/μm2) and 5 controls without neuropathy (29 ± 17 grains/μm2); these differences were not significant (p = 0.31). We then generated a PMP22/MBP ratio for each patient and compared this to the CMTNS. Again there was no significant correlation (r = 0.11; p = 0.74). Taken together, these results did not demonstrate a correlation between PMP22 protein levels and clinical impairment in CMT1A, even when the extent of demyelination was taken into consideration.
CMT neuropathy sub-scores do not correlate with PMP22 expression in CMT1A
The CMTNS is a composite score consisting of symptoms, signs and neurophysiologic results for motor and sensory nerves. To determine whether PMP22 protein expression correlated with any of these individual components we compared PMP22 grain density to individual CMTNS sub-scores. Mild correlations were obtained for sensory symptoms (r = −0.44), motor symptoms in the arms (r = −0.30), ulnar compound muscle action potential amplitudes (r = −0.28) and vibration sensation (r = 0.33). No correlation was obtained for motor symptoms in the arms, arm or leg strength, pin sensation or ulnar sensory nerve action potential amplitudes. There was also no significant correlation between the age of the patients and PMP22 grain densities (r = 0.051).
PMP22 mRNA levels do not significantly correlate with CMTNS
An extra copy of PMP22 demands a regulation of gene expression coordinated temporally and spatially between the three copies of the PMP22 genes in CMT1A (Pareek et al., 1997). However, this coordinated regulation is not programmed or inherited, but new to Schwann cells; thus transcription of PMP22 may also be variable in CMT1A. We utilized real-time PCR to quantify PMP22 mRNA levels in a series of 29 patients with CMT1A and correlated the levels with their CMTNS. PMP22 mRNA levels (normalized to S100 mRNA) were also variable from subject to subject. While there was a trend between mRNA levels and CMTNS (r = 0.33), this correlation did not achieve significance (p = 0.08). Thus, while both protein and mRNA levels varied from patient to patient, there was no demonstrable correlation between PMP22 mRNA or PMP22 protein levels with clinical impairment as measured by the CMTNS (Fig. 3B). Correlation between the PMP22 protein levels and mRNA expression was not possible because the two measurements were performed on two different groups of subjects (an entire biopsy is needed for immuno-electron microscopy or for RNA extraction).
Discussion
We have demonstrated that there is wide variability in the density of PMP22 in dermal myelin in patients with CMT1A but not in HNPP, or subjects without CMT. We have also demonstrated that variability of PMP22 protein or mRNA levels does not correlate with clinical impairment and thus does not explain the observed phenotypic variability of patients with CMT1A, at least when this variability is measured by the CMTNS. We were initially concerned that variable demyelination in sampled internodes from CMT1A patients may have influenced our results. However MBP expression levels did not vary in our patients and MBP levels are normally similar to those of PMP22 in peripheral nervous system myelin (Trapp et al., 2003). If our PMP22 results simply varied with the extent of demyelination we would have expected to find variable expression of MBP as well. Moreover, we have demonstrated previously and in the present study that there is a decreased level of PMP22 in patients with HNPP and heterozygous deletion of chromosome 17p11.2 (Li et al., 2005), similar to that reported in sural nerve biopsies using immuno-electron microscopy (Vallat et al., 1996). This reduction of PMP22 level was also replicated by the same technique in a large family of HNPP patients with a heterozygous missense mutation resulting in a premature termination of PMP22, demonstrating the predicted reduction of PMP22 in myelin (Li et al., 2007). The reduction of PMP22 among all these patients was uniform, not variable. These findings suggest that our method of PMP22 measurement in skin biopsies with immuno-electron microscopy is reliable, and reproduces findings obtained in sural nerve biopsies. Thus, we believe that variable PMP22 over-expression in CMT1A myelin represents a specific property of the disease. We hypothesize that dysregulation of PMP22 expression, rather than the absolute level of increased PMP22 per se, causes neuropathy in CMT1A. It would be important in future studies to test whether degrees of oscillation of PMP22 expression level correlate with disabilities in patients with CMT1A.
As much as 90% of newly translated PMP22 is immediately degraded before the newly synthesized protein leaves the endoplasmic reticulum; only a small portion is targeted to myelin (Pareek et al., 1997). PMP22 that is targeted to myelin is processed through the secretory pathway in which proteins are folded within the endoplasmic reticulum, glycosylated within the Golgi and then incorporated into vesicles that transport them to the myelin sheath as tetramembrane proteins (Snipes et al., 1992; Trapp et al., 2003). The function of PMP22 in myelin remains unknown although a leading hypothesis is that it binds to tetramers of myelin protein zero in a stoichiometric fashion (one PMP22 to four myelin protein zero molecules), helping to stabilize myelin protein zero and compact myelin (D'Urso et al., 1999; Hasse et al., 2004). Processes that disrupt the timing and amounts of myelin proteins into myelin have the potential to disrupt myelination or cause dysmyelination. However, these processes must do more than simply reduce the normal amount of the myelin protein since haploinsufficiency of PMP22 causes HNPP, a distinct disorder from CMT1A and haploinsufficiency of myelin protein zero causes only a mild, late-onset neuropathy in heterozygous myelin protein zero null mice (Martini et al., 1995; Shy et al., 1997). Moreover, our data show that PMP22 levels in myelin continue to be tightly regulated in HNPP; the values cluster at about half of the normal amount as would be predicted by haploinsufficiency.
Another way of considering this issue is in terms of ‘toxic gain-of-function’ compared to ‘loss-of-function’ mutations. Since CMT1A is distinct and typically more severe than HNPP, it cannot result simply from a loss of normal PMP22 function, the cause of HNPP. There must be an additional pathogenic mechanism in CMT1A that may have nothing to do with the normal function of PMP22. Similarly, missense mutations in PMP22 or myelin protein zero may cause very severe neuropathies because of toxic gain-of-function abnormalities. In these circumstances endoplasmic reticulum retention of mutated proteins and activation of processes such as the unfolded protein pathway may contribute to the severity of the neuropathy (Shames et al., 2003; Grandis et al., 2008; Pennuto et al., 2008). Alternatively, in loss-of-function disorders, such as HNPP, the normal trafficking of PMP22 is not disrupted and PMP22 levels remain clustered at the predicted 50% of their normal level. If our hypothesis of variability of PMP22 expression is correct, this would provide a novel approach to treatment strategies; working to stabilize PMP22 levels in myelin rather than focusing exclusively on simply reducing PMP22 levels in patients with CMT1A. The latter approach may even be harmful for those patients with CMT1A with near normal or normal levels of PMP22.
Our results demonstrate that phenotypic variability in CMT1A cannot be explained by variable PMP22 levels in myelin. Whether particular haplotypes or modifier genes contribute to phenotypic variability is an ongoing area of research. The fact that identical twins with CMT1A have demonstrated phenotypic variability (Garcia et al., 1995) suggests that non-genetic as well as genetic factors are likely to contribute. For example, medications such as vincristine are known to exacerbate CMT1A (Weimer and Podwall, 2006) and we have recently shown that CMT1A is often more severe in patients with diabetes mellitus (Sheth et al., 2008). Sorting this out will require careful uniform clinical analysis of many patients in addition to analysis of their DNA.
We appreciate that a correlation between Pmp22 mRNA levels and clinical findings was observed in CMT1A rats (Meyer zu Horste et al., 2007). CMT1A rats are bred into a defined genetic background. In contrast, the genetic background in the human subjects enrolled in our study was mixed. Thus, the presence of modifiers, as discussed above, may contribute to expression differences between rats and patients.
Finally, our results raise the issue of how to interpret PMP22 mRNA levels in clinical trials. Treatment trials of CMT1A mice (Passage et al., 2004) and rats (Sereda et al., 2003; Meyer zu Horste et al., 2007) have demonstrated a reduction of PMP22 mRNA associated with clinical improvement following therapy. Based on these results, PMP22 mRNA levels, obtained from skin biopsies, are outcome measures in current clinical trials in Europe, the UK and the USA designed to interpret the ability of high dose ascorbic acid to slow progression or improve patients with CMT1A. Although our results suggest that PMP22 levels do not correlate directly with severity, this correlation in longitudinal changes remains to be determined. However, our results do suggest that measurements of mRNA and protein variability may become important end-points.
Funding
MDA (MDA4029); NINDS (K08 NS048204).
Glossary
Abbreviations
- CMT1A
Charcot-Marie-Tooth disease type 1A
- CMTNS
CMT Neuropathy Score
- HNPP
Hereditary Neuropathy with liability to Pressure Palsies
- MBP
myelin basic protein
- PMP22
peripheral myelin protein 22
References
- Behse F, Buchthal F, Carlsen F, Knappeis GG. Hereditary neuropathy with liability to pressure palsies. Electrophysiological and histopathological aspects. Brain. 1972;95:777–94. doi: 10.1093/brain/95.4.777. [DOI] [PubMed] [Google Scholar]
- Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–51. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- D'Urso D, Ehrhardt P, Muller HW. Peripheral myelin protein 22 and protein zero: a novel association in peripheral nervous system myelin. J Neurosci. 1999;19:3396–403. doi: 10.1523/JNEUROSCI.19-09-03396.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewings JD, Blumbergs PC, Mukherjee TM, Hallpike JF. Tomaculous neuropathy: hereditary predisposition to pressure palsies. Aust N Z J Med. 1985;15:598–603. [PubMed] [Google Scholar]
- Gabreels-Festen AA, Bolhuis PA, Hoogendijk JE, Valentijn LJ, Eshuis EJ, Gabreels FJ. Charcot-Marie-Tooth disease type 1A: morphological phenotype of the 17p duplication versus PMP22 point mutations. Acta Neuropathol. 1995;90:645–9. doi: 10.1007/BF00318579. [DOI] [PubMed] [Google Scholar]
- Gabreels-Festen A, van Beersum S, Eshuis L, LeGuern E, Gabreels F, van Engelen B, et al. Study on the gene and phenotypic characterisation of autosomal recessive demyelinating motor and sensory neuropathy (Charcot-Marie-Tooth disease) with a gene locus on chromosome 5q23–q33. J Neurol Neurosurg Psychiatry. 1999;66:569–74. doi: 10.1136/jnnp.66.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel JM, Erne B, Pareyson D, Sghirlanzoni A, Taroni F, Steck AJ. Gene dosage effects in hereditary peripheral neuropathy. Expression of peripheral myelin protein 22 in Charcot–Marie–Tooth disease type 1A and hereditary neuropathy with liability to pressure palsies nerve biopsies. Neurology. 1997;49:1635–40. doi: 10.1212/wnl.49.6.1635. [DOI] [PubMed] [Google Scholar]
- Garcia CA, Malamut RE, England JD, Parry GS, Liu P, Lupski JR. Clinical variability in two pairs of identical twins with the Charcot–Marie–Tooth disease type 1A duplication. Neurology. 1995;45:2090–3. doi: 10.1212/wnl.45.11.2090. [DOI] [PubMed] [Google Scholar]
- Grandis M, Vigo T, Passalacqua M, Jain M, Scazzola S, La Padula V, et al. Different cellular and molecular mechanisms for early and late-onset myelin protein zero mutations. Hum Mol Genet. 2008;17:1877–89. doi: 10.1093/hmg/ddn083. [DOI] [PubMed] [Google Scholar]
- Hanemann CO, Stoll G, D'Urso D, Fricke W, Martin JJ, Van Broeckhoven C, et al. Peripheral myelin protein-22 expression in Charcot–Marie–Tooth disease type 1a sural nerve biopsies. J Neurosci Res. 1994;37:654–9. doi: 10.1002/jnr.490370513. [DOI] [PubMed] [Google Scholar]
- Hasse B, Bosse F, Hanenberg H, Muller HW. Peripheral myelin protein 22 kDa and protein zero: domain specific trans-interactions. Mol Cell Neurosci. 2004;27:370–8. doi: 10.1016/j.mcn.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Inoue K, Dewar K, Katsanis N, Reiter LT, Lander ES, Devon KL, et al. The 1.4-Mb CMT1A duplication/HNPP deletion genomic region reveals unique genome architectural features and provides insights into the recent evolution of new genes. Genome Res. 2001;11:1018–33. doi: 10.1101/gr.180401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionasescu VV, Ionasescu R, Searby C, Barker DF. Charcot-Marie-Tooth neuropathy type 1A with both duplication and non-duplication. Hum Mol Genet. 1993;2:405–10. doi: 10.1093/hmg/2.4.405. [DOI] [PubMed] [Google Scholar]
- Jani-Acsadi A, Krajewski K, Shy ME. Charcot-Marie-Tooth neuropathies: diagnosis and management. Semin Neurol. 2008;28:185–94. doi: 10.1055/s-2008-1062264. [DOI] [PubMed] [Google Scholar]
- Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, et al. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. 2000;123(Pt 7):1516–27. doi: 10.1093/brain/123.7.1516. [DOI] [PubMed] [Google Scholar]
- Li J, Bai Y, Ghandour K, Qin P, Grandis M, Trostinskaia A, et al. Skin biopsies in myelin-related neuropathies: bringing molecular pathology to the bedside. Brain. 2005;128:1168–77. doi: 10.1093/brain/awh483. [DOI] [PubMed] [Google Scholar]
- Li J, Ghandour K, Radovanovic D, Shy RR, Krajewski KM, Shy ME, et al. Stoichiometric alteration of PMP22 protein determines the phenotype of hereditary neuropathy with liability to pressure palsies. Arch Neurol. 2007;64:974–8. doi: 10.1001/archneur.64.7.974. [DOI] [PubMed] [Google Scholar]
- Li J, Krajewski K, Lewis RA, Shy ME. Loss-of-function phenotype of hereditary neuropathy with liability to pressure palsies. Muscle Nerve. 2004;29:205–10. doi: 10.1002/mus.10521. [DOI] [PubMed] [Google Scholar]
- Lombardi R, Erne B, Lauria G, Pareyson D, Borgna M, Morbin M, et al. IgM deposits on skin nerves in anti-myelin-associated glycoprotein neuropathy. Ann Neurol. 2005;57:180–7. doi: 10.1002/ana.20364. [DOI] [PubMed] [Google Scholar]
- Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–32. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- Lupski JR, Garcia CA. Molecular genetics and neuropathology of Charcot-Marie-Tooth disease type 1A. Brain Pathol. 1992;2:337–49. doi: 10.1111/j.1750-3639.1992.tb00710.x. [DOI] [PubMed] [Google Scholar]
- Madrid R, Bradley WG. The pathology of neuropathies with focal thickening of the myelin sheath (tomaculous neuropathy), studies on the formation of the abnormal myelin sheath. J Neurol Sci. 1975;25:415–48. [Google Scholar]
- Martini R, Zielasek J, Toyka KV, Giese KP, Schachner M. Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nat Genet. 1995;11:281–6. doi: 10.1038/ng1195-281. [DOI] [PubMed] [Google Scholar]
- Meyer zu Horste G, Prukop T, Liebetanz D, Mobius W, Nave KA, Sereda MW. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann Neurol. 2007;61:61–72. doi: 10.1002/ana.21026. [DOI] [PubMed] [Google Scholar]
- Nelis E, Van Broeckhoven C, De Jonghe P, Lofgren A, Vandenberghe A, Latour P, et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet. 1996;4:25–33. doi: 10.1159/000472166. [DOI] [PubMed] [Google Scholar]
- Nolano M, Provitera V, Crisci C, Stancanelli A, Wendelschafer-Crabb G, Kennedy WR, et al. Quantification of myelinated endings and mechanoreceptors in human digital skin. Ann Neurol. 2003;54:197–205. doi: 10.1002/ana.10615. [DOI] [PubMed] [Google Scholar]
- Pareek S, Notterpek L, Snipes GJ, Naef R, Sossin W, Laliberte J, et al. Neurons promote the translocation of peripheral myelin protein 22 into myelin. J Neurosci. 1997;17:7754–62. doi: 10.1523/JNEUROSCI.17-20-07754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004;10:396–401. doi: 10.1038/nm1023. [DOI] [PubMed] [Google Scholar]
- Pennuto M, Tinelli E, Malaguti M, Del Carro U, D'Antonio M, Ron D, et al. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. 2008;57:393–405. doi: 10.1016/j.neuron.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogendijk JE, Baas F, et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group. Neuromuscul Disord. 1991;1:93–7. doi: 10.1016/0960-8966(91)90055-w. [DOI] [PubMed] [Google Scholar]
- Raeymaekers P, Timmerman V, Nelis E, Van Hul W, De Jonghe P, Martin JJ, et al. Estimation of the size of the chromosome 17p11.2 duplication in Charcot-Marie-Tooth neuropathy type 1a (CMT1a). HMSN Collaborative Research Group. J Med Genet. 1992;29:5–11. doi: 10.1136/jmg.29.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabet A, Li J, Ghandour K, Pu Q, Wu X, Kamholz J, et al. Skin biopsies demonstrate MPZ splicing abnormalities in Charcot-Marie-Tooth neuropathy 1B. Neurology. 2006;67:1141–6. doi: 10.1212/01.wnl.0000238499.37764.b1. [DOI] [PubMed] [Google Scholar]
- Schenone A, Nobbio L, Mandich P, Bellone E, Abbruzzese M, Aymar F, et al. Underexpression of messenger RNA for peripheral myelin protein 22 in hereditary neuropathy with liability to pressure palsies. Neurology. 1997;48:445–9. doi: 10.1212/wnl.48.2.445. [DOI] [PubMed] [Google Scholar]
- Sereda MW, Meyer zu Horste G, Suter U, Uzma N, Nave KA. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A) Nat Med. 2003;9:1533–7. doi: 10.1038/nm957. [DOI] [PubMed] [Google Scholar]
- Shames I, Fraser A, Colby J, Orfali W, Snipes GJ. Phenotypic differences between peripheral myelin protein-22 (PMP22) and myelin protein zero (P0) mutations associated with Charcot-Marie-Tooth-related diseases. J Neuropathol Exp Neurol. 2003;62:751–64. doi: 10.1093/jnen/62.7.751. [DOI] [PubMed] [Google Scholar]
- Sheth S, Francies K, Siskind CE, Feely SME, Lewis R, Shy ME. Diabetes mellitus exacerbates motor and sensory impairment in CMT1A. J Peripher Nerv Sys. 2008;13:299–304. doi: 10.1111/j.1529-8027.2008.00196.x. [DOI] [PubMed] [Google Scholar]
- Shy ME, Arroyo E, Sladky J, Menichella D, Jiang H, Xu W, et al. Heterozygous P0 knockout mice develop a peripheral neuropathy that resembles chronic inflammatory demyelinating polyneuropathy (CIDP) J Neuropathol Exp Neurol. 1997;56:811–21. [PubMed] [Google Scholar]
- Shy ME, Blake J, Krajewski K, Fuerst DR, Laura M, Hahn AF, et al. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology. 2005;64:1209–14. doi: 10.1212/01.WNL.0000156517.00615.A3. [DOI] [PubMed] [Google Scholar]
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- Snipes GJ, Suter U. Molecular anatomy and genetics of myelin proteins in the peripheral nervous system. J Anat. 1995;186(Pt 3):483–94. [PMC free article] [PubMed] [Google Scholar]
- Snipes GJ, Suter U, Welcher AA, Shooter EM. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13) J Cell Biol. 1992;117:225–38. doi: 10.1083/jcb.117.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl N, Harry J, Popko B. Quantitative analysis of myelin protein gene expression during development in the rat sciatic nerve. Brain Res Mol Brain Res. 1990;8:209–12. doi: 10.1016/0169-328x(90)90018-9. [DOI] [PubMed] [Google Scholar]
- Thomas PK, Marques W, Davis MB, Sweeney MG, King RH, Bradley JL, et al. The phenotypic manifestations of chromosome 17p11.2 duplication. Brain. 1997;120:465–78. doi: 10.1093/brain/120.3.465. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Hauer P, Lemke G. Axonal regulation of myelin protein mRNA levels in actively myelinating Schwann cells. J Neurosci. 1988;8:3515–21. doi: 10.1523/JNEUROSCI.08-09-03515.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Pfeiffer SE, Anitei A, Kidd GJ. Cell Biology and myelin assembly. In: Lazzarini RA, editor. Myelin biology and disorders. Vol. 1. San Diego/London: Elsevier Academic Press; 2003. pp. 29–56. [Google Scholar]
- Vallat JM, Sindou P, Preux PM, Tabaraud F, Milor AM, Couratier P, et al. Ultrastructural PMP22 expression in inherited demyelinating neuropathies. Ann Neurol. 1996;39:813–7. doi: 10.1002/ana.410390621. [DOI] [PubMed] [Google Scholar]
- Weimer LH, Podwall D. Medication-induced exacerbation of neuropathy in Charcot Marie Tooth disease. J Neurol Sci. 2006;242:47–54. doi: 10.1016/j.jns.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Wise CA, Garcia CA, Davis SN, Heju Z, Pentao L, Patel PI, et al. Molecular analyses of unrelated Charcot-Marie-Tooth (CMT) disease patients suggest a high frequency of the CMTIA duplication. Am J Hum Genet. 1993;53:853–63. [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa H, Dyck PJ. Uncompacted inner myelin lamellae in inherited tendency to pressure palsy. J Neuropathol Exp Neurol. 1991;50:649–57. doi: 10.1097/00005072-199109000-00009. [DOI] [PubMed] [Google Scholar]
- Yoshikawa H, Nishimura T, Nakatsuji Y, Fujimura H, Himoro M, Hayasaka K, et al. Elevated expression of messenger RNA for peripheral myelin protein 22 in biopsied peripheral nerves of patients with Charcot-Marie-Tooth disease type 1A. Ann Neurol. 1994;35:445–50. doi: 10.1002/ana.410350412. [DOI] [PubMed] [Google Scholar]