

Abstract
The cellular DNA damage response (DDR) is activated by many types of DNA lesions. Upon recognition of DNA damage by sensor proteins, an intricate signal transduction network is activated to coordinate diverse cellular outcomes that promote genome integrity. Key components of the DDR in mammalian cells are the checkpoint effector kinases Chk1 and Chk2 (referred to henceforth as the effector kinases; orthologous to spChk1 and spCds1 in the fission yeast S. pombe and scChk1 and scRad53 in the budding yeast S. cerevisiae). These evolutionarily conserved and structurally divergent kinases phosphorylate numerous substrates to regulate the DDR. This review will focus on recent advances in our understanding of the structure, regulation, and functions of the effector kinases in the DDR, as well as their potential roles in human disease.
Domain organization of the effector kinases
Chk1 and Chk2 contain highly conserved kinase domains but are structurally and functionally distinct. Chk2 has an N-terminal SQ/TQ cluster domain (SCD) and a Forkhead associated domain (FHA) domain, involved in phosphorylation dependent protein-protein interactions, and a kinase domain located near the C-terminus (Figure 1A) [1]. Chk2 is a highly phosphorylated protein with more than 25 phosphorylation sites identified to date [1–4]. The orthologous spCds1 (Figure 1B) is organized in a similar fashion to mammalian Chk2 while scRad53 is more divergent, containing a second FHA domain C-terminal to the centrally located kinase domain (Figure 1C).
Figure 1. The Checkpoint Kinases.
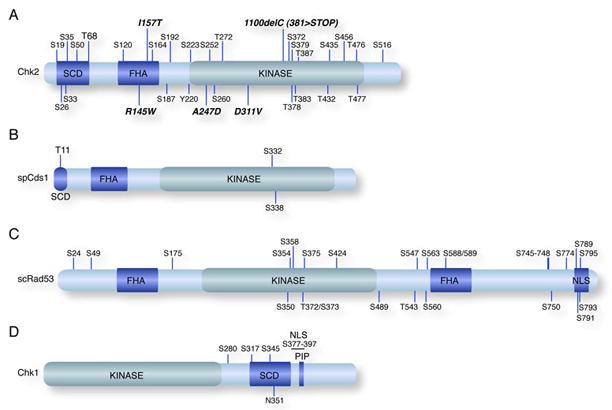
Domain organization of the human Chk1 and Chk2 and the yeast spCds1 and scRad53 kinases are shown. The forkhead associated domains (FHA), SQ/TQ clusters (SCD), and kinase domains (Kinase) are indicated. (A) Mutations in Chk2 identified in human cancers are shown in bold-italic type. Phosphorylation sites identified to date are indicated. (B) spCds1 shares a similar domain organization as human Chk2 with T11 corresponding to the major damage inducible site T68 in Chk2. (C) Schematic of scRad53 domain organization and phosphorylation sites. The putative bipartite nuclear localization signal (NLS) is indicated. (D) Schematic of human Chk1 with domains labeled as in (A).
The domain organization of the mammalian Chk1 proteins and the yeast orthologues appears to be highly conserved. The kinase domain of Chk1 is located in the N-terminus, followed by an SCD, a bipartite nuclear localization signal and a PCNA interacting protein (PIP) motif (Figure 1D) [5]. The C-terminus of Chk1 has been proposed to function as an auto-inhibitory region, as C-terminally truncated isoforms of Chk1 have increased activity, but recent analysis suggests additional complexity as C-terminal regions of spChk1 are required for its activity [6, 7].
Activation of the effector kinases
Sensor proteins are required to recognize damage and trigger the DDR. Damage sensors include the Mre11 complex (Mre11, Rad50, and Nbs1) that recognizes DNA double-strand breaks (DSBs) as well as Rad17 and the Rad9-Hus1-Rad1 (9-1-1) complex that localize rapidly to lesions caused by replication stress [8, 9]. Recognition of damage activates the central transducing kinases of the DDR, Ataxia-telangiectasia mutated (ATM) and ATM and Rad3-related (ATR). ATM and ATR (the orthologues of Tel1/Rad3 in S. pombe and Tel1/Mec1 in S. cerevisiae) are members of the PI-3K like kinase (PIKK) family and promote activation of the effector kinases, in part through the phosphorylation of SQ/TQ residues.
Chk1 and Chk2 activation occurs through distinct mechanisms. Chk1 activation is primarily downstream of ATR in response to genotoxic insults including stalled replication forks, DNA crosslinks, ultraviolet (UV) radiation damage and to a lesser extent, ionizing radiation (IR). ATR activation has recently been reviewed in great detail elsewhere and will be described briefly here [10, 11]. ATR, in complex with the ATR interacting protein (ATRIP), recognizes single stranded DNA (ssDNA) bound by the single-stranded DNA binding protein, Replication Protein A. ATR is then activated through interactions with the Topoisomerase II binding protein (TopBP1) and the 9-1-1 complex that is loaded on DNA by the damage specific clamp loader Rad17 and Replication Factor-C2-5. Active ATR, in the presence of additional factors, phosphorylates Chk1 on at least 2 residues, S317 and S345, associated with its active form.
Chk2 is activated primarily by ATM in response to DSBs (reviewed in [1, 12]). DSB recognition by the Mre11 complex and additional proteins rapidly triggers ATM activation [8]. Once activated, ATM phosphorylates Chk2 on T68 and additional residues in the Chk2 N-terminal SCD domain. Phosphorylation of the SCD domain creates a binding site for the FHA domain of a second Chk2 molecule, bringing the activation loops of the kinase domains into proximity. This multimerization promotes Chk2 autophosphorylation in the catalytic site on T383 and T387, as well as on additional residues, such as S516 and S379, that promote its activity [1, 2, 4]. A similar mode of activation has been proposed for the structurally similar spCds1 and is likely to represent a common mechanism of kinase activation [12, 13].
In response to DSBs, ATM has been proposed to influence the activation and phosphorylation of Chk1 indirectly, through the regulation of DSB break processing and subsequent activation of ATR [14–16]. In addition, ATM, ATR, and another related PIKK, DNA-PKcs, can phosphorylate Chk2 in vitro, and evidence exists for ATM-independent functions of Chk2 [17–22]. These data raise the possibility that significant crosstalk takes place between the ATR-Chk1 and ATM-Chk2 signaling cascades.
Regulation of the checkpoint kinases by Mediator proteins
In addition to ATM and ATR, proteins defined as mediators fine tune the DDR and govern the checkpoint kinases (reviewed in this issue). Mediators are substrates and regulators of both the transducing and effector kinases, promoting their activation, regulating substrate access, and controlling their associations with damaged DNA. The prototypical S. cerevisiae mediator, scRad9, contains domains found in many mediator proteins; dual Brca1 C-terminal (BRCT) domains, a SCD domain, and a Tudor domain, involved in recognizing methylated histones. Mammalian mediators include Nbs1, 53BP1, MDC1, TopBP1, Brca1 and the non-BRCT domain protein Claspin.
Effector kinase activity is controlled through feedback regulation with mediator proteins. The scRad9 SCD is phosphorylated by the transducing kinases and these modifications are recognized by the FHA domains of scRad53 [23]. scRad9-associated scRad53 is preferentially phosphorylated by the transducing kinases, thus promoting autophosphorylation required for its full activation [23–26]. Recent data from our laboratory suggest that interactions also occur between phosphorylated scRad9-SCD and the scRad9-BRCT domain and are the molecular basis of DNA damage-induced scRad9 oligomerization in chromatin [27]. scRad9 oligomerization appears to be dispensable for scRad53 activation, but is required to maintain the activity of scRad53, thus promoting checkpoint maintenance. Once activated, scRad53 can negatively regulate scRad9 oligomerization by phosphorylating the scRad9 BRCT domain and impairing the scRad9-SCD/BRCT interaction. The mammalian mediator proteins MDC1, 53BP1, Nbs1, Brca1 and Claspin influence the regulation of Chk1 and Chk2 [28–33]. Whether the functional interactions described above for scRad9 and scRad53 analogize other those of other mediator and effector proteins is an open question.
The mediator Claspin plays a particularly important role in the regulation of Chk1 through functional and physical associations with Rad17 and Chk1 itself. Depletion of Claspin from X. laevis extracts leads to reduced ATR mediated phosphorylation of Chk1 [34]. A phosphorylation-dependent interaction between Rad17 and Claspin is essential for maintaining Chk1 phosphorylation after HU induced damage [35]. Claspin also interacts with Chk1 in a damage specific manner that requires the ATR-dependent phosphorylation of Claspin as well as Chk1 mediated phosphorylation of Claspin on T916 [36–38].
Post-translational regulation of the effector kinases
Post-translational modifications that affect protein stability and sub-cellular localization of Chk1 and Chk2 also influence their respective activities. Chk1 is a chromatin-associated protein in normally growing cells. Following DNA damage, it is released from chromatin and localizes to the cytoplasm, where a portion localizes to interphase centrosomes [39, 40]. Phosphorylation of Chk1 on S317 is required for chromatin release as well as G2 checkpoint arrest, as S317A mutants and Chk1 tethered to chromatin cannot activate G2/M checkpoint responses [39, 41]. Phosphorylation of both S317 and S345 in Chk1 is required for centrosomal localization while only modification of S345 is required for localization to the cytoplasm. In addition, ATR-mediated phosphorylation of Chk1 promotes its degradation by the proteasome in response to many types of damage [42].
The AKT kinase phosphorylates Chk1 on S280 in response to damage, contributing to its cytoplasmic relocalization as well as mono or diubiquitination following IR treatment [43]. Ubiquitination of Chk2 has also been reported and is controlled by the phosphorylation of S379 and S456 [2, 4]. While both phosphorylation sites are important for Chk2 function, they differentially affect the ubiquitination and stability of Chk2. Mutation of S379 impairs ubiquitination but does not alter the stability of Chk2 while S456 mutation leads to hyper-ubiquitiniation and Chk2 degradation. Although the functional importance of Chk1 and Chk2 ubiquitination remains unclear, it is tempting to speculate that these modifications represent feedback regulation between interacting ubiquitin ligases, such as Mdm4/X and EDD, or the ubiquitin protease USP28 [44–48].
The influence of Chk1 and Chk2 on the DDR
The checkpoint kinases phosphorylate numerous proteins that influence diverse aspects of the DDR to promote genome integrity. Chk1 and Chk2 influence diverse aspects of the DDR, primarily, but not exclusively, via their influences on transcription (Figure 2). While the checkpoint kinases target some overlapping substrates, their functions in the DDR are largely distinct.
Figure 2. The Effector Kinases in the DDR.
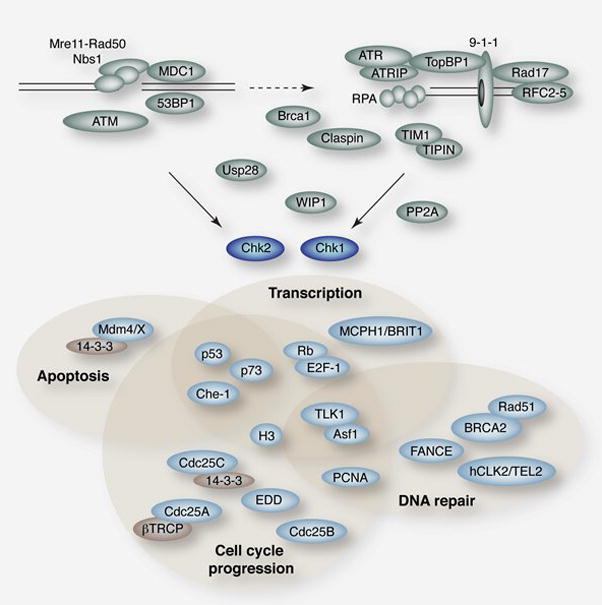
Regulators of effector kinase activation are shown in green. Known substrates and interacting proteins are shown in light blue with some regulatory proteins in tan. Shaded regions indicate some known functions of the proteins and are provided as an organizing principle and are not meant to exclude potential roles of any proteins in additional processes.
Cell cycle checkpoint regulation
The effector kinases play a key role in the regulation of cell cycle checkpoint arrest. In yeast, scRad53 and spCds1 are required to mediate S-phase checkpoint responses to DNA damage while scRad53, scChk1 and spChk1 play prominent roles in arrest at the G2/M boundary. In human and mouse cells, Chk1 is the primary effector of the intra-S and G2/M phase checkpoints, whereas Chk2 plays an accessory role, exerting a partial influence on the intra-S and G1/S checkpoints [49].
Central to their influence on cell cycle checkpoint arrest in fission yeast and mammals, both Chk1 and Chk2 target the Cdc25 family of phosphatases (Cdc25A, B and C in humans) that remove inhibitory phosphorylations from CDKs during normal cell cycles and after DNA damage (reviewed in [50, 51]). The effector kinases negatively regulate the Cdc25s through phosphorylation that controls their binding to 14-3-3 proteins, nucelo-cytoplasmic shuttling, and binding of the SCFβTrcp ubiquitin ligase that targets Cdc25A for degradation by the proteasome to activate the intra-S phase checkpoint [52–55].
SCFβTrcp also plays an important role in the regulation of Chk1 at the G2/M boundary through the modulation of Claspin levels. At the onset of mitosis, Polo like kinase 1 (Plk1) is activated and phosphorylates Claspin in a conserved βTrcp degron motif that leads to its ubiquitinylation by SCFβTrcp and degradation by the proteasome, thus blocking Chk1-dependent inhibition of mitosis [56, 57]. After DNA damage in G2, Claspin is no longer phosphorylated in its SCFβTrcp degron and both Plk1 and Claspin are ubiquitinylated by APC/CCdh1 [58]. While Plk1 is degraded, Claspin remains stable due to the activity of the ubiquitin protease USP28 that prevents Claspin, and many other DDR proteins from degradation by the proteasome [48, 58]. These events culminate in the Chk1-dependent imposition of the G2/M checkpoint.
DNA repair and replication fork maintenance
Chk1 and Chk2 influence the regulation of DNA repair and promote the stability of stalled replication forks through their checkpoint functions, as well as the direct modification of repair proteins. Chk1 interacts with Rad51, as well as members of the Fanconi-anemia (FA) pathway of DNA repair, to govern homologous recombination (HR). siRNA or chemical inhibitors of Chk1 sensitize cells to camptothecin (CPT) and cause increased levels of strand breakage and reduced Rad51 chromatin association and foci formation in response to hydroxyurea (HU) [59]. Chk1 interacts with and phosphorylates Rad51 on T309 and cells expressing mutant Rad51-T309A are sensitive to HU treatment.
Chk1 regulates the FA pathway through interactions with the FANCE, hCLK2/TEL2 and BRCA2 proteins [60, 61]. Chk1 phosphorylates FANCE on two residues (T346 and S374) that promote its degradation and are required for survival after mitomycin-C (MMC) treatment. Knockdown of Chk1 results in higher levels of mono-ubiquitinylated FANCD2 foci in untreated cells through a mechanism that is dependent upon hCLK2/TEL2. Depletion of hCLK2/TEL2 by siRNA results in defects in S-phase checkpoint responses and HR repair, as well as Chk1 instability that is dependent upon its phosphorylation by ATR. These effects are likely due in part to the role of hCLK2/TEL2 in the stabilization of the PIKK kinases [62]. Both Chk1 and Chk2 may further influence the FA pathway and HR by modulating the interactions of Rad51 and the C-terminus of Brca2, that is mutated in some FA subgroups, through the phosphorylation of Brca2 on T3387 [63].
The yeast checkpoint kinases scRad53 and spCds1 play a key role in regulating replication fork stability in response to many types of S-phase damage (Reviewed in [64, 65]). In cells lacking scRad53, stalled forks collapse, compromising genome stability and cell viability. Molecular mechanisms of this regulation remain largely unclear, however, recent studies show that scRad53 phosphorylates Exo1 to prevent it from processing stalled replication forks or exposed telomeres [66, 67]. In addition, scChk1 plays a role in fork stabilization that is independent of scRad53 and the mitotic checkpoint [66].
In metazoans, Chk1 interacts with many replication fork associated proteins including proliferating cell nuclear antigen (PCNA), Claspin, and the TIM1/TIMELESS complex. Chk1 interacts with PCNA through a conserved PIP motif [5]. Mutation of conserved residues in the PIP box results in Chk1 hyperactivity and impaired phosphorylation and chromatin release in response to DNA damage, thus compromising checkpoint regulation. At replication forks stalled by UV lesions or HU, Chk1, Claspin, and TIM1/TIPIN are required for Rad18 mediated ubiquitinylation of PCNA that allows binding of translesion polymerases [68]. It remains unclear if this modification of PCNA also requires its direct interaction with Chk1. TIM1/TIPIN are the closest orthologues of S. cerevisiae Tof1/Swi3 proteins that are required for the maintenance of stalled forks as well as sister chromatid cohesion. Depletion of TIM1 or TIPIN impairs Chk1 phosphorylation in response to stalled forks, potentially through the failure to properly localize Claspin to chromatin [69, 70]. Chk1’s role in regulating replication fork dynamics in response to genotoxic stress may be critical for its role in suppressing fragile site expression [71].
Transcriptional regulation and apoptosis
In the context of a functional DDR, the induction of DNA damage leads to global changes in transcription. To a large extent, this reflects the regulation by the effector kinases of transcriptional activators and repressors that normally govern cell cycle specific transcription. In yeast, cell cycle specific transcription is regulated by the heterodimeric transcription factors SBF and MBF that activate different classes of genes, with MBF controlling those primarily relevant to DNA replication and repair [72]. DNA damage triggers the derepression of MBF through spCds1 mediated phosphorylation of Nrm1 or the MBF subunit Cdc10 allowing the expression of genes that promote cell survival in the presence of genotoxic stress [73, 74].
Additional complexity exists in S. cerevisiae where transcriptional regulation by scRad53 is controlled in part by the Dun1 kinase. The FHA domain of Dun1 binds to diphosphorylated threonines in the scRad53 SCD and scRad53 in turn phosphorylates the catalytic loop of Dun1 leading to its activation [24, 75]. An important target of scRad53/Dun1 is the transcriptional repressor Crt1 that is inactivated by scRad53 and Dun1 dependent hyperphosphorylation to promote survival during replication stress [76].
In metazoans, the E2F/Rb pathway controls cell cycle specific transcription in a manner analogous to SBF/MBF and their repressors Whi5/Nrm1 and both E2F-1 and Rb have been identified as targets of the effector kinases. E2F-1 is phosphorylated by Chk2 on S364, a modification that results in enhanced E2F-1 stability, and Chk1 and/or Chk2 modify S612 of Rb, resulting in an enhanced E2F-1/Rb interaction following DNA damage [77–79]. The effector kinases also regulate apoptosis in response to the deregulation of E2F transcriptional pathways. Overexpression of E2F-1 leads to the activation of proapoptotic genes and apoptotic cell death [80]. Chk2, as well as ATM and the Mre11 complex, are required for the induction of apoptosis by E2F-1 and other oncogenes such as HPV-E7 [81].
Chk2 is also required for IR induced apoptosis and promotes p53-dependent transcriptional responses in many tissues of the mouse [18, 82]. Both Chk1 and Chk2 can phosphorylate p53 on several sites including S20 (S23 in mice) [1]. The stability of p53 is reduced in mice lacking Chk2 and in some cell types of mice harboring a mutation of p53-S23, and both mice show similar defects in IR induced apoptosis [18, 82, 83].
While p53 is a direct target of Chk2 in the regulation of apoptosis, other substrates may also influence the process. Chk1 and Chk2 both phosphorylate the ubiquitin ligase Mdm4/X that targets p53 for degradation. Phosphorylation of Mdm4/X by Chk2 (on S342 and S367) promotes its nuclear retention and degradation through interactions with 14-3-3 proteins and the dissociation of the HAUSP ubiquitin protease [45, 46, 84]. Chk1 also targets the modification of Mdm4/X on S367 after UV damage, promoting the interaction of Mdm4/X with 14-3-3 and stimulating p53 Ptc2 activity [47]. Chk2 further influences p53-dependent transcriptional responses through modification of the Che-1/AATF kinase. Both ATM and Chk2 phosphorylate Che-1 and promote its stability and recruitment to the promoters of p53 responsive genes after DNA damage [85].
Chk2 has been implicated in the regulation of p53-independent apoptosis induced by IR, in part through the modification of the promyelocytic leukemia protein (PML) protein that is important for apoptosis in response to a number of stimuli [86]. Chk2 interacts with and phosphorylates PML on S117 and cells expressing PML mutants in this site fail to mount an efficient apoptotic response after IR treatment [87]. Chk2 is required for an increase in PML nuclear body number in response to radiation, although it remains unclear whether this involves a direct interaction with PML or requires the kinase activity of Chk2 [88].
While Chk2 promotes p53-independent apoptosis, Chk1 appears to suppress it. Depletion or chemical inhibition of Chk1 results in the activation of p53-independent apoptosis in response to IR in zebrafish and human cancer cells [89]. This apoptosis is dependent upon ATM, ATR, and caspase-2 and bypasses increased expression of the apoptosis inhibitor Bcl-2. Precisely how Chk1 normally suppresses these apoptotic pathways remains unclear but its roles in the regulation of cell cycle checkpoints or HR are likely key contributors to the viability of cycling cells following DNA damage.
The activity of Chk1 and Chk2 on Cdc25s, previously well characterized with regards to checkpoint function, has been tied to apoptosis through the regulation of CDK activity on the FOXO1 transcription factor [90]. Cdk1 and Cdk2 phosphorylate FOXO1 on S249 leading to its export from the nucleus, potentially through interactions with 14-3-3 proteins [91]. After DNA damage induced activation of the checkpoint kinases, Cdk2 is inhibited and FOXO1 transits to the nucleus and can activate apoptosis [90].
Chromatin dynamics
One mechanism by which the checkpoint kinases modulate transcriptional responses to genotoxic stress is through the regulation of chromatin. Chk1 phosphorylates histone H3 on T11 to facilitate ultraviolet (UV) radiation induced transcriptional responses [92]. Following exposure to UV, Chk1 is released from chromatin, correlating with a decrease in the phosphorylation of H3 on T11. This impairs the interaction of the histone acetyltransferase GCN5 with H3 at the promoters of cell cycle modulators, such as cyclin-B, resulting in their transcriptional repression.
Chromatin changes are also governed by the checkpoint kinases through the regulation of histone chaperones. scRad53 constitutively associates with phosphorylated Asf1, a histone H3/H4 chaperone, via its N-terminal FHA domain and this association is lost following DNA damage in a Mec1 dependent manner [93, 94]. The interaction between scRad53 and Asf1 is promoted by the presence of the Dun1 kinase [95]. Overexpression of Asf1 can act as a high copy suppressor of HU sensitivity in scRad53 mutant strains, suggesting that the regulation of Asf1 by scRad53 plays an important role in the maintenance of stalled replication forks [25, 93].
A similar signaling axis appears to exist in mammalian cells through interactions between Chk1 and the Tousled like kinase 1 (TLK1). Phosphorylation of TLK1 on S695 by Chk1 following DNA damage results in the inhibition of TLK1 kinase activity that peaks during S-phase [96–98]. Asf1 is a target of TLK1 but it remains unclear how this modification affects its activity in the DDR [99]. Asf1 is required for efficient replication fork progression during S-phase as well as for the acetylation of histone H3-K56 that is required for the resumption of cell cycle progression following DNA repair [100]. Chk1-TLK1-Asf1 signaling may represent a means of coordinating cell cycle checkpoint maintenance with histone dynamics that govern both transcription and DNA replication.
Opposing forces: phosphatases and the effector kinases
In order to recover from checkpoint arrest, cells must disengage checkpoint responses to resume cell cycle progression. While many enzymatic activities regulate this process, phosphatases have emerged as important negative regulators of the effector kinases. Analysis of adaptation revealed a correlation between the dephosphorylation of both scRad53 and scChk1 and cell cycle resumption in the presence of persistent DNA damage [101]. Cells lacking the PP2C-like phosphatases Ptc2 and Ptc3 were unable to adapt, and exhibited prolonged G2 checkpoint arrest and phosphorylation of scRad53 [102]. In addition, overexpression of Ptc2 suppressed the toxicity of a dominant lethal form of scRad53 [103]. Ptc2, as well as an additional PP2A like phosphatase complex Psy2-Pph3, also influence scRad53 control of replication fork progression [104]. Cells lacking both Pph3 and Ptc2 showed slower replication fork restart and increased scRad53 phosphorylation after treatment with MMS [105]. Expression of a dominant negative, kinase dead scRad53 enabled replication fork restart in the absence of Pph3 and Ptc2. scRad53 interacts with both Psy2 and phosphorylated Ptc2 through its the kinase domain and FHA1 domain respectively [102, 104].
spChk1-dependent G2 checkpoint responses are also regulated through associations with phosphatases. A screen to identify overexpressed genes that sensitized G2 cells to MMS treatment identified the PP1 like phosphatase Dis2 [106]. Dis2 is required to dephosphorylate spChk1 following checkpoint induction and cells lacking Dis2 exhibit prolonged spChk1 activity and G2 checkpoint arrest.
In metazoans, several phosphatases have been implicated in the regulation of the checkpoint kinases. The p53 inducible type-2C phosphatase Wip1 (PPM1D) interacts with and inhibits both Chk1 and Chk2 and appears to play an extensive role in counteracting the DDR [107–112]. Depletion of Wip1 leads to enhanced phosphorylation of DDR proteins and more robust G2/M and intra-S phase checkpoint responses after IR or UV treatment [108, 110, 111].
Both Chk1 and Chk2 are also regulated by PP2A and evidence for a complex feedback loop between Chk1 and PP2A has been reported [43, 113]. Chk1 stimulates PP2A activity on itself and pharmacological inhibition of either Chk1 or PP2A results in increased Chk1 phosphorylation on S345 and to a lesser extent S317 [43]. PP2A activity influences many aspects of the damage response, further analysis will be needed to clarify its specific roles in the regulation of the effector kinases [114–116].
The effector kinases and human disease
As lynchpins in the DDR and essential components of genome maintainence pathways, a role for the effector kinases in tumor suppression is not unexpected. Phosphorylated Chk2 and other markers of the DDR have been observed in numerous premalignant and cancerous lesions [117, 118]. These observations and additional data have led to the proposal that the DDR acts as an inducible barrier to tumorigenesis. This is supported by human genetic instability disorders as well as the analysis of mutant mice, including mice lacking Wip1 that show enhanced checkpoint responses and are refractory to oncogene-induced tumorigenesis [111, 119].
Chk2 mutations, commonly 1100delC, have been identified as low penetrance alleles in human prostate and breast cancer (Figure 1A; boldfaced italics)[120]. The 1100delC allele has been modeled in mice and appears to promote genome instability, but thus far information regarding tumorigenesis has not been reported [121]. However, expression of dominant negative Chk2 (Chk2-D347A) from the MMTV promoter results in tumors of the mammary gland and spleen [122].
Deletion of Chk2 in the mouse is insufficient to predispose spontaneous tumors but Chk2 null animals are more susceptible to skin tumors induced by the carcinogen DMBA [18]. In addition, Chk2 null mice harboring mutations in either Brca1 or members of the Mre11 complex, are predisposed to a broad spectrum of tumors [22, 123, 124]. Brca1 is a target of Chk2 kinase activity and mutation of this residue in mice (S971) resulted in increased susceptibility to IR or 1-methyl-1-nitrosourea (MNU) induced tumors as well as uterine hyperplasia [125]. Interestingly, mice expressing p53 with a mutation in the Chk2 phosphorylation site (p53-S23) develop B-cell tumors [83]. As these are not observed in Chk2 null mice, it suggests that additional kinases can modify this residue in the absence of Chk2.
Haploinsufficiency of Chk1 in the mouse results in many phenotypes associated with tumorigenesis, such as impaired cellular proliferation, elevated Cdc25A levels following damage, and genetic instability, but assessment of Chk1’s role in tumorigenesis in the mouse has been complicated by the essential nature of the gene [126–128]. Several somatic mutations of Chk1 have been identified in human tumors but thus far no hereditary mutations have been clearly associated with human disease [49]. Mutations in ATR are associated with human Seckel syndrome, a developmental disorder that results in microcephaly [129]. Microcephalin (MCPH1/BRIT1), a gene mutated in hereditary microcephaly, interacts with Chk1 and influences its transcriptional regulation as well as cell cycle checkpoint induction, suggesting that hypomorphic Chk1 mutations could present themselves in human developmental syndromes in the future [130–132].
Inhibition of both Chk1 and Chk2 has been investigated to augment chemotherapeutic approaches and have been reviewed extensively elsewhere [133–135]. Data from knockout mice would suggest that inhibition of Chk1 would be highly toxic to dividing cells regardless of DNA damage treatment [128]. On the other hand, loss of Chk2 in the mouse leads to a radioresistant phenotype and promotes tumorigenesis in genetic backgrounds where S and G2/M checkpoints are compromised [22, 82, 123, 124]. Whether pharmacological inhibition of Chk2 would recapitulate these phenotypes remains unclear but murine genetic studies suggest that this approach may warrant caution and consideration of the genetic background. However, given the strong conservation of the kinase domains in Chk1 and Chk2, it is likely that current inhibitors target additional kinases, admittedly making predictions of their efficacy based on murine tumor assessment difficult.
Epilogue
Despite the wealth of data available, much remains to be learned about the regulation and influence of the effector kinases on the DDR. Teasing apart the functions of Chk1 and Chk2 in mammalian cells has been complicated by overlapping substrate specificity, redundancy within signaling pathways, and the requirement of Chk1 for cell viability. The generation of knockout mice has been valuable for the analysis of Chk2 in tumorigenesis and the genetic dissection of DDR signaling pathways. The identification of hypomorphic Chk1 alleles that support cell viability would facilitate similar approaches for the analysis of Chk1 function. Future studies, utilizing the diverse model systems available, will be essential to untangle the many contributions of these important regulators of the DDR to the preservation of genome integrity.
Acknowledgments
T.H.S. was supported by an NIH NRSA and T.H.S. and T.U. are Leukemia and Lymphoma Society Special Fellows. J.H.P is supported by the NIH and the Joel and Jean Smilow initiative.
Footnotes
The authors have no conflicts of interest or financial disclosures.
We apologize to those authors whose work on the effector kinases was not cited due to space constraints.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ahn J, Urist M, Prives C. The Chk2 protein kinase. DNA Repair (Amst) 2004;3:1039–1047. doi: 10.1016/j.dnarep.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 2.Kass EM, Ahn J, Tanaka T, Freed-Pastor WA, Keezer S, Prives C. Stability of checkpoint kinase 2 is regulated via phosphorylation at serine 456. J Biol Chem. 2007;282:30311–30321. doi: 10.1074/jbc.M704642200. [DOI] [PubMed] [Google Scholar]
- 3.Gabant G, Lorphelin A, Nozerand N, Marchetti C, Bellanger L, Dedieu A, Quemeneur E, Alpha-Bazin B. Autophosphorylated residues involved in the regulation of human chk2 in vitro. J Mol Biol. 2008;380:489–503. doi: 10.1016/j.jmb.2008.04.053. [DOI] [PubMed] [Google Scholar]
- 4.Lovly CM, Yan L, Ryan CE, Takada S, Piwnica-Worms H. Regulation of Chk2 Ubiquitination and Signaling through Autophosphorylation of Serine 379. Mol Cell Biol. 2008 doi: 10.1128/MCB.00821-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scorah J, Dong MQ, Yates JR, 3rd, Scott M, Gillespie D, McGowan CH. A conserved proliferating cell nuclear antigen-interacting protein sequence in Chk1 is required for checkpoint function. J Biol Chem. 2008;283:17250–17259. doi: 10.1074/jbc.M800369200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katsuragi Y, Sagata N. Regulation of Chk1 kinase by autoinhibition and ATR-mediated phosphorylation. Mol Biol Cell. 2004;15:1680–1689. doi: 10.1091/mbc.E03-12-0874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kosoy A, O’Connell MJ. Regulation of Chk1 by Its C-terminal Domain. Mol Biol Cell. 2008 doi: 10.1091/mbc.E08-04-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene. 2007;26:7741–7748. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- 9.Parrilla-Castellar ER, Arlander SJ, Karnitz L. Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair (Amst) 2004;3:1009–1014. doi: 10.1016/j.dnarep.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 10.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mordes DA, Cortez D. Activation of ATR and related PIKKs. Cell Cycle. 2008;7 doi: 10.4161/cc.7.18.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver AW, Knapp S, Pearl LH. Activation segment exchange: a common mechanism of kinase autophosphorylation? Trends Biochem Sci. 2007;32:351–356. doi: 10.1016/j.tibs.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Xu YJ, Davenport M, Kelly TJ. Two-stage mechanism for activation of the DNA replication checkpoint kinase Cds1 in fission yeast. Genes Dev. 2006;20:990–1003. doi: 10.1101/gad.1406706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K, Savage K, Lukas J, Zhou BB, Bartek J, Khanna KK. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem. 2003;278:14806–14811. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Muse T, Boulton SJ. Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. Embo J. 2005;24:4345–4355. doi: 10.1038/sj.emboj.7600896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 17.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A. 2000;97:10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirao A, Cheung A, Duncan G, Girard PM, Elia AJ, Wakeham A, Okada H, Sarkissian T, Wong JA, Sakai T, De Stanchina E, Bristow RG, Suda T, Lowe SW, Jeggo PA, Elledge SJ, Mak TW. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol Cell Biol. 2002;22:6521–6532. doi: 10.1128/MCB.22.18.6521-6532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goudelock DM, Jiang K, Pereira E, Russell B, Sanchez Y. Regulatory interactions between the checkpoint kinase Chk1 and the proteins of the DNA-dependent protein kinase complex. J Biol Chem. 2003;278:29940–29947. doi: 10.1074/jbc.M301765200. [DOI] [PubMed] [Google Scholar]
- 20.McSherry TD, Mueller PR. Xenopus Cds1 is regulated by DNA-dependent protein kinase and ATR during the cell cycle checkpoint response to double-stranded DNA ends. Mol Cell Biol. 2004;24:9968–9985. doi: 10.1128/MCB.24.22.9968-9985.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Stern DF. Regulation of CHK2 by DNA-dependent protein kinase. J Biol Chem. 2005;280:12041–12050. doi: 10.1074/jbc.M412445200. [DOI] [PubMed] [Google Scholar]
- 22.Stracker TH, Couto SS, Cordon-Cardo C, Matos T, Petrini JH. Chk2 suppresses the oncogenic potential of DNA replication-associated DNA damage. Mol Cell. 2008;31:21–32. doi: 10.1016/j.molcel.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pellicioli A, Foiani M. Signal transduction: how rad53 kinase is activated. Curr Biol. 2005;15:R769–771. doi: 10.1016/j.cub.2005.08.057. [DOI] [PubMed] [Google Scholar]
- 24.Chen SH, Smolka MB, Zhou H. Mechanism of Dun1 activation by Rad53 phosphorylation in Saccharomyces cerevisiae. J Biol Chem. 2007;282:986–995. doi: 10.1074/jbc.M609322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Usui T, Petrini JH. The Saccharomyces cerevisiae 14-3-3 proteins Bmh1 and Bmh2 directly influence the DNA damage-dependent functions of Rad53. Proc Natl Acad Sci U S A. 2007;104:2797–2802. doi: 10.1073/pnas.0611259104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiorani S, Mimun G, Caleca L, Piccini D, Pellicioli A. Characterization of the activation domain of the Rad53 checkpoint kinase. Cell Cycle. 2008;7:493–499. doi: 10.4161/cc.7.4.5323. [DOI] [PubMed] [Google Scholar]
- 27.Usui T, Foster S, Petrini JH. Maintenance of the DNA damage checkpoint requires mediator protein oligomerization. Mol Cell. doi: 10.1016/j.molcel.2008.12.022. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–1438. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 29.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 30.Mochan TA, Venere M, DiTullio RA, Jr, Halazonetis TD. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res. 2003;63:8586–8591. [PubMed] [Google Scholar]
- 31.Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- 32.Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, Alt FW, Chen J. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 33.Minter-Dykhouse K, Ward I, Huen MS, Chen J, Lou Z. Distinct versus overlapping functions of MDC1 and 53BP1 in DNA damage response and tumorigenesis. J Cell Biol. 2008;181:727–735. doi: 10.1083/jcb.200801083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell. 2000;6:839–849. doi: 10.1016/s1097-2765(05)00092-4. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Zou L, Lu T, Bao S, Hurov KE, Hittelman WN, Elledge SJ, Li L. Rad17 phosphorylation is required for claspin recruitment and Chk1 activation in response to replication stress. Mol Cell. 2006;23:331–341. doi: 10.1016/j.molcel.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 36.Kumagai A, Dunphy WG. Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat Cell Biol. 2003;5:161–165. doi: 10.1038/ncb921. [DOI] [PubMed] [Google Scholar]
- 37.Chini CC, Chen J. Repeated phosphopeptide motifs in human Claspin are phosphorylated by Chk1 and mediate Claspin function. J Biol Chem. 2006;281:33276–33282. doi: 10.1074/jbc.M604373200. [DOI] [PubMed] [Google Scholar]
- 38.Chini CC, Wood J, Chen J. Chk1 is required to maintain claspin stability. Oncogene. 2006;25:4165–4171. doi: 10.1038/sj.onc.1209447. [DOI] [PubMed] [Google Scholar]
- 39.Smits VA, Reaper PM, Jackson SP. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol. 2006;16:150–159. doi: 10.1016/j.cub.2005.11.066. [DOI] [PubMed] [Google Scholar]
- 40.Kramer A, Mailand N, Lukas C, Syljuasen RG, Wilkinson CJ, Nigg EA, Bartek J, Lukas J. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat Cell Biol. 2004;6:884–891. doi: 10.1038/ncb1165. [DOI] [PubMed] [Google Scholar]
- 41.Niida H, Katsuno Y, Banerjee B, Hande MP, Nakanishi M. Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol Cell Biol. 2007;27:2572–2581. doi: 10.1128/MCB.01611-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 43.Puc J, Keniry M, Li HS, Pandita TK, Choudhury AD, Memeo L, Mansukhani M, Murty VV, Gaciong Z, Meek SE, Piwnica-Worms H, Hibshoosh H, Parsons R. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7:193–204. doi: 10.1016/j.ccr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 44.Henderson MJ, Munoz MA, Saunders DN, Clancy JL, Russell AJ, Williams B, Pappin D, Khanna KK, Jackson SP, Sutherland RL, Watts CK. EDD mediates DNA damage-induced activation of CHK2. J Biol Chem. 2006;281:39990–40000. doi: 10.1074/jbc.M602818200. [DOI] [PubMed] [Google Scholar]
- 45.Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005;24:3411–3422. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LeBron C, Chen L, Gilkes DM, Chen J. Regulation of MDMX nuclear import and degradation by Chk2 and 14-3-3. EMBO J. 2006;25:1196–1206. doi: 10.1038/sj.emboj.7601032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin Y, Dai MS, Lu SZ, Xu Y, Luo Z, Zhao Y, Lu H. 14-3-3gamma binds to MDMX that is phosphorylated by UV-activated Chk1, resulting in p53 activation. EMBO J. 2006;25:1207–1218. doi: 10.1038/sj.emboj.7601010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang D, Zaugg K, Mak TW, Elledge SJ. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126:529–542. doi: 10.1016/j.cell.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 49.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 50.Karlsson-Rosenthal C, Millar JB. Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006;16:285–292. doi: 10.1016/j.tcb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 51.Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol. 2006;18:185–191. doi: 10.1016/j.ceb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 52.Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- 53.Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, Harper JW. SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–3074. doi: 10.1101/gad.1157503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 55.Kanemori Y, Uto K, Sagata N. Beta-TrCP recognizes a previously undescribed nonphosphorylated destruction motif in Cdc25A and Cdc25B phosphatases. Proc Natl Acad Sci U S A. 2005;102:6279–6284. doi: 10.1073/pnas.0501873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell. 2006;23:319–329. doi: 10.1016/j.molcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 57.Mailand N, Bekker-Jensen S, Bartek J, Lukas J. Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell. 2006;23:307–318. doi: 10.1016/j.molcel.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 58.Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 2008;134:256–267. doi: 10.1016/j.cell.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 60.Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D’Andrea AD. Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2007;27:3098–3108. doi: 10.1128/MCB.02357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Collis SJ, Barber LJ, Clark AJ, Martin JS, Ward JD, Boulton SJ. HCLK2 is essential for the mammalian S-phase checkpoint and impacts on Chk1 stability. Nat Cell Biol. 2007;9:391–401. doi: 10.1038/ncb1555. [DOI] [PubMed] [Google Scholar]
- 62.Takai H, Wang RC, Takai KK, Yang H, de Lange T. Tel2 regulates the stability of PI3K-related protein kinases. Cell. 2007;131:1248–1259. doi: 10.1016/j.cell.2007.10.052. [DOI] [PubMed] [Google Scholar]
- 63.Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene. 2008;27:3977–3985. doi: 10.1038/onc.2008.17. [DOI] [PubMed] [Google Scholar]
- 64.Pasero P, Shimada K, Duncker BP. Multiple roles of replication forks in S phase checkpoints: sensors, effectors and targets. Cell Cycle. 2003;2:568–572. [PubMed] [Google Scholar]
- 65.Branzei D, Foiani M. The Rad53 signal transduction pathway: Replication fork stabilization, DNA repair, and adaptation. Exp Cell Res. 2006;312:2654–2659. doi: 10.1016/j.yexcr.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 66.Segurado M, Diffley JF. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 2008;22:1816–1827. doi: 10.1101/gad.477208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morin I, Ngo HP, Greenall A, Zubko MK, Morrice N, Lydall D. Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J. 2008;27:2400–2410. doi: 10.1038/emboj.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang XH, Shiotani B, Classon M, Zou L. Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev. 2008;22:1147–1152. doi: 10.1101/gad.1632808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Errico A, Costanzo V, Hunt T. Tipin is required for stalled replication forks to resume DNA replication after removal of aphidicolin in Xenopus egg extracts. Proc Natl Acad Sci U S A. 2007;104:14929–14934. doi: 10.1073/pnas.0706347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoshizawa-Sugata N, Masai H. Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J Biol Chem. 2007;282:2729–2740. doi: 10.1074/jbc.M605596200. [DOI] [PubMed] [Google Scholar]
- 71.Durkin SG, Arlt MF, Howlett NG, Glover TW. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene. 2006;25:4381–4388. doi: 10.1038/sj.onc.1209466. [DOI] [PubMed] [Google Scholar]
- 72.Wittenberg C, Reed SI. Cell cycle-dependent transcription in yeast: promoters, transcription factors, and transcriptomes. Oncogene. 2005;24:2746–2755. doi: 10.1038/sj.onc.1208606. [DOI] [PubMed] [Google Scholar]
- 73.de Bruin RA, Kalashnikova TI, Aslanian A, Wohlschlegel J, Chahwan C, Yates JR, 3rd, Russell P, Wittenberg C. DNA replication checkpoint promotes G1-S transcription by inactivating the MBF repressor Nrm1. Proc Natl Acad Sci U S A. 2008;105:11230–11235. doi: 10.1073/pnas.0801106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dutta C, Patel PK, Rosebrock A, Oliva A, Leatherwood J, Rhind N. The DNA Replication Checkpoint Directly Regulates MBF-Dependent G1/S Transcription. Mol Cell Biol. 2008 doi: 10.1128/MCB.00596-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee H, Yuan C, Hammet A, Mahajan A, Chen ES, Wu MR, Su MI, Heierhorst J, Tsai MD. Diphosphothreonine-specific interaction between an SQ/TQ cluster and an FHA domain in the Rad53-Dun1 kinase cascade. Mol Cell. 2008;30:767–778. doi: 10.1016/j.molcel.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 76.Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998;94:595–605. doi: 10.1016/s0092-8674(00)81601-3. [DOI] [PubMed] [Google Scholar]
- 77.Stevens C, Smith L, La Thangue NB. Chk2 activates E2F-1 in response to DNA damage. Nat Cell Biol. 2003;5:401–409. doi: 10.1038/ncb974. [DOI] [PubMed] [Google Scholar]
- 78.Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 2004;18:3041–3054. doi: 10.1101/gad.1221004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Inoue Y, Kitagawa M, Taya Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 2007;26:2083–2093. doi: 10.1038/sj.emboj.7601652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stevens C, La Thangue NB. The emerging role of E2F-1 in the DNA damage response and checkpoint control. DNA Repair (Amst) 2004;3:1071–1079. doi: 10.1016/j.dnarep.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 81.Rogoff HA, Pickering MT, Frame FM, Debatis ME, Sanchez Y, Jones S, Kowalik TF. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol. 2004;24:2968–2977. doi: 10.1128/MCB.24.7.2968-2977.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, Anderson CW, Appella E, Nakanishi M, Suzuki H, Nagashima K, Sawa H, Ikeda K, Motoyama N. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. Embo J. 2002;21:5195–5205. doi: 10.1093/emboj/cdf506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacPherson D, Kim J, Kim T, Rhee BK, Van Oostrom CT, DiTullio RA, Venere M, Halazonetis TD, Bronson R, De Vries A, Fleming M, Jacks T. Defective apoptosis and B-cell lymphomas in mice with p53 point mutation at Ser 23. Embo J. 2004;23:3689–3699. doi: 10.1038/sj.emboj.7600363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pereg Y, Lam S, Teunisse A, Biton S, Meulmeester E, Mittelman L, Buscemi G, Okamoto K, Taya Y, Shiloh Y, Jochemsen AG. Differential roles of ATM- and Chk2-mediated phosphorylations of Hdmx in response to DNA damage. Mol Cell Biol. 2006;26:6819–6831. doi: 10.1128/MCB.00562-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bruno T, De Nicola F, Iezzi S, Lecis D, D’Angelo C, Di Padova M, Corbi N, Dimiziani L, Zannini L, Jekimovs C, Scarsella M, Porrello A, Chersi A, Crescenzi M, Leonetti C, Khanna KK, Soddu S, Floridi A, Passananti C, Delia D, Fanciulli M. Che-1 phosphorylation by ATM/ATR and Chk2 kinases activates p53 transcription and the G2/M checkpoint. Cancer Cell. 2006;10:473–486. doi: 10.1016/j.ccr.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 86.Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, Pandolfi PP. PML is essential for multiple apoptotic pathways. Nat Genet. 1998;20:266–272. doi: 10.1038/3073. [DOI] [PubMed] [Google Scholar]
- 87.Yang S, Kuo C, Bisi JE, Kim MK. PML-dependent apoptosis after DNA damage is regulated by the checkpoint kinase hCds1/Chk2. Nat Cell Biol. 2002;4:865–870. doi: 10.1038/ncb869. [DOI] [PubMed] [Google Scholar]
- 88.Dellaire G, Ching RW, Ahmed K, Jalali F, Tse KC, Bristow RG, Bazett-Jones DP. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR. J Cell Biol. 2006;175:55–66. doi: 10.1083/jcb.200604009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, Kanki JP, Green DR, D’Andrea AA, Look AT. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell. 2008;133:864–877. doi: 10.1016/j.cell.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314:294–297. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- 91.Yuan Z, Becker EB, Merlo P, Yamada T, DiBacco S, Konishi Y, Schaefer EM, Bonni A. Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science. 2008;319:1665–1668. doi: 10.1126/science.1152337. [DOI] [PubMed] [Google Scholar]
- 92.Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, Nakanishi M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008;132:221–232. doi: 10.1016/j.cell.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 93.Hu F, Alcasabas AA, Elledge SJ. Asf1 links Rad53 to control of chromatin assembly. Genes Dev. 2001;15:1061–1066. doi: 10.1101/gad.873201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schwartz MF, Lee SJ, Duong JK, Eminaga S, Stern DF. FHA domain-mediated DNA checkpoint regulation of Rad53. Cell Cycle. 2003;2:384–396. [PubMed] [Google Scholar]
- 95.Lee SJ, Schwartz MF, Duong JK, Stern DF. Rad53 phosphorylation site clusters are important for Rad53 regulation and signaling. Mol Cell Biol. 2003;23:6300–6314. doi: 10.1128/MCB.23.17.6300-6314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sillje HH, Takahashi K, Tanaka K, Van Houwe G, Nigg EA. Mammalian homologues of the plant Tousled gene code for cell-cycle-regulated kinases with maximal activities linked to ongoing DNA replication. EMBO J. 1999;18:5691–5702. doi: 10.1093/emboj/18.20.5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Groth A, Lukas J, Nigg EA, Sillje HH, Wernstedt C, Bartek J, Hansen K. Human Tousled like kinases are targeted by an ATM- and Chk1-dependent DNA damage checkpoint. Embo J. 2003;22:1676–1687. doi: 10.1093/emboj/cdg151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Krause DR, Jonnalagadda JC, Gatei MH, Sillje HH, Zhou BB, Nigg EA, Khanna K. Suppression of Tousled-like kinase activity after DNA damage or replication block requires ATM, NBS1 and Chk1. Oncogene. 2003;22:5927–5937. doi: 10.1038/sj.onc.1206691. [DOI] [PubMed] [Google Scholar]
- 99.Sillje HH, Nigg EA. Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr Biol. 2001;11:1068–1073. doi: 10.1016/s0960-9822(01)00298-6. [DOI] [PubMed] [Google Scholar]
- 100.Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–243. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- 102.Leroy C, Lee SE, Vaze MB, Ochsenbien F, Guerois R, Haber JE, Marsolier-Kergoat MC. PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol Cell. 2003;11:827–835. doi: 10.1016/s1097-2765(03)00058-3. [DOI] [PubMed] [Google Scholar]
- 103.Marsolier MC, Roussel P, Leroy C, Mann C. Involvement of the PP2C-like phosphatase Ptc2p in the DNA checkpoint pathways of Saccharomyces cerevisiae. Genetics. 2000;154:1523–1532. doi: 10.1093/genetics/154.4.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.O’Neill BM, Szyjka SJ, Lis ET, Bailey AO, Yates JR, 3rd, Aparicio OM, Romesberg FE. Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc Natl Acad Sci U S A. 2007;104:9290–9295. doi: 10.1073/pnas.0703252104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Szyjka SJ, Aparicio JG, Viggiani CJ, Knott S, Xu W, Tavare S, Aparicio OM. Rad53 regulates replication fork restart after DNA damage in Saccharomyces cerevisiae. Genes Dev. 2008;22:1906–1920. doi: 10.1101/gad.1660408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.den Elzen NR, O’Connell MJ. Recovery from DNA damage checkpoint arrest by PP1-mediated inhibition of Chk1. EMBO J. 2004;23:908–918. doi: 10.1038/sj.emboj.7600105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, Vande Woude GF, O’Connor PM, Appella E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A. 1997;94:6048–6053. doi: 10.1073/pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–1174. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A, Kondo T, Imamura M, Oishi I, Yoda A, Minami Y. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006;13:1170–1180. doi: 10.1038/sj.cdd.4401801. [DOI] [PubMed] [Google Scholar]
- 110.Shreeram S, Demidov ON, Hee WK, Yamaguchi H, Onishi N, Kek C, Timofeev ON, Dudgeon C, Fornace AJ, Anderson CW, Minami Y, Appella E, Bulavin DV. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006;23:757–764. doi: 10.1016/j.molcel.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 111.Nannenga B, Lu X, Dumble M, Van Maanen M, Nguyen TA, Sutton R, Kumar TR, Donehower LA. Augmented cancer resistance and DNA damage response phenotypes in PPM1D null mice. Mol Carcinog. 2006;45:594–604. doi: 10.1002/mc.20195. [DOI] [PubMed] [Google Scholar]
- 112.Oliva-Trastoy M, Berthonaud V, Chevalier A, Ducrot C, Marsolier-Kergoat MC, Mann C, Leteurtre F. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene. 2007;26:1449–1458. doi: 10.1038/sj.onc.1209927. [DOI] [PubMed] [Google Scholar]
- 113.Dozier C, Bonyadi M, Baricault L, Tonasso L, Darbon JM. Regulation of Chk2 phosphorylation by interaction with protein phosphatase 2A via its B′ regulatory subunit. Biol Cell. 2004;96:509–517. doi: 10.1016/j.biolcel.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 114.Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, Lees-Miller SP, Khanna KK. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004;23:4451–4461. doi: 10.1038/sj.emboj.7600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Petersen P, Chou DM, You Z, Hunter T, Walter JC, Walter G. Protein phosphatase 2A antagonizes ATM and ATR in a Cdk2- and Cdc7-independent DNA damage checkpoint. Mol Cell Biol. 2006;26:1997–2011. doi: 10.1128/MCB.26.5.1997-2011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Margolis SS, Perry JA, Forester CM, Nutt LK, Guo Y, Jardim MJ, Thomenius MJ, Freel CD, Darbandi R, Ahn JH, Arroyo JD, Wang XF, Shenolikar S, Nairn AC, Dunphy WG, Hahn WC, Virshup DM, Kornbluth S. Role for the PP2A/B56delta phosphatase in regulating 14-3-3 release from Cdc25 to control mitosis. Cell. 2006;127:759–773. doi: 10.1016/j.cell.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 118.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 119.Bulavin DV, Phillips C, Nannenga B, Timofeev O, Donehower LA, Anderson CW, Appella E, Fornace AJ., Jr Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–350. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 120.Nevanlinna H, Bartek J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene. 2006;25:5912–5919. doi: 10.1038/sj.onc.1209877. [DOI] [PubMed] [Google Scholar]
- 121.Bahassi el M, Penner CG, Robbins SB, Tichy E, Feliciano E, Yin M, Liang L, Deng L, Tischfield JA, Stambrook PJ. The breast cancer susceptibility allele CHEK2*1100delC promotes genomic instability in a knock-in mouse model. Mutat Res. 2007;616:201–209. doi: 10.1016/j.mrfmmm.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 122.Kwak EL, Kim S, Zhang J, Cardiff RD, Schmidt EV, Haber DA. Mammary tumorigenesis following transgenic expression of a dominant negative CHK2 mutant. Cancer Res. 2006;66:1923–1928. doi: 10.1158/0008-5472.CAN-05-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McPherson JP, Lemmers B, Hirao A, Hakem A, Abraham J, Migon E, Matysiak-Zablocki E, Tamblyn L, Sanchez-Sweatman O, Khokha R, Squire J, Hande MP, Mak TW, Hakem R. Collaboration of Brca1 and Chk2 in tumorigenesis. Genes Dev. 2004;18:1144–1153. doi: 10.1101/gad.1192704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cao L, Kim S, Xiao C, Wang RH, Coumoul X, Wang X, Li WM, Xu XL, De Soto JA, Takai H, Mai S, Elledge SJ, Motoyama N, Deng CX. ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. Embo J. 2006;25:2167–2177. doi: 10.1038/sj.emboj.7601115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim SS, Cao L, Li C, Xu X, Huber LJ, Chodosh LA, Deng CX. Uterus hyperplasia and increased carcinogen-induced tumorigenesis in mice carrying a targeted mutation of the Chk2 phosphorylation site in Brca1. Mol Cell Biol. 2004;24:9498–9507. doi: 10.1128/MCB.24.21.9498-9507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 127.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 128.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev. 2000;14:1439–1447. [PMC free article] [PubMed] [Google Scholar]
- 129.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 130.Xu X, Lee J, Stern DF. Microcephalin is a DNA damage response protein involved in regulation of CHK1 and BRCA1. J Biol Chem. 2004;279:34091–34094. doi: 10.1074/jbc.C400139200. [DOI] [PubMed] [Google Scholar]
- 131.Lin SY, Rai R, Li K, Xu ZX, Elledge SJ. BRIT1/MCPH1 is a DNA damage responsive protein that regulates the Brca1-Chk1 pathway, implicating checkpoint dysfunction in microcephaly. Proc Natl Acad Sci U S A. 2005;102:15105–15109. doi: 10.1073/pnas.0507722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Alderton GK, Galbiati L, Griffith E, Surinya KH, Neitzel H, Jackson AP, Jeggo PA, O’Driscoll M. Regulation of mitotic entry by microcephalin and its overlap with ATR signalling. Nat Cell Biol. 2006;8:725–733. doi: 10.1038/ncb1431. [DOI] [PubMed] [Google Scholar]
- 133.Zhou BB, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer. 2004;4:216–225. doi: 10.1038/nrc1296. [DOI] [PubMed] [Google Scholar]
- 134.Antoni L, Sodha N, Collins I, Garrett MD. CHK2 kinase: cancer susceptibility and cancer therapy - two sides of the same coin? Nat Rev Cancer. 2007;7:925–936. doi: 10.1038/nrc2251. [DOI] [PubMed] [Google Scholar]
- 135.O’Connor MJ, Martin NM, Smith GC. Targeted cancer therapies based on the inhibition of DNA strand break repair. Oncogene. 2007;26:7816–7824. doi: 10.1038/sj.onc.1210879. [DOI] [PubMed] [Google Scholar]