

Abstract
The reconstruction and synthesis of ancestral RNAs is a feasible goal for paleogenetics. This will require new bioinformatics methods, including a robust statistical framework for reconstructing histories of substitutions, indels and structural changes. We describe a “transducer composition” algorithm for extending pairwise probabilistic models of RNA structural evolution to models of multiple sequences related by a phylogenetic tree. This algorithm draws on formal models of computational linguistics as well as the 1985 protosequence algorithm of David Sankoff. The output of the composition algorithm is a multiple-sequence stochastic context-free grammar. We describe dynamic programming algorithms, which are robust to null cycles and empty bifurcations, for parsing this grammar. Example applications include structural alignment of non-coding RNAs, propagation of structural information from an experimentally-characterized sequence to its homologs, and inference of the ancestral structure of a set of diverged RNAs. We implemented the above algorithms for a simple model of pairwise RNA structural evolution; in particular, the algorithms for maximum likelihood (ML) alignment of three known RNA structures and a known phylogeny and inference of the common ancestral structure. We compared this ML algorithm to a variety of related, but simpler, techniques, including ML alignment algorithms for simpler models that omitted various aspects of the full model and also a posterior-decoding alignment algorithm for one of the simpler models. In our tests, incorporation of basepair structure was the most important factor for accurate alignment inference; appropriate use of posterior-decoding was next; and fine details of the model were least important. Posterior-decoding heuristics can be substantially faster than exact phylogenetic inference, so this motivates the use of sum-over-pairs heuristics where possible (and approximate sum-over-pairs). For more exact probabilistic inference, we discuss the use of transducer composition for ML (or MCMC) inference on phylogenies, including possible ways to make the core operations tractable.
Author Summary
A number of leading methods for bioinformatics analysis of structural RNAs use probabilistic grammars as models for pairs of homologous RNAs. We show that any such pairwise grammar can be extended to an entire phylogeny by treating the pairwise grammar as a machine (a “transducer”) that models a single ancestor-descendant relationship in the tree, transforming one RNA structure into another. In addition to phylogenetic enhancement of current applications, such as RNA genefinding, homology detection, alignment and secondary structure prediction, this should enable probabilistic phylogenetic reconstruction of RNA sequences that are ancestral to present-day genes. We describe statistical inference algorithms, software implementations, and a simulation-based comparison of three-taxon maximum likelihood alignment to several other methods for aligning three sibling RNAs. In the Discussion we consider how the three-taxon RNA alignment-reconstruction-folding algorithm, which is currently very computationally-expensive, might be made more efficient so that larger phylogenies could be considered.
Introduction
In 1968, Francis Crick hypothesized that the first ribosome consisted entirely of RNA, without any protein cofactors [1]. A domain structure for this primeval ribosome was recently proposed [2]. To synthesize such a reconstructed ribosome or reconstructions of other evolutionarily significant RNAs such as group II introns [3] or telomerase [4], it will be necessary to develop methods that can predict the sequences and structures of ancient RNAs based on the divergent sequences of their many descendants.
An inspection of RNA alignments, such as those in the RFAM database [5], suggests that an evolutionary model for RNA structure must eventually include multiple layers of detail: point substitutions, covariant substitutions of base-pairs [6],[7], indels [8], local changes in secondary structure such as helix slippage [9], and changes in domain structure [2]. Stochastic context-free grammars (SCFGs), which can efficiently detect the long-range correlations of RNA base-pairing structures, are natural probabilistic models of such phenomena and have been used for ncRNA homology detection [10]–[13], gene prediction [14],[15], folding [16],[17] and alignment [18]–[20].
By analogy with models of substitution processes, which are well-understood [21], we may take the problem of building phylogenetic models of RNA evolution and split it into two halves. The first half is the development of a pairwise model, describing the probability distribution 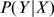 of a descendant (
of a descendant ( ) conditional on its immediate ancestor (
) conditional on its immediate ancestor ( ). In substitution processes, the pairwise model is a conditional substitution matrix. Often (but not always) the pairwise model, representing a finite evolutionary time
). In substitution processes, the pairwise model is a conditional substitution matrix. Often (but not always) the pairwise model, representing a finite evolutionary time  , is derived from an instantaneous model of change over an infinitesimal time interval, i.e., a continuous-time Markov chain (parametrized by a rate matrix). Obtaining the transition probabilities of this chain (via exponentiation of the rate matrix) yields a pairwise model whose parameters are smoothly-varying functions of
, is derived from an instantaneous model of change over an infinitesimal time interval, i.e., a continuous-time Markov chain (parametrized by a rate matrix). Obtaining the transition probabilities of this chain (via exponentiation of the rate matrix) yields a pairwise model whose parameters are smoothly-varying functions of 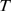 . A pairwise model represents an individual branch of a phylogenetic tree, with
. A pairwise model represents an individual branch of a phylogenetic tree, with 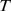 representing the length of that branch.
representing the length of that branch.
The second half of the phylogenetic modeling problem involves extending the model (and related inference algorithms) from a single branch to a complete phylogeny, i.e., from a pairwise model of two sequences to a multiple-sequence model of many sequences. In a typical situation, the sequences at the leaves of the tree are observed but those at internal nodes are not. Questions of interest then include:
What is the likelihood for the observed sequence data?
Can we sample (find the mode, take moments, etc.) from the posterior distribution of the unobserved sequence at the root node?
Can we sample from the posterior of the unobserved sequences at the other internal nodes?
Can we estimate summaries of the evolutionary history, such as the number of substitution events on each branch (for a substitution model), the alignment (for a model which includes indels), or changes in the underlying structure (for a model of RNA structure)?
For substitution models, there has been extensive work focused on answering each of these questions. Given a pairwise substitution model, questions A and B can be answered exactly by Felsenstein's pruning algorithm [22] and question C can be answered by the peeling algorithm (first presented for pedigree analysis by Elston and Stewart [23]). The estimation of evolutionary histories (question D) has been addressed by exact summarization [24] and sampling [25] approaches. Another representation of answers A–C is that the pruning and peeling algorithms (combined) are just the sum-product algorithm on a directed graphical model [26], yielding exact marginal distributions for unobserved variables. Graphical models also suggest general-purpose sampling approaches in addition to the exact sum-product algorithm.
The two halves of the reconstruction problem — developing a pairwise model and then extending it to multiple sequences — are largely independent. Felsenstein's pruning algorithm, for example, is essentially blind to the parametric form of the pairwise substitution model; it just assumes that a substitution matrix is provided for every branch. Subsequent models developed by other researchers can be plugged into the pruning algorithm without modification [27],[28].
We therefore addressed the problem of modeling the indel-evolution of multiple structured RNAs in a similarly-modular fashion by separating the creation of pairwise and multiple-sequence models. In previous work, we addressed the first (pairwise) part of the RNA reconstruction problem by describing a simple continuous-time model of RNA structural evolution [29]. This model corresponded to a Pair SCFG with a time-dependent parametrization which we used to simultaneously align and predict the structure of pairs of related RNAs. The focus of the present work is to solve the second (multiple-sequence) part of the RNA reconstruction problem by giving a general procedure for extending a pairwise model to multiple sequences related by a phylogenetic tree. This process yields a multiple-sequence SCFG, a natural model of the evolutionary relationships between multiple structured RNAs.
The main contributions of this paper are (1) an algorithm that transforms a phylogenetic ensemble of pair grammars, representing models on branches of a phylogenetic tree, into a coherent, multiple-sequence SCFG, (2) dynamic programming (DP) algorithms for performing inference under this multiple-sequence SCFG, and (3) freely-available software implementing algorithms (1) and (2) for the simplified case of a three-taxon star-topology tree. While the idea of composing conditionally-normalized models on trees is intuitive, the resulting models can be very complex, even for simple models of RNA evolution, making (1) necessary. Studies of related indel models have suggested that an implementation of dynamic programming (DP) algorithms on a three-taxon tree is sufficient to draw samples from the posterior distribution of ancestral sequences on more complex tree topologies, using Markov Chain Monte Carlo or MCMC [30]–[32], suggesting that (2) and (3) are, in principle, sufficient for analyzing trees relating many sequences.
We show that our algorithm produces a multiple-sequence grammar which is much more compact than suggested by naive approaches to model construction. We provide analyses of the asymptotic complexities of models constructed using our procedure and provide estimates of the time and memory required to reconstruct the structures of several RNA families for the case of a three-taxon phylogeny, which we have implemented in the program Indiegram. While by these estimates only the smallest sequences currently fit into affordable memory, thereby preventing us from applying our method to many problems of interest, a simulation study suggests that we can hope to accurately reconstruct ancestral structures over long evolutionary time, even in the presence of structural divergence.
In the Discussion, we speculate on algorithmic extensions that may reduce memory requirements, inspired by related work in reconstructing DNA and protein sequences.
Methods
We describe below a general method for constructing a multiple-sequence stochastic grammar for alignment, folding and ancestral reconstruction of RNA, given a phylogenetic tree and a description of the evolutionary process acting along each branch.
Overview
Our problem statement is this: Given a phylogenetic tree relating several structured RNAs and a description of the evolution of a structured RNA along a single branch of the tree (in the form of a Pair SCFG), (1) find the corresponding phylogenetic multiple-sequence grammar and (2) use that grammar to reconstruct, a posteriori , the evolutionary histories of the RNAs. We assume here that the phylogeny, including both the tree topology and branch lengths, is given.
This paper focuses on model construction and inference algorithms rather than the heuristics which will be necessary to make these algorithms fast enough for analysis of many biological datasets. As discussed below, the complexity of general inference algorithms is prohibitively high for many problems of interest. However, this complexity can be significantly reduced by incorporating outside knowledge. For example, if we know the consensus structure of several sequences or their individual structures, then we can constrain our algorithms accordingly. Similarly, we might consider only ancestral structures which are compatible with a given multiple sequence alignment, or a relatively small set of candidate alignments (as in the ORTHEUS program [33]). Such constraints are commonly used by programs for SCFG-based RNA sequence analysis such as QRNA [34], Stemloc [18] and CONSAN [19]. Alignment and structural constraints can be combined [18].
In the following sections we introduce more precise definitions for two-sequence models of RNA structure and outline our algorithms for (1) combining these two-sequence models on a phylogenetic tree and (2) using the composite phylogenetic grammars for inference.
Two-sequence models
We discuss the general problem of creating state-space models of the evolution of related sequences, beginning with models of substitution processes acting at independent sites (as studied in likelihood phylogenetics) and generalizing to models of indels, first in primary sequences and then in sequences with conserved secondary structure.
A stochastic model for the evolution of one sequence (the ancestor, 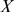 ) into another (the descendant,
) into another (the descendant, 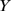 ) over an interval of time (
) over an interval of time (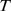 ) can be described by a joint distribution,
) can be described by a joint distribution, 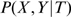 . This joint distribution can be factored,
. This joint distribution can be factored, 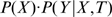 , where
, where 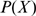 is the marginal distribution over ancestral sequences and
is the marginal distribution over ancestral sequences and 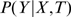 is the conditional distribution over descendant sequences given an ancestral sequence. In terms of phylogenetics, the conditional distribution
is the conditional distribution over descendant sequences given an ancestral sequence. In terms of phylogenetics, the conditional distribution 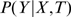 describes the evolution
describes the evolution  along a branch of length
along a branch of length  .
.
It is possible to “multiply” two such models together. More precisely, one multiplies two conditional distributions and sums out the intermediate sequence. Thus, successive evolution along two branches 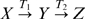 is modeled by the distribution
is modeled by the distribution
and we can sum sequence 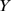 out of this, obtaining the distribution
out of this, obtaining the distribution
for the composite branch 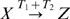 .
.
This formalism underlies likelihood phylogenetics. Working under the independent-sites assumption, 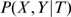 is the
is the 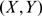 'th element of the joint substitution matrix for a single site and
'th element of the joint substitution matrix for a single site and 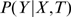 is the corresponding element of the conditional matrix. The conditional matrix is in fact the matrix exponential
is the corresponding element of the conditional matrix. The conditional matrix is in fact the matrix exponential 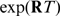 , where
, where 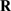 is the substitution rate matrix [24]. Composition of two branches just amounts to a matrix multiplication.
is the substitution rate matrix [24]. Composition of two branches just amounts to a matrix multiplication.
A similar formalism can be used to describe the evolution of whole sequences with indels. Suppose that the joint distribution 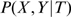 is the distribution modeled by a pair hidden Markov model (Pair HMM) [11], a probabilistic model of the evolution of two sequences under the approximation that only adjacent characters are directly correlated, and the marginal
is the distribution modeled by a pair hidden Markov model (Pair HMM) [11], a probabilistic model of the evolution of two sequences under the approximation that only adjacent characters are directly correlated, and the marginal 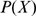 is the distribution of a single-sequence HMM, a probabilistic model of single sequences under the same approximation. The conditional distribution
is the distribution of a single-sequence HMM, a probabilistic model of single sequences under the same approximation. The conditional distribution 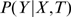 then corresponds to a conditional Pair HMM, a discrete-state machine which transforms one sequence (the input,
then corresponds to a conditional Pair HMM, a discrete-state machine which transforms one sequence (the input, 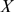 ) into another (the output,
) into another (the output, 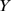 ). Following computational linguists, we call this conditionally-normalized state machine a string transducer or simply a transducer
[35]. Because of its conditional normalization, this state machine is distinct from a standard Pair HMM. A Pair HMM has two outputs
). Following computational linguists, we call this conditionally-normalized state machine a string transducer or simply a transducer
[35]. Because of its conditional normalization, this state machine is distinct from a standard Pair HMM. A Pair HMM has two outputs 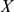 and
and 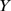 and emits symbols to both of those outputs, while a transducer absorbs symbols from the input
and emits symbols to both of those outputs, while a transducer absorbs symbols from the input 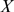 and emits symbols to the output
and emits symbols to the output 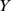 . Despite this distinction, Pair HMMs and transducers share very similar inference algorithms; for example,
. Despite this distinction, Pair HMMs and transducers share very similar inference algorithms; for example, 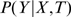 is computed using a direct analogue of the Forward algorithm [11].
is computed using a direct analogue of the Forward algorithm [11].
We extend this formalism to the case of structured RNA as follows. Let 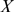 and
and 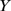 now represent structured RNA sequences or, more precisely, parse trees. A single-sequence SCFG models the marginal
now represent structured RNA sequences or, more precisely, parse trees. A single-sequence SCFG models the marginal 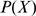 ; a jointly-normalized Pair SCFG [11] models the the joint distribution
; a jointly-normalized Pair SCFG [11] models the the joint distribution 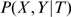 . The conditional distribution
. The conditional distribution 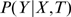 is modeled by a conditionally-normalized Pair SCFG. Following terminology from computational linguistics [36], we call this conditionally-normalized grammar a parse-tree transducer.
is modeled by a conditionally-normalized Pair SCFG. Following terminology from computational linguistics [36], we call this conditionally-normalized grammar a parse-tree transducer.
String transducers are special cases of parse-tree transducers, just as HMMs are special cases of SCFGs. Henceforth, we will drop the distinction between strings and parse trees. We will also refer interchangeably to “states” (in the state-machine representation) and “nonterminals” (in the grammar representation). Likewise, we will refer interchangeably to “state paths” (machines) and “parse trees” (grammars).
Terminology and normalization
Consider the stochastic grammar which generates parse trees from the marginal distribution 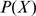 . It is convenient to represent this grammar as a transducer whose input is constrained to be null, i.e. a machine that accepts a dummy (empty) input sequence, and outputs sequence
. It is convenient to represent this grammar as a transducer whose input is constrained to be null, i.e. a machine that accepts a dummy (empty) input sequence, and outputs sequence 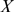 . We refer to this as the singlet transducer. In contrast, the more general type of transducer that absorbs parse trees
. We refer to this as the singlet transducer. In contrast, the more general type of transducer that absorbs parse trees 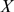 and generates modified parse trees
and generates modified parse trees 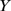 from the conditional distribution
from the conditional distribution 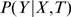 is a branch transducer. By definition, singlet transducers only emit symbols to their output sequence, and use a restricted set of state types. Branch transducers, in contrast, can both emit symbols to their outputs and absorb symbols from their inputs, and so use the full range of state types.
is a branch transducer. By definition, singlet transducers only emit symbols to their output sequence, and use a restricted set of state types. Branch transducers, in contrast, can both emit symbols to their outputs and absorb symbols from their inputs, and so use the full range of state types.
Transducers can have states of type 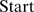 ,
, 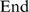 ,
, 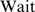 ,
, 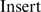 and
and  . The first three state types,
. The first three state types,  ,
,  and
and  , are null: they do not emit or absorb any symbols and are required solely for organizational purposes (see following section). Two types of states can emit and/or absorb symbols,
, are null: they do not emit or absorb any symbols and are required solely for organizational purposes (see following section). Two types of states can emit and/or absorb symbols, 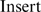 and
and 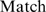 . An
. An 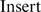 state emits a symbol to the output without absorbing anything. A
state emits a symbol to the output without absorbing anything. A 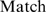 state absorbs a symbol on the input and either emits the same symbol to the output, substitutes a different output symbol, or emits no output symbol at all, the last corresponding to a deletion.
state absorbs a symbol on the input and either emits the same symbol to the output, substitutes a different output symbol, or emits no output symbol at all, the last corresponding to a deletion.
As stated above, the Pair SCFG must be conditionally normalized so that models can be chained together, extending the pairwise model to multiple sequences. The transformation rules are partitioned into co-normalized groups; within each group, the rule probabilities must sum to one. In a jointly-normalized Pair SCFG, each group corresponds to the set of all rules that can be applied to a given nonterminal (i.e., all outgoing transitions from a particular state). In a conditionally-normalized Pair SCFG, in contrast, each co-normalized group includes all rules that can be applied to a given nonterminal for a given set of absorbed symbols.
Multiple-sequence models
We can use the concepts of factoring probability distributions introduced in the two-sequence framework to model the common descent of many homologous sequences. Given a phylogenetic tree and a two-sequence model, we wish to obtain a multiple-sequence SCFG describing the common descent of the observed sequences.
A singlet transducer (which emits, but does not absorb, symbols) lies at the root of the phylogeny and serves as a generative model of the ancestral sequence. To represent the evolution of an ancestral sequence into many descendant sequences, we place a branch transducer on each branch of the phylogeny.
Throughout this paper we frequently refer to two and three-taxon (star) phylogenies. In all cases, the sequence 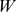 is assumed to be the (unobserved) ancestral sequence and the sequences
is assumed to be the (unobserved) ancestral sequence and the sequences 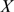 ,
, 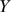 , and
, and 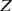 the (observed) extant sequences.
the (observed) extant sequences.
The composition algorithm
While this composition of conditionally-normalized models on a phylogenetic tree is intuitive, in practice building such an ensemble model is challenging due to the sheer number of possible states and transitions of the ensemble model. The maximum possible state space of the ensemble is the Cartesian product of the individual transducer state spaces. If the singlet transducer has  states, each branch transducer has
states, each branch transducer has 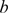 states, and the phylogeny has
states, and the phylogeny has 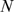 branches, then an upper bound on the number of ensemble states is
branches, then an upper bound on the number of ensemble states is 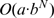 . However, in practice there are many fewer states than suggested by this bound; many state configurations are not reachable. For example, for the tree with two extant sequences and a single parent, the branch transducers above leaves
. However, in practice there are many fewer states than suggested by this bound; many state configurations are not reachable. For example, for the tree with two extant sequences and a single parent, the branch transducers above leaves 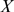 and
and 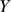 cannot simultaneously be in
cannot simultaneously be in 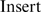 states, as this would correspond to aligning non-homologous (inserted) characters. Similarly, while an upper bound on the number of possible transitions in the transition matrix of the ensemble model is
states, as this would correspond to aligning non-homologous (inserted) characters. Similarly, while an upper bound on the number of possible transitions in the transition matrix of the ensemble model is 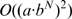 , in practice models never reach this bound, due both to inaccessible configurations, such as the one described above, and the sparseness of transitions between the remaining, accessible configurations.
, in practice models never reach this bound, due both to inaccessible configurations, such as the one described above, and the sparseness of transitions between the remaining, accessible configurations.
While the accessible state space of the ensemble is smaller than that given by the exponential upper bound, it is generally nonetheless too complex to deal with by hand. For example, the simple model of RNA structural evolution described in Results yields an ensemble model of three sequences with 230 states and 1,789 transitions. More realistic models of RNA give rise to even larger ensemble models.
We therefore need an algorithm to efficiently construct the state graph of the ensemble model, consisting of a list of accessible states and the possible transitions between them. By analogy with algorithms for uninformed graph search in artificial intelligence, the transition graph of the ensemble can be constructed by an uninformed depth-first search, where at each step of the search we obtain the next possible ensemble states by changing the state of one or more of the singlet or branch transducers. Beginning with the entire ensemble in state 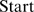 , the depth-first search of states continues until all nodes are in state
, the depth-first search of states continues until all nodes are in state 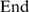 .
.
The allowed transitions of the ensemble can be categorized as follows:
Null Transition: A branch transducer makes a transition into a
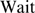 state, with no terminal emission or bifurcation.
state, with no terminal emission or bifurcation.Terminal Emission: A singlet or branch transducer makes a transition into a state of type
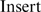 , emitting left and/or right terminal symbols (e.g., a single base or base-pair). These symbols are absorbed by the immediately-descended transducers, which are pushed into states of type
, emitting left and/or right terminal symbols (e.g., a single base or base-pair). These symbols are absorbed by the immediately-descended transducers, which are pushed into states of type 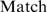 and may themselves emit terminal symbols that will be absorbed by their descendant transducers. This continues down the tree: The terminal symbols are passed from parents to children to grandchildren (albeit possibly being replaced by other terminal symbols as they are propagated down) and they propel branch transducers into
and may themselves emit terminal symbols that will be absorbed by their descendant transducers. This continues down the tree: The terminal symbols are passed from parents to children to grandchildren (albeit possibly being replaced by other terminal symbols as they are propagated down) and they propel branch transducers into  states as they go. Eventually, the cascade of emitted terminal symbols stops when all the symbols have been deleted or when the cascade reaches the leaves of the tree.
states as they go. Eventually, the cascade of emitted terminal symbols stops when all the symbols have been deleted or when the cascade reaches the leaves of the tree.Bifurcation: A singlet or branch transducer makes a transition into a state of type
 that spawns left and/or right nonterminal states. These nonterminals are processed recursively down the tree, just as in a terminal emission (conceptually, a bifurcation is a “nonterminal emission”). As with terminal emissions, absorption of nonterminal emissions propels descendant transducers into
that spawns left and/or right nonterminal states. These nonterminals are processed recursively down the tree, just as in a terminal emission (conceptually, a bifurcation is a “nonterminal emission”). As with terminal emissions, absorption of nonterminal emissions propels descendant transducers into  states, making transitions which may themselves propagate nonterminals further down the tree. A biologically-relevant example of a bifurcation is the insertion of a stem into an ancestral RNA structure, which may then be conserved or deleted in the descendant structures.
states, making transitions which may themselves propagate nonterminals further down the tree. A biologically-relevant example of a bifurcation is the insertion of a stem into an ancestral RNA structure, which may then be conserved or deleted in the descendant structures.End Transition: The singlet transducer at the root makes a transition to the
 state, pushing all the descendant branch transducers into
state, pushing all the descendant branch transducers into 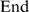 states and terminating the current branch of the parse tree.
states and terminating the current branch of the parse tree.
Co-ordination between the various branch machines is achieved by specifying an ordering on the nodes and by having branch transducers pause in 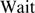 states while waiting to absorb a symbol from the node above. Only one transducer is allowed to make a spontaneous transition at a time. If this transition corresponds to a terminal emission or a bifurcation, then this may force descendant transducers into making reactive transitions.
states while waiting to absorb a symbol from the node above. Only one transducer is allowed to make a spontaneous transition at a time. If this transition corresponds to a terminal emission or a bifurcation, then this may force descendant transducers into making reactive transitions.
The four types of allowed transitions listed above can be formalized as follows. Let the total order on the nodes correspond to any preorder traversal of the tree; thus, “ is ancestral to
is ancestral to  ” is sufficient-but-not-necessary for “
” is sufficient-but-not-necessary for “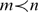 .” Let
.” Let 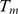 denote the singlet or branch transducer which emits symbols to node
denote the singlet or branch transducer which emits symbols to node  . Transducer
. Transducer 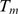 changes state if and only if one of the following three mutually-exclusive conditions holds:
changes state if and only if one of the following three mutually-exclusive conditions holds:
Type 1: Transducer
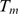 is not in a
is not in a  state, while all its successor transducers
state, while all its successor transducers 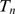 are in
are in 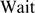 states (where
states (where 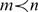 ).
). 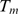 is free to make any transition.
is free to make any transition.Type 2: Transducer
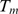 is in a
is in a 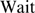 state. Its parent transducer enters a
state. Its parent transducer enters a 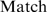 or
or 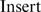 state, emitting a symbol and forcing
state, emitting a symbol and forcing 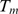 into a
into a 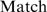 state so it can absorb that symbol.
state so it can absorb that symbol.Type 3: Transducer
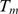 is in a
is in a 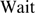 state. Its parent transducer enters the
state. Its parent transducer enters the 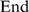 state, forcing
state, forcing 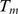 into the
into the  state as well.
state as well.
A notational prescription for the allowed transitions may be found in Text S1.
How the ensemble generates multiple alignments
The possible transitions of the ensemble generate multiple alignments as follows:
The singlet transducer and all branch transducers begin in their respective
 states.
states.Before any residues can appear at the root, the branch transducers all wind back into
 states, via type-1 transitions. This occurs in reverse order (i.e., a postorder traversal of the tree).
states, via type-1 transitions. This occurs in reverse order (i.e., a postorder traversal of the tree).During this initial windback, clade-specific insertions can occur. This process is described in detail at step 9.
With all the branch transducers wound back into
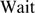 states, the singlet transducer makes a (type-1) transition into an
states, the singlet transducer makes a (type-1) transition into an 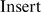 state, emitting a symbol to the sequence at the root node.
state, emitting a symbol to the sequence at the root node.The transducers on outgoing branches from the root then make (type-2) transitions into
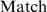 states, either copying the root symbol to their own outputs, substituting it for a different symbol or staying silent (this silence corresponds to a clade-specific deletion; in our formalism, both substitutions and deletions are handled by
states, either copying the root symbol to their own outputs, substituting it for a different symbol or staying silent (this silence corresponds to a clade-specific deletion; in our formalism, both substitutions and deletions are handled by 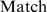 states.)
states.)The transducers on branches one step away from the root then process the symbols which reached them (if any did), followed by transducers on branches two steps away from the root, then three steps, and so on (these can all be regarded as occurring simultaneously, in a single cascading wave of emissions).
Eventually the emitted symbols are propagated, via type-2 transitions, all the way to the tips of the tree (if they survived) or to the nodes where they were deleted (if they did not survive). The wave of type-2 transitions has left a lot of branch transducers in
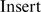 and
and 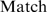 states.
states.The branch transducers then, in postorder, each wind back into
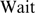 states, just as at step 2. (These windback transitions can be collapsed into a single ensemble transition, as with the emission cascade; however, the windback may be interrupted by clade-specific insertions; see below.)
states, just as at step 2. (These windback transitions can be collapsed into a single ensemble transition, as with the emission cascade; however, the windback may be interrupted by clade-specific insertions; see below.)During the postorder windback, each branch transducer gets an opportunity to generate a new symbol (via type-1 transitions to
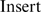 states). (If such a transition to
states). (If such a transition to 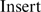 occurs, it corresponds to a clade-specific insertion. This insertion is propagated down the tree via a wave of type-2 transitions, as above, then we go back to step 7.)
occurs, it corresponds to a clade-specific insertion. This insertion is propagated down the tree via a wave of type-2 transitions, as above, then we go back to step 7.)Eventually, the entire ensemble has wound back, so that every transducer is in a
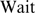 state except the singlet transducer at the root, which is still in an
state except the singlet transducer at the root, which is still in an 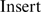 state. At this point, all clade-specific insertions have been processed.
state. At this point, all clade-specific insertions have been processed.The singlet transducer now makes another type-1 transition. If this transition is to an
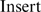 state, the entire cycle begins again: the singlet transducer emits the next symbol at the root, and we go back to step 4.
state, the entire cycle begins again: the singlet transducer emits the next symbol at the root, and we go back to step 4.If, on the other hand, the singlet transducer enters its
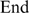 state, then a wave of type-3 transitions drives all the branch transducers into their respective
state, then a wave of type-3 transitions drives all the branch transducers into their respective 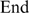 states too, bringing the entire ensemble to a halt.
states too, bringing the entire ensemble to a halt.
Complexity of the transducer ensemble
The total sizes of both the state space and the transition matrix are, in general, dramatically smaller than implied by the exponential upper bounds of 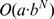 and
and 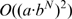 . While we do not have provable bounds on the size of the state space, we have observed that the size of the state space is roughly linear in the number of branches,
. While we do not have provable bounds on the size of the state space, we have observed that the size of the state space is roughly linear in the number of branches, 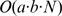 , and the number of transitions is approximately linear in the number of states for several pairwise models, including the pairwise model which we use here. However, these empirical observations are based on a limited class of pairwise models and we do not have theoretical results for how they will generalize to other pairwise models. We do believe, however, that the worst-case exponential bound will be avoided by (1) omitting inaccessible state configurations and (2) eliminating null windback states as described in the following section (which we believe will prevent affine gap penalties from generating exponential growth in the number of states).
, and the number of transitions is approximately linear in the number of states for several pairwise models, including the pairwise model which we use here. However, these empirical observations are based on a limited class of pairwise models and we do not have theoretical results for how they will generalize to other pairwise models. We do believe, however, that the worst-case exponential bound will be avoided by (1) omitting inaccessible state configurations and (2) eliminating null windback states as described in the following section (which we believe will prevent affine gap penalties from generating exponential growth in the number of states).
Therefore, for the models which we have characterized, the search algorithm given above for enumerating all allowed transitions of the ensemble model typically generates 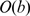 transitions from any given state, thereby creating a very sparse transition matrix of size
transitions from any given state, thereby creating a very sparse transition matrix of size 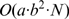 .
.
Inference algorithms for multiple-sequence models
In this section, we describe dynamic programming (DP) algorithms for inferring the alignment, structure and evolutionary history of multiple related RNAs, using the multiple-sequence SCFG we have derived.
The transducer composition algorithm described above constructs a phylogenetic SCFG for both ancestral and extant sequences. A parse tree for this SCFG represents a structural and evolutionary explanation of the extant sequences, including a complete ancestral reconstruction. Consequently, given a set of extant sequences, many of the questions of interest to us can be reduced to searches over, or summarizations of, the set of possible parse trees.
Well-known algorithms already exist for maxing or summing over SCFG parse tree likelihoods. The Cocke-Younger-Kasami (CYK) algorithm performs maximum-likelihood (ML) inference; the Inside algorithm can be used to sum over parse trees or sample them a posteriori; and the Inside-Outside algorithm yields posterior probabilities for individual parse tree nodes [11].
All of these algorithms are, however, complicated (at least in our models) by the existence of “null cycles” in the grammar. A null cycle is a parse tree fragment that is redundant and could be removed, such as a detour through 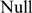 states (
states (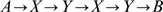 ) that could be replaced by a direct transition (
) that could be replaced by a direct transition (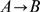 ). Biologically, null cycles correspond to fragments of ancestral sequence that were universally deleted and therefore are unobserved in any of the extant sequences. These unobserved fragments can be unbounded in length (and so, therefore, can the parse tree). Within the CYK, Inside and Outside recursions, this causes cyclic dependencies which cannot be resolved.
). Biologically, null cycles correspond to fragments of ancestral sequence that were universally deleted and therefore are unobserved in any of the extant sequences. These unobserved fragments can be unbounded in length (and so, therefore, can the parse tree). Within the CYK, Inside and Outside recursions, this causes cyclic dependencies which cannot be resolved.
Below we describe a method to eliminate null cycles from the ensemble model by transforming any SCFG to an equivalent acyclic SCFG. We then present multiple-sequence versions of the CYK, Inside and Outside algorithms.
While some sort of null-cycle elimination is often required in order to deal with cyclic dependencies, there are several ways to accomplish this other than the algorithm presented below. A simpler approach (that only works for the CYK algorithm) appears in the computational linguistics literature [37]. We have also developed a heuristic for CYK that simply ignores null cycles as well as an iterative approximation that loops several times over cyclically-dependent cells of the DP matrix until the estimate starts to converge. For conciseness, we have omitted descriptions of these methods, presenting only the exact elimination algorithm.
Exact elimination of null cycles in SCFGs
As noted above, the ensemble grammar contains many rules that can be applied redundantly, together or in isolation, to generate subtrees of the parse tree that do not generate any terminals. This generates cyclic dependencies in the standard DP recursions for inference. In this subsection, we describe how to transform the SCFG so as to eliminate such redundant rules, yielding strictly acyclic DP recursions. This transformation can be applied to any SCFG so as to remove null states and/or bifurcations: the procedure is not restricted to grammars that were generated using our transducer composition algorithm.
We begin by identifying two distinct classes of redundant parse-subtree: empty bifurcations and empty paths. We will eliminate each of these in turn.
An empty bifurcation occurs when a child branch of a bifurcation state transitions to the 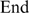 state without emitting any symbols and can be removed from the model by creating an effective direct transition encapsulating the empty bifurcation. For example, we can create an effective direct transition
state without emitting any symbols and can be removed from the model by creating an effective direct transition encapsulating the empty bifurcation. For example, we can create an effective direct transition 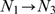 between null states
between null states 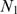 and
and 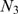 in place of the empty parse-subtree
in place of the empty parse-subtree 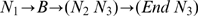 , where
, where 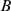 is a bifurcation state with children
is a bifurcation state with children 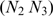 . Bifurcation states are the most computationally-costly part of our models, so it is important to eliminate as many as possible without reducing model expressiveness.
. Bifurcation states are the most computationally-costly part of our models, so it is important to eliminate as many as possible without reducing model expressiveness.
In contrast, an empty path is defined as any parse-subtree without bifurcations that does not emit terminal symbols. If 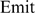 states
states  and
and  are connected in the state graph via
are connected in the state graph via 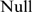 states
states 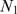 and
and 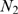 , then the path
, then the path 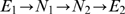 with probability
with probability 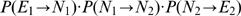 can be replaced by a single direct transition
can be replaced by a single direct transition 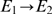 with an identical probability.
with an identical probability.
Empty paths occur in Hidden Markov Models (which are special cases of SCFGs) and independent-sites models (which can be viewed as special cases of HMMs). Conceptually, empty paths can represent histories that are valid according to the model but cannot be resolved by direct observation. Such null events can be real (e.g., ancestral residues that have been deleted in all extant lineages) or they can be artefactual (e.g., transitions between placeholder null states of an HMM).
In our composite model, empty paths occur whenever a series of branch transducers winds back into 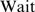 states. Empty bifurcations occur when an entire substructure, present in an ancestor, is deleted in all that ancestor's extant descendants.
states. Empty bifurcations occur when an entire substructure, present in an ancestor, is deleted in all that ancestor's extant descendants.
Empty paths and empty bifurcations are problematic because they can be combined to give finite-probability sequences of rules that transform a nonterminal back into itself, with no observable emissions. We refer to such sequences of rules as null cycles. As noted, null cycles generate cyclic dependencies in the CYK, Inside & Outside algorithms. Our goal is an algorithmic procedure to resolve these dependencies and account for the likelihood of such cycles by exact marginalization.
For simpler models, solutions to this problem are published. Missing (empty) columns in independent-sites models can be accounted for by applying a correction factor 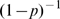 to account for the proportion of columns
to account for the proportion of columns 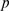 that are unobserved [38]. The slightly more complicated situation of missing emissions in a HMM can be dealt with by summing over all empty paths, yielding a geometric series that is solvable by matrix inversion [39]–[41]. Such algorithms effectively replace the HMM with another HMM that contains no null cycles but is equivalent to the original, in that it models the same probability distribution over sequences. However, these solutions do not easily generalize to SCFGs (which may have empty bifurcations as well as empty paths).
that are unobserved [38]. The slightly more complicated situation of missing emissions in a HMM can be dealt with by summing over all empty paths, yielding a geometric series that is solvable by matrix inversion [39]–[41]. Such algorithms effectively replace the HMM with another HMM that contains no null cycles but is equivalent to the original, in that it models the same probability distribution over sequences. However, these solutions do not easily generalize to SCFGs (which may have empty bifurcations as well as empty paths).
Text S2 includes a complete formal algorithm for exact null-cycle elimination in SCFGs, along with procedures for probabilistically restoring null cycles to sampled parse trees and Inside-Outside expectation counts. Informally, the essence of the algorithm is contained within the following two steps:
separating bifurcations into those which have one or more empty children (and can therefore be represented using transition or termination rules) and those that have two nonempty children;
replacing all empty paths through null states with effective direct transitions between non-null states, obtaining sum-over-paths probabilities by inverting the grammar's transition matrix.
Note that step (i) is unique to SCFGs; step (ii), in contrast, is very similar to the empty-path elimination algorithm for HMMs.
Dynamic programming algorithms for inference
Once we have performed the transformations described above to remove null cycles from the multiple-sequence SCFGs generated by our model-construction algorithm, we can compute likelihoods and sample parse trees using the standard CYK, Inside and Outside algorithms for multiple-sequence SCFGs [11],[42].
The asymptotic time and memory complexities of our inference algorithms are essentially the same as for Sankoff's algorithm [42]: the DP algorithms take memory 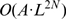 and time
and time 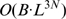 for
for 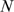 sequences of length
sequences of length 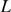 , where
, where 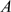 is the number of (accessible) states in the multiple-SCFG and
is the number of (accessible) states in the multiple-SCFG and 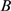 is the number of bifurcations. Note that
is the number of bifurcations. Note that 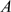 and
and 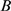 are also dependent on
are also dependent on 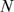 (see “The TKFST model on a three-taxon phylogeny”).
(see “The TKFST model on a three-taxon phylogeny”).
Exact inference on a star phylogeny with 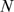 extant sequences therefore has complexities
extant sequences therefore has complexities 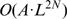 and
and 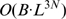 in memory and time (respectively) for a multiple-SCFG with
in memory and time (respectively) for a multiple-SCFG with 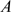 states and
states and 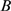 bifurcations. As described earlier, in practice we frequently have expert knowledge (such as a curated multiple alignment) about the structures and/or evolutionary histories of the sequences of interest. We can use this knowledge as a constraint to reduce the accessible volume, and hence the storage requirements, of the DP matrix [18]. The Inside, Outside, and CYK+traceback algorithms for a three-taxon star phylogeny can be constrained using the “fold envelope” concept, which will now be described.
bifurcations. As described earlier, in practice we frequently have expert knowledge (such as a curated multiple alignment) about the structures and/or evolutionary histories of the sequences of interest. We can use this knowledge as a constraint to reduce the accessible volume, and hence the storage requirements, of the DP matrix [18]. The Inside, Outside, and CYK+traceback algorithms for a three-taxon star phylogeny can be constrained using the “fold envelope” concept, which will now be described.
We use the fold envelope concept [29],[43] to constrain the set of structures which our algorithms consider. A fold envelope 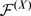 for a sequence
for a sequence 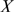 is a set of coordinate pairs satisfying
is a set of coordinate pairs satisfying
| (1) |
We consider a subsequence 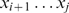 only if the corresponding coordinate pair
only if the corresponding coordinate pair 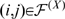 . The unconstrained fold envelope has set equality in Equation 1.
. The unconstrained fold envelope has set equality in Equation 1.
An 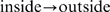 ordering is used for the iteration in the Inside algorithm: Subsequences are ordered such that each successive subsequence contains all previous subsequences in the fold envelope. More precisely, subsequences in
ordering is used for the iteration in the Inside algorithm: Subsequences are ordered such that each successive subsequence contains all previous subsequences in the fold envelope. More precisely, subsequences in 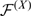 are sorted in the same order as coordinate pairs
are sorted in the same order as coordinate pairs 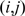 are generated by the iteration
are generated by the iteration 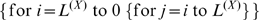 .
.
The Outside algorithm uses the exact reverse of the 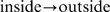 ordering described above; we call this the
ordering described above; we call this the 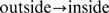 ordering. Subsequences in
ordering. Subsequences in 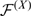 are sorted in the same order as coordinate pairs
are sorted in the same order as coordinate pairs 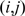 are generated by the iteration
are generated by the iteration 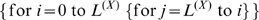 .
.
We frequently refer to subsequences by their index in the fold envelope. The 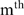 subsequence in
subsequence in 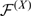 is labeled
is labeled 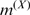 and corresponds to the coordinate pair
and corresponds to the coordinate pair 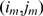 . The index of a pair
. The index of a pair 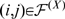 is
is 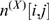 .
.
In order to take full advantage of the reduction in computational complexity offered by restricting our inference algorithms to subsequences contained in the fold envelopes, we must avoid iterating over unreachable combinations of subsequences (unreachable because they are not permitted by the fold envelope constraints). An efficient implementation relies on iterators over subsequences in the fold envelope which are connected by production rules of the ensemble grammar. Inward and outward emission connections for a sequence 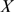 , specifying which subsequence is reachable from a given subsequence
, specifying which subsequence is reachable from a given subsequence 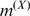 and ensemble state b, are defined as
and ensemble state b, are defined as
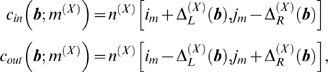 |
where the quantities 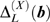 and
and 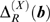 are the lengths of the left and right emissions of the ensemble state b to the sequence X. (Recall that the mth subsequence in
are the lengths of the left and right emissions of the ensemble state b to the sequence X. (Recall that the mth subsequence in  is labeled m
(X) and corresponds to the coordinate pair (im, jm).) The emission connection is undefined if the corresponding subsequence is not in the fold envelope. Inward, outward-left and outward-right bifurcation connections, specifying which subsequences are connected by bifurcation production rules of the ensemble SCFG, are defined for a subsequence n
(X) as
is labeled m
(X) and corresponds to the coordinate pair (im, jm).) The emission connection is undefined if the corresponding subsequence is not in the fold envelope. Inward, outward-left and outward-right bifurcation connections, specifying which subsequences are connected by bifurcation production rules of the ensemble SCFG, are defined for a subsequence n
(X) as
| (2) |
| (3) |
| (4) |
We generally write out explicit subsequence coordinate pairs (i, j) when their usage will make mathematical formulas clearer and fold-envelope labels n (X) when writing pseudocode.
Using the fold envelope formalism, the main iteration over cells in the Inside and CYK matrices can be expressed as three nested loops: one for each sequence, traversing the fold envelope subsequences in inside→outside order. Conversely, the main iteration of the Outside algorithm consists of three nested outside→inside loops.
The Inside algorithm is used to calculate the likelihood of sequences under an ensemble model. It is analogous to the Forward algorithm for HMMs.
The inside probability 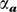 (n
(X), n
(Y), n
(Z)) is the summed probability of the triplet of subsequences (n
(X), n
(Y), n
(Z)) for sequences X,Y,Z under all paths through the model which are rooted in state
(n
(X), n
(Y), n
(Z)) is the summed probability of the triplet of subsequences (n
(X), n
(Y), n
(Z)) for sequences X,Y,Z under all paths through the model which are rooted in state 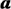 . Figure 1 gives pseudocode for the fold-envelope version of the Inside algorithm. The subroutines calcTransEmitProb, calcLBifurcProb and calcRBifurcProb used in the Inside algorithm are defined below.
. Figure 1 gives pseudocode for the fold-envelope version of the Inside algorithm. The subroutines calcTransEmitProb, calcLBifurcProb and calcRBifurcProb used in the Inside algorithm are defined below.
Figure 1. Algorithm 1.

The constrained Inside algorithm for three sequences X, Y, Z. Ensemble states in the iteration over states are sorted in Inside fill order with Emit states first, then Null states in reverse topological order.
The transition and emission probability calcTransEmitProb(; ·) can be calculated by iterating over ensemble states b which connect the subsequence triplet (n
(X), n
(Y), n
(Z)) to others in the fold envelopes.
Pseudocode for the constrained calculation is given in Figure 2.
Figure 2. Algorithm 2.

Subroutine calcTransEmitProb() for the Inside algorithm. and b are ensemble states; l and r are left and right terminal emissions.
The left-bifurcation probability for an ensemble state bifurcating to two ensemble states, calcLBifurcProb (; 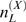 ,
, 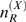 ,
, 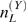 ,
, 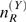 ,
, 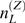 ,
, 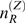 ), is
), is
and the right-bifurcation probability for an ensemble state bifurcating to two ensemble states, calcRBifurcProb (; 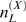 ,
, 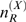 ,
, 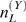 ,
, 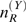 ,
, 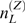 ,
, 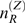 ), is
), is
The boundary condition of the probability of 0-length subsequences is determined by the probability of transitions to End. The termination condition is
where Start is the unique start state of the ensemble grammar and N (X) is the outermost subsequence for sequence X, etc.
Note that we are assuming that the transformations described in “Exact elimination of null cycles in SCFGs” have been performed, such that there are no cycles of Null states as well as no empty bifurcations.
The CYK algorithm is used to calculate the probability of the most-likely state path (or parse) capable of generating the input sequences. It is analogous to the Viterbi algorithm for HMMs.
The CYK algorithm can be obtained from the Inside algorithm by replacing sums over paths through the ensemble model with the max operation. The CYK probability for indices 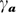 (n
(X), n
(Y), n
(Z)) then represents the probability of the most likely path through the model generating the triplet of subsequences (n
(X), n
(Y), n
(Z)).
(n
(X), n
(Y), n
(Z)) then represents the probability of the most likely path through the model generating the triplet of subsequences (n
(X), n
(Y), n
(Z)).
The resulting CYK algorithm is shown in Figure 3. The subroutine caleTransEmitProb is defined in Figure 4. The subroutines calcLBifurcProb and calcRBifurcProb used in the CYK algorithm are defined as
and
Figure 3. Algorithm 3.

The constrained CYK algorithm for three sequences X, Y, Z. Ensemble states in the iteration over states are sorted in Inside fill order with Emit states first, then Null states in reverse topological order.
Figure 4. Algorithm 4.

Subroutine calcTransEmitProb() for the CYK algorithm. and b are ensemble states; l and r are left and right terminal emissions.
The CYK traceback algorithm, in combination with the CYK algorithm, is used to find the most-likely state path generating the extant sequences (in other words, the maximum-likelihood parse generating the observed data). It is analogous to the Viterbi traceback algorithm for HMMs. Figure 5 gives the constrained CYK traceback algorithm.
Figure 5. Algorithm 5.

The constrained CYK traceback algorithm for three sequences X, Y, Z.
The Outside algorithm is primarily used an an intermediary for calculating nucleotide-level posterior probabilities, e.g. for posterior decoding on the model. It is analogous to the Backward algorithm for HMMs.
The outside probability β b (n (X), n (Y), n (Z)) for an ensemble state b is the summed probability of the sequences X,Y,Z under all paths through the ensemble model which are rooted in the start state of the model, excluding all paths for the triplet of subsequences (n (X), n (Y), n (Z)) which are rooted in the ensemble state b. Figure 6 gives pseudocode for the fold-envelope version of the Outside algorithm. The subroutines calcTransEmitProb, calcLBifurcProb and calcRBifurcProb used in the Outside algorithm are defined below.
Figure 6. Algorithm 6.

The constrained Outside algorithm for three sequences X,Y,Z. Ensemble states a in the iteration over states are sorted in Outside fill order with Emit states first, then Null states in topological order.
As with the Inside and CYK algorithms, the transition and emission probability calcTransEmitProb can be calculated efficiently using the subsequence connections defined earlier (Figure 7). The left-bifurcation probability calcLBifurcProb (b; 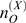 ,
, 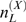 ,
, 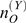 ,
, 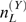 ,
, 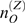 ,
, 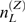 ) is
) is
and the right-bifurcation probability calcRBifurcProb (b; 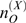 ,
, 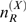 ,
, 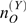 ,
, 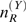 ,
, 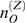 ,
, 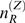 ) is
) is
The boundary condition is just
where N (X) is the outermost subsequence for sequence X, etc.
Figure 7. Algorithm 7.

Subroutine calcTransEmitProb() for the Outside algorithm. and b are ensemble states; l and r are left and right terminal emissions.
Results
Automated grammar construction
We implemented our model construction algorithm on the three-taxon star phylogeny. Given a singlet transducer modeling ancestral structures and a branch transducer modeling structural evolution, our Perl modules generate C++ code for the corresponding jointly-normalized three-sequence (Triplet) SCFG. Any model of structural evolution which can be represented as a Pair SCFG and factored into singlet and branch transducers is permitted as input to the packages, allowing for flexible, automated model design. The available software is described in Text S3.
A simple model of RNA structural evolution
We illustrated our method for building models of structured sequences using a model which was introduced in previous work, the TKF Structure Tree [29], a simplified probabilistic model of the evolution of RNA structure.
The TKF Structure Tree (TKFST) model is based on the Thorne-Kishino-Felsenstein (TKF) model of the stochastic evolution of primary sequences via indel events [44]. In the original TKF model, sequence evolves under a time-homogeneous linear birth-death-immigration process [45]. Single characters (“links”) are inserted with rate 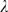 and deleted with rate
and deleted with rate 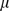 . At equilibrium, sequences obey a geometric length distribution with parameter
. At equilibrium, sequences obey a geometric length distribution with parameter  . Although this model has flaws (e.g., it lacks affine gap penalties, rate heterogeneity and context-dependent mutation rates), it illustrates many of the key ideas used by more sophisticated indel models, notably the possibility for systematic derivation of pairwise alignment automata from first principles via analysis of birth-death processes [44],[46].
. Although this model has flaws (e.g., it lacks affine gap penalties, rate heterogeneity and context-dependent mutation rates), it illustrates many of the key ideas used by more sophisticated indel models, notably the possibility for systematic derivation of pairwise alignment automata from first principles via analysis of birth-death processes [44],[46].
The TKF Structure Tree model is an extension of the TKF model to RNA structure. In this model, loop and stem regions are mutually nested (Figure 8): the parameter 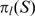 determines the proportion of links within loop sequences that are nested stems, and every stem sequence has a nested loop at the end. Single bases are inserted and deleted in loops with rates
determines the proportion of links within loop sequences that are nested stems, and every stem sequence has a nested loop at the end. Single bases are inserted and deleted in loops with rates 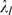 and
and 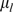 ; similarly, base-pairs are inserted and deleted in stems with rates
; similarly, base-pairs are inserted and deleted in stems with rates 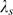 and
and 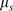 . Both loops and stems have geometric length distributions with parameters
. Both loops and stems have geometric length distributions with parameters 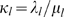 and
and 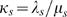 . Insertions of a new stem into an existing loop sequence (or deletions of an existing stem) occur at the same rate as single-base insertions (or deletions) and can model large-scale structural changes (Figure 9).
. Insertions of a new stem into an existing loop sequence (or deletions of an existing stem) occur at the same rate as single-base insertions (or deletions) and can model large-scale structural changes (Figure 9).
Figure 8. The TKF Structure Tree model represents the evolution of RNA structure as nested stem and loop sequences.

The model consists of recursively nested loop sequences (gray, horizontal) and stem sequences (black, vertical). The loops are sequences of unpaired bases and the stems are sequences of covarying base-pairs. Both loop and stem sequences evolve according to the Thorne-Kishino-Felsenstein (TKF) model [44] of molecular evolution. Figure is extended from a similar version in [29].
Figure 9. Evolution of a RNA structure under the TKF Structure Tree model.

The TKF Structure Tree model includes phenomena such as point mutations in loop sequences ( and
and  ), covariant mutations in stem sequences (
), covariant mutations in stem sequences ( ), insertions in loop sequences (
), insertions in loop sequences ( ), insertions in stem sequences (
), insertions in stem sequences ( ), structural insertions (
), structural insertions ( ), and structural deletions (
), and structural deletions ( ). Figure is extended from a similar version in [29].
). Figure is extended from a similar version in [29].
We parametrized the singlet and branch transducers of the TKFST model using estimates reported by a phylo-grammar for RNA secondary structure prediction, PFOLD [16], and an implementation of pairwise alignment for the TKF Structure Tree model, Evoldoer [29]. The equilibrium distributions of unpaired and paired nucleotides of the singlet and branch transducers, as well as the substitution models of unpaired and paired nucleotides of the branch transducers, were derived from the substitution rate matrices of the PFOLD program. These rate matrices, which have proven useful for RNA structure prediction [16],[17],[47], were derived from the Bayreuth tRNA database [48] and the European large subunit rRNA database [49].
This continuous-time model corresponds to a Pair SCFG and as such fits neatly into our modeling framework once the probability distribution is appropriately factored into marginal and conditional distributions (generated by singlet and branch transducers). Tables 1 and 2 show the states and transitions of the singlet transducer (single-sequence SCFG) which generates ancestral sequence under the Structure Tree model. Tables 3 and 4 show the states and transitions of the branch transducer (conditionally-normalized Pair SCFG) which evolves a sequence and structure along a branch of the phylogenetic tree.
Table 1. State types of the singlet transducer (single-sequence SCFG) of the TKF Structure Tree model.
| State | type | absorb |
|
description |

|
|
Start of a loop | ||
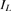
|
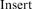
|
|
Single-base emission | |
|
|
Start of a stem | ||
|
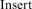
|
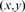
|
Base-pair emission | |

|

|
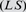
|
Bifurcation |
Singlet transducers can only have states of type 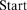 or
or 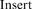 .
.
Table 2. Singlet transducer (single-sequence SCFG) of the TKF Structure Tree model.
| Source→Destinatioon | probability | Source→Destinatioon | probability |
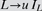
|

|

|
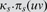
|
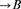
|
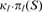
|
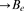
|
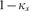
|
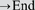
|

|
||
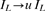
|
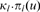
|
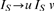
|
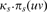
|

|

|
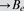
|

|
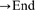
|

|
||
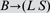
|
1 | ||
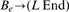
|
1 |
The state types for this model are shown in Table 1. The singlet transducer generates ancestral RNA sequences and structures. We use the notation of formal grammars to represent state transformation rules; for example, the rule 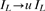 corresponds to (in a Pair HMM) an
corresponds to (in a Pair HMM) an  state
state 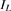 emitting a nucleotide
emitting a nucleotide  and then making a self-transition. Both loop (
and then making a self-transition. Both loop ( and
and 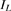 ) and stem (
) and stem (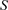 and
and 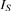 ) sequence evolve as TKF sequences with length parameters
) sequence evolve as TKF sequences with length parameters  and
and 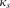 (defined in “A simple model of RNA structural evolution”).
(defined in “A simple model of RNA structural evolution”). 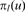 and
and 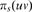 are the equilibrium distributions of unpaired nucleotides
are the equilibrium distributions of unpaired nucleotides  and paired nucleotides
and paired nucleotides 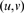 and are normalized such that
and are normalized such that  and
and 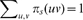 . The bifurcation state
. The bifurcation state 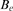 is used to end stem sequences (only loop sequences are allowed to transition to the empty string).
is used to end stem sequences (only loop sequences are allowed to transition to the empty string).
Table 3. State types of the branch transducer (conditionally-normalized Pair SCFG) of the TKF Structure Tree model.
| State | type | absorb | emit | description |

|

|
Start of a loop | ||
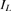
|
|
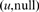
|
Single-base insertion | |
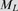
|
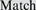
|

|

|
Single-base substitution |
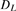
|
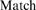
|
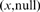
|
Single-base deletion | |
|
|
Wait for next base | ||
|
|
Start of a stem | ||
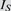
|

|
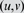
|
Base-pair insertion | |
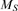
|
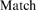
|
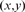
|
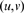
|
Base-pair substitution |
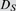
|
|
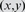
|
Base-pair deletion | |
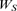
|
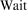
|
Wait for next base-pair | ||
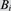
|

|
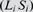
|
Stem insertion | |

|
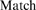
|
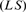
|
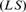
|
Stem conservation |
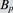
|
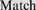
|
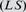
|
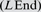
|
Stem deletion |
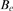
|
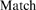
|
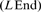
|
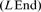
|
Stem extinction |
States which have the same names as states of the singlet transducer in Table 1 are the branch-transducer equivalents of the corresponding singlet-transducer states (e.g., a 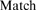 state might be the branch equivalent of an
state might be the branch equivalent of an 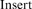 state). States
state). States 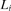 and
and 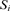 are the
are the 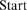 states of a sub-model (not shown) identical in structure to the singlet transducer. They are used to insert a new stem-loop structure.
states of a sub-model (not shown) identical in structure to the singlet transducer. They are used to insert a new stem-loop structure.
Table 4. Branch transducer (conditionally-normalized Pair SCFG) of the TKF Structure Tree model.
| Source→Destinatioon | probability | Source→Destinatioon | probability |

|
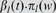
|

|
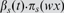
|
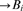
|
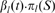
|
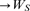
|
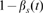
|
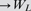
|
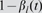
|
||
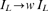
|
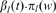
|
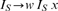
|
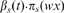
|
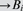
|
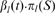
|
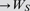
|
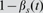
|
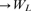
|
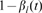
|
||

|
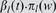
|

|

|
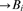
|
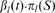
|
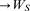
|
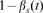
|
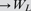
|
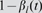
|
||
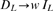
|
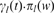
|
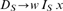
|
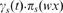
|
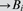
|

|
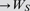
|
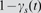
|
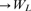
|
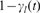
|
||

|
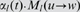
|

|
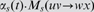
|
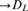
|
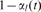
|
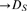
|
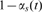
|

|
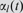
|
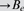
|

|
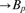
|
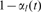
|
||

|

|
||
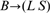
|
1 |
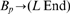
|
1 |
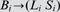
|
1 |
|
1 |
The state types for this model are shown in Table 3. The branch transducer evolves a sequence and structure along a branch of the phylogenetic tree. States 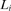 and
and 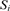 are the
are the 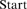 states for a sub-model corresponding to an insertion of a new stem in the descendant sequences; the sub-model (not shown) is identical in structure to the singlet transducer shown in Table 2.
states for a sub-model corresponding to an insertion of a new stem in the descendant sequences; the sub-model (not shown) is identical in structure to the singlet transducer shown in Table 2. 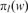 and
and 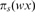 are the equilibrium distributions of, respectively, descendant unpaired nucleotide
are the equilibrium distributions of, respectively, descendant unpaired nucleotide  and descendant paired nucleotides
and descendant paired nucleotides 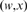 ;
; 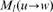 and
and 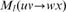 are the conditional distributions (i.e., match probabilities) of a descendant unpaired nucleotide
are the conditional distributions (i.e., match probabilities) of a descendant unpaired nucleotide  given an ancestral unpaired nucleotide
given an ancestral unpaired nucleotide  and descendant paired nucleotides
and descendant paired nucleotides 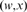 given ancestral nucleotides
given ancestral nucleotides 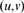 . The functions
. The functions 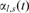 ,
, 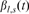 and
and 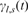 are parametrized by the insertion and deletion rates of the TKFST model and are defined in “A simple model of RNA structural evolution”.
are parametrized by the insertion and deletion rates of the TKFST model and are defined in “A simple model of RNA structural evolution”.
The equilibrium distribution and transition probabilities between states of the TKFST model can be expressed in terms of functions of the evolutionary time along a branch and the insertion and deletion rates 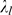 and
and 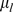 of the model. The length of ancestral sequences is geometric in
of the model. The length of ancestral sequences is geometric in 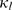 (Table 2), defined as
(Table 2), defined as 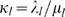 . The three functions
. The three functions 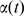 ,
, 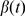 and
and 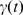 which govern the transition probabilities in Table 4 are defined for loop sequences as
which govern the transition probabilities in Table 4 are defined for loop sequences as
and similarly for stem sequences [29].
The above-described TKFST SCFGs must be transformed slightly before they can be loaded into Indiegram. The grammars are presented in Indiegram format in Text S4.
A few other useful statistics for the TKFST model: the expected number of links in a loop sequence is 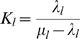 and in a stem sequence
and in a stem sequence 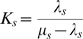 . Since
. Since 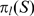 of the links in a loop sequence are nested stems, and since each stem has twice as many nucleotides as it has links (since each link is a base pair), the expected number of bases in a loop sequence is
of the links in a loop sequence are nested stems, and since each stem has twice as many nucleotides as it has links (since each link is a base pair), the expected number of bases in a loop sequence is
The expected number of bases in a stem sequence is
The expected number of bases that are created/removed when a loop-sequence link is inserted/deleted is
The expected number of stems directly rooted in a given loop sequence is 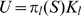 and the expected number of stems directly rooted in, or indirectly descended from, a given loop sequence is
and the expected number of stems directly rooted in, or indirectly descended from, a given loop sequence is 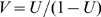 (note that this is also the expected total number of loop sequences indirectly descended from a given loop sequence). Therefore, in the equilibrium structure, the expected number of stems is
(note that this is also the expected total number of loop sequences indirectly descended from a given loop sequence). Therefore, in the equilibrium structure, the expected number of stems is 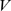 ; of loops,
; of loops, 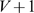 ; of unpaired bases,
; of unpaired bases, 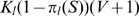 ; and of base-pairs,
; and of base-pairs, 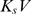 . In a tree with total branch length
. In a tree with total branch length 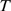 , the expected number of single-base deletions is
, the expected number of single-base deletions is 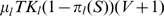 ; of base-pair deletions,
; of base-pair deletions, 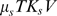 ; and of substructure deletions,
; and of substructure deletions, 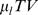 .
.
Assessing TKFST as a model of RNA structure
The TKFST model, like the original TKF model, probably needs refinements in order to accurately model many structural RNAs. For example, it fails to model certain phenomena observed in natural RNA structures (such as base-stacking or tetraloops) and in alignments of those structures (such as helix slippage). We assessed its appropriateness as a model of RNA structural evolution by conducting benchmarks of its capabilities for (1) multiple sequence alignment of structured RNAs, summing over all possible structures, and (2) structure prediction of homologous structured RNAs and comparing its performance to Stemloc (one of the better-performing pairwise SCFGs used for RNA multiple alignment [20]). The results of these benchmarks, reported in Table 5 and Table 6, suggest that TKFST is a useful guide for deriving more complicated models of RNA evolution: while it has relatively poor sensitivity (but high positive predictive value) as a base-pairing predictor, it is competitive with one of the most accurate RNA multiple sequence alignment programs [20].
Table 5. Percentage sensitivity and positive predictive value (Sensitivity/PPV) for pairwise nucleotide-level alignments in the BRalibaseII benchmark.
| U5 | g2intron | rRNA | tRNA | |
| TKFST grammar | 81.6/81.7 | 75.4/75.0 | 91.4/92.6 | 94.6/94.4 |
| Stemloc grammar | 82.6/83.7 | 74.2/74.8 | 92.6/92.8 | 93.2/93.9 |
We compared the performance of the TKFST model for progressive multiple alignment of RNAs against the performance of a grammar with a richer model of RNA structure (Stemloc [18]). Sensitivity is defined as 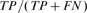 and PPV is defined as
and PPV is defined as 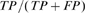 , where TP is the number of true positives (correctly aligned residue pairs), FN is the number of false negatives (residue pairs that should have been aligned but were not) and FP is the number of false positives (residue pairs that were incorrectly aligned). These statistics are summed over all pairs of sequences in the multiple alignment; therefore, “Sensitivity” for pairwise residue alignments is equivalent to the Sum of Pairs Score or SPS [103]. “g2intron” is the RFAM entry Intron_gpII, containing domains V and VI of the Group II intron.
, where TP is the number of true positives (correctly aligned residue pairs), FN is the number of false negatives (residue pairs that should have been aligned but were not) and FP is the number of false positives (residue pairs that were incorrectly aligned). These statistics are summed over all pairs of sequences in the multiple alignment; therefore, “Sensitivity” for pairwise residue alignments is equivalent to the Sum of Pairs Score or SPS [103]. “g2intron” is the RFAM entry Intron_gpII, containing domains V and VI of the Group II intron.
Table 6. Percentage sensitivity and positive predictive value (Sensitivity/PPV) for predicted base-pairs in the BRalibaseII benchmark.
| U5 | g2intron | rRNA | tRNA | |
| TKFST grammar | 37.9/68.0 | 42.1/63.8 | 37.4/66.5 | 70.9/88.3 |
| Stemloc grammar | 74.9/73.9 | 64.3/56.7 | 51.0/59.0 | 74.0/76.4 |
We compared the performance of the TKFST model for structure-prediction accuracy during progressive multiple alignment of RNAs against the performance of a grammar with a richer model of RNA structure (Stemloc [18]). “g2intron” is the RFAM entry Intron_gpII, containing domains V and VI of the Group II intron.
TKFST's poorer performance at base-pairing prediction is likely due to its much-simpler model of RNA structure. The richer grammar, as described in [18], is much more complex than TKFST: excluding the substitution model, it has 14 free parameters (compared to TKFST's 4), uses an affine gap penalty (compared to TKFST's linear gap penalty), and explicitly models structural features such as multiple-branched loops, symmetric/asymmetric bulges, and minimum loop lengths. Unlike TKFST, the richer grammar is structurally unambiguous: a one-to-one mapping exists from structures to parse trees. Although we use the TKFST model as an illustrative example of a Pair SCFG that can be extended with our method, the model is not fundamental to our approach and can be replaced by a different and more realistic pairwise model, such as the Stemloc pairwise SCFG used in these comparisons [20]. We anticipate that further improvements should be possible by reviewing other comparisons of SCFGs at structure prediction, such as the study of [17].
The TKFST model on a three-taxon phylogeny
We used our model-construction algorithm to build the grammar corresponding to the TKFST model acting on a star phylogeny with three (extant) leaf sequences and a single (unobserved) ancestral sequence. We chose this phylogeny for two reasons: (1) it is the simplest extension of the well-studied, standard two-sequence (Pair SCFG) model and (2) algorithms on a phylogeny with three leaves should be sufficient for ergodic sampling of reconstructions on any larger phylogeny, using, e.g., a Gibbs-sampling MCMC kernel [31] or a progressive suboptimal-alignment sampling heuristic [33].
The statistics of the TKFST model on the three-taxon phylogeny illustrate the advantages of our procedure for model construction. While the singlet and branch transducers are relatively simple—the singlet transducer, shown in Table 2, has 7 total states and 2 bifurcation states and the branch transducer, shown in Table 4, has 21 total states and 6 bifurcation states—the ensemble model of three extant sequences is very complex. The naive exponential upper-bound gives a maximal state space of size 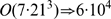 states. Using our uninformed search algorithm, we determined that there are 287 accessible states and 686 possible transitions between these states (compare with the
states. Using our uninformed search algorithm, we determined that there are 287 accessible states and 686 possible transitions between these states (compare with the 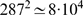 transitions estimated with the exponential calculation). After performing the transformations described in “Exact elimination of null cycles in SCFGs” to eliminate useless windback states, the ensemble model has a reduced state space with 230 states, albeit at the cost of extra transitions, bring the total to 1,789 transitions (here we are trading reduced memory complexity, which is linear in the number of states, for increased time complexity, which is linear in the number of transitions). Note that both before and after the reduction in complexity, the total number of states and transitions are less than the approximate bounds of
transitions estimated with the exponential calculation). After performing the transformations described in “Exact elimination of null cycles in SCFGs” to eliminate useless windback states, the ensemble model has a reduced state space with 230 states, albeit at the cost of extra transitions, bring the total to 1,789 transitions (here we are trading reduced memory complexity, which is linear in the number of states, for increased time complexity, which is linear in the number of transitions). Note that both before and after the reduction in complexity, the total number of states and transitions are less than the approximate bounds of 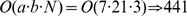 states and
states and 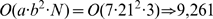 transitions suggested in “The composition algorithm”. Nonetheless, the extreme complexity of the ensemble model, despite the simplicity of the underlying model of RNA structure, makes clear the necessity for automated procedures for model construction. Dataset S1 gives the state space of the ensemble model constructed by the search algorithm and Dataset S2 the reduced model after eliminating windback states; both are in Graphviz format for visualization and show the state of the singlet transducer generating ancestral sequence as well as the states of the branch transducers generating observed sequences.
transitions suggested in “The composition algorithm”. Nonetheless, the extreme complexity of the ensemble model, despite the simplicity of the underlying model of RNA structure, makes clear the necessity for automated procedures for model construction. Dataset S1 gives the state space of the ensemble model constructed by the search algorithm and Dataset S2 the reduced model after eliminating windback states; both are in Graphviz format for visualization and show the state of the singlet transducer generating ancestral sequence as well as the states of the branch transducers generating observed sequences.
We implemented constrained maximum-likelihood inference of the structural alignment and ancestral structure of three extant sequences in a C++ program (Indiegram). For tractability, Indiegram uses the concept of fold envelopes described earlier to limit the fold space considered by the CYK algorithm, permitting structural information for the three extant sequences to be (optionally) supplied as input. If no structural information is supplied, then Indiegram uses a single-sequence SCFG to estimate a set of plausible folds [18], which are used to constrain the CYK algorithm.
The inference algorithms in Indiegram could be further constrained to enforce, for example, a fixed multiple alignment or a consensus structure for extant sequences. While experimentally-determined structures of individual RNAs are relatively rare, curated deep sequence alignments, such as those constructed for ribosomal RNAs [50], are frequently available for characterized RNA families. By constraining the inference algorithms with such sequence alignments, the memory and time complexity of the algorithms could be dramatically reduced. Such constraints can be naturally expressed with “alignment envelopes,” the alignment-space analogue of fold envelopes [18]. However, in this paper we focus on model construction and inference algorithms and postpone exploration of heuristics and constraints of these algorithms for future work.
Reconstructing small RNAs with the TKFST model
While reconstructing large RNAs such as ribosomal subunits is currently computationally-inaccessible without further heuristics to constrain our algorithms, reconstructing small RNAs of biological interest will soon be feasible. Table 7 shows estimates of the memory and time required to reconstruct biologically-interesting subunits of the nanos 3′ translational control element and tRNAs, as well as two small RNAs which show significant structural divergence, the Y RNAs and Group II introns, and therefore promise to be interesting candidates for ancestral reconstruction. The reconstructed structures for three nanos 3′ translational control elements (TCEs) and three tRNAs, which could be analyzed given current computational limitations, can be found at http://biowiki.org/IndieGram; however, the phylogenetic trees relating the tested sequences have short branch lengths, making the reconstruction problem easy by forcing the reconstructed structures to be essentially-identical to those of one of the extant RNAs.
Table 7. Estimates of the memory and time required to reconstruct ancestral structures of three RNAs from several families of biological interest (as reported by Indiegram).
| Family | Sequence lengths | Memory | Time |
| nanos 3′ TCE | 61–64 nt | 3 Gb | 3 min |
| tRNA | 69–73 nt | 11 Gb | 19 min |
| Y RNA | 47–81 nt | 33 Gb | 70 min |
| Group II intron (domains V and VI) | 76–91 nt | 122 Gb | 90 min |
The nanos 3′ translational control element (TCE) sequences are the seed sequences of the corresponding RFAM family [5] and the three tRNA sequences are from the BRalibaseII database [55] (identifiers AB042432.1-14140_14072, Z82044.1-16031_16103 and AC008670.6-83725_83795). The group II intron sequences (identifiers Z00044.1-87253_87177, X57546.1-2817_2907 and X04465.1-2700_2775) are from BralibaseII [55]. The Y RNAs are hY1, hY4, and hY5 from [104]; sequence lengths exclude the conserved stem S1. The time estimates are for a 2.2 GHz AMD Opteron 848 CPU.
Comparison of alignment methods
Guided by our experience with the nanos 3′ TCE and tRNA, where the reconstruction problem was made easy by the presence of a close outgroup, we conducted a simulation study of the dependence of reconstruction accuracy on outgroup branch length, with the further goal of comparing the performance of our reconstruction method (when simulating directly from the model) to simpler reconstruction methods that ignore either structure or phylogeny. (We here use the term “outgroup” loosely to denote the variable-length branch in our three-taxon study, where the other two branches are held at unit length.)
We simulated the evolution of RNAs under the TKFST model along three-taxon phylogenies (with one internal node), where we kept the branch lengths of two sibling species constant and varied the branch length of the outgroup between 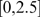 at steps of size
at steps of size 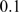 . Parameters used in the simulation were
. Parameters used in the simulation were 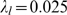 and
and 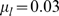 for loop sequence and
for loop sequence and 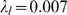 and
and 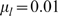 for stem sequence; the probability of a stem insertion was
for stem sequence; the probability of a stem insertion was  . These yielded a mean loop length of 5 bp and a mean stem length of 2.33 bp, with
. These yielded a mean loop length of 5 bp and a mean stem length of 2.33 bp, with  substructure indels per alignment. We selected alignments to reconstruct by requiring that there be at least two ancestral stems, loops of
substructure indels per alignment. We selected alignments to reconstruct by requiring that there be at least two ancestral stems, loops of 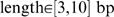 and stems of
and stems of 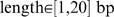 ; to reduce the complexity of our algorithms we additionally required that the sequences have
; to reduce the complexity of our algorithms we additionally required that the sequences have 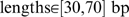 .
.
We then attempted three-way multiple alignment (and, in some cases, reconstruction of the ancestor) using a variety of statistical inference algorithms. We sought insight as to the relative importance of the following factors in reconstructing ancestral RNA: (i) modeling the secondary structure; (ii) modeling the phylogenetic topology & branch lengths; (iii) using posterior-decoding algorithms to maximize the expected alignment accuracy, rather than picking the single most likely alignment [20],[51],[52].
The alignment programs we used in this benchmark were Indiegram (exact ML inference of alignment and ancestral structure, given phylogeny, descendant structures and correct model); Stemloc (a greedy ML heuristic, ignoring phylogeny in favor of a single-linkage clustering of the descendant structures); Stemloc-AMA (a posterior-decoding heuristic, maximizing the alignment's expected accuracy rather than its likelihood); and Handel (ML alignment under various indel models that ignore secondary structure completely). In detail, the reconstruction methods were
Stemloc : the Stemloc program was used to align the three sequences via single-linkage clustering with a Pair SCFG [18]. The structures of the leaf sequences were provided, but not the phylogenetic branch lengths. Instead of modeling a true phylogeny by introducing unobserved ancestral sequences, it just does single-linkage clustering of the observed sequences.
Stemloc-AMA : the Stemloc program was used to align the three sequences in “sequence annealing” mode, a posterior decoding method that attempts to optimize AMA, a sum-over-pairs alignment accuracy metric [20]. The structures of the leaf sequences were provided, but not the phylogenetic branch lengths. This program uses the same underlying pair SCFG as Stemloc, but instead of maximizing likelihood, it attempts to maximize an alignment accuracy metric.
TKF91: with the TKF91 model [44], the Handel package [30],[39],[40] was used to align the three extant sequences and reconstruct the ancestor. The correct phylogenetic tree and branch lengths were supplied (as they were for the Indiegram benchmark). The insertion, deletion and substitution rates for the TKF91 model were set equal to those of the loop submodel of TKFST. This may be understood as a naive sequence-only reconstruction that completely ignores basepair structure (i.e. the stem sub-model of TKFST).
Long Indel: with a single-event trajectory approximation to the long indel model [53], the Handel package was used to align the three extant sequences and reconstruct the ancestor. The correct phylogenetic tree and branch lengths were supplied. The deletion and substitution rates were set equal to those of the loop submodel of TKFST. The mean indel length was set equal to 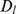 , the mean number of bases that are created/removed by an insertion/deletion in the loop submodel of TKFST; the mean equilibrium sequence length (and thereby the insertion rate) was equal to
, the mean number of bases that are created/removed by an insertion/deletion in the loop submodel of TKFST; the mean equilibrium sequence length (and thereby the insertion rate) was equal to 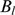 , the mean number of bases in TKFST at equilibrium (“A simple model of RNA structural evolution” has formulae for these quantities in terms of the TKFST rate parameters). This model improves on the previous model (TKF91) by introducing affine gap penalties.
, the mean number of bases in TKFST at equilibrium (“A simple model of RNA structural evolution” has formulae for these quantities in terms of the TKFST rate parameters). This model improves on the previous model (TKF91) by introducing affine gap penalties.
We measured alignment accuracy, under the simplifying assumption that this correlates well with ancestral reconstruction accuracy.
We first consider the perfect alignment rate; that is, the number of times each method gets the alignment exactly correct. Theory predicts that Maximum Likelihood inference, using the correct model and parameters, should be asymptotically optimal (if one only counts perfect guesses). Inspecting Figure 10, we find this to be almost the case; the exception is when the outgroup is very distant and the bins may be undersampled (the departure from prediction that we observe for low-identity alignments is not statistically significant: when the optimal success rate drops below 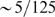 , then 125 trials are probably insufficient to compare two near-optimal methods). We also note that the ML version of Stemloc is near-optimal, despite the Stemloc pair-SCFG being slightly different from the TKFST pair-SCFG in parameterization and structure (e.g. Stemloc 's grammar does not allow insertion/deletion of entire substructures). The ML version of Stemloc is also observed to have a slightly higher perfect alignment rate than the posterior-decoding version (Stemloc -AMA). Finally, we note that the structure-blind models (TKF91 and Long Indel) perform consistently worse than the structure-aware methods; furthermore, both linear (TKF91) and affine (Long Indel) gap-penalties perform equally bad in this test (note that the TKFST model, from which the true alignments were simulated, does not allow long-indel events, which may partly explain why affine gap-penalties do not help in this benchmark).
, then 125 trials are probably insufficient to compare two near-optimal methods). We also note that the ML version of Stemloc is near-optimal, despite the Stemloc pair-SCFG being slightly different from the TKFST pair-SCFG in parameterization and structure (e.g. Stemloc 's grammar does not allow insertion/deletion of entire substructures). The ML version of Stemloc is also observed to have a slightly higher perfect alignment rate than the posterior-decoding version (Stemloc -AMA). Finally, we note that the structure-blind models (TKF91 and Long Indel) perform consistently worse than the structure-aware methods; furthermore, both linear (TKF91) and affine (Long Indel) gap-penalties perform equally bad in this test (note that the TKFST model, from which the true alignments were simulated, does not allow long-indel events, which may partly explain why affine gap-penalties do not help in this benchmark).
Figure 10. Dependence of perfect alignment rate on alignment method and outgroup branch length.

Perfect alignment is when a given alignment program estimates the alignment 100% correctly, with no errors. Simulating the evolution of three structural RNAs under the TKFST model, we investigated the dependency of perfect alignment rate on outgroup branch length. The simulation included two sister species at unit distance from the ancestral sequence, plus one “outgroup” whose branch length  was varied between
was varied between 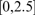 by selecting 25 equally spaced values of
by selecting 25 equally spaced values of  in this range, spaced
in this range, spaced 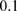 apart. (A unit-length branch here corresponds to one expected substitution per site in loop sequence.) We simulated 25 alignments for each value of
apart. (A unit-length branch here corresponds to one expected substitution per site in loop sequence.) We simulated 25 alignments for each value of  , using TKFST model parameters described in the text. Since the perfect alignment rate is rather low, we further aggregated the
, using TKFST model parameters described in the text. Since the perfect alignment rate is rather low, we further aggregated the  into bins of five; thus, for example, the bin named “
into bins of five; thus, for example, the bin named “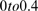 ” includes
” includes 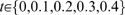 and represents
and represents  trials in total. The perfect alignment rate was measured for various statistical alignment inference procedures. These procedures are described in the text, but may be summarized very briefly as ML under the true model (“Indiegram”); greedy approximate-ML progressive alignment by single-linkage clustering with pair SCFGs (“Stemloc”); sequence annealing, a form of posterior decoding to maximize a sum-over-pairs accuracy metric, using pair SCFGs to get the posterior probabilities (“Stemloc-AMA”); statistical alignment using the TKF91 model, i.e. linear gap-penalties (“TKF91”); and statistical alignment using a long-indel model, i.e. affine gap-penalties (“Long Indel”).
trials in total. The perfect alignment rate was measured for various statistical alignment inference procedures. These procedures are described in the text, but may be summarized very briefly as ML under the true model (“Indiegram”); greedy approximate-ML progressive alignment by single-linkage clustering with pair SCFGs (“Stemloc”); sequence annealing, a form of posterior decoding to maximize a sum-over-pairs accuracy metric, using pair SCFGs to get the posterior probabilities (“Stemloc-AMA”); statistical alignment using the TKF91 model, i.e. linear gap-penalties (“TKF91”); and statistical alignment using a long-indel model, i.e. affine gap-penalties (“Long Indel”).
A subtly different ranking emerges from consideration of the alignment accuracy. In Figure 11, we abandon the all-or-nothing metric of counting only perfect alignments, instead using a metric that shows what proportion of the alignment is correct. Specifically, we plot the Alignment Metric Accuracy (AMA) as a function of outgroup branch length. AMA measures the proportion of residues which are correctly aligned, averaged over all pairs of sequences [54]. Figure 4 reveals that Stemloc-AMA (which attempts to find the alignment with the maximum expected AMA) edges out both Indiegram and the ML version of Stemloc (both of which attempt to find the alignment with the maximum likelihood). These results, compared to the subtly different story told by the perfect alignment rate, underscore the point that benchmark results for alignment methods can depend exquisitely on the choice of accuracy metric. The superiority of ML methods is only assured in terms of perfect alignment rate, and not necessarily other accuracy metrics.
Figure 11. Dependence of alignment metric accuracy on alignment method and outgroup branch length.

We simulated the evolution of three structural RNAs under the TKFST model. The simulation included two sister species at unit distance from the ancestral sequence, plus one “outgroup” whose branch length  was varied between
was varied between 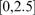 by selecting 25 equally spaced values of
by selecting 25 equally spaced values of  in this range, spaced
in this range, spaced 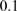 apart. We then simulated 25 alignments for each value of
apart. We then simulated 25 alignments for each value of  , using TKFST model parameters described in the text. The Alignment Metric Accuracy (AMA) is, roughly, the proportion of residues that are correctly aligned, averaged over all pairs of sequences (see [54] for a precise definition; we set the AMA Gap Factor to 1). The AMA between the true alignment and the inferred alignment was measured for various statistical alignment inference procedures. These procedures are described in the text, but may be summarized very briefly as ML under the true model (“Indiegram”); greedy approximate-ML progressive alignment by single-linkage clustering with pair SCFGs (“Stemloc”); sequence annealing, a form of posterior decoding to maximize a sum-over-pairs accuracy metric, using pair SCFGs to get the posterior probabilities (“Stemloc-AMA”); statistical alignment using the TKF91 model, i.e. linear gap-penalties (“TKF91”); and statistical alignment using a long-indel model, i.e. affine gap-penalties (“Long Indel”).
, using TKFST model parameters described in the text. The Alignment Metric Accuracy (AMA) is, roughly, the proportion of residues that are correctly aligned, averaged over all pairs of sequences (see [54] for a precise definition; we set the AMA Gap Factor to 1). The AMA between the true alignment and the inferred alignment was measured for various statistical alignment inference procedures. These procedures are described in the text, but may be summarized very briefly as ML under the true model (“Indiegram”); greedy approximate-ML progressive alignment by single-linkage clustering with pair SCFGs (“Stemloc”); sequence annealing, a form of posterior decoding to maximize a sum-over-pairs accuracy metric, using pair SCFGs to get the posterior probabilities (“Stemloc-AMA”); statistical alignment using the TKF91 model, i.e. linear gap-penalties (“TKF91”); and statistical alignment using a long-indel model, i.e. affine gap-penalties (“Long Indel”).
Taken together, these results suggest that the most important factor distinguishing the various models we have examined is the incorporation of some form of basepair structure: structure-blind Handel (regardless of linear vs affine gap penalty) performs much worse than the structure-aware SCFG methods. Intuitively, this is to be expected: whenever a basepair-aware method aligns one half of a basepair, it gets the other nucleotide correctly aligned for free. In benchmarks of RNA multiple alignment programs, structure-aware scoring schemes routinely outperform structure-blind scoring schemes [55],[56]. Since we know that modeling structure is very important, it's not too surprising that it turns out to be the most important of the factors we considered.
The second most important, amongst the factors we have considered in this experiment, is selection of the most appropriate objective function for the task at hand (c.f. perfect alignment rate vs AMA), followed by use of the correct posterior-decoding algorithm for the chosen objective function (c.f. Stemloc vs Stemloc-AMA). This is a subtle but important point: before deciding exactly what inference algorithm we're going to use to reconstruct ancestral sequences, we need to decide whether we want to maximize (a) the probability that our reconstructed sequence is 100% correct, (b) the expected number of nucleotides that are correctly reconstructed, (c) the expected number of base-pairs that are correctly reconstructed, (d) the expected number of stems that are correctly reconstructed, (e) some other metric. Each of these metrics would require a slightly different inference algorithm.
Lastly, the fine details of the scoring scheme—including branch lengths, substitution scores, gap penalties and so forth—appear to be the least important of the factors we considered, yielding observable differences only when all other aspects of the inference procedure were more-or-less equal. While such details of the model may affect reconstruction quality, they appear to have very minor influence on alignment quality.
Discussion
Following the conception of paleogenetics [57], a large number of synthetic reconstructions of ancestral protein sequences have been reported in the literature [58]–[65]. There is also scientific interest in reconstructing DNA sequences [33], [66]–[71]. Given the importance of the RNA world hypothesis to current discussions of the origin of life [72]–[78], the many modern-day relics of this world [79]–[82] and the recent proposal of a structural model for the primordial ribosome [2], we believe that phylogenetic reconstruction of ancient RNA is a significant problem, deserving of strong bioinformatics support.
The work reported in this paper builds on extensive prior art in the areas of evolutionary modeling and ancestral reconstruction. Reviewing all of this would take several books, but we can note some key references. The reconstruction of ancient sequences was first proposed in 1963 by Pauling and Zuckerkandl [57]; current applications of this idea, mostly using substitution models, are surveyed in the book edited by Liberles [83]. Many algorithms in phylogenetics implicitly reconstruct substitution histories, whether by parsimony [84],[85] or likelihood [22]. There is a substantial body of work to model indels on phylogenies [30], [35], [39], [44], [53], [86]–[92]. Recent work has extended these ideas to the reconstruction of indel histories [93],[94], particularly at the genomic scale [33],[95]. There is also prior work in computational linguistics on the theory of transducers for sequences [96] and parse trees [36],[37],[97],[98] (from which we take the terms “string transducer” and “parse-tree transducer”). We draw on the bioinformatics literature for SCFGs [10],[11],[99], especially Pair SCFGs [14],[18],[19] and phylogenetic SCFGs [16]. In particular, an early example of a pairwise conditional model 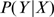 for structure-dependent RNA evolution was given by Eddy et al
[12]. A conditional framework similar to ours in some respects is described by Sakakibara et al
[100]. The dynamic programming inference algorithms for multiple-sequence SCFGs are closely related to the protosequence algorithm of Sankoff [42].
for structure-dependent RNA evolution was given by Eddy et al
[12]. A conditional framework similar to ours in some respects is described by Sakakibara et al
[100]. The dynamic programming inference algorithms for multiple-sequence SCFGs are closely related to the protosequence algorithm of Sankoff [42].
While we have focused on the TKF Structure Tree model in our Results, our model-construction algorithm is applicable to any model of the evolution of secondary structure which can be expressed as a Pair SCFG. Realistic structural and thermodynamic effects—such as base-stacking or loop length distributions—can, in principle, be incorporated. Other phenomena of RNA evolution may prove more difficult: modeling helix slippage with a branch transducer is awkward, let alone more radical changes in structure; pseudoknots, too, are impossible with the models we have described here. Even so, variants of our models could be used for proposing candidate alignments for more accurate scoring by such models.
An implementation of inference algorithms for models on the three-taxon phylogeny is sufficient to construct a MCMC sampling algorithm over many sequences on an arbitrary phylogeny. A sketch of such a sampling algorithm is as follows: at each step of the sampling algorithm, we re-sample the sequence and structure of the ancestral node 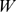 , conditioned on the sequences and structures of
, conditioned on the sequences and structures of 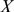 ,
, 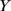 and
and 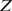 . The structural alignment of all four sequences can change at each step, providing for fast mixing and guaranteeing ergodicity. This move is similar to the sampler proposed by [31] for models with a HMM structure. Note that this, in principle, permits construction of a crude sampler to simultaneously infer phylogeny as well, by proposing and accepting or rejecting changes to the underlying tree as well as the implied structural alignment.
. The structural alignment of all four sequences can change at each step, providing for fast mixing and guaranteeing ergodicity. This move is similar to the sampler proposed by [31] for models with a HMM structure. Note that this, in principle, permits construction of a crude sampler to simultaneously infer phylogeny as well, by proposing and accepting or rejecting changes to the underlying tree as well as the implied structural alignment.
Reconstructing structural changes of large RNAs using the three-way sampling kernel which we have described would require resources far in excess of those currently available; barring the availability of supercomputers with terabytes of memory, such algorithms will only be feasible for short RNAs (Table 7). A promising direction is to consider variations on the three-way sampling kernel, such as the importance-sampling approach described for the TKF model by [32]. This approach first proposes an ancestor 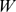 by aligning extant sequence
by aligning extant sequence 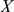 to
to 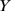 (ignoring
(ignoring 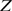 ); then, in a second step, the proposed
); then, in a second step, the proposed 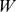 is independently aligned to
is independently aligned to 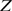 . The proposed three-way alignment and reconstruction is then randomly accepted (or rejected) using a Hastings ratio based on the three-way transducer composition. The complexity of this kernel is the same as the pairwise case; with suitable constraints, this is feasible for RNA grammars on present hardware, at least for ribosomal domains (if not yet whole subunits—although pairwise alignment of those should also be possible soon). The approach of Redelings and Suchard therefore merits future consideration in the context of modeling the evolution of RNAs on a tree.
. The proposed three-way alignment and reconstruction is then randomly accepted (or rejected) using a Hastings ratio based on the three-way transducer composition. The complexity of this kernel is the same as the pairwise case; with suitable constraints, this is feasible for RNA grammars on present hardware, at least for ribosomal domains (if not yet whole subunits—although pairwise alignment of those should also be possible soon). The approach of Redelings and Suchard therefore merits future consideration in the context of modeling the evolution of RNAs on a tree.
An alternative MCMC scheme for sampling RNA phylogeny, structure and alignment was developed for the SimulFold program [101]. SimulFold does not use a strictly normalized probabilistic model, resulting in some oddities in the ways that structure and indels interact (for example, it does not penalize deletion of one half of a basepair). Currently, it is not clear how appropriate SimulFold would be for ancestral reconstruction, although it has several advantages (e.g., explicit treatment of pseudoknots). Of course, MCMC kernels are inherently adaptable to other purposes: the MCMC moves developed for SimulFold may be useful for inference under different models.
This paper focuses on the case where the tree topology is known, but many of the methods which we have described can be extended to the more general case where none of the possible constraints (phylogeny, structure or alignment) are final. For example, the probabilistic framework readily allows us to compare likelihoods of two different phylogenetic trees by constructing a composite transducer for each tree. Thus, the MCMC samplers described above for alignments could, in principal, be extended to phylogenies (albeit at a computational cost).
While MCMC provides the most information about the posterior distribution of evolutionary histories, in practice a maximum likelihood inference may be adequate (and typically much faster). The progressive profiling used by the Ortheus program for reconstructing ancestral genomes is promising [33]. This approach is similar to a progressive multiple alignment algorithm, in that it proceeds via a single postorder (leaf-to-root) traversal of the phylogeny. As each node is visited, a profile is generated for that node, by aligning the profiles of its children to a composite transducer using DP, then sampling a finite number of traceback paths through the DP matrix. The profile is not linear: the sampled paths instead form a reticulate network, a.k.a. a partial order graph [102]. An equivalent of Ortheus for RNA reconstruction should be possible, representing the intermediate profiles using transducers.
Given the excellent performance of Stemloc-AMA 's sequence annealing, particularly when measured using its own scoring metric (AMA), such posterior-decoding methods should also be considered for reconstruction.
In summary, the evolutionary models and algorithms we have described form a systematic theoretical platform on which we can test different optimization and sampling strategies for studying the structural evolution of RNA gene families in detail. Stochastic grammars are powerful tools for this task, although they will not be the only tools we need, particularly as we move towards modeling RNA evolution in greater detail. Our hope is that these algorithms will allow us to test and refine our understanding of RNA evolution by computational reconstruction and (eventually) direct experimental investigation of early ribonucleic machines.
Supporting Information
Formal description of transducer composition algorithm yielding phylogenetic grammar rules
(0.15 MB PDF)
Algorithm for elimination of null cycles and empty bifurcations from general SCFGs
(0.13 MB PDF)
Description of software tools
(0.09 MB PDF)
Example presentation of TKFST inference grammars in Indiegram format
(0.14 MB PDF)
Grammar for three-taxon TKFST model returned by transducer composition algorithm (graphviz dot format)
(0.03 MB TXT)
Grammar for three-taxon TKFST model returned by transducer composition algorithm, after eliminating windback states (graphviz dot format)
(0.06 MB TXT)
Acknowledgments
We thank Jeff Thorne, Jamie Cate, Jennifer Doudna, Jotun Hein, David Haussler, Sean Eddy, Elena Rivas and Eric Westhof for inspirational discussions. We are grateful to Ben Redelings and one anonymous referee for illuminating criticism.
Footnotes
The authors have declared that no competing interests exist.
This work was funded by NIH/NHGRI grant GM076705. RKB was partially supported by a NSF Graduate Research Fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Crick FH. The origin of the genetic code. Journal of Molecular Biology. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 2.Smith TF, Lee JC, Gutell RR, Hartman H. The origin and evolution of the ribosome. Biology Direct. 2008;3:16. doi: 10.1186/1745-6150-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehmann K, Schmidt U. Group II introns: structure and catalytic versatility of large natural ribozymes. Critical Reviews in Biochemistry and Molecular Biology. 2003;38:249–303. doi: 10.1080/713609236. [DOI] [PubMed] [Google Scholar]
- 4.Antal M, Boros E, Solymosy F, Kiss T. Analysis of the structure of human telomerase RNA in vivo. Nucleic Acids Research. 2002;30:912–920. doi: 10.1093/nar/30.4.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Research. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hancock JM, Tautz D, Dover GA. Evolution of the secondary structures and compensatory mutations of the ribosomal RNAs of Drosophila melanogaster. Molecular Biology and Evolution. 1988;5:393–414. doi: 10.1093/oxfordjournals.molbev.a040501. [DOI] [PubMed] [Google Scholar]
- 7.Leontis NB, Stombaugh J, Westhof E. Motif prediction in ribosomal RNAs: Lessons and prospects for automated motif prediction in homologous RNA molecules. Biochimie. 2002;84:961–973. doi: 10.1016/s0300-9084(02)01463-3. [DOI] [PubMed] [Google Scholar]
- 8.Yokoyama T, Suzuki T. Ribosomal RNAs are tolerant toward genetic insertions: evolutionary origin of the expansion segments. Nucleic Acids Research. 2008;36:3539–3551. doi: 10.1093/nar/gkn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hancock JM, Dover GA. ‘compensatory slippage’ in the evolution of ribosomal RNA genes. Nucleic Acids Research. 1990;18:5949–5954. doi: 10.1093/nar/18.20.5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Research. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durbin R, Eddy S, Krogh A, Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press; 1998. [Google Scholar]
- 12.Klein R, Eddy SR. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics. 2003;4 doi: 10.1186/1471-2105-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nawrocki E, Eddy S. Query-dependent banding (QDB) for faster RNA similarity searches. PLoS Computational Biology. 2007;3:e56. doi: 10.1371/journal.pcbi.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rivas E, Eddy SR. A dynamic programming algorithm for RNA structure prediction including pseudoknots. Journal of Molecular Biology. 1999;285:2053–2068. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- 15.Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, et al. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Computational Biology. 2006;2:e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knudsen B, Hein J. Pfold: RNA secondary structure prediction using stochastic contextfree grammars. Nucleic Acids Research. 2003;31:3423–3428. doi: 10.1093/nar/gkg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dowell RD, Eddy SR. Evaluation of several lightweight stochastic context-free grammars for RNA secondary structure prediction. BMC Bioinformatics. 2004;5 doi: 10.1186/1471-2105-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmes I. Accelerated probabilistic inference of RNA structure evolution. BMC Bioinformatics. 2005;6 doi: 10.1186/1471-2105-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dowell RD, Eddy SR. Efficient pairwise RNA structure prediction and alignment using sequence alignment constraints. BMC Bioinformatics. 2006;7 doi: 10.1186/1471-2105-7-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradley RK, Pachter L, Holmes I. Specific alignment of structured RNA: Stochastic grammars and sequence annealing. Bioinformatics. 2008;24:2677–2683. doi: 10.1093/bioinformatics/btn495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Felsenstein J. Inferring Phylogenies. Sinauer Associates, Inc.; 2003. ISBN 0878931775. [Google Scholar]
- 22.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. Journal of Molecular Evolution. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 23.Elston RC, Stewart J. A general model for the genetic analysis of pedigree data. Human Heredity. 1971;21:523–542. doi: 10.1159/000152448. [DOI] [PubMed] [Google Scholar]
- 24.Holmes I, Rubin GM. An Expectation Maximization algorithm for training hidden substitution models. Journal of Molecular Biology. 2002;317:757–768. doi: 10.1006/jmbi.2002.5405. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen R. Mutations as missing data: inferences on the ages and distributions of nonsynonymous and synonymous mutations. Genetics. 2001;159:401–411. doi: 10.1093/genetics/159.1.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pearl J. Proceedings American Association of Artificial Intelligence National Conference on AI. Pittsburgh, PA. : 1982. Reverend Bayes on inference engines: A distributed hierarchical approach. pp. 133–136. [Google Scholar]
- 27.Yang Z. Estimating the pattern of nucleotide substitution. Journal of Molecular Evolution. 1994;39:105–111. doi: 10.1007/BF00178256. [DOI] [PubMed] [Google Scholar]
- 28.Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Molecular Biology and Evolution. 2001;18:691–699. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- 29.Holmes I. A probabilistic model for the evolution of RNA structure. BMC Bioinformatics. 2004;5 doi: 10.1186/1471-2105-5-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmes I, Bruno WJ. Evolutionary HMMs: a Bayesian approach to multiple alignment. Bioinformatics. 2001;17:803–820. doi: 10.1093/bioinformatics/17.9.803. [DOI] [PubMed] [Google Scholar]
- 31.Jensen J, Hein J. Gibbs sampler for statistical multiple alignment. Aarhus, Denmark: Dept. of Theoretical Statistics, University of Aarhus; 2002. Technical Report No. 429. [Google Scholar]
- 32.Redelings BD, Suchard MA. Joint bayesian estimation of alignment and phylogeny. Systematic Biology. 2005;54:401–418. doi: 10.1080/10635150590947041. [DOI] [PubMed] [Google Scholar]
- 33.Paten B, Herrero J, Fitzgerald S, Flicek P, Holmes I, et al. Genome-wide nucleotide level mammalian ancestor reconstruction. Genome Research. 2008;18:1829–1843. doi: 10.1101/gr.076521.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rivas E, Eddy SR. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics. 2001;2 doi: 10.1186/1471-2105-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradley RK, Holmes I. Transducers: an emerging probabilistic framework for modeling indels on trees. Bioinformatics. 2007;23:3258–3262. doi: 10.1093/bioinformatics/btm402. [DOI] [PubMed] [Google Scholar]
- 36.Comon H, Dauchet M, Gilleron R, L̈oding C, Jacquemard F, et al. Tree Automata Techniques and Applications, 2007. pp. 161–182. Available from: http://tata.gforge.inria.fr/, chapter 6 (“Tree Transducers”) Release October, 12th 2007.
- 37.G'ecseg F, Steinby M. 1997. pp. 1–61. Handbook of formal languages, Volume 3: Beyond Words, Springer-Verlag, chapter Tree languages.
- 38.Rivas E, Eddy S. Probabilistic phylogenetic inference with insertions and deletions. PLoS Computational Biology. 2008;4:e1000172. doi: 10.1371/journal.pcbi.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holmes I. Using guide trees to construct multiple-sequence evolutionary HMMs. Bioinformatics. 2003;19(Suppl. 1):i147–157. doi: 10.1093/bioinformatics/btg1019. [DOI] [PubMed] [Google Scholar]
- 40.Holmes I. Phylocomposer and Phylodirector: Analysis and Visualization of Transducer Indel Models. Bioinformatics. 2007;23:3263–3264. doi: 10.1093/bioinformatics/btm432. [DOI] [PubMed] [Google Scholar]
- 41.Lunter G. HMMoC–a compiler for hidden Markov models. Bioinformatics. 2007;23:2485–2487. doi: 10.1093/bioinformatics/btm350. [DOI] [PubMed] [Google Scholar]
- 42.Sankoff D. Simultaneous solution of the RNA folding, alignment, and protosequence problems. SIAM Journal of Applied Mathematics. 1985;45:810–825. [Google Scholar]
- 43.Holmes I, Rubin GM. Pairwise RNA structure comparison using stochastic context-free grammars. 2002. Pacific Symposium on Biocomputing, 2002. [DOI] [PubMed]
- 44.Thorne JL, Kishino H, Felsenstein J. An evolutionary model for maximum likelihood alignment of DNA sequences. Journal of Molecular Evolution. 1991;33:114–124. doi: 10.1007/BF02193625. [DOI] [PubMed] [Google Scholar]
- 45.Kendall DG. On the generalized birth-and-death process. Annals of Mathematical Statistics. 1948;19:1–15. [Google Scholar]
- 46.Feller W. An Introduction to Probability Theory and its Applications, Vol II. New York: John Wiley and Sons; 1971. [Google Scholar]
- 47.Bradley RK, Uzilov AV, Skinner M, Bendana YR, Varadarajan A, et al. Non-coding RNA gene predictors in Drosophila. 2008. Submitted.
- 48.Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Research. 1998;26:148–53. doi: 10.1093/nar/26.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rijk PD, Caers A, Peer YVd, Wachter RD. Database on the structure of large ribosomal subunit RNA. Nucleic Acids Research. 1998;26:183–6. doi: 10.1093/nar/26.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gutell RR. Collection of small subunit (16S and 16S-like) ribosomal RNA structures. Nucleic Acids Research. 1993;21:3051–3054. doi: 10.1093/nar/21.13.3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holmes I, Durbin R. Dynamic programming alignment accuracy. Journal of Computational Biology. 1998;5:493–504. doi: 10.1089/cmb.1998.5.493. [DOI] [PubMed] [Google Scholar]
- 52.Schwartz AS, Pachter L. Multiple alignment by sequence annealing. Bioinformatics. 2007;23:e24–9. doi: 10.1093/bioinformatics/btl311. [DOI] [PubMed] [Google Scholar]
- 53.Miklós I, Lunter G, Holmes I. A long indel model for evolutionary sequence alignment. Molecular Biology and Evolution. 2004;21:529–540. doi: 10.1093/molbev/msh043. [DOI] [PubMed] [Google Scholar]
- 54.Schwartz AS, Myers EW, Pachter L. Alignment metric accuracy. 2006. http://arxiv.org/abs/qbio/0510052.
- 55.Gardner PP, Wilm A, Washietl S. A benchmark of multiple sequence alignment programs upon structural RNAs. Nucleic Acids Research. 2005;33:2433–2439. doi: 10.1093/nar/gki541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilm A, Mainz I, Steger G. An enhanced RNA alignment benchmark for sequence alignment programs. Algorithms for Molecular Biology. 2006;1:19. doi: 10.1186/1748-7188-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pauling L, Zuckerkandl E. Chemical paleogenetics, molecular “restoration studies” of extinct forms of life. Acta Chemica Scandinavica. 1963;17:S9–S16. [Google Scholar]
- 58.Malcolm BA, Wilson KP, Matthews BW, Kirsch JF, Wilson AC. Ancestral lysozymes reconstructed, neutrality tested, and thermostability linked to hydrocarbon packing. Nature. 1990;345:86–89. doi: 10.1038/345086a0. [DOI] [PubMed] [Google Scholar]
- 59.Stackhouse J, Presnell SR, McGeehan GM, Nambiar KP, Benner SA. The ribonuclease from an extinct bovid ruminant. FEBS letters. 1990;262:104–106. doi: 10.1016/0014-5793(90)80164-e. [DOI] [PubMed] [Google Scholar]
- 60.Zhang J, Rosenberg HF. Complementary advantageous substitutions in the evolution of an antiviral RNase of higher primates. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5486–5491. doi: 10.1073/pnas.072626199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gaucher EA, Thomson JM, Burgan MF, Benner SA. Inferring the palaeoenvironment of ancient bacteria on the basis of resurrected proteins. Nature. 2003;425:285–288. doi: 10.1038/nature01977. [DOI] [PubMed] [Google Scholar]
- 62.Thomson JM, Gaucher EA, Burgan MF, De Kee DW, Li T, et al. Resurrecting ancestral alcohol dehydrogenases from yeast. Nature Genetics. 2005;37:630–635. doi: 10.1038/ng1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang BSW, Jönsson K, Kazmi MA, Donoghue MJ, Sakmar TP. Recreating a functional ancestral archosaur visual pigment. Molecular biology and evolution. 2002;19:1483–1489. doi: 10.1093/oxfordjournals.molbev.a004211. [DOI] [PubMed] [Google Scholar]
- 64.Sun H, Merugu S, Gu X, Kang YY, Dickinson DP, et al. Identification of essential amino acid changes in paired domain evolution using a novel combination of evolutionary analysis and in vitro and in vivo studies. Molecular Biology and Evolution. 2002;19:1490–1500. doi: 10.1093/oxfordjournals.molbev.a004212. [DOI] [PubMed] [Google Scholar]
- 65.Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. Crystal structure of an ancient protein: evolution by conformational epistasis. Science. 2007;317:1544–8. doi: 10.1126/science.1142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–10. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 67.Adey NB, Tollefsbol TO, Sparks AB, Edgell MH, Hutchison CA. Molecular resurrection of an extinct ancestral promoter for mouse l1. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:1569–1573. doi: 10.1073/pnas.91.4.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blanchette M, Green ED, Miller W, Haussler D. Reconstructing large regions of an ancestral mammalian genome in silico. Genome Research. 2004;14:2412–2423. doi: 10.1101/gr.2800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Noonan JP, Coop G, Kudaravalli S, Smith D, Krause J, et al. Sequencing and analysis of Neanderthal genomic DNA. Science. 2006;314:1113–1118. doi: 10.1126/science.1131412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gaucher EA. Ancestral sequence reconstruction as a tool to understand natural history and guide synthetic biology: realizing and extending the vision of Zuckerkandl and Pauling. 2007. pp. 20–33. In: Liberles [83], chapter 2.
- 71.Elias I, Tuller T. Reconstruction of ancestral genomic sequences using likelihood. Journal of Computational Biology. 2007;14:216–37. doi: 10.1089/cmb.2006.0101. [DOI] [PubMed] [Google Scholar]
- 72.Marintchev A, Wagner G. Translation initiation: structures, mechanisms and evolution. Quarterly reviews of biophysics. 2004;37:197–284. doi: 10.1017/S0033583505004026. [DOI] [PubMed] [Google Scholar]
- 73.Muller AWJ. Thermosynthesis as energy source for the RNA world: a model for the bioenergetics of the origin of life. Bio Systems. 2005;82:93–102. doi: 10.1016/j.biosystems.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 74.Cavalier-Smith T. Rooting the tree of life by transition analyses. Biology Direct. 2006;1:19. doi: 10.1186/1745-6150-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martin W, Koonin EV. Introns and the origin of nucleus-cytosol compartmentalization. Nature. 2006;440:41–45. doi: 10.1038/nature04531. [DOI] [PubMed] [Google Scholar]
- 76.Forterre P. Three RNA cells for ribosomal lineages and three DNA viruses to replicate their genomes: a hypothesis for the origin of cellular domain. Proceedings of the National Academy of Sciences of the USA. 2006;103:3669–3674. doi: 10.1073/pnas.0510333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yakhnin AV. A model for the origin of protein synthesis as coreplicational scanning of nascent RNA. Origins of life and evolution of the biosphere : the journal of the International Society for the Study of the Origin of Life. 2007;37:523–536. doi: 10.1007/s11084-007-9108-z. [DOI] [PubMed] [Google Scholar]
- 78.Danchin A, Fang G, Noria S. The extant core bacterial proteome is an archive of the origin of life. Proteomics. 2007;7:875–889. doi: 10.1002/pmic.200600442. [DOI] [PubMed] [Google Scholar]
- 79.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 80.Lowe TM, Eddy SR. A computational screen for methylation guide snoRNAs in yeast. Science. 1999;283:1168–1171. doi: 10.1126/science.283.5405.1168. [DOI] [PubMed] [Google Scholar]
- 81.Eddy SR. Non-coding RNA genes and the modern RNA world. Nature Reviews Genetics. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- 82.Mandal M, Boese B, Barrick JE, Winkler WC, Breaker RR. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell. 2003;113:577–586. doi: 10.1016/s0092-8674(03)00391-x. [DOI] [PubMed] [Google Scholar]
- 83.Liberles DA, editor. Ancestral Sequence Reconstruction. Oxford University Press; 2007. [Google Scholar]
- 84.Edwards AWF, Cavalli-Sforza L. The reconstruction of evolution. Annals of Human Genetics. 1963;27:105. [Google Scholar]
- 85.Hendy MD, Penny D. Branch and bound algorithms to determine minimal evolutionary trees. Mathematical Biosciences. 1982;59:277–290. [Google Scholar]
- 86.Hein J, Wiuf C, Knudsen B, Moller MB, Wibling G. Statistical alignment: computational properties, homology testing and goodness-of-fit. Journal of Molecular Biology. 2000;302:265–279. doi: 10.1006/jmbi.2000.4061. [DOI] [PubMed] [Google Scholar]
- 87.Hein J. An algorithm for statistical alignment of sequences related by a binary tree. In: Altman RB, Dunker AK, Hunter L, Lauderdale K, Klein TE, editors. Pacific Symposium on Biocomputing. Singapore: World Scientific; 2001. pp. 179–190. [DOI] [PubMed] [Google Scholar]
- 88.Steel M, Hein J. Applying the Thorne-Kishino-Felsenstein model to sequence evolution on a star-shaped tree. Applied Mathematics Letters. 2001;14:679–684. [Google Scholar]
- 89.Knudsen B, Miyamoto M. Sequence alignments and pair hidden Markov models using evolutionary history. Journal of Molecular Biology. 2003;333:453–460. doi: 10.1016/j.jmb.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 90.Lunter GA, Miklós I, Song YS, Hein J. An efficient algorithm for statistical multiple alignment on arbitrary phylogenetic trees. Journal of Computational Biology. 2003;10:869–889. doi: 10.1089/106652703322756122. [DOI] [PubMed] [Google Scholar]
- 91.Lunter GA, Drummond AJ, Miklós I, Hein J. Nielsen R, editor. Statistical alignment: Recent progress, new applications, and challenges. Statistical methods in Molecular Evolution, Springer Verlag, Series in Statistics in Health and Medicine, chapter 14. 2004. pp. 375–406.
- 92.Satija R, Pachter L, Hein J. Combining statistical alignment and phylogenetic footprinting to detect regulatory elements. Bioinformatics. 2008;24:1236–1242. doi: 10.1093/bioinformatics/btn104. [DOI] [PubMed] [Google Scholar]
- 93.Kim J, Sinha S. Indelign: a probabilistic framework for annotation of insertions and deletions in a multiple alignment. Bioinformatics. 2007;23:289–297. doi: 10.1093/bioinformatics/btl578. [DOI] [PubMed] [Google Scholar]
- 94.Diallo AB, Makarenkov V, Blanchette M. Exact and heuristic algorithms for the indel maximum likelihood problem. Journal of Computational Biology. 2007;14:446–461. doi: 10.1089/cmb.2007.A006. [DOI] [PubMed] [Google Scholar]
- 95.Ma J, Zhang L, Suh BB, Raney BJ, Brian J, et al. Reconstructing contiguous regions of an ancestral genome. Genome Research. 2006;16:1557–1565. doi: 10.1101/gr.5383506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mohri M, Pereira F, Riley M. Weighted finite-state transducers in speech recognition. Computer Speech and Language. 2002;16:69–88. [Google Scholar]
- 97.Rounds WC. Mappings and grammars on trees. Mathematical Systems Theory. 1970;4:257–287. [Google Scholar]
- 98.Thatcher JW. Generalized sequential machine maps. Journal of Computer and System Sciences. 1970;4:339–367. [Google Scholar]
- 99.Sakakibara Y, Brown M, Hughey R, Mian IS, Sjölander K, et al. Stochastic context-free grammars for tRNA modeling. Nucleic Acids Research. 1994;22:5112–5120. doi: 10.1093/nar/22.23.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sakakibara Y. Pair hidden Markov models on tree structures. Bioinformatics. 2003;19(Suppl 1):i232–240. doi: 10.1093/bioinformatics/btg1032. [DOI] [PubMed] [Google Scholar]
- 101.Meyer IM, Miklós I. SimulFold: simultaneously inferring rna structures including pseudoknots, alignments, and trees using a Bayesian MCMC framework. PLoS Computational Biology. 2007;3:e149. doi: 10.1371/journal.pcbi.0030149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee C, Grasso C, Sharlow M. Multiple sequence alignment using partial order graphs. Bioinformatics. 2002;18:452–464. doi: 10.1093/bioinformatics/18.3.452. [DOI] [PubMed] [Google Scholar]
- 103.Thompson JD, Plewniak F, Poch O. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Research. 1999;27:2682–2690. doi: 10.1093/nar/27.13.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Teunissen S, Kruithof M, Farris A, Harley J, Venrooij W, et al. Conserved features of Y RNAs: a comparison of experimentally derived secondary structures. Nucleic Acids Research. 2000;28:610–619. doi: 10.1093/nar/28.2.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Formal description of transducer composition algorithm yielding phylogenetic grammar rules
(0.15 MB PDF)
Algorithm for elimination of null cycles and empty bifurcations from general SCFGs
(0.13 MB PDF)
Description of software tools
(0.09 MB PDF)
Example presentation of TKFST inference grammars in Indiegram format
(0.14 MB PDF)
Grammar for three-taxon TKFST model returned by transducer composition algorithm (graphviz dot format)
(0.03 MB TXT)
Grammar for three-taxon TKFST model returned by transducer composition algorithm, after eliminating windback states (graphviz dot format)
(0.06 MB TXT)