

Abstract
The synthesis of the 6-O-DPC-2-N-methylguanosine (m2G) nucleoside and the corresponding 5′-O-DMT-2′-O-TOM-protected 6-O-DPC-2-N-methylguanosine phosphoramidite is reported [DPC, diphenyl carbamoyl; DMT, 4, 4′-dimethoxytrityl; TOM, [(triisopropylsilyl)oxy]methyl]. The availability of the phosphoramidite allows for syntheses of hairpin RNAs with site-selective incorporation of 2-N-methylguanosine modification. Four 18-nt hairpin RNA analogues representing the 970-loop region (helix 31 or h31; U960–A975) of Escherichia coli 16S rRNA were synthesized with and without modifications in the loop region. Subsequently, stabilities and conformations of the singly and doubly modified RNAs were examined and compared with the corresponding unmodified RNA. Thermodynamic parameters and circular dichroism spectra are presented for the four helix 31 RNA analogues. Surprisingly, methylations in the loop region of helix 31 slightly destabilize the hairpin, which may have subtle effects on ribosome function. The hairpin construct is suitable for future ligand-binding experiments.
1. Introduction
An intriguing question about the structure and function of the ribosome is the role played by the naturally occurring modified nucleotides.1–3 The range of modifications includes methylation on both the nucleoside base and 2′-hydroxyl group of the ribose moiety, pseudouridylation (uridine isomerization), as well as more elaborate alterations.4 Although the rRNA modifications have only been mapped for a small number of organisms, it appears that most, if not all, ribosomes contain modified nucleotides. It has been suggested that the modified nucleotides of the small subunit have important roles in ribosome assembly and protein synthesis, despite the fact that unmodified ribosomes are able to carry out key biological functions in vitro.5–8 The Escherichia coli (E. coli) small subunit RNA (16S rRNA) contains one pseudouridine and ten methylated nucleotides (http://library.med.utah.edu/RNAmods); m7G527, m2G966, m5C967, m2G1207, m4Cm1402, m5C1407, m3U1498, m2G1516, m26A1518, and m26A1519.9–11 In 3D models of the 30S subunit, based on high-resolution X-ray crystal structures, eight of the ten methylated residues cluster in the subunit cleft near the decoding region.3,12,13
The 970 loop (helix 31, h31) of E. coli 16S rRNA is located near the ribosomal P site and therefore believed to be intimately involved in translation.14–17 E. coli h31 contains two modified nucleotides, N2-methylguanosine at position 966 (m2G966) and 5-methylcytidine at position 967 (m5C967) (Figure 1). Among the nucleotides present in h31, residue 965 is the least conserved, with a U present in 52% of known sequences, and A (37%), C (9%) or G (2%) in the remaining sequences.18 In contrast, nucleotide 966 is the most conserved, with G occurring in 97.5% of the sequences.18 Similarly, nucleotide 967 is a C in 85% of the sequences and an A in 13%.18 Mutations at either G966 or C967, as well as loss of methylation at G966, do not affect the growth rate of E. coli;19,20 however, a single-base deletion at C967 leads to a dominant lethal phenotype.19 Recently, a saturation mutagenesis study on the 970 loop revealed that mutations at positions 966 and 969 significantly affect protein synthesis by the ribosome.21 It was also shown that ribosomes with single mutations at positions m2G966 and m5C967 are capable of producing more protein than the wild-type ribosome.21 These studies suggest that modifications in the 970 loop influence the function of the ribosomal machinery.
Figure 1.

The structures of N2-methylguanosine (m2G) and 5-methylcytidine (m5C) are shown. Secondary structure representations of E. coli helix 31 of the small subunit ribosomal RNA with indications of the modification sites and the synthetic RNA hairpins derived from residues 960–975 are shown (unmodified (ECh31UNMOD), fully modified (wild-type or ECh31WT, containing m2G966 and m5C967), m5C967 singly modified (ECh31M5C), and m2G966 singly modified (ECh31M2G)). The terminal G-C base pair (g1-c18) was added to stabilize the short stem region.
There is a considerable amount of structural information for h31 from ribosome X-ray crystal structures.12,15–17,22,23 In E. coli 70S ribosomes, a three-base stacking interaction is observed between residues 966, 967, and 968.23 In the crystal structure of T. thermophilus 30S ribosomes, stacking is observed between those same residues, as well as between positions 965, 969, and 970.22 In addition, the stacked arrangement formed by residues 965, 969, and 970 shows interactions with the ribosomal protein S13.22 Residue G971 makes six hydrogen bonds within a pocket formed by residues 949, 950, 1363, 1364, and 1365 in the 16S rRNA.22 In the ribosome, m2G966 is also protected by the P-site-bound tRNA from chemical probes.24 Direct contacts between residues of the tRNA anticodon loop, the P site, and h31 of 16S rRNA were observed in X-ray crystal structures of the 70S ribosome.15,16 These structures suggest that the anticodon loop of the P-site tRNA is held in place by two stacking interactions; one between m5C1400 of 16S rRNA and G34 of the tRNA, and a second between m2G966 of h31 and ribose 34 of tRNA.16,25 The m22G966 residue is flipped out in the 70S ribosomal complex of T. thermophilus containing a model mRNA and two tRNAs.16 In the E. coli 70S ribosome crystal structure with a tRNA analogue and mRNA, the corresponding m2G966 residue resides in a tight binding pocket with the anticodon and the decoding site, which differs from the 30S structure.22 These data suggest that the 970 loop is dynamic and may be involved in proper positioning of tRNA in the ribosomal P site. Modeling studies by Aduri et al.13 suggested that methylation of residues 966 and 967 increases the surface area for stacking and also improves van der Waals contacts with the hydrophobic portion of Arg128 of the S9 protein in the ribosome.
We were interested in developing a better understanding of the structural and possible stabilizing roles of the modifications in h31 of 16S rRNA. Currently, information is limited regarding the effects of nucleotide modifications on rRNA structure and function. Until recently, the chemical properties of the modified nucleotides had not yet been found to influence specific functional roles in the ribosome.26 Nevertheless, even minor changes in chemical composition of the RNA nucleotides can lead to altered steric properties, hydrogen-bonding interactions, local base-stacking potential, van der Waals interactions, or structural rigidity.2 The goals of this research were to synthesize N2-methylguanosine and its corresponding phosphoramidite, and then utilize the amidite in the syntheses of h31 model sequences. The syntheses were then followed by biophysical studies in order to understand the influence of base methylations on structure and stability of h31 in a systematic manner. The structures and stabilities of four model h31 RNAs were examined by using circular dichroism (CD) and thermal melting studies.
2. Results and discussion
2.1. Synthesis of N2-methylguanosine and the corresponding phosphoramidite
The methylated guanosine nucleoside, m2G, was synthesized by using a combination of several published procedures (Scheme 1). The first step involved the acetylation of guanosine to give compound 1.27 The second and third steps to generate N-methylated intermediate 3 were adapted from Bridson and Reese’s method using a p-thiocresol intermediate.28 The generation of compounds 2 and 3 was accomplished with 75% yields for each step. To avoid solubility problems typically encountered during the phosphoramidite synthesis of guanosine derivatives, we used a strategy that involves protection at the lactam function of the guanine residue according to a procedure devised by Kamimura29 and used by others.30 Diphenylcarbamoyl (DPC) protection and acetyl deprotection under mild basic conditions gave compound 5 in good yield (55%, two steps). In addition to the successful O6-protection of the guanine residue, the presence of DPC improved the solubility and chromatographic purification properties of the resulting derivative. The corresponding phosphoramidite was generated by using a similar approach as Höbartner and coworkers,31 but employing the DPC-protected precursor (compound 5). In our hands, compound 5 gave higher yields in the m2G phosphoramidite (compound 8) synthesis (Supplementary material) than the methods reported previously.31,32 The synthesis could be performed on a reasonably high scale to produce sufficient amounts of amidite 8 for multiple couplings.
Scheme 1.

Synthesis of the 6-O-DPC-2-N-methylguanosine 5: (i) acetic anhydride, DMAP, Et3N, acetonitrile, 0.5 h; (ii) p-thiocresol, acetic acid, formaldehyde, ethanol, reflux; (iii) NaBH4, DMSO, 100 °C; (iv) DPCCl, diisopropylethylamine, pyridine; (v) a) 2 M NaOH, 20 min; b) acetic acid.
2.2. Synthesis and characterization of RNA analogues
Figure 1 shows the RNA analogues representing the 970 stem-loop region (h31) of E. coli 16S rRNA. The numbering system is based on the full-length E. coli rRNA sequence. A terminal G-C base pair was added to each stem (denoted by lower case g-c) in order to stabilize the hairpin structure. Residues in all four constructs are numbered from g1 to c18 for the ends and U960 through A975 for the component representing the natural E. coli h31 sequence. Two h31 RNAs (ECh31UNMOD and ECh31M5C) were obtained using commercial amidites. The two remaining h31 analogues (ECh31M2G and ECh31WT) were synthesized using the 5′-O-DMT-2′-O-TOM-6-O-DPC-2-N-methylguanosine phosphoramidite (compound 8) along with the commercially available amidites. The doubly modified h31 analogue (ECh31WT) represents the wild-type sequence of E. coli 16S rRNA helix 31. The incorporation of m2G and m5C residues was confirmed by MALDI-TOF mass spectrometric analysis of purified full-length RNA and by P1 nuclease digestion and calf intestinal phosphatase treatment of the RNAs, followed by reverse-phase HPLC analysis of the enzyme digest products (see Supplementary material).
2.3. Effects of modifications on the stability of h31
Absorbance versus temperature profiles (melting curves) were obtained at pH 7.0 for all four RNA constructs. They were analyzed in terms of ΔG°37, ΔG°50, ΔH°, ΔS°, and melting temperature (Tm).33,34 The normalized absorbance plots at single RNA concentrations are shown in Figure 2. The melting curves are biphasic for four RNAs (see Supplementary material) with transitions at 0 to 35 °C and 40 to 80 °C. The low temperature transitions were concentration dependent, suggesting the formation of a bimolecular complex, such as a duplex or loop–loop interaction. The higher melting transitions were concentration independent, consistent with unimolecular unfolding of a hairpin structure.35 The corresponding thermodynamic parameters for the four RNAs are given in Table 1. The data indicate slight destabilizing effects (ΔΔG°37 values of 0.2–0.5 kcal/mol) of the modifications on helix 31. The observed order of stability of the RNAs is ECh31UNMOD > ECh31M2G > ECh31M5C ≥ ECh31WT. The RNA samples were analyzed as five different dilutions per experiment and UV melts were also performed in triplicate for each construct. Experiments in K+ buffer (15 mM KCl, 20 mM cacodylic acid, 20 mM Tris [basic form], 0.5 mM Na2EDTA, pH 7.0) gave similar results (Supplementary material).
Figure 2.

Representative normalized UV melting curves of the modified RNAs compared to the unmodified RNA, taken in 15 mM NaCl, 20 mM sodium cacodylate, 0.5 mM Na2EDTA (pH 7.0), are shown. The melting curves for the unmodified RNA (ECh31UNMOD, solid grey lines in panels A – C) are compared to those for the modified RNAs (dashed lines): (A) ECh31WT, (B) ECh31M2G, and (C) ECh31M5C. All of the melting curves were normalized at 95 °C and absorbance measurements were taken at 280 nm.
Table 1.
Thermodynamics of the four RNA analogues
| ΔG°50 (kcal/mol) | ΔG°37 (kcal/mol) | ΔH° (kcal/mol) | ΔS° (cal/K·mol) | Tm (°C) | |
|---|---|---|---|---|---|
| ECh31UNMOD | −0.9a | −2.6 ± 0.1 | −44.1 ± 1.0 | −133.8 ± 3.0 | 57 |
| ECh31M5C | −0.6a | −2.2 ± 0.1 | −40.5 ± 0.9 | −123.3 ± 3.1 | 55 |
| ECh31M2G | −0.7a | −2.4 ± 0.1 | −41.9 ± 1.1 | −127.5 ± 3.2 | 56 |
| ECh31WT | −0.5a | −2.1 ± 0.1 | −37.5 ± 2.2 | −114.4 ± 5.8 | 56 |
Each measurement was taken in triplicate. Best fits were obtained by assuming a hairpin formation. The buffer conditions were 15 mM NaCl, 20 mM sodium cacodylate and 0.5 mM Na2EDTA, pH 7.0.
A conservative estimate of the standard error for ΔG°50 is 3% (± 0.2 kcal/mol).43
The subtle destabilizing effect of the methylations was somewhat unexpected. In X-ray crystal structures of E. coli 70S ribosomes23 and T. thermophilus 30S ribosomes,22 m2G and m5C are involved in a three-base stacking interaction with residue 968. One might expect the methyl groups in the modified nucleosides to facilitate stacking interactions;13 however, both m2G and m5C are destabilizing relative to standard nucleotides G and C within the given sequence context. The presence of the h31 methylations might serve another purpose, such as stabilizing the ribosome through hydrophobic interactions with Arg128 of the S9 protein.13 Furthermore, the slight destabilization caused by the base methylation may facilitate base flipping of m2G966, which has been observed in ribosome crystal structures and likely plays an important role in protein synthesis.16 The small energetic penalty (ΔΔG°37 = 0.5 kcal/mol) would then be overcome by contacts with various ribosome components such as 16S rRNA helices, ribosomal proteins, and tRNA.16 Although the effects on stability may appear to be quite small, this level of destabilization could account for an approximate two-fold difference in binding to a ribosomal RNA component, which may be important for fidelity of protein synthesis.
2.4. Effects of modifications on the secondary structure of h31
The CD spectra of the helix 31 analogues were obtained to analyze the effects of modifications on the folded structure (Figure 3). The data indicate that the unmodified, singly modified, or doubly modified RNAs contain A-form stem regions, and they display similar conformations. They all have peak maxima at 270 nm and minima at 240 nm, similar to other A-form RNAs. A difference spectrum was obtained using Equation 1 in order to determine if the structural changes induced by the modified nucleotides are additive. In Equation 1, the difference spectrum for the singly modified RNAs and unmodified RNA is set equal to the difference spectrum of fully modified (wild-type) and unmodified RNA. If the effects of the modifications on the structure are additive, then the total difference spectrum should be equal to zero (Equation 2).
Figure 3.

CD spectra of the modified and unmodified RNA constructs are shown. Each spectrum is an average of five scans. The CD spectrum of the unmodified analogue (ECh31UNMOD, solid grey line) is shown in panels A–C with overlays (dashed lines) of the ECh31M5C (A), ECh31M2G (B), and ECh31WT (C) RNAs. The molar ellipticities are normalized to RNA concentrations. The total difference spectrum (ECh31M5C + ECh31M2G − ECh31UNMOD − ECh31WT) is shown in panel D (solid black line).
| (1) |
| (2) |
As shown in Figure 3D, the total difference spectrum is close to zero except for slight changes in the lower wavelength range (<260 nm). Therefore, the data are interpreted as the presence of modifications in h31 having subtle effects on the RNA structure. Studies in K+- and Mg2+-containing buffers showed similar results (Supplementary material).
3. Conclusions
Several conclusions can be drawn from these studies with respect to the relative stabilities and structures of RNAs with single and multiple modified nucleotides. Small reproducible differences in the free energy values for the E. coli h31 variants reveal slight destabilizing effects of the modifications on helix 31. A recent X-ray crystal structure of the T. thermophilus 70S ribosome complexed with a model mRNA and two tRNAs revealed that the positions of the 16S rRNA P-site nucleotides in the vacant ribosome superimpose well with those in the tRNA-containing complex, with the exception of m22G966.16 Residue m22G966 (m2G966 in E. coli ribosomes) is flipped out in the crystal structure of the T. thermophilus 70S ribosome containing a model mRNA and two tRNAs (Figure 4A)15,16 or remains stacked in the 30S ribosomal subunit from T. thermophilus crystal structure (Figure 4B).22 The interaction with the anticodon loop of the P-site-bound tRNA appears to be stabilized by stacking interactions involving m22G966 with ribose 34. Hence, the flipped-out base has been suggested to facilitate correct positioning of the tRNA during translation.16,25 Therefore, the slight destabilizing effects of modifications in h31 may be important for facilitating the flipping movement of residue 966; but at the same time, stacking interactions with the tRNA are stabilized through the methyl group. Positioned in the middle of the three stacked bases, the m5C967 residue has a greater destabilizing effect than m2G966 (ECh31M5C vs. ECh31M2G). This result may be due to a greater disruption of stacking by the methylated base of m5C967.
Figure 4.
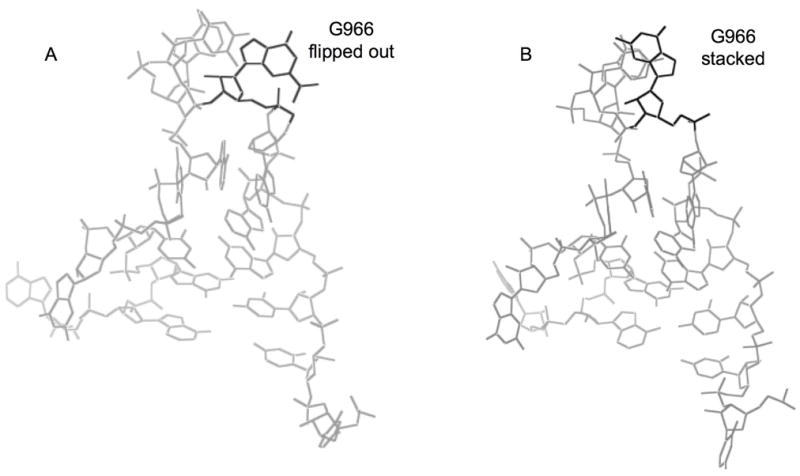
Structures of h31 showing the flipped out (A) and stacked (B) conformations of residue G966 (PDB accession IDs - 2J0015 and 1FJF22).
The CD data indicate that the unmodified, singly modified, and fully modified RNAs all contain A-form stem regions and display similar conformations. Minor differences between the CD spectra of the fully modified and unmodified h31 constructs indicate possible differences in the loop regions. These differences could arise from modification-dependent changes in the loop, such as altered base stacking at positions 966, 967, and 968. The UV melting data reveal, however, that the presence of modifications at specific locations does not influence the ability of the constructs to form stable hairpin loop structures.
The exact functional role of the modifications at positions 966 and 967 of h31 is still unknown. Ribosomes carry out the essential biochemical process of translation, which requires an exquisite array of highly specific interactions between rRNA, mRNA, tRNAs, and ribosomal proteins. Modifications are believed to help fine-tune ribosome function.3 Since proper ribosome function depends on the correct balance of speed and accuracy of tRNA binding, peptide-bond formation and tRNA release, methylations in h31 could play a role in maintaining proper interactions within the ribosome.36,37 Mutational analyses revealed that a loss of methylation at either position 966 or 967, leads to increased protein production by the mutant ribosomes.21 Our data would therefore suggest that methylations destabilize h31 in order to maintain the proper interactions with tRNA, rRNA, or proteins. A lack of modification at residues 966 or 967 in h31 could reduce the ability of base 966 to flip and regulate tRNA affinity, positioning, or accuracy. Thus, it will be of great interest to explore in greater detail the relationship between G966 and C967 methylation and translational fidelity. Furthermore, the availability of a suitable method for synthesizing m2G and its corresponding phosphoramidite (commercially not available) will allow RNAs containing m2G modifications to be generated in sufficiently large quantities for use in additional biophysical and ligand-binding studies.
4. Experimental
4.1. Synthesis of the 6-O-DPC-2-N-methylguanosine
4.1.1. General
Most reagents and solvents were either purchased from Aldrich (St. Louis, MO) or Acros (Morris Plains, NJ) and used as received. Anhydrous pyridine was purchased in a sure-seal bottle from Aldrich. Methylene chloride (CH2Cl2) was distilled over CaH2. Methanol and triethylamine were purchased dry from Aldrich in a sure-seal bottle. Moisture sensitive reactions involved flame-drying equipment (syringes, round-bottom flasks, stir-bars, etc.) under vacuum and performing reactions under dry argon. TLC analyses were accomplished with precoated silica gel (0.25 mm thickness) glass plates. Reactions were monitored by visualizing the TLC plates under UV light and/or by staining with phosphomolybdic acid (PMA) solution (10% w/v in absolute ethanol) and heating with a hot plate. Flash chromatography was performed with silica gel 60 (0.038–0.063 mm). Flash columns were neutralized with 0.5–1% triethylamine (TEA) prior to purification of pH sensitive intermediates/compounds. 1H NMR and 13C NMR spectra were recorded on either a Varian Unity 300, Mercury 400, or Varian Unity 500 spectrometer and referenced to tetramethylsilane as an internal standard. ESI spectra were recorded on a Quattro LC (Bruker Daltonics) in the positive ion mode unless otherwise noted.
4.1.2. 2′, 3′, 5′-Tri-O-acetylguanosine (1)
Compound 1 was generated according to a literature procedure.27
4.1.3. 2′, 3′, 5′-Tri-O-acetyl-2-N-(p-methylphenylthiomethyl)guanosine (2)
This compound was generated using a procedure described in the literature.28
4.1.4. 2′, 3′, 5′-Tri-O-acetyl-2-N-methylguanosine (3)
Compound 3 was prepared using a procedure described in the literature.38
4.1.5. 2-N-Methyl-6-O-(diphenylcarbamoyl)guanosine (5)
To a solution of 2′, 3′, 5′-tri-O-acetyl-2-N-methylguanosine 3 (3.0 g, 7.1 mmol, 1.0 eq) in dry pyridine (50 mL) were added diphenylcarbamoyl chloride (3.45 g, 15 mmol, 2.1 eq) and diisopropylethylamine (1.9 mL, 11.4 mmol, 1.6 eq). The dark brown reaction mixture was then stirred at room temperature for 1 h to obtain 2′, 3′, 5′-tri-O-acetyl-6-O-(diphenylcarbamoyl)-2-N-methylguanosine 4. TLC showed complete conversion to product at this stage of the reaction. The reaction mixture was then diluted with pyridine (15 mL) and EtOH (30 mL). To this solution cooled at 0 °C was added 2 M NaOH (25 mL), which was also precooled to 0 °C. The reaction mixture was stirred for 10 min at 0 °C. Acetic acid (c.a. 5 mL) was then added to neutralize the solution. Extraction with CH2Cl2 followed by chromatography on silica gel afforded 5 after two steps in 55% yield (1.92 g). TLC (CH2Cl2: MeOH, 9:1 v/v): Rf = 0.5; ESI-MS (ES+) calculated for C24H24N6O6 492.1757, found 493.2 (M+H+), 515.2 (M+Na+), 1007.4 (2M+Na+).
4.2. Synthesis of 5′-O-DMT-2′-O-TOM-6-O-DPC-2-N-methylguanosine phosphor-amidite
4.2.1. 5′-O-(4,4′-Dimethoxytrityl)-2-N-methyl-6-O-(diphenylcarbamoyl)guanosine (6)
Compound 5 (0.51 g, 1.05 mmol, 1.0 eq) and 4, 4′-dimethoxytritylchloride (0.39 g, 1.14 mmol, 1.09 eq) were azeotroped three times with toluene for ~5 h. To the dried compound 5, anhydrous pyridine (5 mL) was added. The mixture was stirred at room temperature under Ar atmosphere for 4 h. 4-Dimethylaminopyridine (0.09 g, 0.7 mmol, 0.7 eq) was subsequently added and stirring was continued for 17 h. The reaction was quenched with methanol (1 mL) and evaporated to dryness. The crude residue was dissolved in 50 mL of dichloromethane and washed with 5% sodium bicarbonate followed by saturated sodium chloride. The organic layer was dried over sodium sulfate and filtered. The product was then purified by silica gel chromatography using a solvent mixture of 90% dichloromethane, 9% methanol, 1% triethylamine to give 6 as a light yellow crystalline solid (0.56 g, 67%). TLC (CH2Cl2: MeOH, 9:1 v/v): Rf =0.4; ESI-MS (ES+) calculated for C45H42N6O8 794.3, found 795.7 (M+H+), 303.4 ([(MeO)2Tr]+).
4.2.2. 5′-O-(4,4′-Dimethoxytrityl)-2′-O-[[(triisopropylsilyl)oxy]methyl]-2-N-methyl-6-O-(diphenylcarbamoyl) guanosine (7)
Di-tert-butyltindichloride (0.26 g, 0.846 mmol, 1.2 eq) was added to a solution of dry 1,2-dichloroethane (6.5 mL) containing compound 6 (0.56 g, 0.705 mmol, 1.0 eq) and ethyldiisopropylamine (0.36 mL, 2.82 mmol, 4.0 eq). The reaction mixture was heated to 70 °C for 15 min under refluxing conditions. Then, the mixture was allowed to cool to room temperature. Upon cooling down, the mixture became cloudy and light brown in color. The crude reaction mixture was then stirred with [(triisopropylsilyl)oxy]methylchloride (0.18 mL, 0.776 mmol, 1.1 eq) for 3 h at room temperature. After 3 h, the mixture was evaporated to dryness. The resulting crude residue was dissolved in 20 mL of dichloromethane and washed with saturated sodium bicarbonate followed by saturated sodium chloride. The organic layer was dried over sodium sulfate and filtered. The product was then purified by silica gel column chromatography using a solvent mixture of dichloromethane: methanol (20:1) and triethylamine (0.5%) to give 7 as a light yellow oil (0.49 g, 70%). TLC (CH2Cl2: MeOH, 9:1 v/v): Rf =0.7; ESI-MS (ES+) calculated for C55H64N6O9Si 980.4, found 981.9 (M+H+), 1003.9 (M+Na+), 1019.9 (M+K+).
4.2.3. 5′-O-(4,4′-Dimethoxytrityl)-2′-O-[[(triisopropylsilyl)oxy]methyl]-2-N-methyl-6-O-(diphenylcarbamoyl) guanosine 3′-(2-cyanoethyl diisopropylphosphoramidite) (8)
Compound 7 (0.25 g, 0.25 mmol, 1.0 eq) was dried extensively under vacuum. Then, it was dissolved in 5 mL of anhydrous dichloromethane. Next, N,N-diisopropylethylamine (0.3 mL, 2.5 mmol, 10 eq) and 2-cyanoethyldiisopropylchlorophosphoramidite (0.08 mL, 0.38 mmol, 1.5 eq) were added and the mixture was stirred for 2 h at room temperature. The reaction was quenched with 5% aqueous sodium bicarbonate, and then it was extracted with 2 × 50 mL of dichloromethane. Combined extracts were dried over anhydrous sodium sulfate and evaporated. The crude mixture was purified by silica gel column chromatography (hexane: ethyl acetate, 3:1 and triethylamine, 0.5%) to yield 8 as a white form (0.285 g, 95%). TLC (hexane: EtOAc: Et3N=75%: 24.5%: 0.5%): Rf =0.23; 1H NMR ((CD3)2CO, 400 MHz) (mixture of diastereoisomers) 0.89–1.05 (2m, 60H), 1.18–1.30 (2m, 10H), 1.45 (t, 4H), 2.49 (br.s, 2H), 2.79–2.84 (m, 8H), 3.39–3.41 (m, 2H), 3.66–3.75 (m, 14H), 4.17–4.19 (m, 2H), 4.69 (m, 2H), 5.12–5.14 (m, 8H), 6.11 (d, J = 5.2 Hz, 2H), 6.79–6.83 (m, 10H), 7.17–7.50 (2m, 36H), 8.04 (s, 2H); 13C NMR ((CD3)2CO, 400 MHz) 12.56, 18.04, 18.08, 20.7, 20.75, 24.76, 24.82, 24.89, 24.95, 28.97, 43.82, 43.91, 43.96, 44.06, 55.45, 59.0, 59.16, 59.89, 60.02, 64.39, 72.59, 84.4, 84.7, 87.14, 87.71, 90.03, 90.36, 113.86, 127.54, 128.54, 128.58, 128.91, 128.98, 129.91, 130.85, 130.93, 130.99, 136.57, 136.69, 143.38, 145.96, 151.43, 157.33, 159.58, 160.82, 206.15; 31P NMR ((CD3)2CO, 400 MHz) (mixture of diastereoisomers) 150.96, 151.14; High resolution ESI-MS (ES+) calculated for C64H81N8O10PSi 1180.5583, found 1181.5634 (M+H+), 1203.5439 (M+Na+), 1219.5358 (M+K+).
4.3. RNA oligonucleotide synthesis
The four RNA hairpins used in this study (Figure 1) were chemically synthesized at the W. M. Keck Foundation at Yale University, New Haven, Connecticut, USA. For RNAs containing m2G, 50 μmoles of the corresponding phosphoramidite were provided. The m5C phosphoramidite was purchased from Glen Research. The sequences of the four synthetic RNAs are as follows (U, C, G, A are nucleotides representing the natural E. coli sequence; g, c are added nucleotides to enhance hairpin stability):
5′-gUU CGA UGC AAC GCG AAc-3′ (ECh31UNMOD)
5′-gUU CGA UG(m5C) AAC GCG AAc-3′ (ECh31M5C)
5′-gUU CGA U(m2G)C AAC GCG AAc-3′ (ECh31M2G)
5′-gUU CGA U(m2G)(m5C) AAC GCG AAc-3′ (ECh31WT)
4.4. RNA deprotection and purification
Upon completion of coupling on an automated synthesizer, the CPG-bound RNA was cleaved from the solid support and deprotected with 1:3 (v/v) EtOH/NH4OH and TBAF (tetrabutylammonium fluoride solution, 1 M in THF) as described in the literature.29,39 The RNAs were desalted over Poly-Pak II cartridges (Glen Research), then purified by HPLC on an XTerra MS C18 column (2.5 μm, 10 × 50 mm, Waters) in which the eluent was 0.1 M TEAA (triethylammonium acetate) buffer, pH 7.0, with a 5 – 15% linear gradient of acetonitrile over 25 min at a flow rate of 4.0 mL/min. After HPLC purification, each oligomer was further desalted by ethanol precipitation and dialysis for 3 days against RNase-free, deionized water using a 1000 molecular weight cut-off membrane (Spectra-Por). RNA concentrations were calculated using Beer’s law and a single-stranded extinction coefficient (ε) of 176,900 M−1cm−1.40 The same extinction coefficients were used for guanosine and m2G (1.4 × 104 M−1cm−1 at pH 7.0) and cytidine and m5C (9.1 × 103 M−1cm−1 at pH 7.0).
4.5. UV melting studies
The absorbance versus temperature profiles were obtained on an Aviv 14DS UV-visible spectrophotometer with a five-cuvette thermoelectric controller. Microcuvettes with two different pathlengths, 0.1 and 0.2 cm (60 and 120 μL volumes, respectively), were employed. Each measurement was taken in triplicate. The buffer used in each experiment contained 15 mM NaCl, 20 mM sodium cacodylate, and 0.5 mM Na2EDTA (pH 7.0), unless noted otherwise. Each oligomer was dissolved in a specific volume to yield an absorbance reading just below 2.0 in a 0.1 cm pathlength cuvette. The RNA concentrations were determined from the absorbance values (260 nm) at 95 °C. The absorbance data were collected at 280 nm from 0 to 95 °C with a constant heating rate of 0.5 °C/min.41 Thermodynamic parameters were obtained from the absorbance versus temperature profiles using the MELTWIN v. 3.5 melting curve program.33 This program performs a van’t Hoff analysis, assuming a two-state model for the transition between a native and a denatured (random coil) structure of a hairpin loop.
4.6. Circular dichroism (CD) spectroscopy
CD spectra were obtained on an Applied Photophysics Chirascan circular dichroism spectrometer (220–320 nm) at 25 °C in 15 mM NaCl, 20 mM sodium cacodylate, and 0.5 mM Na2EDTA at pH 7.0. The RNA concentrations were maintained at 2.5–3.0 μM for all CD experiments. Based on the RNA strand concentration, the measured CD spectra were converted to molar ellipticity (Δε),42 which denotes the moles of RNA molecules rather than moles of individual residues present in the sequence.
Supplementary Material
Acknowledgments
We thank Tek Lamichhane, John SantaLucia, and Philip Cunningham for helpful discussions and Lew Hryhorczuk, Andrew Feig, and Norman Watkins for technical assistance. This work was supported by the National Institutes of Health (AI061192).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brimacombe R, Mitchell P, Osswald M, Stade K, Bochkariov D. FASEB J. 1993;7:161. doi: 10.1096/fasebj.7.1.8422963. [DOI] [PubMed] [Google Scholar]
- 2.Agris PF. Prog Nucleic Acid Re. 1996;53:79. doi: 10.1016/s0079-6603(08)60143-9. [DOI] [PubMed] [Google Scholar]
- 3.Decatur WA, Fournier MJ. Trends Biochem Sci. 2002;27:344. doi: 10.1016/s0968-0004(02)02109-6. [DOI] [PubMed] [Google Scholar]
- 4.Chow CS, Lamichhane TN, Mahto SK. ACS Chem Biol. 2007;2:610. doi: 10.1021/cb7001494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunningham PR, Richard RB, Weitzmann CJ, Nurse K, Ofengand J. Biochimie. 1991;73:789. doi: 10.1016/0300-9084(91)90058-9. [DOI] [PubMed] [Google Scholar]
- 6.Denman R, Weitzmann C, Cunningham PR, Negre D, Nurse K, Colgan J, Pan YC, Miedel M, Ofengand J. Biochemistry. 1989;28:1002. doi: 10.1021/bi00429a013. [DOI] [PubMed] [Google Scholar]
- 7.Denman R, Negre D, Cunningham PR, Nurse K, Colgan J, Weitzmann C, Ofengand J. Biochemistry. 1989;28:1012. doi: 10.1021/bi00429a014. [DOI] [PubMed] [Google Scholar]
- 8.Green R, Noller HF. RNA. 1996;2:1011. [PMC free article] [PubMed] [Google Scholar]
- 9.Starr JL, Fefferman R. J Biol Chem. 1964;239:3457. [PubMed] [Google Scholar]
- 10.Ehresmann C, Stiegler P, Mackie GA, Zimmermann RA, Ebel JP, Fellner P. Nucleic Acids Res. 1975;2:265. doi: 10.1093/nar/2.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ehresmann C, Stiegler P, Fellner P, Ebel JP. Biochimie. 1975;57:711. doi: 10.1016/s0300-9084(75)80047-2. [DOI] [PubMed] [Google Scholar]
- 12.Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Science. 2001;292:883. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
- 13.Aduri R, Psciuk BT, Saro P, Taniga H, Schlegel HB, SantaLucia J. J Chem Theory Comput. 2007;3:1464. doi: 10.1021/ct600329w. [DOI] [PubMed] [Google Scholar]
- 14.Doring T, Mitchell P, Osswald M, Bochkariov D, Brimacombe R. EMBO J. 1994;13:2677. doi: 10.1002/j.1460-2075.1994.tb06558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selmer M, Dunham CM, Murphy FV, IV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Science. 2006;313:1935. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 16.Korostelev A, Trakhanov S, Laurberg M, Noller HF. Cell. 2006;126:1065. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 17.Berk V, Zhang W, Pai RD, Cate JH. P Natl Acad Sci U S A. 2006;103:15830. doi: 10.1073/pnas.0607541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du Y, Feng B, Lin N, Madabusi LV, Muller KM, Pande N, Shang Z, Yu N, Gutell RR. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jemiolo DK, Taurence JS, Giese S. Nucleic Acids Res. 1991;19:4259. doi: 10.1093/nar/19.15.4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesnyak DV, Osipiuk J, Skarina T, Sergiev PV, Bogdanov AA, Edwards A, Savchenko A, Joachimiak A, Dontsova OA. J Biol Chem. 2007;282:5880. doi: 10.1074/jbc.M608214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saraiya AA, Lamichhane TN, Chow CS, SantaLucia J, Jr, Cunningham PR. J Mol Biol. 2008;376:645. doi: 10.1016/j.jmb.2007.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brodersen DE, Clemons WM, Jr, Carter AP, Wimberly BT, Ramakrishnan V. J Mol Biol. 2002;316:725. doi: 10.1006/jmbi.2001.5359. [DOI] [PubMed] [Google Scholar]
- 23.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Science. 2005;310:827. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 24.Moazed D, Noller HF. J Mol Biol. 1990;211:135. doi: 10.1016/0022-2836(90)90016-F. [DOI] [PubMed] [Google Scholar]
- 25.Selmer M, Dunham CM, Murphy FVt, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. Science. 2006;313:1935. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 26.Baxter-Roshek JL, Petrov AN, Dinman JD. PLoS ONE. 2007;2:e174. doi: 10.1371/journal.pone.0000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuda A, Shinozaki M, Suzuki M, Watanabe K, Miyasaka T. Synthesis-Stuttgart. 1986:385. [Google Scholar]
- 28.Bridson PK, Reese CB. Bioorg Chem. 1979;8:339. [Google Scholar]
- 29.Kamimura T, Tsuchiya M, Urakami K, Koura K, Sekine M, Shinozaki K, Miura K, Hata T. J Am Chem Soc. 1984;106:4552. [PubMed] [Google Scholar]
- 30.Koizumi M, Akahori K, Ohmine T, Tsutsumi S, Sone J, Kosaka T, Kaneko M, Kimura S, Shimada K. Bioorg Med Chem Lett. 2000;10:2213. doi: 10.1016/s0960-894x(00)00432-7. [DOI] [PubMed] [Google Scholar]
- 31.Höbartner C, Kreutz C, Flecker E, Ottenschlager E, Pils W, Grubmayr K, Micura R. Monatsh Chem. 2003;134:851. [Google Scholar]
- 32.Boudou V, Langridge J, Aerschot AV, Hendrix C, Millar A, Weiss P, Herdewijn P. Helv Chim Acta. 2000;83:152. [Google Scholar]
- 33.McDowell JA, Turner DH. Biochemistry. 1996;35:14077. doi: 10.1021/bi9615710. [DOI] [PubMed] [Google Scholar]
- 34.SantaLucia J, Jr, Kierzek R, Turner DH. Science. 1992;256:217. doi: 10.1126/science.1373521. [DOI] [PubMed] [Google Scholar]
- 35.Tinoco I, Jr, Sauer K, Wang JC. Physical chemistry, principles and applications in biological sciences. Prentice-Hall; Englewood Cliffs, NJ: 1985. [Google Scholar]
- 36.Gromadski KB, Daviter T, Rodnina MV. Mol Cell. 2006;21:369. doi: 10.1016/j.molcel.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 37.Fahlman RP, Dale T, Uhlenbeck OC. Mol Cell. 2004;16:799. doi: 10.1016/j.molcel.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 38.Kemal, Reese CB. Synthesis. 1980;12:1025. [Google Scholar]
- 39.Pitsch S, Weiss PA, Jenny L, Stutz A, Wu X. Helv Chim Acta. 2001;84:3773. [Google Scholar]
- 40.Richards EG. Handbook of biochemistry and molecular biology. CRC Press; Cleveland, OH: 1975. p. 596. [Google Scholar]
- 41.SantaLucia J., Jr . In: Spectrometry and Spectrofluorometry. Gore MG, editor. Oxford University Press; Oxford, UK: 2000. p. 329. [Google Scholar]
- 42.Cantor CR, Schimmel PR. Biophysical chemistry. W. H. Freeman and Co; San Francisco, CA: 1980. p. 412. Part II. [Google Scholar]
- 43.SantaLucia J, Jr, Turner DH. Biopolymers. 1997;44:309. doi: 10.1002/(SICI)1097-0282(1997)44:3<309::AID-BIP8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.