

Abstract
In this review we consider the physiological effects of endogenous and pharmacological levels of nitrite under conditions of hypoxia. In humans, the nitrite anion has long been considered as metastable intermediate in the oxidation of nitric oxide radicals to the stable metabolite nitrate. This oxidation cascade was thought to be irreversible under physiological conditions. However, a growing body of experimental observations attests that the presence of endogenous nitrite regulates a number of signaling events along the physiological and pathophysiological oxygen gradient. Hypoxic signaling events include vasodilation, modulation of mitochondrial respiration, and cytoprotection following ischemic insult. These phenomena are attributed to the reduction of nitrite anions to nitric oxide if local oxygen levels in tissues decrease. Recent research identified a growing list of enzymatic and non-enzymatic pathways for this endogenous reduction of nitrite. Additional direct signaling events not involving free nitric oxide are proposed. We here discuss the mechanisms and properties of these various pathways and the role played by the local concentration of free oxygen in the affected tissue.
Keywords: Nitrite, vasodilation, ischemia/reperfusion, nitric oxide, hypoxia
1. INTRODUCTION
The effects of nitrite anions on mammalian physiology have now been investigated for over a century after the first report (1) of its vasodilating effect at pharmacological concentrations. Its reactivity towards hemoglobin had also been recognized early in connection with toxicity of methemoglobinemia (2, 3). However, both phenomena were thought to require supraphysiological levels of nitrite that are unlikely to occur under normal conditions. Indeed, the anion was examined extensively in aortic ring bioassay studies as early as 1952 and shown to activate soluble guanylate cyclase in 1978, but the low potency of nitrite in such oxygenated assays suggested that this molecule would only be of pharmacological rather than physiological relevance (4–6). Therefore, endogenous nitrite was relegated to the status of a passive intermediate in the oxidation cascade from the nitric oxide radical (NO, also often written as NO•) towards nitrate (6). It has only been recently realized that even low concentrations of nitrite are vasodilating in vivo when applied in combination with low concentrations of oxygen (6–10). As such, nitrite was seen to be cytoprotective against ischemic damage in a wide range of tissues (8–10). Since then a number of clinical observations and animal studies have shown that the nitrite-mediated protection involved the reduction of nitrite and subsequent release of free NO radicals as oxygen tensions decreased. It shows the formation of NO by endogenous pathways other than nitric oxide synthases (NOS). These enzymes synthesize NO from L-arginine and require oxygen as essential cofactor. Therefore, at physiological pH ~ 7.4 nitrite seems inert under normoxic conditions, but starts to act as a source of NO if oxygen levels in the tissue drop below a certain threshold (10,11).
Various mechanisms for nitrite reduction have now been identified. Direct uncatalyzed reduction requires protonation and is very slow except at extremely acidic conditions as found in the stomach (12, 13) or urine (14). While acidification under ischemia is rather mild (in rabbit muscle pH remained above 6 (15)), pH values as low as 5.5 in the ischemic heart may promote this pathway during cardiac ischemia and reperfusion (16). Several mammalian enzymes, such as xanthine oxidase (XO), cytochrome C oxidase, and even endothelial NO synthase, have been found to reduce nitrite under hypoxia, even though the normoxic functions of these enzymes are very different. (9, 17–21). The deoxygenated states of hemoglobin and myoglobin have now been characterized as allosterically regulated nitrite reductases (22–27). Each of these enzymes has its own oxygen threshold for activation of nitrite reduction. We propose that as oxygen levels fall deeper and deeper from physiological hypoxia within blood vessels and tissue to pathological hypoxia in the setting of ischemia-reperfusion injury, additional reduction mechanisms are being successively activated to provide a graded generation of NO. Therefore, the various mechanisms may operate in a cooperative fashion.
In this context it should be noted that various human tissues show widely different rates of oxygen consumption as well as sensitivity to hypoxia.(28). Loss of oxygen implies loss of function after ca 10 seconds in brain, 4 min in heart and 2 hrs in skeletal muscle. Irreversible ischemic damage occurs in brain after several minutes, ca 15 min in heart and 6 hrs in skeletal muscles (28). Furthermore, the enzymatic composition varies significantly with tissue type. Accordingly, we may expect that the significance of the endogenous mechanisms for nitrite reduction varies with tissue type. For example, in the heart two pathways appear to reduce significant quantities of nitrite to NO, namely deoxymyoglobin (deoxyMb) and XO (23, 24). In blood, deoxyhemoglobin (deoxyHb) definitely plays an important role (9,25, 26). Sensitivity of a given tissue type to oxygen deficiency also varies between mammalian species. This state of affairs reminds us that extrapolation of results from animal studies to human clinical context is not necessarily justified.
The concept of hypoxic signaling is often strictly associated with the activation of hypoxia inducible transcription factors by falling oxygen levels in tissues. However, the imposition of hypoxia has more effects, in particular by changing the balance between the various NO metabolites in the tissue. In this, nitrite was recently found to play a crucial role. This review aims to cover the current status of nitrite physiology with special emphasis on the effects of hypoxia.
2. NITRITE LEVELS IN MAMMALIAN TISSUES
In mammalian physiology, three sources of nitrite have been identified. The first significant source is dietary nitrite ingested from food. Baked goods and cereals, beets, corn, spinach and turnip greens are major sources of nitrite (2.0 to 4.0 mg/kg food). The nitrite content of our food may be natural or artificially enhanced to suppress growth of toxic bacteria like botulism. Cured meats in particular form a dietary source of nitrite. The second source is nitrite released during the reduction of nitrate. Considerable quantities of nitrate enter our body via consumption of nitrate rich vegetables like spinach, lettuce or beetroot. Small quantities of nitrate may also be present in fish and dairy products such as cheese. Nitrate reductase enzymes are found in commensal bacteria (29, 30) in the mouth or intestines. Such bacterial nitrate reductases contribute significantly to the endogenous nitrite pool of the host organism (30). However, it has been reported (17, 31) that nitrate may be reduced back to free NO by XO under hypoxic conditions. Recently, XO was implicated in a slow in vivo reduction of nitrate under normoxia as well (32). The above two dietary sources dominate the third, endogenous, source of nitrite: oxidation of endogenous NO radicals to nitrite. Although this oxidation is very slow in vitro (33), the reaction is significantly faster within the blood of mammals and humans (34). So far, two mechanisms are known to accelerate this oxidation. First, apolar molecules like NO or oxygen partition preferably into lipid of protein fractions of low polarity (cf table I). In cells, these low polarity fractions acquire greatly enhanced local concentrations, and the oxidation reaction is accelerated by several orders of magnitude since it is second order in [NO] concentration (35). The second mechanism involves the copper-storage plasma enzyme ceruloplasmin (Cp). This enzyme catalyzes the oxidation of NO to NO+ which is rapidly hydrolysed to nitrite. In a mouse model, endogenous Cp has been shown to act as a significant catalyst for such oxidation of NO to nitrite and Cp deficient mice had significantly lower levels of plasma nitrite (36) than wild type mice.
Table I.
Ratios of partitioning between water and apolar fractions, for selected small neutral molecules (1D = 3.336 ·10−30 Cm). For comparison, the dipole moment of water is 1.85 D.
| molecule | Dipole moment | Solubility In H2O at20 ° C | Partitioning factor Apolar/water | Apolar fraction | reference |
|---|---|---|---|---|---|
| NO• | 0.159 D | 2.1 mM | 9.7 | Bulk cyclohexane | (37) |
| NO• | 0.159 D | 2.1 mM | 9.2 | Bulk n-hexane | (37) |
| O2 | -- | 1.4 mM | 3 | DMPC bilayers | (38) |
| CO2 | -- | 39.2 mM | 1.7 | Bulk olive oil | (39) |
| CO2 | -- | 39.2 mM | 0.95 | Lecithin bilayers | (39) |
| CO2 | -- | 39.2 mM | 1.6 | Red cell membrane | (40) |
| H2O2 | 2.0 D | unlimited | 0.09 | octanol |
Depending on the conditions, all three sources have been shown to contribute significantly to the endogenous nitrite pool in man. Measured nitrite levels in whole blood or plasma show significant variability due to differences in dietary habits, lifestyle (eg tobacco consumption) and physical exercise prior to testing. In fasting humans, almost all nitrite in the vascular circulation was shown to originate from NO released by nitric oxide synthases (NOS) (41). In resting humans, various values for plasma nitrite and nitrate concentrations were reported: [NO2−] = 0.15 – 0.20 μM and [NO3−] = 14.4 ± 1.7 μM (42–44). Ref (11) quotes values of 0.1 – 0.5 μM for plasma nitrite, and 20 – 50 μM for plasma nitrate. Ref (45) quotes higher plasma values as compiled in table II. This table shows the concentration of various nitrogen oxides in the blood circulation of adults on a normal diet and illustrates the arterial to venous gradient in the nitrite levels. This gradient is significantly enhanced under exercise (45). The artery-to-vein nitrite gradient was confirmed in whole blood as well (arterial 176 ± 10 nM to venous 143 ± 7 nM) (44). Plasma nitrite and nitrate may be lowered by about 50 % by dietary restriction (45), and are affected by the level of physical activity prior to measurement (11, 45). These correlations may contribute to the considerable variation in nitrite levels reported by various studies.
Table II.
Concentration of various nitrogen oxide species in plasma from the blood circulation of resting adults on a normal diet (adapted from ref (45)). HMW-SNO and LMW-SNO are S-nitrosothiols of high and low molecular weight, respectively. SNO-Hb is S-nitrosated hemoglobin. Lower values of plasma nitrite were reported in (44), with arterial plasma nitrite of 176 ± 10 nM, and venous plasma nitrite of 143 ± 7 nM.
| species | Artery | Vein |
|---|---|---|
| Plasma nitrate (μM) | 40.7 ± 4.5 | 41.3 ± 4.5 |
| Plasma nitrite (nM) | 540 ± 74 | 466 ± 79 |
| Plasma LMW-SNO | bd | bd |
| Plasma HMW-SNO (nM) | 45 ± 15 | 63 ± 13 |
| SNO-Hb ¶ (nM) | 161 ± 42 | 142 ± 29 |
bd: below detection limit of ca 25 nM
concentration of SNO-Hb in the red cell fraction.
Nitrite levels inside erythrocytes are higher than those in plasma (44, 48). For humans, ref (44) reports 121 nM for plasma and 288 nM for the RBC compartment, respectively. These values result in an average nitrite concentration of 176 nM for human whole arterial blood. Similarly, in Wistar rats (48), the RBC compartment had more than twofold higher nitrite than plasma (680±60 nM in RBC vs. 290±50 nM in plasma). Given that the hematocrit comprises between 40 % (children) and 50 % (adult males) of total blood volume, the erythrocytes contribute the largest nitrite pool in whole blood.
It should be noted that nitrite concentrations in human plasma tend to be significantly lower than in plasma from rodents, although considerable variations were reported here also: The plasma nitrite of 1.6 μM in wild type mice is depressed to 0.74 μM in mutants lacking eNOS (46). Even higher values of plasma nitrite of 20 μM were reported for male CD-1 mice (47). Slightly lower plasma nitrite of 10 μM was reported for Sprague-Dawley rats (47). For Wistar rats, considerably lower plasma nitrite of 0.3 μM has been reported (48), thereby falling in the range for human plasma.
The plasma levels should be distinguished from endogenous nitrite content of various tissues. In Wistar rats, the nitrite content of heart, liver, kidney and lung was 0.5 – 0.8 μM, whereas brain and aortic tissue had significantly higher concentrations (1.7 ± 0.3 μM and 22 ± 9 μM, respectively) (48).
Such average nitrite levels may be significantly altered in a number of situations: During pregnancy, the plasma nitrite levels of women are markedly lower. This phenomenon was observed in normotensive as well as preeclamptic pregnancies (49, 50). Nitrite levels show considerable variation between individuals and are significantly affected by dietary habits (43). Circulating nitrite may be significantly enhanced in individuals suffering from an infection (153–154). Interestingly, a recent study (51) of circulating NO metabolites in Tibetan highlanders, a population well adapted to environmental hypoxia associated with high altitudes, reported plasma nitrite levels of approximately 10 μM. This value exceeds 50-fold the plasma nitrite in humans living at sea-level. These high concentrations of circulating nitrite were associated with increased basal blood flow and increases in exhaled NO levels, and were found in healthy individuals without overt signs of inflammation or low blood pressure.
The nitrite concentrations listed above describe the normal situation encountered in humans. These values should be compared with the quantity considered lethal: The US Food and Drug Administration considers a dose of 22 mg sodium nitrite/kg as “fatal” for adults due to the complications arising from excessive methemoglobinemia. Assuming homogenous distribution throughout the body, this dose corresponds to an average concentration of ca 320 μM nitrite. This is roughly three orders of magnitude higher than the physiological nitrite levels found in humans (see Table II). It should be mentioned that lower dosages apply for infants, who are more susceptible and vulnerable than adults.
It has been observed that bolus infusions of nitrite decay with a half life of several minutes (11). This decay of nitrite is dominated by the reaction with oxy-hemoglobin to methemoglobin and nitrate (cf section 9A). However, alternative endogenous reaction pathways are known, like the formation of paramagnetic dinitrosyl-iron complexes (cf section 9B). As discussed in this review, more pathways become operational at low oxygen levels. Therefore, nitrite is more than just an intermediate in the oxidation cascade leading from NO to nitrate.
Finally, we mention that nitrate, the endpoint of the, cascade, may also be reduced back to free NO by XO (17, 31). In absence of oxygen, this interesting mammalian enzyme is capable of reducing a range of nitrogen compounds (organic nitrates, nitroglycerin, nitrate and nitrite). This reaction will be discussed further in section 7.C.
3. OXYGEN LEVELS IN MAMMALIAN TISSUES
Before discussing hypoxia, we should clarify the nomenclature. The simplest classification requires that we distinguish at least four degrees of oxygenation in mammalian tissues: Hyperoxia, normoxia, hypoxia, and full anoxia. These expressions should be clearly distinguished from hypoxemia, that refers to a specific deficiency in arterial oxygen concentration (for example due to shunts in the blood circulation). It seems tempting to identify the onset of hypoxia in tissues with the activation of hypoxia induced factors (HIF) (52, 53). However, this choice is ambiguous as the threshold for HIF response shows plasticity and may readjust adaptively to ambient oxygen levels (54, 55). For an alternative definition we note that normoxic arterial blood has a free oxygen concentration of [O2] ~100 – 130 μM, and venous blood has 35 – 40 μM. These boundaries seem conserved throughout the mammalian kingdom. The tissues surrounding these blood vessels have slightly lower oxygen levels, as required for gradient driven oxygen diffusion into the tissue. Perivenous tissues have ca 20 – 30 μM. Given these values, we define hyperoxia as [O2] > 130 μM, and normoxia as the interval spanned by arterial blood and perivenous tissue, i.e. [O2] = 130 – 20 μM. We take hypoxia as the interval between perivenous tissues and the level where mitochondrial respiration is reduced due to oxygen deficiency, i.e. the interval between 20 and 2 μM. For still lower oxygen concentrations mitochondrial respiration is compromised and the intracellular metabolic pathways are profoundly affected. For the purpose of this review, we will refer to this range of 0 – 2 μM free oxygen as anoxia (cf table III). These boundaries between the above definitions should be compared with a range of critical oxygen concentrations that were found to have high relevance for mammalian physiology. A selection is listed in table IV.
Table III.
proposed definitions for oxygen status of tissues. [O2] refers to the concentration of free oxygen. An oxygen pressure of 1 Torr = 1 mm Hg corresponds to a free oxygen concentration [O2] = 1.32 μM in H2O at 37 °C.
| Status of tissue | [O2] | pO2(Torr) |
|---|---|---|
| hyperoxia | > 130 μM | 100 |
| normoxia | 20 –130 μM | 15 – 100 |
| hypoxia | 2 – 20 μM | 1.5 – 15 |
| anoxia | < 2 μM | < 1.5 |
Table IV.
Selection of free oxygen concentrations with relevance for human physiology. Note that half-loading points of Hb and Mb in animals may be different (eg rats have Hb with P50~35 – 40 Torr). The partial oxygen pressure pO2 is expressed in a range of units, like 1 Torr = 1 mm Hg =0.13 % O2. When in equilibrium, a partial pressure of 1 Torr O2 implies that aqueous buffers contain a free oxygen concentration of 1.32 μM (37 °C) or 1.84 μM (20 °C).
| [O2] (μM) | pO2 (Torr) | |
|---|---|---|
| 286 | 160 | in cell cultures at 20° C under atmosphere of 760 Torr with 21 % oxygen |
| 214 | 160 | in cell cultures at 37° C under atmosphere of 760 Torr with 21 % oxygen |
| 130 | 98 | oxygen pressure in normoxic arterial blood |
| 35–40 | 27 – 31 | oxygen pressure in normoxic venous blood |
| 35 | 27 | half-loading point of human adult Hb at 37° C in whole blood (in presence of CO2, variations due to the Bohr effect) |
| 20 | 15 | normal O2 level in non-exercised muscle (58) |
| 20–50 | 15 – 38 | HIF-α subunits are stabilized and start to accumulate, HIF heterodimers activate hypoxia-sensitive genes (62, 53). HIF trigger levels adapt to local oxygen levels (54, 55) |
| 10 | 7.6 | L-Arg → L-Citrulline conversion by eNOS is progressively slowed if [O2] < 10 μM (63) |
| 4 | 3.0 | half-loading point of human Mb at 37° C |
| 2 | 1.5 | oxygen deficiency becomes rate-limiting for mitochondrial respiration (58, 64) |
Table V compiles a selection of published oxygen levels as measured with a variety of techniques in mammalian tissues. As expected, all tissues in the table are indeed normoxic according to our definition. The renal medulla occupies the lower end of the normoxic range, attesting to the low blood flow through medullar tissue needed to maintain osmotic gradients for the process of urinary concentration (56). It should also be noted that within certain tissue, such as skeletal mucle, measured oxygen levels suggest a sharp oxygen gradient in blood along the vascular tree, with decreases in oxygen partial pressure in blood from arteries to muscularized capillaries ranging from 130 μM down to 25 μM (57). These oxygen gradients span the P50 of hemoglobin, the oxygen tension at which hemoglobin is 50% saturated with oxygen. Human hemoglobin A has P50 of 27 Torr or 35.6 μM oxygen (cf table IV). The oxygen in the perivascular tissues is not homogenously distributed: Significant radial oxygen gradients were found along directions perpendicular to the (micro)vessel. The steepest gradients were found around arterioles, less so in the capillary bed, and lowest near venules (57). These oxygen gradients became steeper when the metabolic rate increased.
Table V.
selection of partial oxygen pressures measured in normoxic tissues of adult mammals. 1 Torr = 1 mm Hg
| animal | tisue | pO2 (Torr) | method | reference |
|---|---|---|---|---|
| rat | kidney, periportal | 70 | electrochemical | (65) |
| rat | kidney, perivenous | 35 | electrochemical | (65) |
| rat | renal cortex | 50 | electrochemical | (56) |
| rat | renal medulla | 15 | electrochemical | (56) |
| rat | liver surface | 70 – 77 | 19F NMR | (66, 67) |
| rat | brain, periarteriolar | 75 | Fluorescence quenching | (68) |
| rat | cerebral cortex | 35 ± 10 | EPR linewidth | (69, 70) |
| rat | spleen | 80±20 | 19F NMR | (67) |
| rat | lung | 73±20 | 19F NMR | (67) |
| mouse | renal cortex | 23 | EPR linewidth | (71) |
| mouse | renal medulla | 15 | EPR linewidth | (71) |
| rabbit | resting muscle | 21 | Microprobe catheter | (15) |
| pig | arterial blood | 75 | 19F NMR | (72) |
| pig | venous blood | 37 | 19F NMR | (72) |
| pig | liver | 20 | 19F NMR | (72) |
| pig | spleen | 25 | 19F NMR | (72) |
| pig | working heart | 35 – 63 | electrochemical | (73) |
| dog | LV-heart | 5 – 20 | electrochemical | (74) |
| human | arterial blood | 90 | electrochemical | |
| human | venous blood | 40 | electrochemical | |
| human | Resting muscle | 24 | Microprobe catheter | (75) |
| human | Working muscle | 8 | Microprobe catheter | (75) |
Table V compiles a range of experimental concentrations of free oxygen for humans and a selection of values from animal studies. We note that important exceptions to these numbers occur: In humans, fetal arterial pO2 is only 38 Torr (~50 μM), and reaches only half the adult value. Strongly exercised muscle may become essentially anoxic in any mammal (57,58). Oxymetric imaging has shown that tumours often contain a distinctly hypoxic core (59). It should be noted that cell cultures are commonly grown in a controlled atmosphere containing 5 % CO2 and 21 % O2, equivalent to pO2=150 Torr. In this atmosphere, the medium contains ca 200 μM oxygen and should be considered hyperoxic according to our classification. This effect may significantly modulate the metabolism and response of cultured cells (60). Finally, the oxygen levels in tissues should be regarded as coarse grained averages over a lengthscale of at least 100 μm. It is expected that significant oxygen gradients exist on smaller lengthscales. For example, it is expected that oxygen preferentially accumulates in low polarity compartments like protein (eg albumin) of lipid membranes. The partition factors for oxygen and NO were reported (38, 61) as 3–4 and 9–10 respectively. Such preferential partitioning in the lipid and protein compartments may enhance local concentrations. It is known that this phenomenon significantly accelerates reactions like the oxidation of NO by oxygen (61). In addition, mitochondria act as the main sink of oxygen, and significant intracellular oxygen gradients are expected to exist. Therefore, the usual assumption that intracellular oxygen levels are equal to the coarse grained average concentrations in tissues is unlikely to hold generally.
The preceding discussion considers the oxygen levels in various tissues. It should be noted that the consumption of oxygen varies significantly with tissue type as well. In resting humans, oxygen consumption per weight of heart tissue is ca threefold higher than in brain (28). Under exercise, the oxygen consumption by heart may increase by an order of magnitude (28), although the average oxygen levels fall significantly (table V). The oxygen consumption in tissues and the effects of hypoxia are discussed in (28, 76) and many textbooks.
4. EVIDENCE THAT NITRITE IS A PHYSIOLOGICAL AND THERAPEUTIC VASODILATOR
Nitrite has very low potency in isolated aortic ring preparations (4) or as a direct activator of soluble guanylate cyclase (5, 78). Therefore, it was not considered as a putative physiological signalling molecule or vasodilatory molecule until very recently. Prior to 2000 it was found that nitrite levels in the human forearm circulation dropped from artery-to-vein, suggesting a consumption of nitrite across this circulation (45) An increase in nitrite consumption with exercise stress suggested that nitrite might be metabolized in vivo under exercise. Indeed, under low pH conditions even physiological concentrations of nitrite elicited clear vasodilatory effects in isolated aortic rings (79). Studies using inhaled NO gas in humans further supported this observation as the apparent “endocrine” effect of inhaled NO on forarm blood flow was associated with increases in plasma nitrite (80). Despite overwhelming conventional thought that nitrite was not a vasodilator at μM concentration in vivo (81, 6), Cosby et al.(26) infused nitrite at concentrations of 200 μM and 2.5 μM into the human forearm and observed significant vasodilation associated with partial deoxygenation of the Hb. During exercise stress vasodilation was observed at nitrite concentrations far lower than 900 nM. Recently, a significant drop in human mean arterial blood pressure was elicited by nitrite concentrations as low as 350 nM. The drop in blood pressure correlated with a concomitant increase in circulating metHb (82).
Although often compared on a molar basis, the total doses applied in the above studies differed substantially due to different durations of nitrite infusion (despite the same concentrations used), and vasodilatation was observed after some delay. Thus, the most likely explanation for this apparent discrepancy is the conversion of nitrite into longer-lived NO-metabolites with the latter accounting for most of the vasodilator effects. Obvious candidates are S-nitrosated thiol residues or nitrosylated species like the dinitrosyl-iron complexes discussed in section 9B.. For a comprehensive review of such longer-lived NO-metabolites cf (33). S-nitroso hemoglobin (SNO-Hb) in particular has been scrutinized for its vasodilating properties and its formation upon exposure to nitrite has been confirmed (83, 84). This does not exclude the possibility that the in vivo vasodilating effects of nitrite are associated with the formation of free NO, as suggested by the formation of NO-Fe2+Hb (ferrous nitrosyl-hemoglobin). The formation of HbNO increases as hemoglobin oxygen saturation drops, suggesting that circulating deoxygenated hemoglobin (deoxyHb) act as a nitrite reductase under hypoxia (26). This chemical biology will be discussed in detail later (cf section 7.A).
A recent study by Maher and colleagues has now confirmed that nitrite is a hypoxic vasodilator in humans (85). In these studies intrabrachial infusions of nitrite were performed under inhalation of either normal air (21 % O2) or hypoxic gas containing 12 % O2. Under normoxia, high doses of nitrite (3.14 μmol/L·min) were needed to induce significant decreases of forearm venous tone. When inhaling hypoxic gas, tenfold lower doses of 314 nmol/L·min induced significant vasodilation and increased the brachial blood flow (85). This human model demonstrates that hypoxia enhances significantly the potency of nitrite in the arterial circulation, thereby fulfilling a fundamental requirement for a putative mediator of hypoxic vasodilation.
A growing number of studies now confirm that nitrite is a potent vasodilator in vivo. Nitrite-dependent vasodilation has been demonstrated in the mouse, rat, dog, sheep, primate and human circulation (11, 26, 86–90). In most of these studies the nitrite-dependent vasodilation coincides with NO formation in the red blood cells.
In rabbits, the imposition of acute hypoxia drastically depressed the concentration of exhaled NO. This effect was attributed to the inhibition of NOS enzymes by oxygen deficiency. Infusion of nitrite greatly enhanced exhaled NO under hypoxic conditions only (91). In lambs, the inhalation of nebulized nitrite achieved pulmonary vasodilation under hypoxic conditions (88). The vasodilating response was strongly reminiscent of the reaction when small quantities of NO gas are added to the breathed air. These observations are consistent with potentiation of the nitrite vasodilation by hypoxia. Significantly, vasodilation by nebulized nitrite was far smaller under hyperoxic conditions (100% oxygen and thromboxane infusion to generate pulmonary hypertension). The results suggest that inhalation of nitrite have therapeutic potential for treatment of human neonatal pulmonary hypertension.
Before concluding this section it should be mentioned that hypoxic vasodilation may arise via alternative pathways unrelated to nitrite: Deoxygenation induces a release of ATP from RBC’s which stimulates P2Y (purigenic) receptors and endothelial NOS ((22) and references therein). The magnitude of this vasodilating response varies with species, rabbit being particularly sensitive (22). The contribution of the ATP mediated pathway may be distinguished from other pathways by inhibition of the purigenic receptor, or by application of apyrase, a plasma-membrane enzyme that catalyzes the hydrolysis of ATP to AMP and inorganic phosphate. In addition, numerous studies have confirmed that smooth muscle cells per se have intrinsic pathways for hypoxia induced relaxation (cf (92) and references therein).
5. NITRITE PROTECTS AGAINST ISCHEMIA/REPERFUSION INJURY: EARLY OBSERVATIONS IN ANIMAL MODELS AND CLINICAL SETTING
In vitro studies of perfused Langendorff hearts have shown protection of myocardial tissue against ischemia-reperfusion injury by nitrite. This effect has been attributed to the generation of NO from nitrite by XO (9) or by myoglobin (8, 23, 24).
Isolated perfused pig lungs exhaled significantly larger quantities of NO gas if nitrite was added to the perfusate, an oxygenated but blood-free Krebs buffer with albumin. Concomitantly, the pulmonary vascular resistance was markedly reduced, suggesting the release of NO radicals under these conditions (90).
Most in vivo studies of nitrite have been carried out in rodents. Analogous to the study of nitrite as a vasodilator, very early studies were focused on effects of higher doses of nitrite, which induce methemoglobinemia and secondary hypoxia. In one such study, nitrite was found to protect mice against the effects of ionizing radiation (93). More recent studies published since 2005 have revealed a striking low-dose effect of nitrite (high potency) on limiting cellular injury, necrosis and apoptosis after prolonged ischemia and reperfusion. Cytoprotection of mouse and rat tissue was reported in heart (8,9) and liver (8) at low nitrite dosages. Cytoprotection was optimal with intraperitoneal injection of only 48 nmol nitrite per mouse, thereby raising the plasma nitrite concentration from basal 0.6 μM to 0.7 μM. Renal tissues of rat show mixed results for ischemia-reperfusion injury: (94) reports that administration of nitrite during reperfusion improved renal function and reduced histological damage of the affected tissue. Part of this effect was attributed the activity of XO enzymes. In contrast, another recent study showed no protection of nitrite therapy against renal ischemia in rats (95). Possibly, this negative result is related to the unusually low oxygen status of normally perfused kidney tissue (cf table V) or unusual mechanisms of anion clearance in renal cells.
Nitrite was reported to cross the blood-brain barrier (89). As such, it may be a promising candidate for the clinical treatment of stroke. In ischemia-reperfusion of rat brain, therapeutic intravenous infusion of 0.5 μmol nitrite per animal during the reperfusion stage improved cerebral blood flow, promoted functional recovery and reduced infarct size (96). Significant improvement was already found at tenfold lower dose of 0.05 μmol nitrite. This dose corresponds to an enhancement of blood nitrite concentration by only 1 μM. The neuroprotective effects of nitrite were cancelled by coadministration of the NO scavenger C-PTIO (96). In contrast, no improvement was reported in rat brain when nitrite was administered as coadjuvant in a stroke therapy with recombinant tissue plasminogen activator (rtPA) (97). Studies on primates showed that nitrite therapy was beneficial in treatment of severe cerebral artery vasospasm after subarachnoidal artery hemorrhage (89).
Studies in human models, though far fewer, have generally confirmed these observations. In human volunteers, the bloodflow through the forearm was measured with plethysmography, and NO metabolites in the bloodstream were monitored. Exercise caused a significant arterial to venous gradient of nitrite in the bloodstream and suggested that significant quantities of nitrite are consumed in exercising tissues where the local oxygen level has fallen to low values (45). Table V shows that the oxygen levels in working muscle fall to rather low values. More recently, forearm plethysmographic studies have shown that the administration of nitrite prior to ischemic stress improves the post-ischemic blood flow in the affected area (98).
Recent studies of inhaled NO gas suggest that the formation of nitrite in blood accounts for vasodilation and cytoprotection of non-pulmonary organs. Cannon and colleagues (80) found that inhaled NO gas at a dose of 80 ppm would produce vasodilation of the human forearm circulation at rest, during regional NO synthase inhibition, and under exercise stress. They found that the levels of plasma nitrite significantly increased in this inhalational protocol. More recently, Lang and colleagues (99) gave inhaled NO gas to humans undergoing orthotopic liver transplantation. They found a twofold increases in plasma nitrite during NO gas inhalation, with prominent artery-to-vein gradients of the nitrite concentration. This was associated with improved liver function (reduced coagulopathy) and reduced liver injury. Although not all effects of inhaled NO may be mediated by nitrite, these studies suggest that the beneficial effects of nitrite on ischemia-reperfusion injury may translate to humans.
The mechanism of nitrite-dependent cytoprotection after ischemia/reperfusion appears highly complex and apparently involves multiple pathways. It is known that free NO enhances expression and binding activity of HIF-1α protein. S-nitrosation inhibits the transcriptional activity of inflammatory transcription factors like activator protein 1 (AP1) or nuclear factor κB (NF-κB). NO has significant anti-inflammatory effect by inhibiting the expression of a wide range of adhesion molecules on the cellular membrane, thereby suppressing leukocyte adhesion to the endothelium and diapedesis (extravasation of leukocytes). Other protective mechanisms operate at the mitochondrial level and involve the modulation of reactive oxygen species generation from the mitochondrion. It has recently been shown (100) that exposure to nitrite leads to S-nitrosation of critical thiols on mitochondrial complex I. The modification was observed in vitro as well as in isolated mitochondria and inhibits the activity of complex I. This decrease in electron transfer through complex I effectively decreases the leakage of reactive oxygen species from the mitochondrion and protects downstream complexes (complexes II–IV) as well as other mitochondrial proteins against damage from ischemia-reperfusion. This protection also prevents the release of cytochrome c from the mitochondrion and inhibits the initiation of the mitochondrial apoptotic pathway. More details of the effect of nitrite on mitochondrial activity will be given in section 7.F.
The above considerations relate to the cytoprotective action of nitrite against acute ischemia-reperfusion injury. However, many clinical problems involve chronic rather than acute tissue ischemia due to local inadequacy of perfusion. This raises the question whether nitrite be beneficial for chronic ischemia as well. Recently, it was shown (101) that chronic nitrite therapy provides local enhancement of angiogenic activity and tissue perfusion in those regions where vascular perfusion was defective. The phenomenon was studied in the murine hind-limb ischemia model and was mediated by free NO radicals as the effects were abolished by application of the NO scavenger C-PTIO.
6. PHYSIOLOGICAL EFFECTS OF NITRITE OTHER THAN NO RELEASE
The majority of the physiological responses discussed above are compatible with a reaction sequence where nitrite is first reduced to free NO radicals, and the latter acts as the true effector of the observed response. In addition to this NO-mediated pathway, nitrite anions are known to elicit a number of physiological reponses that do not appear to involve reduction. The best known example is the rapid reaction of nitrite with oxy-hemoglobin to nitrate and methemoglobin. It is known that dietary intake of large doses of nitrite may cause significant and even lethal methemoglobinemia (102) in rodents and humans. This complicated autocatalytic reaction also generates nitrogen dioxide and ferrylhemoglobin as catalytic intermediates. Although studied for over a century, the mechanism for this reaction is still controversial. The reaction will be discussed in more detail below (section 9.A.). In addition, nitrite has been reported to inhibit a number of mammalian enzymes. In the presence of halide anions like Cl− or F−, catalase was reported to be inhibited by supraphysiological doses of nitrite (103). This mechanism may explain the early observations (103 and references therein) that nitrite protects hydrogen peroxide against destruction by catalase in hemolysates and intact erythrocytes. Additionally, concentrations of 50 – 100 μM nitrite inhibit (104) the enzymatic activity of myeloperoxidases and thereby prevent consumption of hydrogen peroxide by this enzyme. The toxicity of this phenomenon has been demonstrated by enhancement of neutrophil-induced DNA strand breakage in cultured epithelial cells (104). At supraphysiological mM concentrations, nitrite has also been shown to inhibit the enzymatic activity of arginase in vitro and in harvested murine and rat macrophages (105).
A recent study shows potent activation of estrogen receptor-alpha in breast cancer cells by physiological amounts of nitrate or nitrite (106). The authors suggested that nitrite was the active compound since the nitrate effects were abolished by pharmacological inhibition of nitrate reduction.
Earlier studies in rats have demonstrated that the profile of NO metabolites in vivo changed rapidly during brief intervals of global hypoxia (48). Within minutes the tissue concentrations of S-nitrosothiols and nitrosyl-heme increased at the expense of nitrite and suggested that under hypoxia tissue nitrite serve as an extravascular pool of NO.
Subsequent experiments (107) investigated the effects of nitrite in normally breathing rats, i.e. under normal physiological oxygen tension. On a timescale of minutes, the administration of nitrite led to an increase of S-nitrosothiols and nitrosyl-heme, increase in cGMP, and inhibition of cytochrome P450,. On a longer timescale of 24 hr, alterations were noted in the expression of heat shock proteins in a range of organs. Intriguingly, these effects were not inhibited by NO scavengers like oxy-Hb in vitro or C-PTIO in vivo, and suggest that nitrite may act as a signalling molecule in its own right. It should be noted that these experiments were performed at normal physiological oxygen levels.
On a pharmacological level, nitrite has beneficial effects as antidote against acute poisoning by cyanide or hydrogen sulphide (H2S). In both cases, the beneficial effect of nitrite is mediated by the formation of copious quantities of methemoglobin (cf next section), and the latter acts as the actual scavenger of cyanide or H2S (108).
7. ENDOGENOUS PATHWAYS FOR REDUCTION OF NITRITE TO NO
A: Deoxy hemoglobin (deoxyHb)
In 1937 J. Brooks (109) showed that deoxygenated hemoglobin (deoxyHb, Fe2+Hb) reacts with nitrite to form equimolar concentrations of methemoglobin (metHb, Fe3+Hb) and ferrous nitrosyl hemoglobin (NO-Fe2+Hb). In 1981, Michael Doyle and colleagues investigated the mechanism and kinetics of this reaction (110). From the pH dependence of the reaction they hypothesized that nitrous acid HNO2 be involved. They proposed the following set of reactions:
| (1) |
| (2) |
| (3) |
However, Doyle reported a complicated stoichiometery where the ratio metHb: NO-Fe2+Hb was about 5:2. However, in 2005 it was demonstrated that traces of oxygen have a pronounced effect on the stoichiometry of the reaction, in particular on the balance between metHb and NO-Fe2+Hb. A truly 1:1 ratio is only achieved with careful exclusion of all oxygen from the samples (111, 112). Remaining traces of oxygen lead to formation of oxyHb which reacts very rapidly with free NO to metHb and nitrate (see section 9.A). This additional pathway favors formation of metHb, and increases its yield over that of NO-Fe2+Hb. The kinetics of the truly anoxic reactions was carefully studied (111,112), and the results were quite surprising:
Equation 2 predicts pseudo-first order kinetics in [Fe2+Hb] when nitrite is in excess. However, the experimental timecurves of [Fe2+Hb] suggest zero-order kinetics and actually have sigmoidal character, with the rate being slowest at the beginning and end of the reaction (fig 1A). The sigmoidal character of the kinetic trace is evident when the instantaneous rate is plotted as a function of time (fig 1B). Interestingly, the reaction is fastest about halfway through the reaction. The surprising kinetics of this reaction are explained by the freedom of Hb tetramers to exist in two quaternary conformations (R or T geometry) that have differing binding affinities and reaction rates for small ligands like oxygen or nitrite. Unliganded ferrous heme in the relaxed (high oxygen affinity) R-state quaternary form reacts with nitrite approximately sixty times faster than heme in the tense form (low oxygen affinity, T-state) (113). In absence of ligands, the T-state is more stable than R-state. Accordingly, the Hb tetramers exist in T-state with low reactivity just before the nitrite is added. Therefore, the nitrite consumption remains slow although the number of vacant ferrous heme sites for nitrite binding is highest. As the reaction proceeds, the number of vacant ferrous heme sites is depleted which slows the reaction down. However, some of the Hb tetramers are converted to the R-state due to formation of metHb and NO- Fe2+Hb. This conversion to highly reactive R-state accelerates the reaction. The balance of these two counteracting influences results in a sigmoidal kinetics as shown in fig. 1B. We note that consumption of one nitrite molecule results in the formation of one ferric heme (metHb) and one NO-Fe2+Hb, introducing an element of autocatalysis in the reaction, since one nitrite affects the conformation of two Hb tetramers.
Figure 1.

Kinetics of the reaction between human deoxyHb (50 μM) and nitrite (10 mM) at pH 7.4 and 37° C. (A) UV/VIS absorption spectra were deconvoluted to determine the percentage of each species as a function of time. DeoxyHb is observed to form equal amounts of metHb and iron-nitrosyl-Hb. Deviation from first order behavior is evident in the curve for decay of deoxyHb, having a sigmoidal shape. (B) The instantaneous rate of the reaction shown in panel A where the negative of the slope of the decay curve for deoxyHb is plotted as a function of time. (C) Nitrite (10 mM) was reacted with Hb (50 μM) at various oxygen tensions. The initial rate of the reaction is plotted. (A, B from (114), C from (111) with permission).
The hypothesis of allosteric autocatalysis predicts that the rate of the reaction between nitrite and hemoglobin be dependent on the oxygen tension. As the Hb oxygen saturation is increased, the number of available ferrous hemes decreases (slowing the reaction down), but the number of available ferrous hemes that are in the R-state increases (speeding the reaction up). This prediction was experimentally confirmed in Fig 1C, where the initial reaction rate shows a sigmoidal dependence on oxygen level. The figure confirms that the highest initial reaction rate occurs for oxygen levels near the half-loading point P50 of human Hb (111).
This finding has three important implications for physiology:
Hemoglobin functions as a mammalian nitrite reductase whose activity is controlled by the ambient oxygen level. The highest rate of reduction is reached for [O2] ~ 35 μM, corresponding to P50 of human adult Hb (cf table IV).
Because hemoglobin deoxygenates under physiological conditions, hemoglobin shows nitrite reductase activity over a wide range of oxygen tensions that are higher than the oxygen tensions required to reduce nitrite by other enzymes, except XO (cf fig 10). Fig. 2 illustrates the allosteric nature of the hemoglobin nitrite reductase activity.
Because the R state of hemoglobin is the fastest nitrite reductase (highest bimolecular rate constant for nitrite reduction is in the R-state tetramer) the formation of NO from the nitrite-hemoglobin reaction should be most efficient during rapid deoxygenation from artery to vein. Under these conditions the R-state (oxygenated) tetramer releases oxygen to expose deoxygenated heme sites on R-state molecules (R3 and R2 tetramers) (7).
Figure 2.

Oxygen-dependence of nitrite reductase activity of hemoglobin. As oxygen tension increases the amount of deoxyHb (blue trace) decreases while the amount of R-state Hb (and free hemes in R-state Hb tetramers, red trace) increases. The nitrire reductase activity (gray) depends on both of these factors and is maximal at the P50. It should be noted that this figure is merely illustrative and the precise oxygen tension dependencies are more complex yet give a similar result. From (7) with permission.
The above describes the reaction of nitrite with deoxyHb. However, in presence of oxygen an alternative pathway exists: nitrite also reacts with oxyHb to form metHb and nitrate. Since Hb is partially saturated in vivo, Grubina et al (114) studied the reaction of nitrite with Hb at varying oxygen tensions and found that the reactions with oxyHb and deoxyHb proceed simultaneously under these conditions. At the beginning of the reaction, deoxyHb is consumed much faster than oxyHb. In fact, the reaction of nitrite with deoxyHb partially inhibits the reaction of nitrite with oxyHb. The autocatalytic phase of the oxyHb reaction is inhibited by the presence of deoxyHb and products of the deoxyHb/nitrite reaction, namely NO-Fe2+Hb. Interestingly, intermediates in the oxyHb/nitrite reaction, probably NO2•, oxidize NO-Fe2+Hb and release NO in a process called oxidative denitrosylation (114). These results demonstrate that the reaction of nitrite with oxyHb is limited under physiological conditions, yet its occurrence can also facilitate NO release from NO-Fe2+Hb, provided that there is sufficient compartmentalization of these chemistries within the RBC. More details on the reaction of nitrite with oxyHb will be given in section 9.A.
The above reactions were investigated in vitro. The three implications for mammalian physiology were also tested in more biological models like aortic ring bioassays (22, 26, 87). In this context a careful distinction should be made between the vasodilating effects of free Hb and intact RBC’s. Deoxygenation of RBC’s induces the release of ATP. This by itself may cause vasodilation by activation of purinergic P2γ receptors and eNOS (22). This alternative pathway is independent of nitrite.
It has been noted that the combination of nitrite with RBC is particularly effective to elicit hypoxic vasodilation. RBCs in the presence of 2 μM nitrite resulted in vessel relaxation at much higher oxygen tensions than nitrite or RBCs alone (26). These vasodilations do not require high nitrite levels: It has been shown that 200 nM nitrite could relax rat and rabbit thoracic aortic rings in the presence (but not in the absence) of 25 μM deoxyHb (22). The vasodilating potency of nitrite infusions was assessed in humans in Ref (82). It was found that enhancement of venous nitrite by only 300 nM enhanced the forearm blood flow significantly. Such values are in the normal physiological range of blood nitrite (cf section 2.).
A number of observations suggest that this vasodilation be mediated by free NO radicals: Firstly, Nitrite/RBC-dependent vasorelaxation was shown to coincide with increased cGMP levels, and could be inhibited by the NO scavenger C-PTIO (22). Moreover, the efficiency of vasorelaxation was shown to have the same dependence on oxygen tension as the nitrite reductase activity of human Hb, as shown in Fig 1C and illustrated in Fig. 2 (22). A further indication of the release of free NO was the inhibition of mitochondrial respiration by nitrite in the presence, but not in the absence, of RBCs. These data strongly suggests that Hb in RBC release sufficient NO from nitrite to achieve vasodilation under physiological conditions.
A major conceptual obstacle to NO signalling via nitrite reduction by Hb in RBC is the low probability that NO escape from the intracellular compartment. Numerical simulations of the diffusion process suggest that NO cannot leave the RBC since the reaction with oxyHb is very fast (diffusion limited (110, 115–119). From the experimental lifetime and diffusion rate, it is estimated that the diffusion length of NO is of the order of 0.02 μm. This distance is far smaller than the cellular dimensions. Therefore, free NO radicals could not escape the cell (120). Computer simulations predicted that even with a high therapeutic dose of nitrite (200 μM), only 0.1 picomolar of NO generated from the reaction of nitrite with Hb would reach the smooth muscle cells at steady state; this quantity is far below the activation threshold for vasodilation (121).
The explanation for this paradox may lie in the nature of the species escaping from the RBC: it is possible that it is not NO per se, but some more stable intermediate neutral species, such as N2O3. This highly polar molecule easily reacts with water, but is thought to be a major agent for S-nitrosation of thiol residues in biological systems (cf chapter 1 of (33)). It was proposed that this intermediate could diffuse out of the RBC release free NO in the extracellular space, possibly via S-nitrosated intermediates (121, 122). Interestingly, a mechanism for N2O3 formation from the nitrite/deoxyHb reaction was recently proposed (123). This nitrite reductase/anhydrase activity of hemoglobin is illustrated in Fig 3. A key player in this mechanism is nitrite bound metHb. Surprisingly, it was found that nitrite-metHb has no signal in electron paramagnetic resonance (EPR), indicating a peculiar electronic structure that, by density functional theory calculations, was shown to include some Fe2+-NO2• character (123). This species reacts quickly with NO to form N2O3. As suggested by the illustration in Fig. 3, metHb can be formed by the reaction of nitrite with deoxyHb and/or the reaction with oxyHb. NO is also formed by the deoxyHb/nitrite reaction as well as by oxidative denitrosylation at various levels of oxygen. Thus, initially, metHb could build up under high oxygen tension. Subsequently, as the oxygen tension is lowered, N2O3 would be produced. The mechanism is illustrated in Fig. 3. Together with the discussion above, it suggests that Hb is an allosterically controlled nitrite reductase/anhydrase and the overall stoichiometry of the set of reactions is
Figure 3.

The N2O3 forming reaction of nitrite and hemoglobin may regulate the export of NO from the erythrocyte. Hemoglobin deoxygenation (purple) occurs preferentially at the submembrane of the red blood cell as it traverses the arteriole. Nitrite reacts with deoxyHb to metHb and free NO. Much of this NO binds to hemes of deoxyHb or reacts with oxyHb to form nitrate and metHb. MetHb binds nitrite to form an adduct with some Fe2+-NO2 character (Hb- ). This species reacts quickly with NO to N2O3 which can diffuse out of the red cell, later forming NO or extracellular S-nitrosothiols. Low molecular weight nitrosothiols may contribute to exportable vasodilatory activity. (from (123) with permission).
| (4) |
demonstrating the catalytic nature of the ferrous Hb protein.
In order for nitrite-metHb to react with NO, it must compete with reactions of oxyHb and deoxyHb. The relative yields of these three competing pathways depends on reaction rates, starting concentrations and cellular compartment. These kinetic challenges cannot be surmounted by the rate of the nitrite-metHb/NO reaction alone, however the reaction can be inefficient. Since NO has high potency as a vasodilator (EC50 ~ 5 nM), very little nitrite must be reduced and only small quantities of NO need to escape the red cell to exert physiological effects. Future work will elucidate the nature and extent of any intraerythrocytic compartmentalization that could promote the formation of N2O3.
B: Myoglobin
The monomer myoglobin (Mb) is a small (17.6 kDa) but important intracellular oxygen binding heme protein. Under basal conditions the tissue is well oxygenated and Mb predominantly exists in the oxygenated state (cf table IV). Therefore, it has long been accepted as an intracellular oxygen store. In exercising skeletal muscle and in the beating heart, Mb serves as a short-term oxygen reservoir, tiding the muscle over from one contraction to the next. Similarly, Mb is expressed in high concentrations in skeletal muscle of mammals and humans adapted to high altitudes (126). More controversially, a potential role of Mb in intracellular oxygen diffusion within muscle cells has been considered (124, 127–129).
The Mb concentration various between mammalian species (124) and between tissue type: Mb is highly expressed in type I and IIa skeletal muscle fibers, in cardiac and tongue muscles, and to a lesser extent in smooth muscle cells (124, 129). In human cardiac tissue, Mb levels are ca 200 – 300 μmol/kg wet tissue, whereas skeletal muscles reach concentrations of ca 400 – 500 μmol/kg wet tissue (124). The Mb contents of different skeletal muscles in man and rats are tabulated in ref (129). In diving mammals, the Mb concentrations reach ca 2 mmol/kg wet tissue. This value is about tenfold higher than in terrestrial mammals and serves as an O2 store contributing to the extension of diving time (125).
The role of Mb in vivo has been investigated by comparison of wild type mice with Mb deficient mutants (Mb−/−) (130, 131). In these mutants, multiple compensatory mechanisms were activated to compensate for the loss of the oxygen-storage and –transport function of Mb. These included increased capillary density, elevated hemoglobin levels, increased coronary flow, and a switch in cardiac substrate utilization from fatty acid to glucose (132). In addition, oxyMb is a highly effective scavenger of free NO radicals. Therefore, the NO status of cardiac tissue was investigated in this mutant.
Whereas the role of endogenous NO for myocardial function is still a subject of significant controversy, a it was postulated (133) that the effect of NO depends on its concentration: A positive inotropic effect at low concentrations and a negative one at higher concentrations. Similar concentration-dependent effects of NO play a role in the modulation of transduction of the parasympathetic effects of cholinergic stimulation, in attenuation of oxygen consumption, and in apoptosis of cardiomyocytes.
Depending on ambient oxygen level, Mb acts either as an NO-scavenger under normoxic conditions or as a nitrite-reductase under conditions of hypoxia and ischemia (134, 24) (cf. Fig 4). Because Mb must be at least partially deoxygenated to act as nitrite-reductase, the latter reaction pathway can become significant only when the oxygen level falls below the P50 of myoglobin (ca 3 Torr, equivalent to a free oxygen concentration of ca 4 μM, cf table IV). In contrast, the detrimental effects of overproduction of NO radicals in heart tissue may be prevented by the dioxygenase function of oxygenated myoglobin (135, 136). Mb efficiently protects the respiratory chain against nitrosative stress from NO radicals (137). Experiments in Mb knockout mice confirmed (134, 136) that the presence of Mb has a significant effect on NO levels in cardiac tissue: In hearts from Mb−/− mice, endogenous and exogenous NO were more effective in the regulation of coronary tone and myocardial contractility. It suggests that the hearts of Mb−/− mutants have higher NO levels than the hearts from WT mice.
Figure 4.

Depending on ambient oxygen, myoglobin acts as a dioxygenase or as a nitrite-reductase. Under normoxia, oxymyoglobin acts as an NO-scavenger, protecting the mitochondria from inhibition by NO) (left). Under hypoxia, myoglobin changes its function from a dioxygenase to a nitrite-reductase. Now it converts nitrite to free NO (right), regulating mitochondrial respiration and myocardial function.
The oxygen levels in the left ventricle of beating dog hearts were measured electrochemically (74) and show a wide distribution of values between zero and venous levels (cf table V). The distribution suggests that certain regions of ventricular tissue have very low oxygen pressures below 5 Torr, offering the physiological prerequisite for the role of Mb as a functionally relevant nitrite-reductase. The oxygen binding curve of Mb is a hyperbolic curve with a half-loading pressure of P50 ~2.75 Torr (cf table IV). This value is an order of magnitude lower than P50 of the sigmoid-shaped binding curve of Hb. It allows Mb to take up oxygen from Hb and to load and unload oxygen in the range of the pO2 values that occur within the cell. Although oxyMb limits NO bioavailability in tissues due to its rapid reaction with NO, under hypoxic conditions, the myoglobin-dependent nitrite reduction may provide a mechanism by which NO is generated to regulate the physiological functions under conditions where the arginine to citrulline conversion by NOS is oxygen-limited. The redox potential of Mb is lower than R-state Hb and deoxyMb was found to reduce nitrite and generate NO at a faster rate than deoxyHb (112) with a bimolecular rate constant of 12 (Ms)−1 at 37°C (24). In isolated cardiomyocytes the nitrite reductase activity of deoxyMb releases NO in proximity to mitochondria and regulates mitochondrial respiration through cytochrome c oxidase (24).
Recent studies showed (134) that this NO interacts reversibly with myocytic cytochromes and down-regulates cardiac mitochondrial activity. This leads to a reduction in oxygen consumption and consecutively also of cardiac contractility (134). Cardiac contractile function and energy metabolism are actively downregulated, when coronary blood supply is critically reduced. On acute restriction of coronary artery inflow, the contractile function of the ischemic region is rapidly decreased and the oxygen consumption is reduced. This dampens the fall in high energy phosphates and over time even can restore myocardial energy balance. This adaptive response is referred to as “short-term hibernation” (138).
A very similar response of the metabolic system was observed upon nitrite infusion in mice. These infusions led to a marked decrease in phosphocreatine (PCr), together with an increase in inorganic phosphate and a reduction of the available driving force for all energy-consuming processes (ΔGATP) (134). Simultaneously, the infusion of nitrite reduced the synthesis and utilization of ATP. By implication,, the reduction of endogenous nitrite to NO by deoxyMb may be significant for such “short-term hibernation” as observed upon restriction of coronary arterial flow. The presented experiments were carried out under hypoxic perfusion conditions which cause Mb to be deoxygenated by about 50%. Severe low-flow ischemia certainly can lower tissue pO2 even further, thereby further augmenting the ability of deoxyMb to form NO from nitrite (138).
These data suggest that the mechanisms may be relevant under physiologic conditions and at physiological nitrite levels. Typical endogenous nitrite levels in Wistar rats are ca 0.3 μM in plasma, ca 0.8 μM in cardiac tissue, and up to 20 μM in aortic tissue (48) (for a more comprehensive discussion of endogenous nitrite levels, cf section 2.). Although high extracellular concentrations of nitrite (10 to 100 μmol/L) were required to elicit the biological response, it is the intracellular concentration of nitrite which is of critical importance for the reaction with deoxyMb. Pretreatment of animals with the NOS-inhibitor L-NIO decreased cytosolic nitrite by appromimately 70%, and perfusion with concentrations ≥ 10μmol/L nitrite was required to replenish the myocytic levels to the range of untreated controls (138). Obviously, comparatively high extracellular nitrite concentrations have to be applied under our experimental conditions to mimic the in vivo conditions with unrestricted activity of NOS and unlimited availability of its substrate L-arginine (the latter was deliberately not supplemented with the perfusion buffer). Together, these data suggest that endogenous levels of intracellular nitrite be sufficient to affect cardiac function upon imposition of hypoxia.
Recent results (139) showed that deoxyMb acted as a functional nitrite reductase that generatedf NO and downregulated cellular respiration. This beneficial cascade is a cytoprotective response to cardiac ischemia-reperfusion (I/R) injury. Myoglobin was found responsible for nitrite-dependent NO generation and cardiomyocyte protein iron-nitrosylation. Nitrite reduction to NO by myoglobin dynamically inhibits cellular respiration and limits reactive oxygen species generation and mitochondrial enzyme oxidative inactivation after I/R injury. In vivo administration of nitrite reduced myocardial infarction by 60 % in myoglobin+/+ mice, whereas similar administration of nitrite had no protective effects in myoglobin−/−mutants. These data support an emerging paradigm that myoglobin subserves a critical function as an intrinsic nitrite reductase that regulates responses to cellular hypoxia and reoxygenation.
The preceding discussion considered the relevance of deoxyMb for cardiac tissue. The same mechanism could also contribute to hypoxic vasodilation described for the human circulation (26). The oxygenation state of Mb has been studied in vivo with 1H NMR spectroscopy. The deoxy fraction of Mb in skeletal muscle of healthy humans was found to be 9 % at rest (140). Upon exercise (50–60 % of maximum work rate) the deoxyMb fraction increased to about 50% corresponding to an intracellular pO2 less than 5 Torr (141). According to our definitions of table III, this oxygen pressure represents deep hypoxia.
The exact role and the potential impact of deoxyMb as a nitrite-reductase in physiology and pathophysiology remain an important area for future studies.
C: Xanthine Oxidase (XO)
Xanthine oxidase (XO) is a ubiquitous enzyme in mammalian cells that is involved in the catabolism of purine and pyrimidines, oxidizing hypoxanthine to xanthine and xanthine to uric acid. XO also reduces oxygen to superoxide (O2•−) and hydrogen peroxide (H2O2) and is one of the key enzymes responsible for superoxide-mediated cellular injury. Interestingly, XO has structural similarity to some bacterial nitrate or nitrite reductases (142).
It has been established that XO can reduce nitrite to NO (17, 31, 142–144). It was shown that NADH (17, 142) and xanthine (144) can donate electrons to XO and catalyze the reduction of nitrite. The kinetics of the anaerobic reaction were subsequently studied with EPR spectroscopy, chemiluminescence NO analyzers, and NO electrodes (31). Each of the typical reducing substrates for xanthine, 2,3-dihydroxybenz-aldehyde (DBA), and NADH can act as electron donors to support this XO-mediated nitrite reduction. Moreover, the reaction was inhibited by oxypurinol, a specific ligand for reduced Mo4+ as in the catalytic site of XO. It suggests that reduced XO was the direct electron donor to nitrite, with nitrite binding and reduction occurring at the molybdenum site (31). Whereas NADH-stimulated NO generation was inhibited by the flavin modifier DPI, NO generation stimulated by xanthine or DBA was unaffected. Thus, whereas xanthine or DBA directly reduce the molybdenum center, NADH initially reduces the flavin, which subsequently transfers electrons to the molybdenum.
The binding constant of nitrite was found as Km = 2.4 ± 0.2 mM, and did not depend on the substrate (NADH, xanthine, or 2,3-dihydroxybenz-aldehyde). The three substrates were distinguished by markedly different binding constants: The Km = 878 μM for NADH, 1.5 μM for xanthine, 35 μM for DBA (all in the presence of 1 mM nitrite (31)). Although xanthine was the most efficient substrate for XO-catalyzed nitrite reduction, excessive xanthine inhibited the release of NO (31, 145, 146)..
Nitrite reduction to NO occurs at the molybdenum site, with either NADH or xanthine serving as reducing substrates. Diphenyleneiodonium (DPI), which acts at the FAD site, inhibited XO dependent nitrite reduction by NADH but not from xanthine. This suggests that NADH donates electrons to FAD, and then electrons are transported back to reduce the Mo that in turn reduces nitrite to NO. When xanthine or aldehydes are the electron donors, both XO reduction (by xanthine or aldehydes) and oxidation (by nitrite) takes place at the Mo site of the enzyme. This explains why only oxypurinol could inhibit XO dependent NO formation.
It has been reported that hydroxylation of the purine and aldehyde substrates takes place via a base-catalyzed mechanism and that substrate must be protonated for hydroxylation (147). The rate of XO reduction by purine and aldehydes greatly increases when the pH is increased from 6.0 to 8.0, and this increased rate of XO reduction will lead to an increased rate of nitrite reduction. However, acidification from pH 8.0 to 6.0 accelerated XO-catalyzed nitrite reduction (cf table VI). It suggests that nitrite reduction takes place via an acid-catalyzed mechanism, presumably due to nitrite protonation. HNO2 concentration increases when the pH decreases, and it could be the direct binding substrate of XO. Although lowering pH would decrease the rate of XO reduction by reducing substrates, it would greatly accelerate the oxidation of XO by nitrite/HNO2.
Table VI.
Effect of pH and substrate on the release of NO by 0.02 mg/ml XO. The nitrite concentration is fixed at 1 mM. The rates of release are in nmol·mg−1·s−1
| pH | 6.0 | 7.4 | 8.0 |
|---|---|---|---|
| Xanthine (10 μM) | 2.15 ± 0.10 | 1.87 ± 0.09 | 0.34 ± 0.03 |
| NADH (1mM) | 0.70 ± 0.05 | 0.30 ± 0.03 | 0.11 ± 0.01 |
| DBA (0.1mM) | 1.96 ± 0.10 | 0.76 ± 0.05 | 0.45 ± 0.04 |
The levels of tissue nitrite and enzyme reducing substrates have a critical role in controlling the reaction. Nitrite is the limiting substrate, given the high value of Km ~2.5 mM. This number exceeds typical tissue levels of nitrite by at least 2 orders of magnitude (cf section 2). Therefore, any enhancement of tissue nitrite by, for example, activation of constitutive or inducible NOS in inflammatory conditions, dietary sources, pharmacological sources, or bacterial sources, could all modulate this pathway of NO generation (148–155). This pathway also requires a reducing substrate, such as NADH or xanthine. Xanthine was the most effective substrate, triggering NO generation under anaerobic conditions with a Vmax 4-fold higher than that of NADH (31) and Km ~ 1.5 μM (31). Excess xanthine, above 20 μM, results in prominent inhibition. If particularly high levels of xanthine accumulate, this pathway would be inhibited, and perhaps this may serve a regulatory role to prevent overproduction of NO. Under anaerobic conditions, XO reduces nitrite to NO at the molybdenum site of the enzyme with xanthine, NADH, or aldehyde providing the necessary electrons. It makes XO an alternative source of NO under ischemic conditions when NO production from NOS is impaired.
While XO-mediated reduction of nitrite and nitrate occurs under conditions of limited tissue perfusion and resulting acidosis, questions remain regarding whether XO-mediated NO generation also occurs in the presence of oxygen. In mammalian organs under normoxic resting conditions, the O2 concentration ranges from ca 130 μM) in arterial blood to ca 50 μM in the myocardium (cf tables IV and V). During mild hypoxia, myocardial O2 levels drop below 20 μM (cf table III). Therefore, studies have been performed to measure the magnitude and kinetics of XO-mediated NO formation under different oxygen tensions (156).
All three typical reducing substrates of XO induced release of NO under hypoxia; however, their kinetics are quite different in the presence of molybdenum-site binding substrates xanthine or DBA, compared with that of the FAD-site binding substrate NADH. With xanthine or DBA as reducing substrates, the rate of NO production followed typical Michaelis-Menten kinetics, with oxygen acting as a strong competitive inhibitor. Under aerobic conditions, with xanthine or DBA as reducing substrates, XO-mediated NO production is less than the 10% of NO production under anaerobic conditions (156). With the FAD site binding reducing substrate, NADH, as electron donor, XO-mediated NO production is maintained at more than 70% of anaerobic levels. With NADH, under aerobic conditions, XO-mediated nitrite reduction did not follow Michaelis-Menten kinetics. NADH serves as electron donor to XO at the FAD site, the same site as that for oxygen binding, whereas nitrite reduction takes place at the molybdenum site of the enzyme (156). With NADH as reducing substrate, XO-mediated NO generation may occur through two processes as shown in Fig 5.
Figure 5.
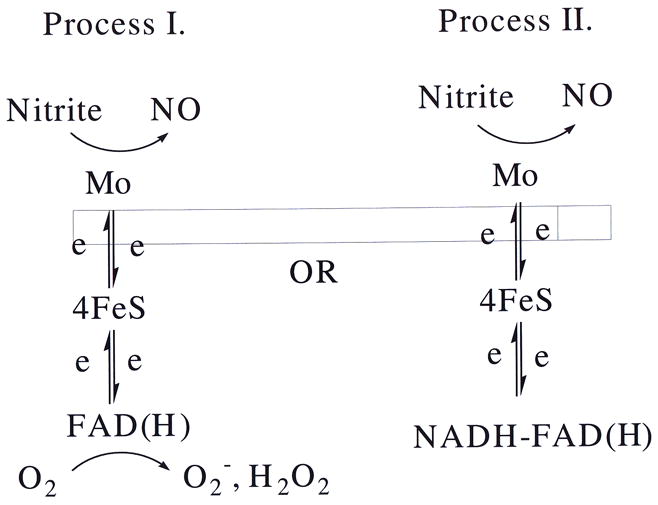
Depending on the substrate, nitrite may be reduced to free NO at the Molybdenum site of the XO enzyme. Process I is progressively inhibited by oxygen. Process II continues to operate even under normoxia.
In Process I, XO starts in reduced state. With FAD site free, XO can pass its electron to either oxygen or nitrite. Thus, under aerobic conditions, oxygen is a strong competitive inhibitor to reduction of nitrite. Therefore, Process I is inhibited by the presence of oxygen. Process II is different in that the FAD site is occupied by the NADH, and remains inaccessible to oxygen. Meanwhile, at the molybdenum site, XO-mediated nitrite reduction is unaffected. Process II should not be strongly affected by the presence of oxygen. In Process II, under aerobic conditions, less than 30% of the nitrite reductase activity of XO is inhibited, which suggests that most nitrite reduction happens while the FAD site is occupied by NADH.
NADH is necessary for many biochemical reactions within the body and is found in every living cell. Typical concentrations are 50 μg NADH/gram brain tissue (ca 75 μM), and 90 μg NADH/gram heart (ca 135 μM). With molybdenum-site binding electron donors xanthine or DBA, nitrite reduction is greatly inhibited by the presence of oxygen, whereas with NADH, XO-mediated NO generation remains at more than 70% of anaerobic levels. This makes NADH the major electron donor for XO-catalyzed NO production under aerobic conditions.
Interestingly, DPI, the inhibitor of FAD site-related function, greatly increased NO generation under aerobic conditions with xanthine or DBA as reducing substrate. It is known that oxypurinol blocks the binding of xanthine, DBA, and nitrite, whereas DPI inhibits the reduction of XO by NADH. With xanthine or DBA as reducing substrates, the presence of DPI inhibits XO-mediated oxygen reduction at the FAD side and thus increases the capability of the enzyme for nitrite reduction at the molybdenum site. Both the reduction of nitrite and the oxidation of xanthine and DBA take place on the molybdenum site of XO. The potential effects of DPI in stimulating NO generation from XO should be taken into account when DPI is used in biological systems, especially when high concentrations of nitrite are present.
Normoxic superoxide generation from XO depends on pH (147), and is maximized at alkaline conditions (pH 8 – 9). In contrast, anaerobic XO-mediated NO generation accelerates tenfold when pH values fall from 8.0 to 6.0. With lower pH, a more rapid increase of XO-mediated NO generation rate was observed under aerobic conditions than under anaerobic conditions. This would be expected, because under aerobic conditions, the acidification would significantly increase XO-mediated nitrite reduction and simultaneously decelerate the competitive reaction of oxygen reduction (147), thus facilitating NO generation under aerobic conditions.
Above, it was noted that XO may simultaneously release NO and superoxide. These radicals react rapidly to the potent oxidant peroxynitrite. The risk from peroxynitrite seems acute when nitrite levels are enhanced as, for example, under inflammation or pharmacological treatment with organic nitrates or NO-donors. However, several in vivo reaction pathways provide protection: First, most peroxynitrite should be removed by the rapid reaction with CO2 (157–159) or by ubiquitous physiological scavengers like urate or NADH (160, 161). In addition, superoxide levels are kept low by superoxide dismutase (SOD), that efficiently catalyzes the dismutation of superoxide radicals to oxygen and hydrogen peroxide. Therefore, the availability of free NO is dependent upon the local activity of SOD (162).
These results suggest that in presence of oxygen, only NADH can significantly sustain XO-catalyzed NO production. During ischemia, the myocardial NADH/NAD+ concentration ratio can increase more than 10-fold (163), xanthine levels rise to the 10–100 μM, with nitrite levels of about 10 μM (154, 155), and the low oxygen pressure and acidosis greatly facilitate XO-mediated NO generation and limit superoxide production. The magnitude of XO-mediated NO generation can approach that of the maximal NO production from NOS (31). Even with mild to moderate levels of hypoxia, as can occur with subtotal coronary lesions or regional ischemia in the presence of collateral flow, this process would be stimulated. Indeed, XO activity is up-regulated during hypoxia (164–166) with increasing acidosis (147), and with atherosclerosis. In patients with coronary artery disease, endothelium-bound XO activity is increased twofold (167).
In summary, XO-mediated NO generation may be supported by a range of reducing substrates. Interestingly, the NADH consuming reaction is not blocked by admission of oxygen. The NO release from XO is modulated by oxygen tension, pH, and the local concentrations of nitrite and reducing substrate.
Aldehyde oxidase is another molybdenum containing flavoenzyme with high sequential homology to xanthine oxidase. This enzyme is expressed in many mammalian tissues, and was recently shown to contribute significantly to the anoxic reduction of nitrite in rat tissue homogenates (168).
The preceding discussion considered the role of xanthine oxidase under hypoxia. Interestingly, it was recently shown that xanthine oxidase also contributes to a slow reduction of nitrate in normoxic tissues (32). Intraperitoneal injection of nitrate enhanced circulating nitrite levels in normoxic mice on a slow timescale of an hour. Applications of selective inhibitors implied that xanthine oxidase contributes significantly to this reaction. In accordance, pretreatment with nitrate attenuated the increase in systemic blood pressure caused by NOS inhibitors and enhanced blood flow during post-ischemic reperfusion. It suggests that mammalian xanthine oxidase mediates nitrate reduction in regulation of nitrite and NO homeostasis (32).
D: Cytochrome P450 (CYP)
Cytochrome P450 (CYP) refers to a very large superfamily of heme proteins with over 7800 different members currently known. They are found in all eukaryotes, and most prokaryotes (169). CYP’s catalyze a vast variety of different reactions, but all share the characteristic catalytic site in the form of a heme with an axial thiolate ligand derived from a nearby cysteine residue. Because of the vast variety of reactions catalyzed by CYPs, the activities and properties of the many CYPs differ in many aspects. The resting state of the protein is ferric Fe3+. For the catalytic cycle, the heme is reduced by electrons supplied by a variety of other proteins like cytochrome P450 reductase (CPR), ferredoxins, or cytochrome b5. Electron transfer from the redox partner to CYP is a key step in the CYP catalytic cycle. Bacterial and mitochondrial CYP receive electrons from a small soluble iron-sulfur protein, whereas the redox partner for mammalian microsomal CYP is a FAD/FMN-dependent NADPH-CPR (170). In CPR, FAD serves as an electron acceptor from NADPH, whereas FAD serves as an electron acceptor from NADPH (171).
The most common reaction catalyzed by CYP is that of a monooxygenase. This reaction is unselective and accepts a wide range of target substrates:
| (5) |
Denitrification was long believed to be restricted to the bacteria (172), according to the reaction
| (6) |
However, in 1989, Shoun and co-workers observed that CYP from the fungus Fusarium oxysporum was specifically induced upon exposure to nitrate and nitrite (173). This observation led to the finding of denitrifying activity in the fungus. With NADH as the direct electron donor, CYP can catalyze a chain of reduction: from nitrate to nitrite, nitrite to nitric oxide, and nitric oxide to dinitrogen oxide (N2O) (173–176)
| (7) |
Mammalian CYP’s are involved in the metabolism of many drugs and dietary substances, and in the synthesis of steroid hormones and other extracellular signaling lipids. CYP from mammalian liver can reduce nitrite as first demonstrated more than thirty years ago (177, 178). EPR spectroscopy confirmed CYP-mediated nitrite reduction by detection of paramagnetic ferrous nitrosyl-heme complexes (179). Subsequent studies detected release of free NO from rat liver CYP or human recombinant CYP (171).
Although it has been reported that the vascular biotransformation of nitroglycerin (GTN) is mediated by CYP (180–183), research (171) on GTN biotransformation indicated that rat liver microsomal CYP can not catalyze the reduction of GTN per se. Instead, rat liver CYP can serve as a nitrite reductase and generate NO radicals from NO2− (171). In the presence of NADPH (100 μM), addition of 20 μM or 40 μM nitrite triggered the release of ~ 2.8 nM.min−1 or ~ 4.5 nM.min−1 NO from microsomes (2 mg/ml). The CYP inhibitor clotrimazole (5 μM) greatly inhibited the generation of NO from nitrite. These results confirm that CYP reduces nitrite to NO in rat liver microsomes. Kinetic studies revealed that NO generation from nitrite reduction contributed less than 10% of total NO generated in the decomposition of GTN in vivo.
All studies to date of NO generation from CYP-mediated nitrite reduction have been performed under anaerobic conditions. This leaves a need to further characterize this process in the presence of small quantities of oxygen. Under aerobic conditions, molecular oxygen will be bound and split by the reduced heme iron of CYP; furthermore, many CYP substrates such as steroids, fatty acids and xenobiotics will compete with nitrite for reaction with CYP under aerobic conditions. Thus, at this time while there is an absence of published data, one would expect that CYP-mediated nitrite reduction would be inhibited by oxygen under aerobic conditions and also by CYP substrates. Future studies will be needed to characterize the precise effects of oxygen on the process of CYP-mediated nitrite reduction.
Generation of NO from nitrite by human recombinant CYP was also studied. With the presence of 100 μM NADPH, addition of 100 μM nitrite triggered an NO release of ~ 7 nM NO/min from 0.1 mg/ml CYP 2B4. NO generation from nitrite was increased with increasing nitrite concentrations or CYP concentration in the reaction mixture. Furthermore, this NO generation derived from nitrite was strongly inhibited by the heme inhibitor cyanide.
It was proposed (160) that nitrite reacts with CYP in its reduced ferrous form resulting in formation of NO-(Fe3+) heme. The nitrosyl ligand is rather weakly bound and may be released as free NO or re-trapped forming the far more stable NO-(Fe2+) heme. The proposed reaction sequence is as shown below.
| (8) |
The final step in this scheme is the reduction to ferrous state by CPR. In the presence of excess nitrite, significant quantities of free NO have been detected. This suggests that CYP-mediated nitrite reduction might be a source of NO in vivo (171).
Under physiologic conditions CPR was proposed to cycle between the 1- and 3-electron reduced states with NADPH or NADH as electron donor (184, 185). CPR prefers NADPH as electron donor and its affinity for NADPH is more than ten times higher than NADH (171). Electron transfer occurs from CPR to CYP and thus completes the catalytic cycle.
The resting state of CYP is oxidized ferric Fe3+. Under ischemic conditions most CYP would be reduced to the ferrous state with the increased NADPH in the tissues and much lower oxygen levels. This ferrous-CYP can be a source of NO in the tissues with a generation rate = [NO-Fe2+ CYP]·Koff
| (9) |
However, ferrous CYP can also bind free NO as a ligand to the heme. The first order binding rate is Kon. · [Fe2+CYP] · [NO].
| (10) |
Whether CYP acts as a sink or source of free NO depends on Kon, Koff, CYP redox-state, NO, and nitrite concentrations. The kinetics and pathophysiological roles of CYP reaction with nitrite remains unclear and needs further investigation.
In conclusion, CYP can reduce nitrite and produce NO. Although most previous studies have been performed under anaerobic conditions, the reaction would also proceed in the presence of small quantities of oxygen. Spectrophotometric studies have also shown that NO binds to ferric and ferrous CYP and can inhibit the normal catalytic cycle of CYP (186). Similarly, reaction of nitrite with CYP leads to formation of paramagnetic ferrous mononitrosyl complexes (NO-Fe2+CYP) complexes as shown by EPR spectroscopy (179). Such ferrous mononitrosyl complexes are quite stable (33, 187) and thus cause reversible inhibition of CYP. Thus, CYP can be a significant source of nitrite reduction to NO and in addition nitrite also inhibits CYP function in drug metabolism. Further studies will be required to provide a more detailed characterization of the role and importance of CYP in the process of nitrite reduction under aerobic conditions and in vivo.
E: Endothelial nitric oxide synthase (eNOS)
The endothelial isoform of NOS releases NO radicals from L-arginine with the consumption of 1.5 NADPH equivalents and two oxygen molecules per NO formed. This aerobic catalysis requires the presence of the cofactors Ca2+-Calmodulin and tetrahydrobiopterin (BH4), and is tightly regulated via a combination of mechanisms (cytosolic Ca2+-Calmodulin, (de)phosphorylation, (de)palmitoylation, and intracellular relocalization between the Golgi system and membrane calveolae within the endothelial cells (188)). Deficiency of arginine or BH4 causes “uncoupling” (189) where the oxygenase domain of eNOS releases superoxide radicals (O2•−) instead of NO (190). Oxygen deficiency is known to halt the L-arginine cycle if the oxygen levels fall below a threshold level of ca [O2] ~ 10 μM (63). However, eNOS is not wholly inactivated by the absence of oxygen. Instead, in the presence of nitrite, it switches to a novel nitrite reductase activity which releases NO (18).
The formation of free NO radicals was observed with three independent techniques carried out simultaneously in the same argon-purged optical cell: First, UV/VIS absorption of the heme group showed reduction of the heme to ferrous state by addition of NADPH, and subsequently nitrosylation of a significant fraction of the available heme. Second, electrochemical detection with an NO sensitive electrode showed the release of significant quantities of free NO radicals into the anoxic solution. Third, using EPR spectroscopy and iron-dithiocarbamate complexes as NO traps (33, 187, 191), significant quantities of gaseous NO were detected in the purging argon flow leaving the reaction vessel. The ferrous mononitrosyl iron-dithiocarbamate complex NO-Fe2+-(DETC)2 is paramagnetic (192, 193) and the shape of its EPR spectrum is sensitive to isotopic labeling of the nitrogen with the stable 15N isotope (cf chapter 18 of (33)). In this way it was proven that the observed 15NO was released from the 15N-nitrite anions and not from the reaction of 14N-arginine with residual traces of oxygen (18). In absence of eNOS, no NO was detected, and the pH was stabilized at neutral 7.4 so that the release of NO from acidic reduction of nitrite could be ruled out.
The anoxic reaction was initiated by administration of NADPH into the buffered solution of eNOS repleted with necessary cofactors and nitrite. The new reaction has some similarities to the aerobic arginine pathway in that it also requires electron injection from NADPH to the flavin domain of the enzyme, and the presence of Ca2+/calmodulin to afford intramolecular electron transport towards the heme (190). Other aspects are very distinct, however: the reduction of nitrite is slowed but not halted by the removal of BH4 and it consumes neither arginine nor oxygen. Comparison of purified WT eNOS and its isolated oxygenase domain shows that nitrite reduction is achieved at the oxygenase domain of the protein (18). Upon readmission of oxygen, eNOS reverts to the normal L-arginine pathway, and the enzyme appeared fully functional after several such argon/oxygen cycles. Such repeated regeneration of the nitrite reductase capacity by cyclic admission of oxygen provides eNOS with an “emergency” NO release in acute anoxia..
The effect of anoxia was studied further in cultured BEND3 cells (19). These immortalized murine brain microvascular endothelial cells express only the endothelial isoform of NOS (194). These cells were grown to confluence corresponding to ca 7.5 ± 0.5 × 106 cells in a monolayer. Confluence and cell count are important parameters for eNOS activity (188) and were verified by inspection with a stereomicroscope. The NO production in these cell cultures was studied using NO trapping with iron-dithiocarbamate complexes. In this accumulating method, the yield of paramagnetic NO-Fe2+-DETC adducts (MNIC) increases with time and is determined with EPR spectroscopy after termination of the experiment. The yields from trapping for 20 min are collected in table VII. The basal (i.e. unstimulated) NO production was measured by trapping for 20 min under the controlled atmosphere with 5 % CO2 and 20 % O2. From table IV, the corresponding free oxygen concentration in the medium was ca 200 μM. This value is normally used for experiments on cultured cells but corresponds to hyperoxia in our classification of table III.
Table VII.
Yields of MNIC adducts (in pmol) in the cellular fractions of a single 75 cm2 flask of cultured confluent endothelial cells. Trapping proceeded for 20 min at 37° C. The second row gives the preincubation times τinc of the supplements. During preincubation the cells were kept at 37° C in an atmosphere with 5% CO2 and 20% O2. (adapted from(19), with permission)
| Basal unstimulated | CaI (5 μM) | NLA (57 μM) | L-NAME (50 μM) | Imidazole (10 mM) | Oxypurinol (100 μM) | NaNO2 (250 μM) | |
|---|---|---|---|---|---|---|---|
| τinc | -- | -- | 20′ | 2′ | 1′ | 20′ | 20′ |
| In presence of oxygen | 110±8 | 400±30 | † | 51± 8 | 85± 10 | n.d. | 116±8 |
| 20 min of anoxia | 160± 10 | n.d. | † | 61±8 | 80±10 | 170± 10 | 154±8 |
below detection limit of ca 10 pmole.
As expected, the basal yield of ca 110 pmol MNIC per flask may be cancelled by coincubation with NOS inhibitors like L-arginine-methyl-ester (L-NAME) or Nω-nitro-L-arginine (NLA), thereby proving that the NO results from enzymatic activity of eNOS. Upon stimulation with the calcium ionophore A23187, the yield was enhanced nearly fourfold to 400 pmol MNIC. These cell cultures were then subjected to acute anoxia by replacing the culture medium with deoxygenated medium and flushing the flasks with argon before closing the top. The imposition of anoxia reduced the oxygen concentration of the medium to a value below the detection limit of ca 2 μM of a Clark electrode. This limit is comfortably lower than the oxygen threshold of the L-arginine pathway of eNOS cf table IV). Although the L-arginine pathway was halted, significant quantities of NO were being released from these anoxic cell cultures. The anoxic yield of ca 160 pmol MNIC was significantly higher than the basal yield of 110 pmol in presence of oxygen. Therefore, deprivation of oxygen actually enhances the release of NO from this endothelial cell line. However, the anoxic NO yields remain in the normoxic regulatory range between basal and fully stimulated yield (110 resp 400 pmol MNIC). Fig 6 shows the kinetics of the anoxic reaction as a function of time. After imposition of anoxia, the MNIC yield increased roughly proportional with time for twenty minutes, and levelled off at an asymptotic value of ca 200 pmol MNIC per flask. The samples could be kept at 37°C for over an hour without significant loss of MNIC adducts. Trypan blue staining did not show enhanced mortality of cells after exposure to anoxia up to 30 min.
Figure 6.

Kinetics of NO formation in a single flask of cultured BEND3 cells under anoxia and normoxia. The MNIC yields are given as function of incubation time of the trapping experiment. The anoxic values (solid squares) were taken from (19) with permission. The accuracy of the MNIC yield is ca 10 %.
When considering these yields, five potential sources of the NO should be considered: intracellular nitrite, extracellular nitrite, nitrate, arginine or endogenous S-nitrosothiols. Extracellular nitrite could be ruled out since the yields were unaffected by addition of 250 μM nitrite to the medium. Arginine is ruled out as a substrate since its oxidation requires oxygen. Although some residual oxygen may still be found after imposition of anoxia, it is inconceivable that the arginine pathway could remain functional at greatly increased rate for over twenty minutes. Hypoxic reduction of nitrate to nitrite and subsequently to NO by xanthine oxidase enzymes (17, 31, 142–144) could be ruled out because the yields were unaffected by addition of the inhibitor oxypurinol. These results show that in this particular model the xanthine oxidase enzyme does not significantly contribute to the observed NO release. S-nitrosothiols were also ruled out because the anoxic NO production could be inhibited with NOS inhibitors L-NAME and LNA. This leaves intracellular nitrite as the probable source of NO. The intracellular nitrite levels were estimated by the Griess colorimetric assay on cell lysates. The data confirmed that the intracellular nitrite levels rapidly fell after imposition of anoxia from an initial concentration of ca 20 μM to below the detection threshold of ca 6 μM and suggests that depletion of intracellular nitrite causes the cessation of NO release after ca 30 min (cf Fig 6). Significantly, the magnitude of anoxic NO release was intermediate between the basal and fully stimulated yields and remained in the benign range of normoxic physiological regulation, far below the cytotoxic levels found in septic shock or allograft rejection.
It was recently found that eNOS is the only NOS isoform capable of reducing nitrite and releasing free NO radicals (18, 195). This gives eNOS a special significance when considering the effects of ischemia on various tissues. Anoxic NO release from reduction of nitrite by eNOS may be a plausible explanation for the plethora of animal and clinical studies showing a protective role of eNOS in the early stages of ischemia (196, 197).
F: Mitochondrial respiratory chain
Several years ago, mitochondrial cell fractions were found to release NO radicals from nitrite (179, 198). The activity involved two distinct components of the mitochondrial respiratory chain, namely complex III (198) and cytochtome c oxidase (complex IV) (179). Published literature shows that the results obtained for complexes III and IV were carried out at different ranges of external nitrite concentrations. At [NO2−] < 50 μM, complex III reduced nitrite. At [NO2−] > 300 μM, cytochrome oxidase was found to be the main source of nitrite reductase activity in the mitochondria (21, 179). A critical question is how nitrite enters the cells and gains access to mitochondria since the NO2− anion carries charge and should not diffuse freely through membranes. It was recently pointed out that a small fraction of nitrite anions is protonated even at physiological pH, and may cross model lipid membranes as neutral molecule HNO2 (199). The situation is less clear for actual physiological membranes. Castello and coauthors have shown that only some 10% of nitrite added to isolated yeast mitochondria is internalized into the mitochondrial matrix (21). Thus, equilibrium concentrations of nitrite available for mitochondrial nitrite reductase activity may be significantly lower than cytoplasmic nitrite concentrations. This may explain that nitrite transport to mitochondrial nitrite reductase seems facilitated by low pH.
The sites where nitrite reduction occurs were identified by applying selective inhibitors of the respiratory chain (198). Rotenone inhibits the electron transfer from complex I to the Q-cycle. Application of rotenone to mitochondrial fractions inhibited NO generation from nitrite by 60%. Thenoyltrifluoroacetone (TTFA) is an inhibitor of complex II. In the presence of succinate, the substrate of complex II, TTFA inhibited NO formation by 40%. In this experiment, reversed electron flow to complex I was suppressed by the presence of rotenone (198). Myxothiazol is an inhibitor of electron transfer from the Q-cycle to complex III. Irrespective of the substrate supplying reducing equivalents to the respiratory chain, myxothiazol completely inhibited release of NO. In contrast, antimycin A, an inhibitor of electron transfer at the oxidant site of the bc1 complex, did not influence the formation of NO. Thus it was determined that the site of reduction is localized between myxothiazol sensitive and antimycine A sensitive components of the respiratory chain (Fig 7). This part of the respiratory chain includes cytochromes b562, b566, and the ubisemiquinone-radical bound to complex III (Fig 7). Myxothiazol is highly specific for mitochondria and completely inhibits nitrite reduction. Therefore, it can be used to estimate the contribution of complex III to the total nitrite reductase activity in different organs. A recent study (86) confirmed inhibition of nitrite reduction by myxothiazol. The inhibition was complete in heart and intestinal homogenates, but only partial in liver homogenates. It shows that liver tissue contains an additional source of nitrite reductase activity. After separating the various fractions of liver tissue, significant nitrite reductase activity was found in the microsomal fractions as well (200). Within these microsomal fractions, the nitrite reductase activity was attributed to cytochrome P450 enzymes in the endoplasmatic reticulum (cf section 7.D.).
Figure 7.

Possible sites of nitrite reduction in mitochondria at complexes III and IV of the respiratory chain. Abbreviations: R - Rotenone, M - Myxothiazol, A - Antimycin A, T - Thenoyltrifluoroacetone, CN – Cyanide, UQ – Ubiquinone, UQH2 – ubiquinol, Q•−- semiquinone radical anion, Cyt – Cytochrome.
It has been shown that at millimolar concentrations nitrite reacts with the oxygen binding site of cytochrome c oxidase (complex IV) rather than with the cytochrome c binding site of the enzyme (201). The NO is a product of this reaction. It requires the presence of the substrates of both complexes I and II and is inhibited by antimycin A, myxothiazol, KCN, and carbon monoxide (21). It has been suggested that Cytochromes aa3 (complex IV) catalyse the reduction of NO2− to NO (21, 179) under anaerobic conditions. Under normal conditions this site reduces oxygen to water.
Under normal physiological conditions, the mitochondrial complex III releases a significant quantity of superoxide radicals. It is known that NO reacts very rapidly with superoxide to form peroxynitrite (ONOO−). It seems that NO and superoxide are generated at different sites of complex III. The release of superoxide radicals is dramatically increased by antimycin A (202, 203), but antimycin A does not influence the reduction of NO2− to NO (198). One can expect that nitrite reductase activity of complex III is deleterious yielding peroxynitrite if NO and superoxide are generated simultaneously in the same segment of respiratory chain. However, significant release of NO occurs only under hypoxic conditions and the generation of superoxide requires oxygen. In addition, it has been shown that nitrite can also modulate the mitochondrial respiratory chain during anoxia by S-nitrosation of complex I. This modification decreases its activity and attenuates the release of reactive oxygen species from the mitochondrial chain (100). Therefore it is unlikely that nitrite reductase activity significantly contributes to the formation of peroxynitrite. However, this point has not yet been addressed in detail.
It is important to consider the levels of oxygen and pH values in tissues. The physiological oxygen levels in resting tissues remain slightly below those of venous blood (cf tables IV and V). At these oxygen levels, the release of NO from nitrite must be very small or even zero as the quantity of nitrosyl-hemoglobin (NO-Fe2+Hb) remains below the detection threshold of EPR (204). The effect of pH was studied in homogenates prepared from rat intestine. At low pH values the nitrite reductase activity of intestinal homogenate was elevated due a slight increase in mitochondrial nitrite reductase activity and a far stronger increase in the rate of nitrite reduction by low molecular weight reducing agents (86). This is in line with publications confirming that acidic reduction of nitrite become more significant at low pH values (16, 199).
The NO released by the mitochondria cannot be distinguished from that provided by other sources in the tissue. Therefore, the mitochondrial nitrite reductase activity is difficult to separate from other mechanisms such as deoxyHb or deoxyMb (see above). To avoid direct contact with blood the nitrite was administered into intestinal lumen (86). Nitrite infusion in intestinal lumen results in the formation of NO-Fe2+Hb complexes in blood and a simultaneous drop in blood pressure at a threshold concentration of 10 μM of NO-Fe2+Hb complexes (86). This mechanism is expected to operate independently of the route of nitrite administration. Recently, it has also been suggested that the mechanism for nitrite-induced vasorelaxation is largely intrinsic to the vessel and that under hypoxia physiological nitrite concentrations are sufficient to induce NO-mediated vasodilation (205). These data suggest the existence of additional mechanisms releasing NO from nitrite, possibly mediated by eNOS (see section 7.E.).
Once released from the mitochondrial respiratory chain, NO can act in a variety of ways: NO and its metabolites like peroxynitrite (206) are known to inhibit mitochondrial respiration via binding to the enzymes of the respiratory chain (207, 208). Protracted inhibition of the respiratory chain by NO has been shown to trigger apoptosis and, ultimately, cell death in rat thymocytes (209). Since mitochondria reduce nitrite to NO one can expect that nitrite should inhibit mitochondrial respiration as well. It has long been known that well-oxygenated mitochondrial enzymes are inhibited by very high nitrite concentrations exceeding 0.3 mM (210). In contrast, at low oxygen pressures, mitochondrial respiration is inhibited by far lower concentrations of nitrite (211). This observation once again confirms that hypoxia significantly promotes the release of NO from nitrite in biological systems.
8: INORGANIC NITRATE AS A SOURCE OF NO
As stated in the section 2, humans have plasma nitrite in the range of 50–500 nM which makes it a major circulating NO storage pool (42–44). A potentially even larger pool would be nitrate (plasma levels 20–40 μM) provided that this anion could first be reduced to nitrite. However, it was generally believed that nitrate is not metabolized in our bodies and consequently this anion was thought to be biologically inert. This long-held dogma was recently challenged by findings as discussed in more detail below. Via our diet we are continuously exposed to considerable amounts of nitrate and many vegetables are particularily rich in this anion (30). After absorption in the gut, dietary nitrate mixes with nitrate from endogenous sources (oxidation of NO generated by NO synthases) and is actively taken up by the salivary glands and concentrated in saliva. Salivary nitrate levels are 10–20 fold higher than those in plasma. Commensal bacteria in the oral cavity then reduce nitrate to nitrite and when swallowed this nitrite can form NO and other reactive nitrogen oxides locally in the gastric lumen as discussed above (12, 212).
In this review numerous pathways for systemic reduction of nitrite to bioactive NO were described. This gives high relevance to the question whether some of the nitrite generated locally in the gut can survive and reach the systemic circulation. Indeed, it was recently shown that plasma nitrite increases greatly after ingestion of inorganic nitrate and this increase was due to enterosalivary reduction of nitrate (43) (fig 8).
Figure 8.

Many vegetables including spinach, lettuce and beetroot are extremely rich in inorganic nitrate. Ingested nitrate from dietary sources is rapidly absorbed in the small intestine and in the circulation it mixes with endogenous nitrate from the NO pathway. While much of the circulating nitrate is eventually excreted in urine, up to 25% is actively extracted by the salivary glands and concentrated in saliva. In the mouth commensal anaerobic bacteria effectively reduce nitrate to nitrite by the action of nitrate reductase enzymes. Until recently this entero-salivary circulation of nitrate and bacterial reduction to nitrite has received attention only because of nitrites ability to form potentially carcinogenic nitrosamines. However, the link between dietary nitrate and gastric cancer is still very uncertain despite more than 40 years of active research. More recently it was shown that in the acidic stomach nitrite is spontaneously decomposed to form nitric oxide and other bioactive nitrogen oxides which serve to regulate important physiological functions. Gastric NO helps to kill ingested pathogens and also protects the gastric mucosa against luminal aggressors via stimulation of mucosal blood flow and mucus generation. Moreover, much nitrite is also absorbed intact into the ciculation and can convert back to bioactive NO in blood and tissues. This enterosalivary nitrate circulation and serial reduction to nitrite and NO explains the recently described reduction in blood pressure seen after dietary supplementation with nitrate. It has been suggested (216) that the well-known cardioprotective effects of a diet rich in vegetables is related to their high nitrate content.
This finding implied that dietary nitrate could form bioactive NO systemically with concomitant effects such as vasodilation. Larsen and colleagues recently tested this idea in healthy young volunteers (213). In a double-blind placebo-controlled crossover evaluation of dietary nitrate supplementation, they found an increase in plasma nitrite from 138 to 219 nM after intake of nitrate. Significantly, there was a concomitant drop in resting blood pressure, an effect consistent with formation of vasodilatory NO. The nitrate dose corresponded to the amount found in 150–250 g of a nitrate-rich vegetable such as spinach, lettuce or beetroot. Remarkably, the reduction in blood pressure was similar to that described in healthy controls consuming a diet rich in fruits and vegetables in the well-known Dietary Approaches to Stop Hypertension (DASH) trial (214). Interestingly, ingestion of beetroot juice led to a marked decrease in blood pressure in healthy individuals (215). The effect on blood pressure correlated with peak levels of plasma nitrite and was abolished by spitting, demonstrating the importance of the enterosalivary conversion of nitrate to nitrite to achieve the observed effects (215). Several new pre-clinical studies now also indicate that dietary levels of nitrite and nitrate provide significant protection against cardiac ischemia-reperfusion injury (24, 217) and gastric injury (218–220), respectively.
The effects of dietary nitrate seem to extend beyond vasoregulation. Larsen et al showed recently that nitrate reduces the oxygen cost during submaximal exercise in healthy subjects (213). That is, for a given work load, the amount of oxygen required was less if the person had added nitrate to his nitrate-restricted diet for three days prior to the test. This highly surprising effect occurred without an accompanying increase in lactate concentration, indicating that the energy production had become more efficient. Although the exact mechanism for this effect remains to be elucidated, a likely initial step is the formation of nitrite. Indeed, circulating nitrite levels had increased after the dietary nitrate intervention and this nitrite was consumed during the exercise. As discussed in section 7.B., nitrite can interact with deoxymyoglobin to form NO which in turn is known to interact with mitochondrial respiratory chain enzymes (24, 221–223). In the Larsen study it was speculated that such interactions could lead to less proton leakage over the inner mitochondrial membrane (reduced mitochondrial uncoupling) which would leave more protons available for ATP production.
The above has shown that nitrate cannot be considered as stable and inert when administered through the digestive tract. This should be kept in mind in preclinical studies on the therapeutic effects of nitrite, where the nitrate anion is often assumed to be an inert control substance. In addition, standard animal chow typically contains considerable amounts of nitrate; a fact that is normally not considered in the study designs. It was recently shown that tissue levels of nitrite were markedly reduced when standard rodent chow was changed to a special diet low in nitrate/nitrite (107). Interestingly, this dietary change had effects on NO related signaling pathways including guanylate cyclase activity and gene expression.
As stated above, oral bacteria play a central role in bioactivation of dietary nitrate by effectively reducing salivary nitrate to nitrite. A recent study by Jansson and colleagues now surprisingly shows that inorganic nitrate can be reduced to nitrite also by mammalian enzymes (32). Administration of nitrate to germ-free mice resulted in marked increases in circulating nitrite levels. Importantly, these effects were seen under normoxic conditions. Experiments with various pharmacological inhibitors suggest that xanthine oxidase catalyses normoxic nitrate reduction but the presence of other pathways was also indicated. Nitrate pretreatment attenuated the increase in systemic blood pressure caused by NO synthase inhibition and enhanced blood flow during post-ischemic reperfusion, suggesting a role for mammalian nitrate reduction in regulation of nitrite and NO homeostasis.
With all the new information on nitrate described here it is strongly advised that nitrate should not been used as a negative control when studying the biological and therapeutic role of nitrite. This is especially important when using higher doses and in long-term experiments in awake animals when the enterosalivary circulation of nitrate comes into play.
Taken together, as illustrated in the many examples above, dietary nitrate is bioactivated in vivo to form nitrite and then likely NO. In this context nitrate may be considered a “prodrug”, which produces a sustained delivery of nitrite to the systemic circulation following enterosalivary circulation (the t1/2 for plasma nitrate is 5–6 h). The possibility of fueling NOS-independent NO production by dietary interventions may have important implications for public health, in particular cardiovascular disease. The central role of commensal bacteria in bioactivation of nitrate is intriguing and suggests a symbiotic host-microbial relationship involved in regulation of cardiovascular function. Future clinical studies will elucidate if nitrate can offer a nutritional approach to prevention and treatment of disease.
9: OTHER IN-VIVO REACTIONS OF NITRITE
A: The reaction with oxy-hemoglobin (oxyHb)
While the reactions of nitrite with deoxyHb have received significant recent attention, most previous work was focused on the reaction of nitrite with oxyHb as a potential consequence of nitrite poisoning (224–227). This work has recently been reviewed (228). It is important, given the renewed focus on nitrite/hemoglobin biochemistry, that this reaction is not forgotten and is placed into its correct context. Nitrite reacts with oxyHb to generate metHb (ferric hemoglobin Fe3+-Hb) and nitrate (NO3−) as the only discernable end products. However, the reaction kinetics reveal that the reaction is far more complex than just a simple bimolecular reaction with a single rate-limiting step. If nitrite is in excess over hemoglobin, the reaction follows an autocatalytic time course. In other words, the reaction speeds up as a function of time. This type of reaction is usually indicative of a free-radical chain reaction which contains a branching step. While there have been several mechanisms postulated for this reaction, the reaction sequence below appears to conform to most experimental data (229–231).
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
| (16) |
In these equations HX denotes the amino acid residue on the hemoglobin that participates in the formation of the ferryl Hb radical.
Using free radical reaction terminology, this process can be divided into initiation, propagation and termination steps. Equations 11 and 12 represent the initiation process, which consists of the slow generation of hydrogen peroxide and metHb, which then react to form the ferryl-radical hemoglobin, thus initiating a new chain reaction. The chain reaction is described by Equations 13 to 16. The first two steps of the chain reaction consist of two sequential oxidations of ferryl radical hemoglobin (′X −[Hb(FeIVO)]2+) and ferryl hemoglobin (HX −[Hb(FeIVO)]2+), respectively, by nitrite, each generating a nitrogen dioxide radical. It is interesting to note that Equations 13 to 15 represent a typical peroxidase cycle, and peroxidases such as myeloperoxidase and horseradish peroxidase have been shown to catalyze similar chemistry. Equation 16 is the last step in the chain reaction. It encompasses the chain-branching step that is required for autocatalysis. In this step, the nitrogen dioxide, generated in Equations 13 and 14 reacts with oxyHb to generate ferryl radical Hb and nitrate. Consequently, each nitrogen dioxide can initiate a new chain of oxidation, and each ‘link’ of the chain will generate two nitrogen dioxides. This explains why the reaction will continuously accelerate until all oxyHb is consumed unless nitrogen dioxide is consumed by alternative pathways. Equation 16 represents one such termination reaction where the nitrogen dioxide dimerizes into N2O4. Another pathway is the rapid reaction with NOto N2O3. These reactions represent real sinks for NO2since N2O4 rapidly hydrolyzes to nitrate and nitrite and N2O3 is also quite reactive as a nitrosating species (these reactions and their rate constants are reviewed in detail elsewhere (cf chapter 1 of (33)). Other termination pathways can be envisioned that include the reaction of nitrogen dioxide with protein residues to form a protein radical which can then react with a second nitrogen dioxide. Similar mechanisms have been invoked for the formation of 3-nitrotyrosine during peroxidase-mediated oxidation of nitrite (232).
This mechanism described above is based on recent kinetic simulation of experimental data (229) and is different from previously published mechanisms (230, 231) in two important ways. First, Equation 11 does not generate nitrogen dioxide but only hydrogen peroxide, and second, Equation 15 does not require the intermediacy of hydrogen peroxide. These changes completely uncouple the roles of nitrogen dioxide and hydrogen peroxide in the reaction scheme. Hydrogen peroxide now acts only as an initiating species and nitrogen dioxide is only a propagating radical. Without such uncoupling of roles, kinetic models failed to quantitatively describe the experimental data.
Direct evidence for the involvement of hydrogen peroxide derives from the inhibitory effects of catalase (110). Interestingly, catalase will inhibit the reaction only when added before significant acceleration occurs. In contrast, catalase is ineffective when added during the rapid propagation phase (229). This indicates that hydrogen peroxide is only important for the initiation of chain propagation and not for propagation itself. The formation of ferryl and ferryl radical hemoglobin intermediates has been confirmed by spectrophotometry and EPR spectroscopy respectively (114, 230, 233). The unambiguous identification of nitrogen dioxide as an intermediate in this reaction is always a significant experimental challenge due both to its inherent reactivity and instability. Moreover, there are no specific scavengers of this free radical. Although NO2has long been invoked as an intermediate (and it is difficult to envisage a mechanism that does not involve NO2•), evidence for its involvement is sparse. Using immuno spin-trapping, a technique in which protein radicals are trapped by spin-trapping agents and subsequently detected by immunological techniques, it was shown (233) that the reaction between nitrite and oxyHb (or oxyMb) results in the formation of protein radicals. The protein radicals occurred on both the heme protein and on a secondary bystander protein (in this case bovine serum albumin was used). This differs significantly from the reaction between hydrogen peroxide and metHb, where no radical formation was detected on bystander proteins (233). These data suggest that the reaction with nitrite involves an oxidizing intermediate that can damage a secondary protein target. It was also shown (229) that C-PTIO can inhibit the propagation reaction. Although C-PTIO is widely employed as a nitric oxide scavenger (234), it has been shown to be a very efficient scavenger of NO2as well (235) and is most likely functioning through this mechanism.
Allosteric effectors of oxygen binding exhibit interesting effects on the kinetics of the reaction between nitrite and oxyHb. For example, inositol hexaphosphate lowers the oxygen affinity and shifts the oxygen-binding curve to higher oxygen pressures. Administration of inositol slows nitrite-mediated oxyHb oxidation (236) whereas blockade of the β-93 cystiene residue counteracts this inhibition (237). This effect has parallels in the effects of T-state and R-state stabilizers on the reaction of nitrite with deoxyHb (111).
The above considered the effects of nitrite on Hb in the presence of the high levels of oxygen found in the atmosphere. However, this review is mainly devoted to the effects of nitrite under hypoxic or even completely anoxic conditions (cf introduction). When Hb is partially saturated with oxygen, nitrite reacts preferentially with the deoxygenated hemes to form metHb and NO, which will then bind to a vacant ferrous heme to form NO-Fe2+Hb, or react with oxyHb to form metHb and nitrate. Surprisingly, deoxyHb is consumed more rapidly than oxyHb (114) in this reaction. If it is assumed that oxygen is in rapid equilibrium with its heme binding sites, and that the fraction of oxygen-bound hemes (i.e. oxyHb/(oxyHb+deoxyHb)) is a reflection of oxygen affinity at each time point, then the more rapid oxidation of deoxyHb must mean that the oxygen affinity of the hemoglobin is increasing during the course of the reaction. This is likely due to the formation of metHb and NO-Fe2+Hb, both of which can increase the oxygen affinity of associated ferrous subunits within a tetramer (112). The reaction exhibits autocatalytic kinetics only after the deoxyHb has been fully consumed. This suggests that small quantities of deoxyHb and nitrosyl-Hb inhibit the propagation of radical reactions (114). Interestingly, lowering oxygen tension slows the progression of the reaction between nitrite and oxyHb, even in the absence of an observable increase in deoxyHb (229). Once the reaction enters the propagation phase, oxidants are generated (presumably hydrogen peroxide and NO2•) that are able to liberate NO that is bound to ferrous hemes (114). While the physiological relevance of this observation is unclear, it demonstrates that hemoglobin has the intrinsic capacity to both generate NO-Fe2+-Hb and release NO during its reactions with nitrite.
An understanding of the kinetic rate of this reaction allows a very rough quantification of the importance of this reaction in vivo at both basal and supplemented nitrite levels. Kinetic analysis has determined that the rate limiting step of Equation 11 has a rate constant of the order of 0.5 to 1 (Ms)−1. This is comparable to the rate constant for the reaction between nitrite and T-state deoxyHb, and about 50 times slower than the reaction between nitrite and R-state deoxyHb. Since whole blood has an average [Hb] ~ 8 mM, the above reaction rate of 1 (Ms)−1 predicts that nitrite in fully oxygenated whole blood has a half life of ca 125 s. This lifetime will be further shortened by additional reaction pathways for removal of nitrite. The experimental half-life of nitrite in the human circulation is ca 1 – 2 min (11). These numbers show that the reaction with oxyHb may be significant in vivo (note that the half life depends on experimental details: for whole blood drawn in a test tube at room temperature, the half-life of nitrite was reported ca 10 min (44)). The products of reaction 11 are easily handled within the red cell. Hydrogen peroxide is consumed by both catalase and glutathione peroxidase and so the initiation of the radical chain reaction will be greatly suppressed. In addition, metHb is reduced back to the ferrous (Fe2+-Hb) form by methemoglobin reductase. Unless the rate of hydrogen peroxide formation from equation 11 exceeds the ability of the red cell to consume it, then the reaction will proceed at the rate limit of equation 11 and will never become autocatalytic. In addition, even if a fraction of hydrogen peroxide escapes destruction and participates in reaction 12, small molecule antioxidants such as glutathione, ascorbate and urate will inhibit the radical propagation steps by reducing protein radicals and ferryl species. Consequently, except for severe nitrite poisoning, the reaction rate of nitrite with oxyHb will likely be limited by reaction 11.
B: Formation of dinitrosyl-iron complexes
Exposure of normoxic cultured cells or mammalian tissues to high levels of NO usually leads to formation of significant quantities of dinitrosyl iron complexes (DNIC). These paramagnetic complexes form a class characterized by a dinitrosyl-iron structure (NO+)2-Fe+-(L−)2. Here L− denotes a thiolate or other anionic ligand. DNIC with thiol-containing ligands were first discovered in cells and tissues during the 1960’s by EPR spectroscopy. The details and symmetry (axial or rhombic) of the EPR signal depend slightly on ligand type, but the g-factors remain in the range g⊥~2,040. g| |~2,014. gav= 2.03 (so-called 2.03 signal). The spectroscopic properties of these paramagnetic DNIC (S=½) have been extensively reviewed in chapter 2 of (33).
Several studies in mouse and rat models have shown that prolonged dietary administration of nitrite also leads to the formation of such endogenous DNIC. For example, after 3–5 days of addition of 0.3% sodium nitrite to drinking diets significant amounts of DNIC could be detected in mouse tissue, liver in particular (238, 239). After 5 days, the content of DNIC in liver tissue constituted ~10 nmoles per 1 g of wet tissue (238). In other organs, e.g., heart, lungs and spleen, DNIC remained below the EPR detection limit of ca 100 pmol/gram. In the kidney, DNIC content was 5 times lower than in the liver. The appearance of a significant quantity of DNIC implies the enhancement of free NO. The formation of DNIC correlates well with independent reports of enhanced nitrosylation of blood hemoglobin after oral administration of nitrite to rats (85). In addition, DNIC is an effective NO donor when injected into animals and participates in a complicated chemical equilibrium (240) with S-nitrosothiols, thiols, and loosely bound iron from the labile iron pool (cf chapter 11 of (33)). Of note, the observed formation of DNIC does not require the imposition of hypoxia. The complexes are formed in normoxic conditions, where the animals breathe ambient air. However, the administration of dietary nitrite could lead to formation of some metHb in blood and diminish the capacity for vascular oxygen transport (cf section 9.A). The extent of metHb was not quantified in these experiments.
The quantity of endogenous labile iron seems a limiting factor in the formation of DNIC: The yields of DNIC could be enhanced threefold by co-supplementation of 0.02% iron citrate complexes (iron:citrate = 1:5) together with the nitrite in the drinking water (238, 239). The nature of the iron at the center of these complexes was investigated by isotopic substitution of 56Fe for its isotope 57Fe in the nitrite-rich drinking diet (238). Such substitution did not influence the quantity of DNIC in liver tissues, but caused significant linebroadening of the EPR signal, particularly, in the g⊥~2,040 component. The broadening showed that the DNIC complexes contained the 57Fe isotope. Subsequent treatment of the extracted tissues with gaseous NO caused a threefold increase in the DNIC content (as in the case of mice kept on a “water + nitrite + 56Fe diet). The EPR linewidth of the 57Fe-DNIC signal was significantly decreased towards that characteristic of the 56Fe- DNIC. These results show that exogenous iron, when co-administered with nitrite via drinking water, dominates the formation of DNIC formation in vivo. Significant quantities of DNIC with endogenous (intracellular) iron are formed only upon exposure of tissue biopts or homogenates to gaseous NO.
The sources of this endogenous iron remain hotly debated by practitioners in the field. It seems to involve loosely bound iron from the so called labile iron pool (LIP). Many types of intracellular iron sulfur clusters (ISC) are known to form DNIC when disrupted by free NO radicals (chapter 5 of (33)). The data show that iron from drinking water is a preferred source for DNIC formation in vivo. It suggests that this process does not involve penetration of free NO into liver cells and subsequent binding to intracellular free 56Fe. More likely, dinitrosyl (NO)2-57Fe moieties are formed in the extracellular compartment. At early stages, these DNIC are probably low-molecular weight and involve anionic ligands of low molecular weight. Such small DNIC are quite mobile (as evidenced by the Redfield narrowing of the EPR spectrum at room temperature (239)) and may be capable of entering the intracellular compartment in intact form. The uptake of dinitrosyl moieties into the interior of the cells is evidenced by the subsequent formation of protein-bound 57Fe-DNIC localized in the intracellular compartment. Due to the slow exchange with thiols on proteins, a population of protein-bound DNIC with high molecular weight is subsequently formed. Such exchange between DNIC populations of low and high molecular weight has been extensively studied with EPR spectroscopy (239, chapter 2 of (33)).
This hypothetical mechanism of DNIC formation in animal tissues is consistent with the observations when mice, rats or rabbits are injected with solutions of preformed low-molecular DNIC with thiol and non-thiol ligands (33, 239).
We conclude with the question why DNICs are observed in tissues only when levels of nitrite or NO are artificially enhanced. The release of some NO from nitrite may be caused by protonation in local acid environments (241) (e.g. in the digestive tract, see preceding section), or by several enzymatic pathways under hypoxic conditions. Such pathways were discussed above. In our opinion, a small pool of DNIC is always present in systems containing labile iron, thiols, S-nitrosothiols and NO. After all, these compounds are always connected via the reaction equilibria described in (240). Tissues do satisfy this requirement, but the DNIC levels probably remain below the detection limit of EPR spectroscopy (ca 100 pmol DNIC/g tissue) under normal physiological conditions. It remains unclear at this stage whether such a small pool of DNIC have physiological relevance in mammals. Administration of DNIC to cultured Reuber H35 hepatoma cells induced significant expression of a range of heat shock proteins (242). The role of DNIC is firmly established by now as vasodilator (33), as effective mediator for S-nitrosation of thiols and active participant in the reaction equilibria of thiols (240). Whether endogenous DNIC can also act as a direct effector per se (for example as NO donor or modulator of gene expression or apoptosis) remains to be seen.
C. Binding to human estrogen receptor α(ERα)
It was recently reported (106) that nitrite anions act similar to estradiol on the growth and gene expression of cultured human MCF-7 breast cancer cells. The administration of low (1 μM) nitrite dosages promoted cell growth, induction of mRNA of progesterone receptors and the pS2 promoter. Nitrite also promoted recruitment of the exogenously expressed ERα to the pS2 promoter. These effects are very similar to those observed by administration of the estradiol hormone. These findings may have significance for the etiology of breast cancer, where estrogens like estradiol are known to play an important role. A similar effect of nitrate could be inhibited by the oxidoreductase inhibitor diphenyleneiodonium (DPI). This inhibition suggests that nitrate be active indirectly after conversion to nitrite by oxidoreductases like xanthine oxidase (cf section 7. C). The dose dependence revealed that the nitrite competes with estradiol for binding to ERα. It suggests that nitrite anions interact directly with the ligand-binding domain of ERα (106). The close homology of ERα to the other ERβ class of estrogen receptors suggests that nitrite may activate other steroid receptors as well.
10: DISCUSSION: THE LEVEL OF OXYGEN DETERMINES THE PATHWAY FOR REDUCTION OF NITRITE
Section 7. gave an overview over the various enzymatic mediators that have been found to reduce nitrite to NO. Of these pathways, only XO seems seems active at any degree of oxygenation via process II (cf Fig. 5). The other mechanisms share the common feature of being activated by low oxygen levels (cf table III and Fig 9). Their reduction pathways become significant only when the local oxygen level falls below a certain threshold.
Figure 9.

Schematic representation of the ranges of oxygenation where the various pathways for reduction of nitrite are active. For cytochrome P450 no definite data exist yet, but this pathway is likely to function only in near complete absence of oxygen (cf section 7. D). The two different reaction processes for XO were explained in fig 5. The vertical lines indicate the boundaries between anoxia, hypoxia, normoxia and hyperoxia as defined in table III.
The numbers in table IV show that the threshold for deoxyHb distinguishes itself from the others: At a value of [O2] ~ 30 μM, deoxyHb already starts to reduce nitrite. Such oxygen levels are near the lower end of the normoxic regime. The remaining mechanisms are activated in the range of 20 - 2 μM, a range we classified as “hypoxia”. DeoxyHb is also special in that it is confined to the blood stream only. Although whole blood is a very hostile environment for NO, the rapid scavenging of free NO by oxyHb makes improbable that the release of NO by deoxyHb could contribute significantly to the activation of guanylate cyclase in the smooth muscle cells as long as a sizable fraction of Hb remains oxygenated. Therefore, under normoxic conditions, endothelial NO production determines vascular tone. even though some nitrite is already being reduced to NO in the bloodstream. The situation changes if oxygen levels fall further and the tissues become hypoxic. The fraction of deoxyHb rises and accelerates the release of NO from the RBC. Simultaneously the fraction of oxyHb falls, survivability of NO improves and helps to maintain the blood flow. Upon deep hypoxia, the additional mechanisms are successively activated until, finally, oxygen starts to be the limiting factor for the rate of mitochondrial respiration as well ([O2] < 2 μM, cf table III). However, the role of nitrite is not determined by oxygen levels alone. The properties of the affected tissues are also highly relevant as the enzymatic effectors of nitrite reduction are not expressed to the same extent in all tissues. For example, the myoglobin mechanism should have higher relevance in myocardial tissues than blood vessels that lack Mb (244). The eNOS mechanism should affect the direct periphery of the endothelium. In addition, eNOS has been identified recently in membranes of human RBC by means of microscopy (245) and Western blotting (246). The mitochondrial mechanism and xanthine oxidase should have particularly high relevance for liver tissue etc. But XO has also been identified on the outer membrane of human epithelial and endothelial cells (247) as well as RBC’s (246). In vascular tissues, therefore, deoxyHb, eNOS and XO appear colocalized and the hypoxic response may involve a combined effect of these mediators. This problem was recently addressed in ref (246), where it was concluded that deoxyHb and eNOS dominate the nitrite reductase activity in the circulation under physiological pH. Given the variability in composition of various tissues,, the relevance of a given endogenous nitrite pool will depend very much on the severity of hypoxia, local pH and the enzymatic composition of the affected tissue.
This review is concerned with endogenous pathways for the reduction of nitrite to NO radicals under hypoxia. Little has been said about the downstream effects elicited by such “hypoxic NO”. It is certain that the significance of such NO goes far beyond just vasodilation or regulation of mitochondrial respiration. In recent years, it has become clear that the presence of NO does affect the hypoxic response of cells or tissues. This is partially mediated by modulation of gene expression by NO itself, partially via interference with the cellular apparatus for hypoxic signalling (243). For example, inhibition of mitochondrial respiration by NO was reported to redirect the remaining oxygen to prolyl hydroxylases (248). The activation of these hydroxylases prevents accumulation of the Hypoxia Inducible Factor 1α (HIF1α), and inhibits the normal hypoxic cellular response (248). Phrased otherwise, the accumulation of HIF1α is affected by NO via the status of the mitochondrial respiratory chain. Such indirect secondary effects should be kept in mind when considering “the effect of nitrite under hypoxia”. Such an attractively simple “effect” is not likely to exist at all. Instead, one has to face a basic phenomenon of nitrite reduction where the reaction itself will usually involve several reaction pathways operating simultaneously, with the outcome depending on the tissue type and profile of the enzymes expressed. It really is a highly complex phenomenon even within the stringent boundary conditions on acidity, temperature etc as exist in mammalian physiology. While this complexity may seem a deterrent for research at first, it may ultimately offer new therapeutic opportunities (108, 249–252). Precisely this complexity raises the prospect that nitrite therapy will find a very wide range of useful applications.
11 PROSPECTS AND IMPLICATIONS FOR CLINICAL PRACTICE
The preceding sections have discussed various pathways for the reduction of nitrite to free NO radicals. We note that these pathways are operational in mammalian tissues if oxy gen levels fall to hypoxic or anoxic levels (for the definition of these thresholds, cf. section 3). These pathways seem significant for human physiology: A growing corpus of experimental and statistical data confirms that nitrite mediates a succession of signaling events as oxygen levels fall from physiological levels to zero. Therefore, individual nitrite signalling pathways can be easily “activated” and deactivated in a “switch mode” fashion, depending on local oxygen level. Importantly, these signalling cascades can be (de)activated independently from blood flow because of the abundance of nitrite in tissues, organs, plasma and blood cells (cf section 2).
The hypoxic release of NO from nitrite has potential for clinical applications in any disease where not only hypoxia but also an altered NO bioavailability is relevant. This opens a very wide field for research into potential diagnostic and therapeutic benefits of nitrite in a clinical setting in the near future (108, 249–252). Among the many different topics, therapeutic benefits of nitrite seem particularly promising in four systems: Support of systemic blood flow, protection against ischemia-reperfusion injury, promotion of endothelial reconstruction after injury, and effects on the gastrointestinal tract.
Support of systemic blood flow under hypoxia
The nitrite-dependent regulation of vascular tone and thus blood flow under hypoxia seems clinically relevant at the level of the conduit arteries and the microcirculation. This point is evidenced by the following examples:
normal conduit arteries are not as exposed to reversible hypoxia as microvessels. But during vasospasm, in particular in the cerebral circulation due to its unique innervation and susceptibility to hypercontraction of the vascular smooth muscles, a significant drop of oxygen tension may occur. In primates it has been shown that infusion of nitrite selectively dilates spastic cerebral arteries (89) without any dilatation further downstream. This points to the possibility that nitrite might be tested in clinical trials as a therapeutic to selectively reverse cerebral vasospasm in patients without concomitant drop in systemic blood pressure as seen with other NO donors.
Human plasma nitrite shows arterial-venous gradients at rest (cf section 2). These gradients become more pronounced under exercise. The gradients indicate consumption of nitrite during passage through the microcirculation and along the natural oxygen gradient within the vasculature (44, 45, 57). The concomitant generation of NO was not assayed but seems plausible. Plasma levels of nitrite post exercise were independent predictors of stress endurance and power in healthy subjects. This suggests that nitrite play a role in the hemodynamic adaption during exercise (253). After exercise, plasma nitrite is generally enhanced over basal values. However, in healthy volunteers the enhancement by exercise is less pronounced as age increases. Most importantly, the enhancement by exercise is strongly reduced in patients with coronary artery disease (254). These findings not only point towards the significance of nitrite dependent regulation of blood flow during stress and hypoxia but also may open new avenues in the diagnosis of endothelial dysfunction in patients via measurement of nitrite “reserve” at rest and during exercise.
During crisis in sickle cell disease local areas of hypoxia occur within the circulation due to obstruction of blood flow. The obstructions manifest impaired rheologic properties of the red blood cells. NO and also the NOS substrate L-arginine have been shown to improve rheologic function of red blood cells (255). Although untested, nitrite might be potentially useful to selectively provide NO in an oxygen-dependent fashion to those sites to improve red blood cell function, prevent further lysis of red blood cells, and thus improve blood flow (88, 255).
Protection against damage from ischemia-reperfusion
Ischemia is defined as a hypoxic condition with no blood flow. There is a graded overlap to “low flow” hypoxia. Under both conditions oxygen levels are in the very low range close to anoxia (cf section 3, table III). Prolonged oxygen deficiency is known to cause irreversible damage to tissues, with the brain being particularly sensitive to hypoxia (28). Reperfusion after ischemia might induce acute additional organ damage due to the formation of reactive oxygen species (ROS) upon reoxygenation. On a longer timescale, inflammatory processes contribute to ischemic damage. The mechanism of nitrite-dependent cytoprotection after ischemia/reperfusion appears highly complex and apparently involves multiple pathways. The inflammatory reaction involving endothelial activation and diapedesis of leukocytes is inhibited by NO. The latter acts as antagonist to expression of cell adhesion molecules on the endothelial membrane. As a result, leukocyte adhesion to the endothelium is strongly suppressed (256, 257). It is known that free NO enhances the expression and binding activity of HIF-1α protein (258). NO also protects by suppressing the abnormal proliferation of vascular smooth muscle cells (259). It was noted above that nitrite as well as NO enhance the level of S-nitrosothiols in the extra- and intracellular compartments (33). Although neither nitrite nor NO per se are effective S-nitrosating species in vitro, the presence of transition metal ions may catalytically accelerate the S-nitrosation of thiols in biological systems (33). Such enhancement of S-nitrosation may have high significance as this modification inhibits the transcriptional activity of inflammatory transcription factors like AP1 and NF-κB, thereby helping to maintain the endothelial cells in quiescent state. S-nitrosation of the sarcolemmal ryanodine receptor in cardiac muscle is necessary for the normal receptor function (260). S-nitrosation of thiol residues in N-ethylmaleimide-sensitive factor (NSF) regulates granule exocytosis from activated platelets (261). Platelet activity itself is downregulated in presence of free NO as well as S-nitrosothiols. All these various pathways combine to dampen the inflammatory response.
Nitrite is easily reduced to NO during ischemia and might act antagonistically on various ROS related signalling cascades and thus ameliorate the damage from ischemia-reperfusion. This could be of major importance in acute myocardial infarction or stroke, in particular during the resuscitation stage. The beneficial effect of nitrite has been demonstrated in several experimental models of global hypoxia/ischemia (cf examples quoted in section 5). Further randomized controlled trials are under way to substantiate this hypothesis derived from various ex vivo, in situ and in vivo models of ischemia-reperfusion. Closely related to this area are the potential applications of nitrite in preservation of excised organs during transplant operations.
Endothelial reconstruction
Another promising field could be vascular healing, in particular the reconstruction of damage to the endothelial lining of the vascular tree. If free oxygen concentrations fall below ca. 10 μM, the normal L-Arginine pathway of eNOS is impaired due to oxygen starvation (cf. section 7. E.). However, eNOS starts to release free NO from nitrite. NO has been shown to promote the mobilization and function of endothelial progenitor cells (EPC) derived from the bone marrow (262, 263). Conversely, endogenous NOS inhibitors like asymmetric dimethylarginine (ADMA) were found to suppress the mobilization, differentiation and function of EPC (264). The same correlations are observed in diabetic or age-related endothelial dysfunction (265). The number of EPC circulating in blood correlates positively with endothelial function (266, 267) and can be used to quantify cardiovascular risk. Intriguingly, in a clinical setting, several recent studies (268, 269) noted a correlation between EPC count and colony formation on the one hand, and plasma nitrite concentrations on the other.
Stimulation of angiogenesis and arteriogenesis
Local occlusion of blood vessels leads to chronic ischemia in the downstream tissues. So far, no drugs are available that promote new vessel formation (angiogenesis) selectively in the affected regions only (252). Although many angiogenic compounds are known (like VEGF, angiotensin II, or losartan) they share the problem that their action is systemic rather than local and that vascularization is promoted in healthy tissues as well. Local rather than systemic angiogenic activity was recently reported (101) for nitrite. Protracted administration of nitrite significantly promoted local angiogenesis and and tissue perfusion in a hind-limb ischemia mouse model. These preliminary results look particularly promising in that the effect was obtained with low and safe doses of nitrite.
Immunity and gastrointestinal system
Substantial quantities of nitrite are ingested daily via nutrition. Part of this nitrite derives from reduction of nitrate by commensal bacteria in the mouth and upper gastrointestinal tract (cf section 8). Subsequent reduction of the nitrite in the stomach regulates the function of the mucosal barrier and the blood flow within the stomach lining (270). A recent study reported potent gastroprotective effects of dietary nitrate in a rat model of NSAID-induced gastric ulcers (32). Further down the gastrointestinal tract, nitrite is subject to extensive metabolic signalling by commensal bacteria resident in bowels (271). As yet, little is known about the interaction of nitrite with these bacteria and its interaction with various nutrients. Such pathways might have relevance for inflammatory bowel diseases and aspects of innate immunity. Patients subjected to artificial ventilation have reduced NO/nitrite levels in the upper gastrointestinal tract as this upper part is bypassed by the ventilating tubes (272). Although unconfirmed as yet, it has been speculated that such anomalous nitrite levels affect immunity in critical ill and artificially ventilated patients in hypoxic regions of the gastrointestinal tract and the circulation (272).
Outlook and clinical significance
So far, the majority of data on effects of nitrite under hypoxic conditions were collected from animal models. However, preliminary studies in humans seem to confirm the importance of nitrite for regulation of vascular tone and support of blood flow under conditions of oxygen deficiency. Clearly, prevention of cerebral vasospasm, improvement of pulmonary ventilation in respiratory syndromes, functional preservation of transplanted organs, endothelial reconstruction and adaptation to chronic hypoxia are all targets of high clinical relevance. In the near future larger clinical trials will emerge to assess the significance of nitrite signalling in human pathology and disease. The authors hope that this review will stimulate and contribute to future research in this promising area.
Acknowledgments
M. Feelisch acknowledges support from the Medical Research Council (Strategic Appointment Scheme).
N. Hogg acknowledges support from NIH grant GM55792 from the National Institute of General Medicine.
D. Kim-Shapiro acknowledges support from NIH grants HL05891 and HL078706.
T. Rassaf and M. Kelm are supported by grants from the DFG (RA 969/4-1 to TR and KE 405/5-1 to MK).
S. Shiva acknowledges generous support by NIH grants HL058091 and HL078706, and by the Division of Intramural Research of the National Heart, Lung and Blood Institute.
A. Vanin thanks the Russian Foundation for Basic Research (grants 04-05-49383 and 07-04-1350ofi_c) and the French ECONET 2005 program (to Anny Slama-Schwok, dossier 10275).
We thank prof. A. J. Rabelink (Leiden) for careful reading of the manuscript and helpful comments.
L1ST OF ABBREVIATIONS
- ADMA
asymmetric dimethylarginine, endogenous NOS inhibitor
- AMP
adenosine monophosphate, also called 5′-adenylic acid
- AMP
adenosine monophosphate, also called 5′-adenylic acid
- AP1
activator protein-1, inflammatory transcription factor
- ATP
adenosine triphosphate
- BEND3
immortalized murine brain microvascular endothelial cell line
- BH4
tetrahydrobiopterin
- cGMP
cyclic guanosine monophosphate
- Cp
ceruloplasmin
- C-PTIO
carboxy-2-phenyl-4,4,5,5,-tetramethylimidazoline-1-oxyl 3-oxide
- CPR
cytochrome P450 reductase
- CYP
cytochrome P450
- DBA
2,3-dihydroxybenz-aldehyde
- DETC
diethyldithiocarbamate ligand
- DNIC
dinitrosyl iron complex
- DPI
diphenylene iodinium
- eNOS
endothelial isoform of nitric oxide synthase
- EPR
electron paramagnetic resonance
- EPC
endothelial progenitor cell
- ERα
estrogen receptor α
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- GTN
glycerol trinitrate, nitroglycerin
- Hb
hemoglobin
- HbNO
nitrosylated haemoglobin
- HIF-1
hypoxia inducible factor-1, a heterodimer with subunits HIF-1α and HIF-1β
- iNOS
inducible isoform of nitric oxide synthase
- I/R
ischemia-reperfusion
- ISC
iron-sulfur clusters
- LIP
labile iron pool
- LNA
Nω-nitro-L-arginine, NOS inhibitor
- L-NAME
L-arginine methyl ester, NOS inhibitor
- L-NIO
N-iminoethyl-L-ornithine, NOS inhibitor
- Mb
myoglobin
- MCF-7
human breast adenocarcinoma cell line
- metHb
methemoglobin (ferric Fe3+-Hb)
- metMb
metmyoglobin (ferric Fe3+-Mb)
- MGD
N-methyl-D-glucamine dithiocarbamate ligand
- MNIC
mononitrosyl iron complex
- mRNA
messenger ribonucleic acid
- NADH
nicotinamide adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NF-κB
nuclear factor κB
- nNOS
neuronal isoform of nitric oxide synthase
- NOS
nitric oxide synthase
- NSAID
non-steroidal anti-inflammatory drugs
- NSF
N-ethylmaleimide-sensitive factor
- PCr
phosphocreatine
- Q-cycle
mitochondrial reaction sequence for ATP generation
- RBC
red blood cell
- rtPA
recombinant tissue plasminogen activator
- SNO-Hb
S-nitroso hemoglobin
- SOD
superoxide dismutase
- TTFA
thenoyl trifluoro-acetone (inhibitor of mitochondrial complex II)
- UQ
ubiquinone
- VEGF
vascular endothelial growth factor
- WT
wild type
- XO
xanthine oxidase
Biographies
Ernst van Faassen received his PhD in theoretical physics from Utrecht University in 1985. After a postdoc at Maryland University he joined the Biophysics group in Utrecht where he applied a variety of spectroscopic techniques to biological systems, in particular membranes and cultured cells. Through contacts at the University Hospital he became interested and very involved in the physiological role of oxygen radicals and nitric oxide in the human vasculature. He is manager of the EPR-ENDOR facility of the Debye Institute of Utrecht University.
Soheyl Bahrami is associate professor for Medical Chemistry and Biochemistry at the Medical University of Vienna since 1994. He received his PhD in 1984 from the Technical University of Vienna for studies in biochemistry. In 1981 he joined the Ludwig Boltzmann Institute for Experimental and Clinical Traumatology in Vienna and is presently the head of the group for shock research of the Ludwig Boltzmann Institute.
Martin Feelisch received his PhD in pharmacology from the Heinrich-Heine University in Dusseldorf in 1988. Following research positions in both academia and industry in the US and Europe, he currently holds the Chair of Experimental Medicine and Integrative Biology at the University of Warwick, UK, He has a long-standing interest in cardiovascular pharmacology, drug metabolism, disease mechanisms, and the chemical biology of nitric oxide (NO). His recent research interests focus on the physiological significance of nitros(yl)ation reactions and the role of NO storage forms in blood and tissues; biomarkers of cardiovascular disease; mechanisms of adaptation to hypoxia; hypoxic signalling and altitude physiology; and systems-based approaches to assess the role of nitrite, nitrate and other NO-related products in vivo.
Neil Hogg earned his PhD from Essex University in 1993. He became postdoc at the Medical College of Wisconsin where he is currently associate professor. His research interest is in the molecular mechanisms of nitric oxide signalling, in particular via formation and transport of S-nitrosothiols and the interactions between NO and heme proteins.
Malte Kelm received his MD and PhD at CologneUniversity. After Postdocs in Düsseldorf and London, he specialized in Internal Medicine, Angiology and Cardiology at Heinrich Heine University, Düsseldorf. He developed a strong research interest in vascular biology and the role and reaction pathways of endothelium derived nitric oxide. In 2005 he became chairman of the Department of Cardiology, Pulmonary Diseases and Vascular Medicine at Aachen University hospital. His recent research interest are on cardiac imaging and monitoring for cardiac interventions.
Daniel Kim-Shapiro received his PhD in biophysics from the University of California, Berkeley, in 1993. He is a Professor of Physics and the Harbert Family Distinguished Chair, and holds appointments in the departments of Molecular Medicine, Molecular Genetics and Biomedical Engineering at Wake Forest University. Earlier in his career, Dr Kim-Shapiro served in the Peace Corps (1984 –1986) as a college-level physics instructor at L’Institut Superieur du Pedagogie, in Zaire. Currently, he investigates the effects of nitrite and nitric oxide and in biological systems, especially in relation to sickle cell and other blood or cardiovascular diseases.
Andrey V. Kozlov received his PhD in 1986 from the 2nd Moscow Medical School for studies in Biophysics and his habilitation in pharmacology and toxicology at the University of Veterinary Medicine (Vienna, Austria) in 2003. He has a strong interest in applications of electron paramagnetic resonance spectroscopy in biomedical research. More recently, he has focused upon the metabolism of nitric oxide and oxygen radicals. Presently, he is a head of the group for „molecular base of organ failure“ at the Ludwig Boltzmann Institute for Experimental and” Clinical Traumatology in Vienna.
Haitao Li received his PhD in Molecular and cellular Biophysics. He is now research assistant professor of the Davis Heart and Lung Research Institute and Division of Cardiovascular Medicine, Department of Internal Medicine, The Ohio State University College of Medicine. His research interest is focused on the enzymatic mechanisms for generation of nitric oxide (NO) and oxygen free radicals, and on their role in signal transduction, cellular injury, and diseases.
Jon O. Lundberg received his M.D. at the Karolinska Institute (KI), Stockholm, Sweden, in 1993. He is currently professor of pharmacology at the KI. His interest is in the biology of nitric oxide and related nitrogen oxides. In 1994 he and his colleagues discovered an NO-synthase-independent pathway for NO generation in humans resulting from the reduction of inorganic nitrite. His main focus is on characterizing NO-synthase-independent NO generation from nitrate and nitrite and clarifying its role in health and disease.
Ron P. Mason received his PhD in Physical Chemistry from the University of Wisconsin. After several years at the department of Medicine and Pathology at Minneapolis he joined NIEHS at Research Triangle Park, NC, in 1978. He now heads the Free Radical Metabolism Group at the Laboratory of Pharmacology. He is well known for his work on radical reactions and the role of transition metal ions in biological fluids. His current projects aim at in-vivo detection of free radicals and the identification of biomarkers of oxidative stress.
Hans Nohl graduated in medicine and biochemistry from the Universities of Giessen, Freiburg and Paris. He received doctoral degrees in medical sciences (1969) and in natural sciences (1979) from the University of Munich; After his habilitation in biochemical pharmacology and toxicology at the Technical University of Munich, he was appointed full professor and chairman of the Institute of Pharmacology and Toxicology at the Veterinary University of Vienna in 1984. Since 2002 he is chairman of the research group “Basic Research in Pharmacology and Toxicology”. His research concentrates on mitochondrial bioenergetics, and the role of ubiquinone, nitric oxide and reactive oxygen species in human health.
Tienush Rassaf studied medicine at Düsseldorf University and the Texas Heart Institute. He received his MD and PhD from the Heinrich Heine University. After postdocs in Düsseldorf and Louisiana, he specialized in Internal Medicine and Cardioogy and now is Scientific Group Leaader and attending for Cardiology at the University Hospital Aachen. His research is on cardiac function, especially under hypoxic or ischemic conditions, and studies the interactions and modifications of cardiac proteins like myoglobin.
Alexandre Samouilov acquired his PhD in chemistry from Saint Petersburg Technological Institute, Russia in 1988. During the postdoctoral fellowship at Johns Hopkins University heworked on detection and characterization of various paramagnetic substances in vivo and in vitro using EPR techniques. During this time he became fascinated by the biochemistry of nitric oxide and its related compounds, especially nitrite. Currently, he is an assistant professor of the Ohio State University and investigates pathways for NOS independent nitric oxide production.
Anny Slama-Schwok obtained her PhD in Physical Chemistry at the Hebrew University in in1986. As a postdoc at the Collège de France with Jean-Marie Lehn she developed artificial photonuclease for single-stranded DNA photocleavage. After several years as assistant professor at the Hebrew University she joined the french institute for medical research INSERM and studied the molecular regulations of the NO-synthases using ultrafast optical spectroscopy. Her recent interests involve the physiological role of nitrite, development of nanotriggers for photoactivation of NOS activity and smart drugs targeting influenza pathogens.
Sruti Shiva received her PhD from the University of Alabama Birmingham in 2000. She is currently a postdoctoral fellow in the Pulmonary and Vascular Medicine Branch at the National Institutes of Health. Her research focuses on the role of reactive nitrogen species in the regulation of mitochondrial function, particularly during hypoxia and ischemia/reperfusion.
Anatoly Vanin studied Chemical Physics and Biophysics at Lomonossow University in Moscow. During his studies he became interested in biological applications of EPR spectroscopy, in particular for the properties of endogenous iron complexes. A large part of his work was devoted to the formation and physiological role of nitrosylated iron, in particular of dinitrosyl-iron complexes. Since 1989 he is Head of Laboratory at the Semenov Institute of Chemical Physics of the Russian Academy of Sciences in Moscow, and he has made this lab a world center for study of the reaction pathways of NO in biological systems.
Eddie Weitzberg received his M.D. at Uppsala University, Sweden, in 1982 and is presently professor and senior consultant at the Department of Anesthesiology and Intensive Care, at the KI. His research is focused on endothelium-derived factors and their importance in regulation of cardiovascular function with a special focus on inflammation and sepsis. In 1993 he and his colleagues showed that exhaled NO is increased in asthmatics, and in 1994 they discovered an NO-synthase-independent pathway for NO production in humans. Recent research areas involve NO-synthase-independent formation of NO from inorganic nitrate and nitrite, endothelial dysfunction in the intensive care unit and the role of endothelin in sepsis.
Jay Zweier is Director of the Davis Heart & Lung Research Institute, Professor and Lumley Chair in Medicine at the Ohio State University. He received baccalaureate degrees in Physics and Mathematics at Brandeis University. After his PhD in Biophysics at the Albert Einstein College of Medicine, he completed his MD at the University of Maryland and fellowship in Cardiology at Johns Hopkins. After joining the faculty at Johns Hopkins his research focused on the role of free radicals and nitric oxide in cardiovascular disease. He has worked for over two decades to develop innovative technology and techniques for magnetic resonance based detection of free radicals. He published extensively in the fields of cardiovascular research, free radical biology and magnetic resonance techniques.
Mark Gladwin received his M.D. from the University of Miami Honors Program in Medical Education in 1991. After completing his internship and chief residency at the Oregon Health Sciences University in Portland, Ore., he joined the NIH in 1995 as a critical care fellow in the Clinical Center. After a clinical fellowship in pulmonary medicine in Seattle he returned to NIH as a research fellowship at the Critical Care Medicine Department. At NIH, he is currently Chief of the Pulmonary and Vascular Medicine Branch, within the Intramural Research Division of the NHLBI. His research is concentrated on vascular medicine, hypoxia, and the role of nitrite in the human vasculature.
References
- 1.Reichert ET, Mitchell SW. On the physiological action of potassium nitrite. Am J Med Sci. 1880;156:158–180. [Google Scholar]
- 2.Bruningfann C, Kaneene J. The effects of nitrate, nitrite and N-nitroso compounds on human health – a review. Vet Human Toxicol. 1993;35:521– 538. [PubMed] [Google Scholar]
- 3.Weiss S, Wilkins R, Haynes F. The nature of circulatory collapse induced by sodium nitrite. J Clin Invest. 1937;16:73–84. doi: 10.1172/JCI100840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furchgott R, Bhandrakom S. Reactions of strips of rabbit aorta to epinephrine, isopropylarterenol, sodium nitrite and other drugs. J Pharmacol Exp Ther. 1953;108:129–143. [PubMed] [Google Scholar]
- 5.Mittal C, Arnold W, Murad F. Characterization of protein inhibitors of guanylate cyclase activation from rat heart and bovine lung. J Biol Chem. 1978;253:1266–1271. [PubMed] [Google Scholar]
- 6.Lauer T, Preik M, Rassaf T, Strauer B, Deussen A, Feelisch M, Kelm M. Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc Natl Acad Sci USA. 2001;98:12814– 12819. doi: 10.1073/pnas.221381098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gladwin M, Schechter A, Kim-Shapiro D, Rakesh R, Hogg N, Shiva S, Cannon R, Kelm M, Wink D, Espey M, Oldfield E, Pluta R, Freeman B, Lancaster J, Feelisch M, Lundberg J. The emerging biology of the nitrite anion. Nature Chem Biol. 2005;1:308–314. doi: 10.1038/nchembio1105-308. Corrigendum ibid. 2006, 2: 110. [DOI] [PubMed] [Google Scholar]
- 8.Duranski MR, Greer J, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet S, Wang Y, Kevil C, Gladwin M, Lefer D. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232– 1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci USA. 2004;101:13683– 13688. doi: 10.1073/pnas.0402927101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dezfulian C, Raat N, Shiva S, Gladwin M. Role of the anion nitrite in ischemia-reperfusion cytoprotection and therapeutics. Cardiovasc Res. 2007;75:327– 338. doi: 10.1016/j.cardiores.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lundberg J, Weitzberg E. NO generation from nitrite and its role in vascular control. Arterioscler Thromb Vasc Biol. 2005;25:915– 922. doi: 10.1161/01.ATV.0000161048.72004.c2. [DOI] [PubMed] [Google Scholar]
- 12.Lundberg J, Weitzberg E, Lundberg J, Alving K. Intragastric nitric oxide production in humans: measurement in expelled air. Gut. 1994;35:1543– 1546. doi: 10.1136/gut.35.11.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKnight G, Smith L, Drummond R, Duncan C, Golden M, Benjamin N. Chemical synthesis of nitric oxide in the stomach from dietary nitrate in humans. Gut. 1997;40:211–214. doi: 10.1136/gut.40.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lundberg J, Carlsson S, Engstrand L, Morcos E, Wiklund N, Weitzberg E. Urinary nitrite: more than a marker of infection. Urology. 1997;50:189– 191. doi: 10.1016/S0090-4295(97)00257-4. [DOI] [PubMed] [Google Scholar]
- 15.Troitzsch D, Vogt S, Abdul-Khaliq H, Moosdorf R. Muscle tissue oxygen tension and oxidative metabolism during ischemia and reperfusion. J Surg Res. 2005;128:9– 14. doi: 10.1016/j.jss.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 16.Zweier J, Wang P, Samouilov A, Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nature Med. 1995;1:804– 809. doi: 10.1038/nm0895-804. [DOI] [PubMed] [Google Scholar]
- 17.Millar T, Stevens C, Benjamin N, Eisenthal R, Harrison R, Blake D. Xanthine oxidoreductase catalyses the reduction of nitrates and nitrite to nitric oxide under hypoxic conditions. FEBS Lett. 1998;427:225– 228. doi: 10.1016/s0014-5793(98)00430-x. [DOI] [PubMed] [Google Scholar]
- 18.Gautier C, van Faassen E, Mikula I, Martasek P, Slama-Schwok A. Endothelial nitric oxide synthase reduces nitrite anions to NO under anoxia. Biochem Biophys Res Commun. 2006;341:816– 821. doi: 10.1016/j.bbrc.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 19.Vanin A, Bevers L, Slama-Schwok A, van Faassen E. Nitric oxide synthase reduces nitrite to NO under anoxia. Cell Mol Life Sci. 2007;64:96– 103. doi: 10.1007/s00018-006-6374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nohl H, Staniek K, Kozlov A. The existence and significance of a mitochondrial nitrite reductase. Redox Rep. 2005;10:281– 286. doi: 10.1179/135100005X83707. [DOI] [PubMed] [Google Scholar]
- 21.Castello P, David P, McClure T, Crook Z, Poyton R. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions:implications for oxygen sensing and hypoxic signalling in eukaryotes. Cell Metab. 2006;3:277– 287. doi: 10.1016/j.cmet.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 22.Crawford JT, Scott Isbell T, Huang Z, Shiva S, Chacko B, Schechter A, Darley-Usmar V, Kerby J, Lang J, Kraus D, Ho C, Gladwin M, Patel R. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2006;1007:566–574. doi: 10.1182/blood-2005-07-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rassaf T, Flögel U, Drexhage C, Hendgen-Cotta U, Kelm M, Schrader J. Nitrite reductase function of deoxymyoglobin: oxygen sensor and regulator of cardiac energetics and function. Circ Res. 2007;100:1749– 1754. doi: 10.1161/CIRCRESAHA.107.152488. [DOI] [PubMed] [Google Scholar]
- 24.Shiva S, Huang Z, Grubina R, Sun J, Ringwood L, MacArthur P, Xu X, Murphy E, Darley-Usmar V, Gladwin M. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ Res. 2007;100:654–661. doi: 10.1161/01.RES.0000260171.52224.6b. [DOI] [PubMed] [Google Scholar]
- 25.Nagababu E, Ramasamy S, Abernethy D, Rifkind J. Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin-mediated nitrite reduction. J Biol Chem. 2003;278:46349–46356. doi: 10.1074/jbc.M307572200. [DOI] [PubMed] [Google Scholar]
- 26.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation, Nat. Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 27.Huang Z, Shiva S, Kim-Shapiro D, Patel R, Ringwood L, Irby C, Huang K, Ho C, Hogg N, Schechter Am Gladwin M. Enzymatic function of haemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest. 2005;115:2099–2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ward J. Oxygen delivery and demand. Surgery. 2006;24:354– 360. [Google Scholar]
- 29.Spiegelhalder B, Eisenbrand G, Preussmann R. Influence of dietary nitrate on nitrite content of human salive: possible relevance to in vivo formation of N-nitroso compounds. Food Cosmet Toxicol. 1976;14:545– 548. doi: 10.1016/s0015-6264(76)80005-3. [DOI] [PubMed] [Google Scholar]
- 30.Lundberg J, Weitzberg E, Benjamin N. Nitrate, bacteria and human health. Nat Rev Microbiol. 2004;2:593– 602. doi: 10.1038/nrmicro929. [DOI] [PubMed] [Google Scholar]
- 31.Li H, Samouilov A, Liu X, Zweier J. Characterization of the magnitude and kinetics of xanthine-oxidase catalyzed nitrate reduction. Biochem. 2003;42:1150– 1159. doi: 10.1021/bi026385a. [DOI] [PubMed] [Google Scholar]
- 32.Jansson E, Huang L, Malkey R, Govoni M, Nihlen C, Olsson A, Stensdotter M, Petersson J, Holm L, Weitzberg E, Lundberg J. A mammalian functional nitrate reductase that regulates nitrite and nitric oxide homeostasis. Nature Chem Biol. 2008 doi: 10.1038/nchembio.92. in press. [DOI] [PubMed] [Google Scholar]
- 33.van Faassen E, Vanin A, editors. Radicals for life: The various forms of nitric oxide. Elsevier; Amsterdam: 2007. [Google Scholar]
- 34.Rassaf T, Preik M, Kleinbongard P, Lauer T, Heiss C, Strauer B, Feelisch M, Kelm M. Evidence for in vivo transport of bioactive nitric oxide in human plasma. J Clin Invest. 2002;109:1241– 1248. doi: 10.1172/JCI14995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Miller M, Joshi M, Thomas D, Lancaster J. Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc. Natl. Acad. Sci USA. 1998;95:2175– 2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shiva S, Wang X, Xu X, Yuditskaya S, Annavaijjhala V. Ceruloplasmin is a NO oxidase and nitrite synthase that determines endocrine NO homeostasis. Nat Chem Biol. 2006;2:486– 493. doi: 10.1038/nchembio813. [DOI] [PubMed] [Google Scholar]
- 37.Shaw A, Vosper A. Solubility of nitric oxide in aqueous and nonaqueous solvents. J Chem Soc Faraday Trans 1. 1977;73:1239– 1244. [Google Scholar]
- 38.Subczynski W, Hyde J. Concentration of oxygen in lipid bilayers using a spin-label method. Biophys J. 1983;41:283– 286. doi: 10.1016/S0006-3495(83)84439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simon S, Gutknecht J. Solubility of carbon dioxide in lipid bilayer membranes and organic solvents. Biochim Biophys Acta. 1980;596:352– 358. doi: 10.1016/0005-2736(80)90122-4. [DOI] [PubMed] [Google Scholar]
- 40.Power G, Stegall H. Solubility of gases in human red blood cell ghosts. J Appl Physiol. 1970;29:145–149. doi: 10.1152/jappl.1970.29.2.145. [DOI] [PubMed] [Google Scholar]
- 41.Rhodes P, Leone A, Francis P, Struthers A, Moncada S. The L-arginine-NO pathway is the major source of plasma nitrite in fasted humans. Biochem Biophys Res Commun. 1995;209:590– 596. doi: 10.1006/bbrc.1995.1541. [DOI] [PubMed] [Google Scholar]
- 42.Rassaf T, Kelm M, Feelisch M. Concomitant presence of N-nitroso and S-nitroso protein in human plasma. Free Rad Biol Med. 2003;33:1590– 1596. doi: 10.1016/s0891-5849(02)01183-8. [DOI] [PubMed] [Google Scholar]
- 43.Lundberg JO, Govoni M. Inorganic nitrate is a possible source for systemic generation of nitric oxide. Free Radic Biol Med. 2004;37:395–400. doi: 10.1016/j.freeradbiomed.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 44.Dejam A, Hunter C, Pelletier M, Hsu L, Machado R, Shiva S, Power G, Kelm M, Gladwin M, Schechter A. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood. 2005;106:734– 739. doi: 10.1182/blood-2005-02-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gladwin M, Shelhamer J, Schechter A, Pease-Fye M, Waclawei M, Panza J, Oguibene F, Cannon R. Role of circulating nitrite and S-nitrosohemoglobin in the regulation of the regional blood flow in humans. Proc Natl Acad Sci USA. 2000;97:11482–11487. doi: 10.1073/pnas.97.21.11482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gödecke A, Decking Z, Ding J, Hirshenhain J, Bidmon H, Gödecke S, Schrader J. Coronary hemodynamics in endothelial NO synthase knockout mice. Circ Res. 1998;82:186– 194. doi: 10.1161/01.res.82.2.186. [DOI] [PubMed] [Google Scholar]
- 47.Tracey WR, Tse J, Carter G. Lipopolysaccharide-induced changes in plasma nitrite and nitrate concentrations in rats and mice. Pharmacological evaluation of nitric oxide synthase inhibitors. J Pharmacol Exp Ther. 1995;272:1011– 1015. [PubMed] [Google Scholar]
- 48.Bryan N, Rassaf T, Maloney R, Rodriguez C, Saijo F, Rodriguez J, Feelisch M. Cellular targets and mechanisms of nitros(yl)ation: an insight into their nature and kinetics in vivo. Proc Natl Acad Sci. 2004;101:4308– 4313. doi: 10.1073/pnas.0306706101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hata T, Hashimoto M, Kanenishi K, Akiyama M, Yanagihara T, Masumura S. Maternal circulating nitrite levels are decreased in both normal normotensive pregnancies and pregnancies with preeclampsia. Gynecol Obstet Invest. 1999;48:93– 97. doi: 10.1159/000010147. [DOI] [PubMed] [Google Scholar]
- 50.Sikkema J, van Rijn B, Franx A, Bruinse H, de Roos R, Stroes E, van Faassen E. Placental superoxide is increased in preeclampsia. Placenta. 2001;22:304– 308. doi: 10.1053/plac.2001.0629. [DOI] [PubMed] [Google Scholar]
- 51.Erzurum S, Ghosh S, Janocha A, Xu W, Bauer S, Bryan N, Tejero J, Hemann C, Hille R, Stuehr D, Feelisch M, Beal C. Higher blood flow and circulating NO products offset high-altitude hypoxia among Tibetans. Proc Natl Acad Sci USA. 2007;104:17593–17598. doi: 10.1073/pnas.0707462104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaelin WG. The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem Biophys Res Commun. 2005;338:627– 638. doi: 10.1016/j.bbrc.2005.08.165. [DOI] [PubMed] [Google Scholar]
- 53.Haase V. Hypoxia inducible factors in the kidney. Am J Physiol Renal Physiol. 2006;271:F1172– F1180. doi: 10.1152/ajprenal.00071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schofield C, Ratcliffe P. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2006;5:343– 354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 55.Khanna S, Roy S, Maurer M, Ratan R, Sen C. Oxygen sensitive reset of HIF transactivation response: Prolyl hydroxylases tune the biological normoxic set point. Free Rad Biol Med. 2006;40:2147– 2154. doi: 10.1016/j.freeradbiomed.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brezis M, Rosen S. Hypoxia of the renal medulla – its implications for disease. New Engl J Med. 1995;332:647– 655. doi: 10.1056/NEJM199503093321006. [DOI] [PubMed] [Google Scholar]
- 57.Tsai A, Johnson P, Intaglietta M. Oxygen gradients in the microcirculation. Physiol Rev. 2003;83:933– 963. doi: 10.1152/physrev.00034.2002. [DOI] [PubMed] [Google Scholar]
- 58.Marcinek D, Ciesielski W, Conley K, Schenkman K. Oxygen regulation and limitation to cellular respiration in mouse skeletal muscle in vivo. Am J Physiol Heart Circ Physiol. 2003;285:H1900– H1908. doi: 10.1152/ajpheart.00192.2003. [DOI] [PubMed] [Google Scholar]
- 59.Krishna M, English S, Yamada K, Yoo J, Murugesan R, Devasahayam N, Cook J, Goldman K, Ardenkjaer-Larsen J, Subramanian S, Mitchell J. Overhauser-emhanced magnetic resonance imaging for tumor oxymetry: coregistration of tumor anatomy and tissue oxygen concentration. Proc Natl Acad Sci USA. 2002;99:2216– 2221. doi: 10.1073/pnas.042671399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Atkuri K, Herzenberg L, Herzenberg L. Culturing at atmospheric oxygen levels impacts lymphocyte function. Proc Natl Acad Sci. 2004;102:3756– 3759. doi: 10.1073/pnas.0409910102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu X, Miller M, Joshi M, Thomas D, Lancaster J. Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc Natl Acad Sci USA. 1998;95:2175– 2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang B, Semenza G, Bauer C, Marti H. Hypoxia inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol Cell Physiol. 1995;271:C1172– C1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- 63.Abu-Soud HM, Ichimori K, Presta A, Stuehr DE. Electron transfer, oxygen binding and nitric oxide feedback inhibition in endothelial nitric-oxide synthase. J Biol Chem. 2000;275:17349– 17357. doi: 10.1074/jbc.M000050200. [DOI] [PubMed] [Google Scholar]
- 64.Richmond K, Shonat R, Lynch R, Johnson P. Critical pO2 of skeletal muscle in vivo. Am J Physiol Heart Circ Physiol. 1999;277:H1831– H1840. doi: 10.1152/ajpheart.1999.277.5.H1831. [DOI] [PubMed] [Google Scholar]
- 65.Kietzmann T, Jungermann K. Modulation by oxygen of zonal gene expression in liver studied in primary rat hepatocyte culture. Cell Biol Toxicol. 1997;13:243– 255. doi: 10.1023/a:1007427206391. [DOI] [PubMed] [Google Scholar]
- 66.Li J, French B, Wu Y, Venkatesh R, Montgomery R, Bardag F, Kitto J, Wrench S. Liver hypoxia and lack of recovery after reperfusion at high blood alcohol levels in the intragastric feeding model of alcohol liver disease. Exp Molec Pathol. 2004;77:184– 192. doi: 10.1016/j.yexmp.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 67.Fishman J, Joseph P, Floyd T, Mukherji B, Sloviter H. Oxygen-sensitive 19F NMR imaging of the vascular system in vivo. Mag Reson Imag. 1987;5:279– 285. doi: 10.1016/0730-725x(87)90005-1. [DOI] [PubMed] [Google Scholar]
- 68.Kimura S, Matsumoto K, Mineura K, Itoh T. A new technique for the mapping of oxygen tension on the brain surface. J Neurol Sci. 2007;258:60– 68. doi: 10.1016/j.jns.2007.02.032. [DOI] [PubMed] [Google Scholar]
- 69.Dunn J, Swartz H. In vivo electron paramagnetic resonance oxymetry with particulate materials. Methods. 2003;30:159– 166. doi: 10.1016/s1046-2023(03)00077-x. [DOI] [PubMed] [Google Scholar]
- 70.Liu K-J, Basic G, Hoopes P, Jiang J, Du H, Ou L-C, Dunn J, Swartz H. Assessment of cerebral pO2 by EPR oximetry in rodents: effects of anesthsia, ischemia and breathing gas. Brain Res. 1995;685:91– 98. doi: 10.1016/0006-8993(95)00413-k. [DOI] [PubMed] [Google Scholar]
- 71.James P, Bacic G, Grinberg O, Goda F, Dunn J, Jackson S, Swartz H. Endotoxin induced changes in intrarenal pO2, measured by in vivo electron paramagnetic resonance oximetry and magnetic resonance imaging. Free Rad Biol Med. 1996;21:25–34. doi: 10.1016/0891-5849(95)02221-x. [DOI] [PubMed] [Google Scholar]
- 72.Thomas S, Pratt R, Millard R, Samaratunga R, Shiferaw Y, McCoron A, Tan K-K. In vivo pO2 imaging in the porcine model with perfluorocarbon F-19 NMR at low field. Magn Reson Imag. 1996;14:103– 114. doi: 10.1016/0730-725x(95)02046-v. [DOI] [PubMed] [Google Scholar]
- 73.Walfridsson H, Lund N. Tissue oxygen pressure in normal myocardium and across the border zone during coronary artery occlusion in the pig. Effects of different arterial oxygen pressures. Basic Res Card. 1990;85:467– 480. doi: 10.1007/BF01931493. [DOI] [PubMed] [Google Scholar]
- 74.Lösse B, Schuchard S, Niederle N. The oxygen pressure histogram in the left ventricular myocardium of the dog. Pfluegers Arch. 1975;356:121– 132. doi: 10.1007/BF00584292. [DOI] [PubMed] [Google Scholar]
- 75.Bylund-Fellenius A, Walker P, Elander A, Holm S, Holm J, Schersten T. Energy metabolism in relation to oxygen partial pressure in human skeletal muscle during execise. Biochem J. 1981;200:247– 255. doi: 10.1042/bj2000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brahimi-Horn M, Pouyssegur J. Oxygen, a source of life and stress. FEBS lett. 2007;581:3582– 3591. doi: 10.1016/j.febslet.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 77.Kelm M. Nitric oxide metabolism and breakdown. Biochim Biophys Acta. 1999;1411:273– 289. doi: 10.1016/s0005-2728(99)00020-1. [DOI] [PubMed] [Google Scholar]
- 78.Vleeming W, van de Kuil A, te Biesbeek J, Meulenbelt J, Boink A. Effect of nitrite on blood pressure in anaesthized and free-moving rats. Food Chem Toxicol. 1997;35:615–619. doi: 10.1016/s0278-6915(97)00015-x. [DOI] [PubMed] [Google Scholar]
- 79.Modin A, Bjorne H, Herulf M, Alving K, Weitzberg E, Lundberg JO. Nitrite-derived nitric oxide: a possible mediator of ‘acidic-metabolic’ vasodilation. Acta Physiol Scand. 2001;171:9–16. doi: 10.1046/j.1365-201X.2001.00771.x. [DOI] [PubMed] [Google Scholar]
- 80.Cannon RO, 3rd, Schechter AN, Panza JA, Ognibene FP, Pease-Fye ME, Waclawiw MA, Shelhamer JH, Gladwin MT. Effects of inhaled nitric oxide on regional blood flow are consistent with intravascular nitric oxide delivery. J Clin Invest. 2001;108:279–287. doi: 10.1172/JCI12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McMahon TJ. Hemoglobin and nitric oxide. N Engl J Med. 2003;349:402–405. author reply 402–405. [PubMed] [Google Scholar]
- 82.Dejam A, Hunter C, Tremonti C, Pluta R, Hon Y, Grimes G, Partovi K, Pelletier M, Oldfield E, Cannon R, Schechter A, Gladwin M. Nitrite infusion in humans and nonhuman primates: endocrine effects, pharmacokinetics, and tolerance formation. Circulation. 2007;116:1821–1831. doi: 10.1161/CIRCULATIONAHA.107.712133. [DOI] [PubMed] [Google Scholar]
- 83.Angelo M, Singel DJ, Stamler JS. An S-nitrosothiol (SNO) synthase function of haemoglobin that utilizes nitrite as a substrate. Proc Natl Acad Sci USA. 2006;103:8366–8371. doi: 10.1073/pnas.0600942103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: role of nitric oxide and S-nitrosohemoglobin. Physiol Rev. 2005;67:99– 145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- 85.Maher AR, Milsom AB, Gunaruwan P, Abozguia K, Ahmed I, Weaver RA, Thomas P, Ashrafian H, Born G, James P, Frenneaux M. Hypoxic modulation of exogenous nitrite-induced vasodilation in humans. Circulation. 2008;117:670–677. doi: 10.1161/CIRCULATIONAHA.107.719591. [DOI] [PubMed] [Google Scholar]
- 86.Tsuchiya K, Kanematsu Y, Yoshizumi M, Ohnishi H, Kirima K, Izawa Y, Shikishima M, Ishida T, Kondo S, Kagami S, Takiguchi Y, Tamaki T. Nitrite is an alternative source of NO in vivo. Am J Heart Circ Physiol. 2005;288:H2163– H2170. doi: 10.1152/ajpheart.00525.2004. [DOI] [PubMed] [Google Scholar]
- 87.Kozlov A, Constantino G, Sobhian B, Szalay L, Umar F, Nohl H, Bahrami S, Redl H. Mechanisms of vasodilation induced by nitrite instillation in intestinal lumen: possible role of haemoglobin. Antiox Redox Signal. 2005;7:515–521. doi: 10.1089/ars.2005.7.515. [DOI] [PubMed] [Google Scholar]
- 88.Hunter C, Dejam A, Blood A, Shields H, Kim-Shapiro D, Machade R, Tarekegn S, Mulla N, Hopper A, Schechter A, Power G, Gladwin M. Inhaled nebulized nitrite is a hypoxia selective sensitive NO-dependent pulmonary vasodilator. Nat Med. 2004;10:1122– 1127. doi: 10.1038/nm1109. [DOI] [PubMed] [Google Scholar]
- 89.Pluta R, Dejam A, Grimes G, Gladwin M, Oldfield E. Nitrite infusions prevent delayed cerebral vasospasm in a primate model of subarachnoid hemorrhage. JAMA. 2005;293:1477– 1484. doi: 10.1001/jama.293.12.1477. [DOI] [PubMed] [Google Scholar]
- 90.Demoncheaux A, Higenbottam T, Foster P, Borland C, Smith A, Marriott H, Bee D, Akamine S, Davies M. Circulating nitrite anions are a directly acting vasodilator and are donors for nitric oxide. Clin Sci. 2002;102:77– 83. [PubMed] [Google Scholar]
- 91.Agvald P, Adding C, Artlich A, Persson M, Gustafsson L. Mechanisms of nitric oxide generation from nitroglycerin and endogenous sources during hypoxia in vivo. Brit J Pharmacol. 2002;135:373– 382. doi: 10.1038/sj.bjp.0704489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gauthier K. Hypoxia-induced vascular smooth muscle relaxation: Increased ATP sensitive K+ efflux or decreased voltage-sensitive Ca2+ influx? Am J Physiol Heart Circ Physiol. 2006;291:H24– H25. doi: 10.1152/ajpheart.00260.2006. [DOI] [PubMed] [Google Scholar]
- 93.Hasegawa A, Landahl H. Studies on the radioprotective action of sodium nitrite in mice. Rad Res. 1965;24:636– 649. [PubMed] [Google Scholar]
- 94.Tripatara P, Patel N, Webb A, Rathod K, Lecomte F, Mazzon E, Cuzzocrea S, Yaqoob M, Ahluwalia A, Thiemermann C. Nitrite-derived nitric oxide protects the rat kidney against IR injury in vivo: Role for xanthine oxidoreductase. J Am Soc Nephrol. 2007;18:570–580. doi: 10.1681/ASN.2006050450. [DOI] [PubMed] [Google Scholar]
- 95.Basireddy M, Isbell T, Teng X, Patel R, Agarwal A. Effects of sodium nitrite on ischemia reperfusion injury in the rat kidney. Am J Physiol Renal Physiol. 2006;290:F779– F786. doi: 10.1152/ajprenal.00334.2005. [DOI] [PubMed] [Google Scholar]
- 96.Jung KH, Chu K, Ko SY, Lee ST, Sinn DI, Park DK, Kim JM, Song EC, Kim M, Roh JK. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke. 2006;37:2744– 2750. doi: 10.1161/01.STR.0000245116.40163.1c. [DOI] [PubMed] [Google Scholar]
- 97.Schatlo B, Henning E, Pluta R, Latour L, Golpayegani N, Merrill M, Lewin N, Chen Y, Oldfield E. Nitrite does not provide additional protection to thrombolysis in a rat model of stroke with delayed reperfusion. J Cer Blood Flow Metab. 2007;28:482– 489. doi: 10.1038/sj.jcbfm.9600542. [DOI] [PubMed] [Google Scholar]
- 98.Berg A, Tremonti C, Vasquez V, Shiva S, Gladwin M, Sack M. Nitrite augments post-ischemic vascular function – A putative therapeutic preconditioning mimetic. Nitric Oxide. 2006;14:A48–A56. poster 116. [Google Scholar]
- 99.Lang J, Teng X, Chumley P, Crawford J, Isbell T, Patel R. Inhaled NO accelerates restoration of liver function in adults following orthotopic liver transplantation. J Clin Invest. 2007;117:2583– 2591. doi: 10.1172/JCI31892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shiva S, Sack M, Greer J, Duranski M, Ringwood L, Burwell L, Wang X, MAcArthurs P, Shoja A, Raghavachari N, Calvert J, Brookes P, Lefer D, Gladwin M. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kumar D, Branch B, Patillo C, Hood J, Thoma S, Simpson S, Illum S, Arora N, Chidlow J, Langston W, Teng X, Lefer D, Patel R, Kevil C. Chronic sodium nitrite therapy augments ischemia-induced angiogenesis and arteriogenesis. Proc Natl Acad Sci USA. 2008;105:7540– 7545. doi: 10.1073/pnas.0711480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kohn M, Melnick R, Frank Y, Portier C. Pharmacokinetics of sodium-nitrite induced methemoglobinemia in the rat. Drug Metab Disp. 2002;30:676– 683. doi: 10.1124/dmd.30.6.676. [DOI] [PubMed] [Google Scholar]
- 103.Titov V, Petrenko Yu. Nitrite-catalase interaction as an important element of nitrite toxicity. Biochem (Moscow) 2002;68:627–633. doi: 10.1023/a:1024609624652. [DOI] [PubMed] [Google Scholar]
- 104.Knaapen A, Schins R, Borm P, van Schooten F. Nitrite enhances neutrophil-induced DNA strand breakage in pulmonary epithelial cells by inhibition of myeloperoxidase. Carcinogenesis. 2005;26:1642– 1648. doi: 10.1093/carcin/bgi116. [DOI] [PubMed] [Google Scholar]
- 105.Hrabak A, Bajor T, Temesi A, Meszaros G. The inhibitory effect of nitrite, a stable product of nitric oxide (NO) formation, on arginase. FEBS Lett. 1996;390:203– 206. doi: 10.1016/0014-5793(96)00659-x. [DOI] [PubMed] [Google Scholar]
- 106.Veselik D, Divekar S, Dakshanamurthy S, Storchan G, Turner J, Graham K, Huang L, Stoica A, Martin M-B. Activation of estrogen receptor-α by the anion nitrite. Cancer Res. 2008;68:3950– 3958. doi: 10.1158/0008-5472.CAN-07-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bryan NS, Fernandez BO, Garcia-Saura MF, Bauer SM, Milsom AB, Rassaf T, Maloney R, Bharti A, Rodriguez J, Feelisch M. Nitrite is signalling molecule and regulator of gene expression in mammalian tissue. Nat Chem Biol. 2005;1:290–297. doi: 10.1038/nchembio734. [DOI] [PubMed] [Google Scholar]
- 108.Butler A, Feelisch M. Therapeutic uses of inorganic nitrite and nitrate. From the past to the future. Circulation. 2008;117:2151– 2159. doi: 10.1161/CIRCULATIONAHA.107.753814. [DOI] [PubMed] [Google Scholar]
- 109.Brooks J. The action of nitrite on haemoglobin in the absence of oxygen. Proc Royal Soc Med. 1937;123:368–382. [Google Scholar]
- 110.Doyle M, Pickering R, de Weert T, Hoekstra J, Pater D. Oxidation of nitrogen oxides by bound dioxygen in hemoproteins. J Biol chem. 1981;256:12393– 12398. [Google Scholar]
- 111.Huang Z, Shiva S, Kim-Shapiro D, Patel R, Ringwood L, Irby C, Huang K, Ho C, Hogg N, Schechter A, Gladwin M. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest. 2005;115:2099– 2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang K, Keszler A, Patel N, Patel R, Gladwin M, Kim-Shapiro D, Hogg N. The reaction between nitrite and deoxyhemoglobin: Reassessment of the reaction kinetics and stochiometry. J Biol Chem. 2005;280:31126–31131. doi: 10.1074/jbc.M501496200. [DOI] [PubMed] [Google Scholar]
- 113.Gladwin M, Raat N, Shiva S, Dezfulian C, Hogg N, Kim-Shapiro DB, Patel RP. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signalling, cytoprotection, and vasodilation. Am J Physiol Heart Circ Physiol. 2006;291:H2026–H2035. doi: 10.1152/ajpheart.00407.2006. [DOI] [PubMed] [Google Scholar]
- 114.Grubina R, Huang Z, Shiva S, Joshi M, Azarov I, Basu S, Ringwood L, Jiang A, Hogg N, Kim-Shapiro D, Gladwin M. Concerted nitric oxide formation and release from the simultaneous reactions of nitrite with deoxy- and oxy-hemoglobin. J Biol Chem. 2007;282:12916– 12927. doi: 10.1074/jbc.M700546200. [DOI] [PubMed] [Google Scholar]
- 115.Eich R, Li T, Lemon D, Doherty D, Curry S, Aitken J, Mathews A, Johnson K, Smith R, Phillips G, jr, Olson J. Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry. 1996;35:6976–6983. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- 116.Herold S, Exner M, Nauser T. Kinetic and mechanistic studies of the NO-mediated oxidation of oxymyoglobin and oxyhemoglobin. Biochemistry. 2001;40:3385–3395. doi: 10.1021/bi002407m. [DOI] [PubMed] [Google Scholar]
- 117.Huang KT, Han TH, Hyduke DR, Vaughn MW, van Herle H, Hein TW, Zhang C, Kuo L, Liao JC. Modulation of nitric oxide bioavailability by erythrocytes. Proc Natl Acad Sci USA. 2001;98:11771–11776. doi: 10.1073/pnas.201276698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim-Shapiro D. Hemoglobin-nitric oxide cooperativity: Is NO the third respiratory ligand? Free Rad Biol Med. 2004;36:402–412. doi: 10.1016/j.freeradbiomed.2003.10.030. [DOI] [PubMed] [Google Scholar]
- 119.Morris RJ, Gibson QH. The role of diffusion in limiting the rate of ligand binding to hemoglobin. J Biol Chem. 1980;255:8050–8053. [PubMed] [Google Scholar]
- 120.Kim-Shapiro D, Schechter A, Gladwin M. Unraveling the reactions of nitric oxide, nitrite and hemoglobin in physiology and therapeutics. Arterioscl Thromb Vasc Biol. 2006;26:697–705. doi: 10.1161/01.ATV.0000204350.44226.9a. [DOI] [PubMed] [Google Scholar]
- 121.Jeffers A, Xu X, Cho M, Hogg N, Patel RP, Gladwin MT, Kim-Shapiro DB. Hemoglobin mediated nitrite activation of soluble guanylyl cyclase. Comp Physiol Biochem. 2005;A142:130–135. doi: 10.1016/j.cbpb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 122.Robinson JM, Lancaster JR., Jr Hemoglobin-mediated, hypoxia-induced vasodilation via nitric oxide: mechanism(s) and physiologic versus pathophysiologic relevance. Am J Respir Cell Mol Biol. 2005;32:257– 261. doi: 10.1165/rcmb.F292. [DOI] [PubMed] [Google Scholar]
- 123.Basu S, Grubina R, Huang J, Conradie J, Huang Zhi, Jeffers Anne, Jiang A, He X, Azarov I, Seibert R, Mehta1 A, Patel R, King B, Hogg N, Ghosh A, Gladwin M, Kim-Shapiro D. Catalytic generation of N2O3 by a concerted nitrite reductase and anhydrase activity of hemoglobin. Nat. Chem. Biol. 2007;3:785–794. doi: 10.1038/nchembio.2007.46. [DOI] [PubMed] [Google Scholar]
- 124.Wittenberg JB, Wittenberg BA. Myoglobin function reassessed. J Exp Biol. 2003;206:2011–2020. doi: 10.1242/jeb.00243. [DOI] [PubMed] [Google Scholar]
- 125.Guyton GP, Stanek KS, Schneider RC, Hochachka PW, Hurford WE, Zapol DG, Liggins GC, Zapol WM. Myoglobin saturation in free-diving Weddell seals. J Appl Physiol. 1995;79:1148–1155. doi: 10.1152/jappl.1995.79.4.1148. [DOI] [PubMed] [Google Scholar]
- 126.Gimenez M, Sanderson RJ, Reiss OK, Banchero N. Effects of altitude on myoglobin and mitochondrial protein in canine skeletal muscle. Respiration. 1977;34:171–176. doi: 10.1159/000193811. [DOI] [PubMed] [Google Scholar]
- 127.Wittenberg JB. Myoglobin-facilitated oxygen diffusion: role of myoglobin in oxygen entry into muscle. Physiol Rev. 1970;50:559–636. doi: 10.1152/physrev.1970.50.4.559. [DOI] [PubMed] [Google Scholar]
- 128.Taylor DJ, Matthews PM, Radda GK. Myoglobin-dependent oxidative metabolism in the hypoxic rat heart. Respir Physiol. 1986;63:275–283. doi: 10.1016/0034-5687(86)90095-2. [DOI] [PubMed] [Google Scholar]
- 129.van Beek-Harmsen BJ, Bedekam MA, Feenstra HM, Visser FC, van der Laarse WJ. Determination of myoglobin concentration and oxidative capacity in crystat sections of human and rat skeletal muscle fibres and rat cardiomyocytes. Histochem Cell Biol. 2004;121:335– 342. doi: 10.1007/s00418-004-0641-9. [DOI] [PubMed] [Google Scholar]
- 130.Garry DJ, Ordway GA, Lorenz JN, Radford NB, Chin ER, Grange RW, Bassel-Duby R, Williams RS. Mice without myoglobin. Nature. 1998;395:905–908. doi: 10.1038/27681. [DOI] [PubMed] [Google Scholar]
- 131.Gödecke A, Flögel U, Zanger K, Ding Z, Hirchenhain J, Decking UK, Schrader J. Disruption of myoglobin in mice induces multiple compensatory mechanisms. Proc Natl Acad Sci USA. 1999;96:10495–10500. doi: 10.1073/pnas.96.18.10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Flögel U, Laussmann T, Gödecke A, Abanador N, Schäfers M, Fingas CD, Metzger S, Levkau B, Jacoby C, Schrader J. Lack of myoglobin causes a switch in cardiac substrate selection. Circ Res. 2005;96:e68–75. doi: 10.1161/01.RES.0000165481.36288.d2. [DOI] [PubMed] [Google Scholar]
- 133.Rassaf T, Poll LW, Brouzos P, Lauer T, Totzeck M, Kleinbongard P, Gharini P, Andersen K, Schulz R, Heusch G, Mödder U, Kelm M. Positive effects of nitric oxide on left ventricular function in humans. Eur Heart J. 2006;27:1699–1705. doi: 10.1093/eurheartj/ehl096. [DOI] [PubMed] [Google Scholar]
- 134.Rassaf T, Flögel U, Drexhage C, Hendgen-Cotta U, Kelm M, Schrader J. Nitrite reductase function of deoxymyoglobin: oxygen sensor and regulator of cardiac energetics and function. Circ Res. 2007;100:1749–1754. doi: 10.1161/CIRCRESAHA.107.152488. [DOI] [PubMed] [Google Scholar]
- 135.Ishibashi T, Hamaguchi M, Kato K, Kawada T, Ohta H, Sasage H, Imai S. Relationship between myoglobin contents and increases in cyclic GMP produced by glyceryl trinitrate and nitric oxide in rabbit aorta, right atrium and papillary muscle. Naunyn Schmiedebergs Arch Pharmacol. 1993;347:553– 561. doi: 10.1007/BF00166750. [DOI] [PubMed] [Google Scholar]
- 136.Flögel U, Merx MW, Gödecke A, Decking UK, Schrader J. Myoglobin: A scavenger of bioactive NO. Proc Natl Acad Sci USA. 2001;98:735–740. doi: 10.1073/pnas.011460298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Brown GC. Nitric oxide as a competitive inhibitor of oxygen consumption in the mitochondrial respiratory chain. Acta Physiol Scand. 2000;168:667–674. doi: 10.1046/j.1365-201x.2000.00718.x. [DOI] [PubMed] [Google Scholar]
- 138.Heusch G, Schulz R, Rahimtoola SH. Myocardial hibernation: a delicate balance. Am J Physiol Heart Circ Physiol. 2005;288:H984–999. doi: 10.1152/ajpheart.01109.2004. [DOI] [PubMed] [Google Scholar]
- 139.Hendgen-Cotta U, Merx M, Shiva S, Schmitz J, Stefanie Becher S, Klare J, Steinhoff H-J, Goedecke A, Schrader J, Gladwin M, Kelm M, Rassaf T. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2008 doi: 10.1073/pnas.0801336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Richardson RS, Duteil S, Wary C, Wray DW, Hoff J, Carlier PG. Human skeletal muscle intracellular oxygenation: the impact of ambient oxygen availability. J Physiol. 2006;571:415– 424. doi: 10.1113/jphysiol.2005.102327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Richardson RS, Newcomer SC, Noyszewski EA. Skeletal muscle intracellular pO2 assessed by myoglobin desaturation: response to graded exercise. J Appl Physiol. 2001;91:2679– 2685. doi: 10.1152/jappl.2001.91.6.2679. [DOI] [PubMed] [Google Scholar]
- 142.Zhang Z, Naughton D, Winyard PG, Benjamin N, Blake DR, Symons MC. Generation of nitric oxide by a nitrite reductase activity of xanthine oxidase: a potential pathway for nitric oxide formation in the absence of nitric oxide synthase activity. Biochem Biophys Res Comm. 1998;249:767–772. doi: 10.1006/bbrc.1998.9226. erratum in Biochem Biophys Res Comm 1998; 251:667. [DOI] [PubMed] [Google Scholar]
- 143.Zhang Z, Naughton DP, Blake DR, Benjamin N, Stevens CR, Winyard PG, Symons MC, Harrison R. Human xanthine oxidase converts nitrite ions into nitric oxide (NO) Biochem Soc Trans. 1997;25:524S. doi: 10.1042/bst025524s. [DOI] [PubMed] [Google Scholar]
- 144.Godber BL, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, Harrison R. Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J Biol Chem. 2000;275:7757– 7763. doi: 10.1074/jbc.275.11.7757. [DOI] [PubMed] [Google Scholar]
- 145.Hille R, Stewart RC. The inhibition of xanthine oxidase by 8-bromoxanthine. J Biol Chem. 1984;259:1570–1576. [PubMed] [Google Scholar]
- 146.Rubbo H, Radi R, Prodanov E. Substrate inhibition of xanthine oxidase and its influence on superoxide radical production. Biochim Biophys Acta. 1991;1074:386–391. doi: 10.1016/0304-4165(91)90089-y. [DOI] [PubMed] [Google Scholar]
- 147.Godber BLJ, Doel JJ, Sapkota GP, Blake DR, Stevens CR, Eisenthal R, Harrison R. Reduction of Nitrite to Nitric Oxide Catalyzed by Xanthine Oxidoreductase. J Biol Chem. 2000;275:7757–7763. doi: 10.1074/jbc.275.11.7757. [DOI] [PubMed] [Google Scholar]
- 148.Evans T, Carpenter A, Silva A, Cohen J. Inhibition of nitric oxide synthase in experimental gram-negative sepsis. J Infect Dis. 1994;169:343–349. doi: 10.1093/infdis/169.2.343. [DOI] [PubMed] [Google Scholar]
- 149.Evans TJ, Buttery LD, Carpenter A, Springall DR, Polak JM, Cohen J. Cytokine-treated human neutrophils contain inducible nitric oxide synthase that produces nitration of ingested bacteria. Proc Natl Acad Sci USA. 1996;93:9553–9558. doi: 10.1073/pnas.93.18.9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Farrell AJ, Blake DR. Nitric oxide. Ann Rheum Dis. 1996;55:7– 20. doi: 10.1136/ard.55.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Farrell AJ, Blake DR, Palmer RM, Moncada S. Increased concentrations of nitrite in synovial fluid and serum samples suggest increased nitric oxide synthesis in rheumatic diseases. Ann Rheum Dis. 1992;51:1219–1222. doi: 10.1136/ard.51.11.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hukkanen M, Hughes FJ, Buttery LD, Gross SS, Evans TJ, Seddon S, Riveros-Moreno V, Macintyre I, Polak JM. Cytokine-stimulated expression of inducible nitric oxide synthase by mouse, rat, and human osteoblast-like cells and its functional role in osteoblast metabolic activity. Endocrin. 1995;136:5445–5453. doi: 10.1210/endo.136.12.7588294. [DOI] [PubMed] [Google Scholar]
- 153.Torre D, Ferrario G, Speranza F, Martegani R, Zeroli C. Increased levels of nitrite in the sera of children infected with human immunodeficiency virus type 1. Clin Infect Dis. 1996;22:650–653. doi: 10.1093/clinids/22.4.650. [DOI] [PubMed] [Google Scholar]
- 154.Torre D, Ferrario G, Speranza F, Orani A, Fiori GP, Zeroli C. Serum concentrations of nitrite in patients with HIV-1 infection. J Clin Pathol. 1996;49:574–576. doi: 10.1136/jcp.49.7.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Torre D, Zeroli C, Ferrario G, Pugliese A, Speranza F, Orani A, Casari S, Bassi P, Poggio A, Carosi GP, Fiori GP. Levels of nitric oxide, gamma interferon and interleukin-12 in AIDS patients with toxoplasmic encephalitis. Infection. 1999;27:218–220. doi: 10.1007/BF02561533. [DOI] [PubMed] [Google Scholar]
- 156.Li H, Samouilov A, Liu X, Zweier JL. Characterization of the effects of oxygen on xanthine oxidase-mediated nitric oxide formation. J Biol Chem. 2004;279:16939–16946. doi: 10.1074/jbc.M314336200. [DOI] [PubMed] [Google Scholar]
- 157.Lymar S, Hirst J. Rapid reaction between peroxynitrite ion and carbon dioxide. Implications for biological activity. J Am Chem Soc. 1995;177:8867– 8868. [Google Scholar]
- 158.Denicola A, Freeman B, Trujillo M, Radi R. Peroxynitrite reaction with carbon dioxide/bicarbonate: Kinetics and influence on peroxynitrite mediated oxidations. Arch Biochem Biophys. 1996;333:49– 58. doi: 10.1006/abbi.1996.0363. [DOI] [PubMed] [Google Scholar]
- 159.Meli R, Nauser T, Latal P, Koppenol WH. Reaction of peroxynitrite with carbon dioxide: intermediates and determination of the yield of CO3•− and NO2•. J Biol Inorg Chem. 2002;7:31–36. doi: 10.1007/s007750100262. [DOI] [PubMed] [Google Scholar]
- 160.Lee CI, Liu X, Zweier JL. Regulation of xanthine oxidase by nitric oxide and peroxynitrite. J Biol Chem. 2000;275:9369– 9376. doi: 10.1074/jbc.275.13.9369. [DOI] [PubMed] [Google Scholar]
- 161.Kirsch M, de Groot H. Reaction of peroxynitrite with reduced nicotinamide nucleotides, the formation of hydrogen peroxide. J Biol Chem. 1999;274:24664– 24670. doi: 10.1074/jbc.274.35.24664. [DOI] [PubMed] [Google Scholar]
- 162.Lynch SM, Frei B, Morrow JD, Roberts LJ, 2nd, Xu A, Jackson T, Reyna R, Klevay LM, Vita JA, Keaney JF., Jr Vascular superoxide dismutase deficiency impairs endothelial vasodilator function through direct inactivation of nitric oxide and increased lipid peroxidation. Arter Thromb Vasc Biol. 1997;17:2975–2981. doi: 10.1161/01.atv.17.11.2975. [DOI] [PubMed] [Google Scholar]
- 163.Salem JE, Saidel GM, Stanley WC, Cabrera ME. Mechanistic model of myocardial energy metabolism under normal and ischemic conditions. Ann Biomed Engin. 2002;30:202–216. doi: 10.1114/1.1454133. [DOI] [PubMed] [Google Scholar]
- 164.Hassoun PM, Yu FS, Shedd AL, Zulueta JJ, Thannickal VJ, Lanzillo JJ, Fanburg BL. Regulation of endothelial cell xanthine dehydrogenase xanthine oxidase gene expression by oxygen tension. Am J Physiol. 1994;266:L163–171. doi: 10.1152/ajplung.1994.266.2.L163. [DOI] [PubMed] [Google Scholar]
- 165.Kayyali US, Donaldson C, Huang H, Abdelnour R, Hassoun PM. Phosphorylation of xanthine dehydrogenase/oxidase in hypoxia. J Biol Chem. 2001;276:14359–14365. doi: 10.1074/jbc.M010100200. [DOI] [PubMed] [Google Scholar]
- 166.Poss WB, Huecksteadt TP, Panus PC, Freeman BA, Hoidal JR. Regulation of xanthine dehydrogenase and xanthine oxidase activity by hypoxia. Am J Physiol. 1996;270:L941–946. doi: 10.1152/ajplung.1996.270.6.L941. [DOI] [PubMed] [Google Scholar]
- 167.Spiekermann S, Landmesser U, Dikalov S, Bredt M, Gamez G, Tatge H, Reepschlager N, Hornig B, Drexler H, Harrison DG. Electron spin resonance characterization of vascular xanthine and NAD(P)H oxidase activity in patients with coronary artery disease: relation to endothelium-dependent vasodilation. Circulation. 2003;107:1383–1389. doi: 10.1161/01.cir.0000056762.69302.46. [DOI] [PubMed] [Google Scholar]
- 168.Li H, Cui H, Kundu T, Alzawahra W, Zweier J. Nitric oxide production from nitrite occurs primarily in tissues, not in blood. J Biol Chem. 2008;283:17855– 17863. doi: 10.1074/jbc.M801785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nelson DR, Kamataki T, Waxman DJ, Guengerich FP, Estabrook RW, Feyereisen R, Gonzalez FJ, Coon MJ, Gunsalus IC, Gotoh O. The P450 superfamily: update on new sequences, gene mapping, accession numbers, early trivial names of enzymes, and nomenclature DNA. Cell Biol. 1993;12:1–51. doi: 10.1089/dna.1993.12.1. [DOI] [PubMed] [Google Scholar]
- 170.Nakahara K, Tanimoto T, Hatano K, Usuda K, Shoun H. Cytochrome P-450 55A1 (P-450dNIR) acts as nitric oxide reductase employing NADH as the direct electron donor. J Biol Chem. 1993;268:8350–8355. [PubMed] [Google Scholar]
- 171.Li H, Liu X, Cui H, Chen YR, Cardounel AJ, Zweier JL. Characterization of the mechanism of cytochrome P450 reductase-cytochrome P450-mediated nitric oxide and nitrosothiol generation from organic nitrates. J Biol Chem. 2006;281:12546– 12554. doi: 10.1074/jbc.M511803200. [DOI] [PubMed] [Google Scholar]
- 172.Knowles R. Denitrification. Microbiol Rev. 1982;46:43–70. doi: 10.1128/mr.46.1.43-70.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Shoun H, Suyama W, Yasui T. Soluble, nitrate/nitrite-inducible cytochrome P-450 of the fungus, Fusarium Oxysporum. FEBS Lett. 1989;244:11–14. doi: 10.1016/0014-5793(89)81151-2. [DOI] [PubMed] [Google Scholar]
- 174.Zumft WG. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev. 1997;61:533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tomura D, Obika K, Fukamizu A, Shoun H. Nitric oxide reductase cytochrome P-450 gene, CYP 55, of the fungus Fusarium oxysporum containing a potential binding-site for FNR, the transcription factor involved in the regulation of anaerobic growth of Escherichia coli. J Biochem. 1994;116:88–94. doi: 10.1093/oxfordjournals.jbchem.a124508. [DOI] [PubMed] [Google Scholar]
- 176.Kizawa H, Tomura D, Oda M, Fukamizu A, Hoshino T, Gotoh O, Yasui T, Shoun H. Nucleotide sequence of the unique nitrate/nitrite-inducible cytochrome P-450 cDNA from Fusarium oxysporum. J Biol Chem. 1991;266:10632–10637. [PubMed] [Google Scholar]
- 177.Raikhman LM, Annaev BB. Nature of the free-radical states in microsomes. Biofizika (Moscow) 1971;16:1135–1137. [PubMed] [Google Scholar]
- 178.Duthu GS, Shertzer HG. Effect of nitrite on rabbit liver mixed-function oxidase activity. Drug Metab Dispos. 1979;7:263–269. [PubMed] [Google Scholar]
- 179.Reutov VP, Sorokina EG. NO-synthase and nitrite-reductase components of nitric oxide cycle. Biochemistry (Moscow) 1998;63:874–884. [PubMed] [Google Scholar]
- 180.Bennett BM, McDonald BJ, St James MJ. Hepatic cytochrome P-450-mediated activation of rat aortic guanylyl cyclase by glyceryl trinitrate. J Pharmacol Exp Ther. 1992;261:716–723. [PubMed] [Google Scholar]
- 181.Delaforge M, Servent D, Wirsta P, Ducrocq C, Mansuy D, Lenfant M. Particular ability of cytochrome P-450 CYP3A to reduce glyceryl trinitrate in rat liver microsomes: subsequent formation of nitric oxide. Chem Biol Interact. 1993;86:103–117. doi: 10.1016/0009-2797(93)90115-f. [DOI] [PubMed] [Google Scholar]
- 182.Servent D, Delaforge M, Ducrocq C, Mansuy D, Lenfant M. Nitric oxide formation during microsomal hepatic denitration of glyceryl trinitrate: involvement of cytochrome P-450. Biochem Biophys Res Commun. 1989;163:1210–1216. doi: 10.1016/0006-291x(89)91106-6. [DOI] [PubMed] [Google Scholar]
- 183.Feelisch M, Kotsonis P, Siebe J, Clement B, Schmidt H. The soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a] quinoxalin-1-one is a nonselective heme protein inhibitor of nitric oxide synthase and other cytochrome P-450 enzymes involved in nitric oxide donor bioactivation. Mol Pharmacol. 1999;56:243–53. doi: 10.1124/mol.56.2.243. [DOI] [PubMed] [Google Scholar]
- 184.Kurzban GP, Howarth J, Palmer G, Strobel HW. NADPH-cytochrome P-450 reductase. Physical properties and redox behavior in the absence of the FAD moiety. J Biol Chem. 1990;265:12272–12279. [PubMed] [Google Scholar]
- 185.Vermilion JL, Ballou DP, Massey V, Coon MJ. Separate roles for FMN and FAD in catalysis by liver microsomal NADPH-cytochrome P-450 reductase. J Biol Chem. 1981;256:266–277. [PubMed] [Google Scholar]
- 186.Khatsenko OG, Gross SS, Rifkind AB, Vane JR. Nitric oxide is a mediator of the decrease in cytochrome P450-dependent metabolism caused by immunostimulants. Proc Natl Acad Sci USA. 1993;90:11147–11151. doi: 10.1073/pnas.90.23.11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vanin A, Huisman A, van Faassen E. Iron dithiocarbamates as spin trap for nitric oxide detection: Pitfalls and successes. Meth Enzymol. 2002;359:27– 42. doi: 10.1016/s0076-6879(02)59169-2. [DOI] [PubMed] [Google Scholar]
- 188.Govers R, Rabelink TJ. Cellular regulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol. 2001;280:F193– F206. doi: 10.1152/ajprenal.2001.280.2.F193. [DOI] [PubMed] [Google Scholar]
- 189.Stroes E, Rabelink T, van Faassen E. Vascular Protection: Molecular Mechanisms, novel therapeutic Principles and Clinical Applications. Ch. 3. Taylor and Francis; 2002. Uncoupling of endothelial nitric oxide synthase: A molecular basis for atherosclerosis. [Google Scholar]
- 190.Stroes E, Hijmering M, van Zandvoort M, Wever R, Rabelink T, van Faassen E. Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett. 1998;438:161– 164. doi: 10.1016/s0014-5793(98)01292-7. [DOI] [PubMed] [Google Scholar]
- 191.Ohnishi S. Measurement of NO using electron paramagnetic resonance spectroscopy. Meth Mol Biol. 1996;100:129– 153. doi: 10.1385/1-59259-749-1:129. [DOI] [PubMed] [Google Scholar]
- 192.Vanin A, Poltorakov A, Mikoyan V, Kubrina L, van Faassen E. Why iron-dithiocarbamates ensure detection of nitric oxide in cells and tissues. Nitric Oxide. 2006;15:295– 311. doi: 10.1016/j.niox.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 193.Vanin A, Bevers L, Mikoyan V, Poltorakov A, Kubrina L, van Faassen E. Reduction enhances yields of nitric oxide trapping in biological systems. Nitric Oxide. 2007;16:71– 81. doi: 10.1016/j.niox.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 194.Bevers LM, Braam B, Post J-A, van Zonneveld A-J, Rabelink T, Koomans H, Verhaar M, Joles J. Tetrahydrobiopterin but not L-arginine decreases NO synthase uncoupling in cells expressing high levels of endothelial NO synthase. Hypertension. 2006;47:193– 94. doi: 10.1161/01.HYP.0000196735.85398.0e. [DOI] [PubMed] [Google Scholar]
- 195.Mikula I, Durocher S, Martasek P, Mutus B, Slama-Schwok A. doi: 10.1042/BJ20080987. submitted. [DOI] [PubMed] [Google Scholar]
- 196.Endres M, Laufs U, Lia JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci. 2004;27:283– 289. doi: 10.1016/j.tins.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 197.Pozo-Navas B, Stessel H, Wolkart G, Brunner F. Role of myocardial nitric oxide in diabetic ischemia-reperfusion dysfunction: Studies in mice with myocyte-specific overexpression of endothelial nitric-oxide synthase. J Pharmacol Exp Theor. 2006;319:729– 738. doi: 10.1124/jpet.106.107854. [DOI] [PubMed] [Google Scholar]
- 198.Kozlov AV, Staniek K, Nohl H. Nitrite reductase activity is a novel function of mammalian mitochondria. FEBS Lett. 1999;454:127–130. doi: 10.1016/s0014-5793(99)00788-7. [DOI] [PubMed] [Google Scholar]
- 199.Samouilov A, Woldman YY, Zweier JL, Khramtsov VV. Magnetic resonance study of the transmembrane nitrite diffusion. Nitric Oxide. 2007;16:362–370. doi: 10.1016/j.niox.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kozlov AV, Dietrich B, Nohl H. Various intracellular compartments cooperate in the release of nitric oxide from glycerol trinitrate in liver. Br J Pharmacol. 2003;139:989–997. doi: 10.1038/sj.bjp.0705323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Paitian NA, Markossian KA, Nalbandyan RM. The effect of nitrite on cytochrome oxidase. Biochem Biophys Res Commun. 1985;133:1104–1111. doi: 10.1016/0006-291x(85)91250-1. [DOI] [PubMed] [Google Scholar]
- 202.Nohl H, Gille L, Kozlov A, Staniek K. Are mitochondria a spontaneous and permanent source of reactive oxygen species? Redox Rep. 2003;8:135–141. doi: 10.1179/135100003225001502. [DOI] [PubMed] [Google Scholar]
- 203.Staniek K, Gille L, Kozlov AV, Nohl H. Mitochondrial superoxide radical formation is controlled by electron bifurcation to the high and low potential pathways. Free Rad Res. 2002;36:381–387. doi: 10.1080/10715760290021225. [DOI] [PubMed] [Google Scholar]
- 204.Piknova B, Gladwin M, Schechter A, Hogg N. Electron paramagnetic resonance analysis of nitrosylhemoglobin in humans during NO inhalation. J Biol Chem. 2005;280:40583– 40588. doi: 10.1074/jbc.M506292200. [DOI] [PubMed] [Google Scholar]
- 205.Dalsgaard T, Simonsen U, Fago A. Nitrite-dependent vasodilation is facilitated by hypoxia and is independent of known NO-generating nitrite reductase activities. Am J Physiol. 2007;292:H3072–H3077. doi: 10.1152/ajpheart.01298.2006. [DOI] [PubMed] [Google Scholar]
- 206.for a recent review on physiological effects of peroxynitrite cf. Pacher P, Beckman J. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315– 424. doi: 10.1152/physrev.00029.2006.
- 207.Brown GC, Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim Biophys Acta. 2004;1658:44– 49. doi: 10.1016/j.bbabio.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 208.Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- 209.Bustamante J, Bersier G, Romero M, Badin RA, Boveris A. Nitric oxide production and mitochondrial dysfunction during rat thymocyte apoptosis. Arch Biochem Biophys. 2000;376:239–247. doi: 10.1006/abbi.2000.1716. [DOI] [PubMed] [Google Scholar]
- 210.Walters CL, Burger IH, Jewell GG, Lewis DF, Parke DV. Mitochondrial enzyme pathways and their possible role during curing. Z Lebensm Unters Forsch. 1975;158:193–203. doi: 10.1007/BF01261559. [DOI] [PubMed] [Google Scholar]
- 211.Nohl H, Staniek K, Kozlov AV. The existence and significance of a mitochondrial nitrite reductase. Redox Rep. 2005;10:281–286. doi: 10.1179/135100005X83707. [DOI] [PubMed] [Google Scholar]
- 212.Benjamin N, O’Driscoll F, Dougall H, Duncan C, Smith L, Golden M, McKenzie H. Stomach NO synthesis. Nature. 1994;368(6471):502. doi: 10.1038/368502a0. [DOI] [PubMed] [Google Scholar]
- 213.Larsen F, Weitzberg E, Lundberg J, Ekblom B. Effect of dietary nitrate on oxygen cost during exercise. Acta Physiol. 2007;191:59– 66. doi: 10.1111/j.1748-1716.2007.01713.x. [DOI] [PubMed] [Google Scholar]
- 214.Appel LJ, Moore TJ, Obarzanek E, Vollmer WM, Svetkey LP, Sacks FM, Bray GA, Vogt TM, Cutler JA, Windhauser MM, Lin PH, Karanja N. A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med. 1997;336:1117–24. doi: 10.1056/NEJM199704173361601. [DOI] [PubMed] [Google Scholar]
- 215.Webb AJ, Patel N, Loukogeorgakis S, Okorie M, Aboud Z, Misra S, Rashid R, Miall P, Deanfield J, Benjamin N. Acute Blood Pressure Lowering, Vasoprotective, and Antiplatelet Properties of Dietary Nitrate via Bioconversion to Nitrite. Hypertension. 2008;51:784– 790. doi: 10.1161/HYPERTENSIONAHA.107.103523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lundberg J, Feelisch M, Bjorne A, Jansson A, Weitzberg E. Cardioprotective effects of vegetables: Is nitrate the answer? Nitric Oxide. 2006;15:359– 362. doi: 10.1016/j.niox.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 217.Bryan NS, Calvert JW, Elrod JW, Gundewar S, Ji SY, Lefer DJ. Dietary nitrite supplementation protects against myocardial ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2007;104:19144–19149. doi: 10.1073/pnas.0706579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Miyoshi M, Kasahara E, Park AM, Hiramoto PK, Minamiyama Y, Takemura S, Sato EF, Inoue M. Dietary nitrate inhibits stress-induced gastric mucosal injury in the rat. Free Rad Res. 2003;37:85–90. doi: 10.1080/1071576021000086632. [DOI] [PubMed] [Google Scholar]
- 219.Jansson EA, Petersson J, Reinders C, Sobko T, Bjorne H, Phillipson M, Weitzberg E, Holm L, Lundberg JO. Protection from nonsteroidal anti-inflammatory drug (NSAID)-induced gastric ulcers by dietary nitrate. Free Rad Biol Med. 2007;42:510–508. doi: 10.1016/j.freeradbiomed.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 220.Jobgen WS, Fried SK, Fu WJ, Meininger C, Wu G. Regulatory role for the arginine–nitric oxide pathway in metabolism of energy substrates. J Nutr Biochem. 2006;17:571–588. doi: 10.1016/j.jnutbio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 221.Bolaños JP, Peuchen S, Heales SJR, Land JM, Clark JB. Nitric oxide-mediated inhibition of the mitochondrial respiratory chain in cultured astrocytes. J Neurochem. 1994;63:910–916. doi: 10.1046/j.1471-4159.1994.63030910.x. [DOI] [PubMed] [Google Scholar]
- 222.Brown GC, Cooper C. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295– 298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 223.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 224.Doyle MP, Pickering RA, Dykstra RL, Nelson CL, Boyer RF. Involvement of peroxide and superoxide in the oxidation of hemoglobin by nitrite. Biochem Biophys Res Commun. 1982;105:127–132. doi: 10.1016/s0006-291x(82)80020-x. [DOI] [PubMed] [Google Scholar]
- 225.Gamgee A. Researches on the blood. On the action of nitrites on the blood. Philos Trans R Soc London. 1868;158:589–625. [Google Scholar]
- 226.Kosaka H, Imaizumi K, Tyuma I. Mechanism of autocatalytic oxidation of oxyhemoglobin by nitrite. An intermediate detected by electron spin resonance. Biochim Biophys Acta. 1982;702:237–241. doi: 10.1016/0167-4838(82)90508-8. [DOI] [PubMed] [Google Scholar]
- 227.Rodkey FL. A mechanism for the conversion of oxyhemoglobin to methemoglobin by nitrite. Clin Chem. 1976;22:1986–1990. [PubMed] [Google Scholar]
- 228.Kim-Shapiro DB, Gladwin MT, Patel RP, Hogg N. The reaction between nitrite and hemoglobin: the role of nitrite in hemoglobin-mediated hypoxic vasodilation. J Inorg Biochem. 2005;99:237–246. doi: 10.1016/j.jinorgbio.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 229.Keszler A, Piknova B, Schechter AN, Hogg N. The reaction between nitrite and oxyhemoglobin: A mechanistic study. J Biol Chem. 2008;283:9615–9622. doi: 10.1074/jbc.M705630200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Kosaka H, Imaizumi K, Imai K, Tyuma I. Stoichiometry of the reaction of oxyhemoglobin with nitrite. Biochim Biophys Acta. 1979;581:184–188. doi: 10.1016/0005-2795(79)90235-6. [DOI] [PubMed] [Google Scholar]
- 231.Lissi E. Autocatalytic oxidation of hemoglobin by nitrite: A possible mechanism. Free Rad Biol Med. 1998;24:1535–1536. doi: 10.1016/s0891-5849(98)00004-5. [DOI] [PubMed] [Google Scholar]
- 232.van der Vliet A, Eiserich JP, Halliwell B, Cross CE. Formation of reactive nitrogen species during peroxidase-catalyzed oxidation of nitrite. A potential additional mechanism of nitric oxide-dependent toxicity. J Biol Chem. 1997;272:7617–7625. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 233.Keszler A, Mason RP, Hogg N. Immuno spin-trapping of hemoglobin and myoglobin radical derived from nitrite-mediated oxidation. Free Rad Biol Med. 2006;40:507–515. doi: 10.1016/j.freeradbiomed.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 234.Joseph J, Kalyanaraman B, Hyde JS. Trapping of nitric oxide by nitronyl nitroxides: an electron spin resonance investigation. Biochem Biophys Res Commun. 1993;192:926–934. doi: 10.1006/bbrc.1993.1504. [DOI] [PubMed] [Google Scholar]
- 235.Goldstein S, Russo A, Samuni A. Reactions of PTIO and Carboxy-PTIO with NO, NO2, and O3−. J Biol Chem. 2003;278:50949–50955. doi: 10.1074/jbc.M308317200. [DOI] [PubMed] [Google Scholar]
- 236.Tomoda A, Matsukawa S, Takeshita M, Yoneyama Y. Effect of inositol hexaphosphate on hemoglobin oxidation by nitrite and ferricyanide. Biochem Biophys Res Commun. 1977;74:1469–1474. doi: 10.1016/0006-291x(77)90607-6. [DOI] [PubMed] [Google Scholar]
- 237.Mansouri A. Oxidation of human hemoglobin by sodium nitrite--effect of beta-93 thiol groups. Biochem Biophys Res Commun. 1979;89:441–447. doi: 10.1016/0006-291x(79)90649-1. [DOI] [PubMed] [Google Scholar]
- 238.Vanin AF, Kiladze SV, Kubrina L. N Incorporation of non-heme iron into the dinitrosyl complexes in the mouse liver in vivo. Biofizika (Moscow) 1978;23:474–478. [PubMed] [Google Scholar]
- 239.Vanin AF, Varich V. Ja Nitrosyl non-heme iron complexes in animal tissues. Studia Biophysica. 1981;86:175–185. [Google Scholar]
- 240.Vanin A, Papina A, Serezhenkov V, Koppenol W. The mechanisms of S-nitrosothiol decomposition catalyzed by iron. Nitric Oxide. 2004;10:60– 73. doi: 10.1016/j.niox.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 241.Mikoyan VD, Kubrina LN, Khachatryan GN, Vanin A. Nitrite protonation as a necessary stage in the generation of nitric oxide from nitrite in biological systems. Biofizika (Moscow) 2006;51:968– 975. [PubMed] [Google Scholar]
- 242.Wiegant F, Malyshev I, Kleschyov A, van Faassen E, Vanin A. Dinitrosyl iron complexes with thiol containing ligands and S-nitroso-D, L-penicillamine as inductors of heat shock protein synthesis in H35 hepatoma cells. FEBS Lett. 1999;455:179– 182. doi: 10.1016/s0014-5793(99)00806-6. [DOI] [PubMed] [Google Scholar]
- 243.Hellwig-Burgel T, Stiehl DP, Wagner AE, Metzen E, Jelkmann W. Review: hypoxia-inducible factor-1 (HIF-1): a novel transcription factor in immune reactions. J Interferon Cytokine Res. 2005;25:297–310. doi: 10.1089/jir.2005.25.297. [DOI] [PubMed] [Google Scholar]
- 244.Brunori M. Nitric oxide, cytochrome-c oxidase and myoglobin. Trends Biochem Sci. 2001;26:21– 23. doi: 10.1016/s0968-0004(00)01698-4. [DOI] [PubMed] [Google Scholar]
- 245.Kleinbongard P, Schulz R, Rassaf T, Lauer T, Dejam A, Jax T, Kumara I, Gharini P, Kabanova S, Ozuyaman B, Schnurch H, Godecke A, Weber A, Robenek M, Robenek H, Bloch W, Rosen P, Kelm M. Red blood cells express a functional endothelial nitric oxide synthase. Blood. 206;341:816–821. doi: 10.1182/blood-2005-10-3992. [DOI] [PubMed] [Google Scholar]
- 246.Webb A, Milsom A, Rathod KS, Chu WL, Qureshi S, Lovell M, Lecomte F, Perrett D, Raimondo C, Khoshbin A, Ahmed Z, Uppal R, Benjamin N, Hobbs A, Ahluwalia A. Mechanisms underlying erythrocyte and endothelial nitrite reduction to nitric oxide in hypoxia; Role for xanthine oxidoreductase and endothelial nitric oxide synthase. Circ Res. 2008;103:957– 964. doi: 10.1161/CIRCRESAHA.108.175810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Rouquette M, Page S, Bryant R, Benboubetra M, Stevens C, Blake D, Wish W, Harrison R, Tosh D. Xanthine oxidoreductase is asymmetrically localized on the outer surface of human endothelial and epithelial cells in culture. FEBS Lett. 1998;426:397–401. doi: 10.1016/s0014-5793(98)00385-8. [DOI] [PubMed] [Google Scholar]
- 248.Hagen T, Taylor C, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: Effect on HIF1α. Science. 2005;302:1975– 1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 249.Lundberg JO, Weitzberg E, Gladwin M. The nitrate – nitrite – nitric oxide pathway in physiology and therapeutics. Nature Rev Drug Discov. 2008;7:156– 167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 250.Lefer DJ. Nitrite therapy for protection against ischemia-reperfusion injury. A J Physiol Renal Physiol. 2006;290:F777– F778. doi: 10.1152/ajprenal.00470.2005. [DOI] [PubMed] [Google Scholar]
- 251.Pluta R. Sodium nitrite as a therapeutic agent for central nervous system disorders. Surg Neurosci. 2006;66:5–10. doi: 10.1016/j.surneu.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 252.Mazzone M, Carmeliet P. A lifeline for suffocating tissues. Nature. 2008;453:1194–1195. doi: 10.1038/4531194a. [DOI] [PubMed] [Google Scholar]
- 253.Rassaf T, Lauer T, Heiss C, Balzer J, Mangold S, Leyendecker T, Rottler J, Drexhage C, Meyer C, Kelm M. Nitric oxide synthase-derived plasma nitrite predicts exercise capacity. Br J Sports Med. 2007;41:559– 673. doi: 10.1136/bjsm.2007.035758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Rassaf T, Heiss C, Hendgen-Cotta U, Balzer J, Matern S, Kleinbongard P, Lee A, Lauer T, Kelm M. Plasma nitrite reserve and endothelial function in the human forearm circulation. Free Rad Biol Med. 2006;41:295–301. doi: 10.1016/j.freeradbiomed.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 255.Mack A, Kato G. Sickle cell disease and nitric oxide: A paradigm shift? Int J Biochem Cell Biol. 2006;38:1237– 1243. doi: 10.1016/j.biocel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kubes P, Suzuki M, Granger D. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88:4651– 4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Lefer D, Jones S, Girod W, Baines A, Grisham M, Cockrell A. Leukocyte-endothelial cell interactions in nitric oxide syntahse deficient mice. Am J Physiol. 1999;276:H1943– H1950. doi: 10.1152/ajpheart.1999.276.6.H1943. [DOI] [PubMed] [Google Scholar]
- 258.Kimura H, Weisz A, Kurashima Y, Hashimoto k, Ogura T, d’Acquisto F, Addeo R, Makuuchi M, Esumi H. Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: Control of hypoxia inducible factor-1 activity by nitric oxide. Blood. 2000;95:189– 197. [PubMed] [Google Scholar]
- 259.Napoli C, de Nigris F, Williams-Ignarro S, Pignalosa O, Sica V, Ignarro L. Nitric oxide and atherosclerosis: an update. Nitric Oxide. 2006;15:265– 279. doi: 10.1016/j.niox.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 260.Xu l, Eu J, Meissner G, Stamler J. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosation. Science. 1998;279:234– 237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 261.Matsushita K, Morrell C, Cambien B, Yang S, Yamakuchi M, Bao C. Nitric oxide regulates exocytosis by S-nitrosation of N-ethylmaleimide-sensitive factor. Cell. 2003;115:139. doi: 10.1016/s0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 263.Özüyaman B, Ebner P, Kleinbongard P, Jax T, Gödecke A, Kelm M, Kalka C. Nitric oxide differentially regulates proliferation and mobilization of EPC but not of hematopoietic stem cells. Thromb Haemostas. 2005;94:770–772. doi: 10.1160/TH05-01-0038. [DOI] [PubMed] [Google Scholar]
- 264.Thum T, Tsikas D, Stein S, Schultheiss M, Eigenthaler M, Anker S, Poole-Wilson P, Ertl G, Bauersachs J. Suppression of endothelial progenitor cells in human coronary artery disease by the endogenous nitric oxide synthase inhibitor asymmetric dimethylarginine. J Am Coll Cardiol. 2005;46:1693– 1701. doi: 10.1016/j.jacc.2005.04.066. [DOI] [PubMed] [Google Scholar]
- 265.Heiss C, Keymel S, Niesler U, Ziemann J, Kelm M, Kalka C. Impaired progenitor cell activity in age-related endothelial dysfunction. J Am Coll Cardiol. 2005;45:1441–1448. doi: 10.1016/j.jacc.2004.12.074. [DOI] [PubMed] [Google Scholar]
- 266.Shantsila E, Watson T, Lip G. Endothelial progenitor cells in cardiovascular disorders. J Am Coll Cardiol. 2007;49:741– 751. doi: 10.1016/j.jacc.2006.09.050. [DOI] [PubMed] [Google Scholar]
- 267.Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher A, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nature Medicine. 2003;9:1370– 1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 268.Paul JD, Powell TM, Thompson M, Benjamin N, Carlow A, Annavajjhala V, Shiva S, Dejam A, Gladwin M, McCoy J, Zalos G, Press B, Murphy M, Hill JM, Csako G, Waclawiw MA, Cannon RO., 3rd Endothelial progenitor cell mobilization and increased intravascular nitric oxide in patients undergoing cardiac rehabilitation. J Cardiopulm Rehabil Prev. 2007;27:65–73. doi: 10.1097/01.HCR.0000265031.10145.50. [DOI] [PubMed] [Google Scholar]
- 269.Michaud SE, Dussault S, Haddad P, Groleau J, Rivard A. Circulating endothelial progenitor cells from healthy smokers exhibit impaired functional activities. Atheroscl. 2006;187:423– 432. doi: 10.1016/j.atherosclerosis.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 270.Bjorne H, Petersson J, Phillipson, Weitzberg E, Holm L, Lundberg J. Nitrite in saliva increases gastric mucosal flow and mucus thickness. J Clin Invest. 2004;113:106–114. doi: 10.1172/JCI200419019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Sobko T, Huang L, Midtvedt T, Norin E, Gustafsson L, Norman M, Jansson A, Lundberg JO. Generation of NO by probiotic bacteria in the gastrointestinal tract. Free Rad Biol Med. 2006;41:985–991. doi: 10.1016/j.freeradbiomed.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 272.Bjorne H, Govoni M, Tornberg DC, Lundberg JO, Weitzberg E. Intragastric nitric oxide is abolished in intubated patients and restored by nitrite. Crit Care Med. 2005;33:1722–1727. doi: 10.1097/01.ccm.0000171204.59502.aa. [DOI] [PubMed] [Google Scholar]