

Abstract
We have previously shown that p107, a member of the retinoblastoma (Rb) cell cycle regulatory family, has a unique function in regulating the pool of neural precursor cells. As the pool of progenitors is regulated by a limiting supply of trophic factors, we asked if the Rb/E2F pathway may control the size of the progenitor population by regulating the levels of growth factors or their receptors. Here, we demonstrate that fibroblast growth factor 2 (FGF2) is aberrantly upregulated in the brains of animals lacking Rb family proteins and that the gene encoding the FGF2 ligand is directly regulated by p107 and E2F3. Chromatin immunoprecipitation assays demonstrated that E2F3 and p107 occupy E2F consensus sites on the FGF2 promoter in the context of native chromatin. To evaluate the physiological consequence of FGF2 deregulation in both p107 and E2F3 mutants, we measured neural progenitor responsiveness to growth factors. Our results demonstrate that E2F3 and p107 are each mediators of FGF2 growth factor responsiveness in neural progenitor cells. These results support a model whereby p107 regulates the pool of FGF-responsive progenitors by directly regulating FGF2 gene expression in vivo. By identifying novel roles for p107/E2F in regulating genes outside of the classical cell cycle machinery targets, we uncover a new mechanism whereby Rb/E2F mediates proliferation through regulating growth factor responsiveness.
Cell cycle genes have been found to play an important role in brain development, with numerous molecules regulating the G1/S transition having been shown to regulate neural precursor proliferation (reviewed in reference 38). Perhaps the most important regulators of the G1/S transition are the retinoblastoma protein (Rb) and its closely related family member p107. Rb is a pivotal regulator of neural precursor proliferation and the timing of cell cycle withdrawal. For example, Rb has been shown to regulate terminal mitosis of neuroblasts in the central and peripheral nervous systems and retina (7, 18, 34, 35). Furthermore, recent evidence has emerged indicating that Rb itself is capable of regulating diverse cellular processes in the nervous system beyond proliferation. Roles for Rb have been indicated in laminar patterning of the cortex and neuronal migration (17; reviewed in reference 38). These studies highlight the importance of Rb in regulating neural cell populations. In contrast to Rb, little is known about the role of p107. While its role was originally thought to overlap with and compensate for that of Rb (29), distinct functional differences in tissues such as muscle, chondrocytes, and adipocytes, have emerged, suggesting otherwise (10, 28, 51). We have recently shown that p107 plays a unique role, one distinct from Rb, in regulating neural precursor cell numbers in the developing and adult brain (60). p107 null neural precursor cells have an enhanced capacity for self-renewal and, consistent with this, exhibit expanded populations of both precursors and progenitors. While we have previously demonstrated that the increased self-renewal capacity and neural precursor numbers are due, in part, to an upregulation of the Notch-Hes signaling pathway (61), the mechanisms that sustain the increased population are still unknown.
The E2F family of transcription factors, comprised of E2F1 to E2F8, are key Rb/p107-interacting targets best known for their role in promoting cell cycle progression (reviewed in reference 59). Accumulating in vitro and in vivo evidence, however, suggests that E2Fs are capable of regulating expression of a broad spectrum of genes and diverse physiological processes (reviewed in reference 39). In vitro, microarray studies examining changes in gene expression in response to various models of deregulated E2F expression have each identified groups of overlapping novel target genes with well-characterized roles in differentiation, development, and migration (3, 12, 25, 33, 41, 43, 68). Chromatin immunoprecipitation (ChIP)-on-chip studies have localized E2Fs to a number of gene promoters unrelated to cell cycle (1, 2, 6, 26, 47, 64, 65). In vivo, E2Fs have been implicated in a number of distinct aspects of nervous system development. E2F4 has been shown to regulate development of the ventral telencephalon through a genetic interaction with the Sonic hedgehog pathway (50), while E2F1 and E2F3 have been implicated in mediating neural precursor proliferation (11, 37). Intriguingly, in vivo models are emerging to suggest that Rb family members interact with E2Fs to mediate novel functions in nervous system development. For example, Rb has been shown to interact with both E2F3 and E2F1 to mediate neural precursor proliferation and cell cycle exit (8, 37). Additionally, Rb has been shown to mediate neural migration and differentiation, in a manner beyond cell cycle regulation, uniquely through E2F3 (8, 37). Given the emerging importance of the role of the Rb/E2F pathway in nervous system development, it is likely that the key interacting factors for p107-mediated neurogenesis are E2F family proteins.
A key mechanism regulating the pool of neural precursor and progenitor cells is a limiting supply of trophic factors. While the fibroblast growth factor (FGF) superfamily of ligands and receptors are known for having multiple roles in the nervous system (reviewed in references 13 and 48), the predominant factor regulating neural precursor proliferation and survival is thought to be FGF2 (21, 27, 58). FGF2 expression is highest in the ventricular zone (VZ) and subventricular zone, and germ line deficiency of FGF2 results in a significant reduction in the number of proliferating progenitor cells during development, which in turn leads to a decrease in the number of cortical neurons (14, 45, 58). Furthermore, ventricular microinjection of FGF2 during neurogenesis resulted in an expansion of the progenitor population and a corresponding increase in the number of cortical neurons (58). Finally, FGF2 is also known to be required for the proliferation and survival of neural stem cells and is expressed at all stages of development. Little, however, is known regarding the factors that regulate FGF2 expression within this population throughout development.
Given our previous results that have shown that p107 is an important regulator of the neural precursor population (60, 61), we asked if Rb/E2F pathway components play a role in regulating the pool of FGF2-responsive neural progenitors. Here, we demonstrate that (i) both E2F3 and p107 are mediators of FGF2 growth factor responsiveness in neural precursors, and (ii) p107 regulates FGF2 gene transcription through repression of E2F3-mediated activation. These results demonstrate that p107 regulates the pool of FGF2-responsive neural progenitors by directly regulating FGF2 gene transcription in vivo. Furthermore, by demonstrating novel roles for p107/E2F3 in regulating transcription of genes outside of the classical cell cycle machinery targets, these results broaden the scope of Rb/E2F pathway function to include regulation of growth factor responsiveness.
MATERIALS AND METHODS
Mice.
Both germ line p107-deficient mice (28) and germ line E2F3-deficient mice (31) were maintained on a mixed SV-129 and C57BL/6 background. Telencephalon-specific Rb-deficient mice were generated by crossing floxed Rb-F19 (36, 62) and Foxg1-cre (23) mice. Animals were genotyped according to standard protocols with previously published primers for p107 (28), E2F3 (31), and Rb and Cre (17, 18). For embryonic time points, the time of plug identification was counted as embryonic day 0.5 (E0.5). All experiments were approved by the University of Ottawa's Animal Care ethics committee and adhere to the Guidelines of the Canadian Council on Animal Care.
Tissue fixation and cryoprotection.
Pregnant female and adult male mice were euthanized with a lethal injection of sodium pentobarbitol. Embryos were dissected and fixed overnight in 4% paraformaldehyde (PFA) in 1× phosphate-buffered saline (PBS), pH 7.4, cryoprotected in sequential solutions of 12,16 and 22% sucrose in 1× PBS followed by embedding in OCT medium (TissueTek 4583), and frozen on liquid N2. Adult male mice were perfused with 1× PBS followed by cold 4% PFA, and brains were removed. The brains were postfixed overnight in 4% PFA, cryoprotected in 22% sucrose in 1× PBS, and frozen. Sections were collected as 14-μm coronal cryosections on Superfrost Plus slides (12-550-15; Fisher Scientific).
In situ hybridization and immunohistochemistry.
Nonradioactive in situ hybridization and digoxigenin probe labeling was performed according to previously described protocols (63). Riboprobes for FGF1, FGF2, FGF receptor 1 (FGFR-1), and FGFR-2 were provided by Flora Vaccarino (42, 58). To assess cells in mitotic M phase, phosphohistone H3 labeling was performed with rabbit polyclonal anti-phosphohistone H3 (anti-PH3) (1:100, 06-570; Upstate Biotechnology) as previously described (18). To assess cell death, both terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) (in situ end labeling kit, 03 333 566 001; Roche Applied Science) and active caspase-3 (AC-3) (1:500, 559565; BD Pharmingen) immunohistochemistry combined with Hoechst nuclear staining was performed according to standard protocols (18). Labeled cells were quantified throughout the telencephalic hemisphere from eight matched sections per embryo.
Neural progenitor cultures.
Pregnant mice were euthanized at gestation day 14.5 and 16.5. Embryos were removed from their embryonic sacs, and cortices were dissected. Individual cortices were mechanically dissociated, and cortical explants were plated in defined media containing Dulbecco's modified Eagle's medium (DMEM)-F12 medium and N2 medium in polyornithine-coated four-well dishes. Explants were cultured for 22 h in the presence of 10 ng/ml FGF2 or 10 ng/ml epidermal growth factor (EGF) or were left untreated. BrdU was added to each well 6 h prior to fixation. Cultures were fixed for 15 min in 4% PFA and then treated sequentially for 10 min each in 2 N HCl and 0.01 M NaB4O7 followed by BrdU immunohistochemistry (anti-mouse BrdU, 1:100, BD Biosciences, San Jose, CA) and Hoechst nuclear staining. Total cells and BrdU-positive cells were counted in 8 to 10 microscope fields per well. Rates of proliferation were obtained by calculating the proportion of BrdU-positive cells relative to the total cell number. Increase in proliferation in response to FGF2 addition was calculated for each genotype by dividing the difference in proliferation rates between the control and the sample with FGF2 added over the initial proliferation rate without FGF2. Reproducibility of quantification was verified by a second investigator in a blinded manner. Cell death was measured by Hoechst labeling as previously described (60). For Hoechst labeling, dead cells were quantified within proliferating clumps and identified by the characteristic nuclear morphology of chromatin condensation.
FGF2 dose response neural progenitor culture.
For the dose response curve, neuroepithelia from E14.5 embryos were isolated and cultured as neurospheres as previously described (49, 56, 60). Cells were plated at 10,000 cells/ml at the following concentrations of FGF-2: 0.05 ng/ml, 0.2 ng/ml, 0.6 ng/ml, 2 ng/ml, 10 ng/ml, and 20 ng/ml. For each embryo at each concentration, six replicate wells were counted.
qRT-PCR.
Quantitative real-time PCR (qRT-PCR) assays were performed with embryonic neural progenitors cultured as neurospheres derived from E14.5 wild-type and p107 null embryos. Total RNA was isolated using the Trizol method (Invitrogen, Carlsbad, CA) from neurospheres after 6 days in vitro. A SuperScript III Platinum SYBR green One-Step qRT-PCR kit (Invitrogen, Carlsbad, CA) was used to amplify FGF2 and S12 mRNAs from 50 ng of isolated total RNA. The primers designed spanned intronic regions and therefore discriminated between genomic and cDNA. The primer sequences utilized were FGF2 forward (5′-GGCTGCTGGCTTCTAAGTGT-3′) and reverse (5′-CCGTTTTGGATCCGAGTTTA-3′) and S12 forward (5′-GGAAGGCATAGCTGCTGG-3′) and reverse (5′-CCTCGATGACATCCTTGG-3′). RT-PCR was done using a Rotor-Gene RG-3000 (Corbett Research, Sydney, Australia). Values obtained for FGF2 expression levels were normalized to the S12 level as an internal control. At least four embryos from two litters of each genotype were analyzed by two-tailed Student's t test, with a P value of <0.05.
Luciferase reporter assay.
Putative E2F consensus sites were identified by Mulan software analysis. The FGF2 promoter (1,200-bp) sequence was amplified from mouse genomic DNA by PCR and inserted into a pGL3-basic luciferase reporter construct (Promega). The forward primer was 5′-TCCTGGCCTTAACCCTTTCTGTCG-3′, and the reverse primer was 5′-TAGGGGCCGCGTCTTAGTG-3′. The FGF2 luciferase vector was transfected with E2F3, E2F4, and/or p107 or pRb expression vectors into HEK-293A cells by standard calcium phosphate precipitation (53) or Lipofectamine 2000, according to the manufacturer's instructions (Invitrogen). Two micrograms of pMLV-LacZ or 250 ng of pCMV-Renilla was cotransfected with each sample to control for transfection efficiency, in calcium phosphate and Lipofectamine reactions, respectively. For calcium phosphate transfections, a 4-methylumbelliferyl-d-galactoside (MUGS) assay was performed to standardize the transfection reaction, and luciferase activity was assessed according to standard procedures (20). For Lipofectamine transfections, a Dual-Luciferase reporter assay system (Promega) was used to standardize the transfection reaction and assess the luciferase activity. One-way analysis of variance was performed on the means of results from three different experiments followed by a Tukey's post hoc test to detect differences in the means at a P value of <0.05.
ChIP protocol.
ChIP assays were performed as previously described (43). Briefly, neurosphere cultures were prepared from neuroepithelia from E14.5 embryos as previously described (46, 53, 56). Proliferating neurospheres were triturated, cross-linked with formaldehyde, lysed, sonicated, and centrifuged at 14,000 × g to remove cellular debris. Each immunoprecipitation was performed using 2 μg of antibody. Antibodies against E2F3 (sc-878) and p107 (sc-317x), as well as normal rabbit immunoglobulin G (sc-2027), were obtained from Santa Cruz Biotechnology. Immunocomplexes were captured using protein A/G9 Sepharose beads and washed extensively, and cross-links were reversed overnight, followed by treatment with RNase A at 37°C for 1 h and proteinase K at 65°C for 30 min. The purified DNA was examined by PCR using primers designed around the E2F consensus sites at bp −110 and the region encompassing the +17 and +148 E2F sites in exon 1. The PCR primer sequences used are as follows: for the bp −110 site, forward primer 5′-CCCGGGCCGTTGTACACT-3′ and reverse primer 5′-GCTGCCAGCCTCCCAGTC-3′; for the bp +17 and +148 sites in exon 1, forward primer 5′-CAGCGGCATCACCTCGCTTCC-3′ and reverse primer 5′ GGCGAGGGAGCAGTGTGGTC-3′; and for the negative site at bp −3,611, forward primer 5′-ACAAAACAGGCTGGGACACT-3′ and reverse primer 5′-ACTCGGAAACAGGACACCAT-3′. Shown in the figures are representative results of at least three independent experiments.
Sequence alignment.
The 5′ region of the mouse FGF2 locus containing the intergenic region, the untranslated region, and exon 1 was aligned to the human orthologue using ECR Browser (http://ecrbrowser.dcode.org). Minimal sequence similarity was set to 70% in order to identify evolutionarily conserved regions. The conserved region encompassing exon1 was imported into Mulan (http://mulan.dcode.org) to locate potential conserved E2F binding sites. All sites identified in Mulan were manually examined for both human and mouse for their similarity to the consensus (TTTSSCGC) and nonconsensus (BKTSSCGS) motifs (46, 66).
EMSA.
Electrophoretic mobility shift assays (EMSA) were performed on total protein extracts from primary neural precursor cells as described previously (5), with the following modifications. Total cell protein was extracted in lysis buffer (50 mM HEPES, 250 mM KCl, 0.1% NP-40, 0.4 mM sodium orthovanadate, 0.1 mM EGTA, 0.1 mM EDTA, 10% glycerol, 0.2 mg/ml phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, and protease inhibitors [Roche]) and assayed by using the Bradford method (Bio-Rad protein assay reagent, 500-0006). A 15-μg aliquot of lysate was incubated with an excess of 32P-labeled double-stranded DNA probe (70,000 cpm/0.2 ng of DNA) containing a single E2F binding site (5′-GGATTTAAGTTTCGCGCCCTTTCTCAA-3′). The binding reaction (25 μl) was carried out at room temperature for 20 min in binding buffer (20 mM HEPES [pH 7.6], 4% Ficoll, 2.5% MgCl2, 40 mM KCl, 0.1 mM EGTA, 0.5 mg/ml acetylated bovine serum albumin, and 0.5 mM dithiothreitol). To control for binding specificity, a 10-fold excess of unlabeled wild-type oligonucleotide was added to the binding reaction and incubated for 20 min before the addition of labeled probe. To identify the composition of the complexes, tissue culture supernatant or purified antibody was added to the reaction mixture. Complexes were resolved on a 5.0% gel run for 4 h, dried, and visualized by autoradiography. The tissue culture supernatant containing the monoclonal pRb antibody 21C9 was a gift from David Cobrinik (54). All other antibodies (E2F3 [sc878 and sc878x], E2F4 [sc 6851], and p107 [sc318]) were purchased from Santa Cruz Biotechnology, Inc.
RESULTS
Rb family mediates FGF2 expression in vivo.
In our previous studies, we have demonstrated distinct roles for Rb family members in regulating the pool of neural precursor cells. Specifically, we have shown that p107 regulates the pool of neural precursors and progenitors by regulating self-renewal (60). While the increased population is, in part, a consequence of p107-mediated regulation of Hes-1 gene expression, what sustains the increased population of progenitors remains a question (61). Given the importance of FGF2 in promoting the survival and proliferation of the progenitor population, we asked whether p107 may be regulating components of the FGF signaling pathway. To address this question, we examined the expression of FGF ligands and receptors in wild-type and p107-deficient brains at E14.5 and E18.5. No differences were observed in the expression levels of the receptors FGFR1 and FGFR2, or the FGF1 ligand, during the course of development (results not shown). In contrast, higher levels of FGF2 were observed for p107−/− versus wild-type brains at E14.5 and E18.5 (Fig. 1). While FGF2 is expressed in a gradient manner throughout the cortex, this increase was observed specifically in the VZ, where neural progenitors reside.
FIG. 1.

p107 deficiency results in increased FGF2 expression. (A) In situ hybridization was performed on E14.5 and E18.5 coronal brain sections with riboprobes for FGF2. At both E14.5 and E18.5, higher levels of FGF2 mRNA were observed in p107−/− brains specifically within the VZ. At E14.5, higher levels of FGF2 mRNA were observed for conditional (Cond.) Rb mutants; however, in contrast to the case for p107 deficiency, this increase was observed not only within the VZ but also across the developing cortex. Shown here are representative images from experiments performed in triplicate. Scale bar = 250 μm. (B) qRT-PCR displaying mRNA expression in neurospheres cultured for 6 days in vitro from E14.5 wild-type and p107 null embryos. FGF2 expression was normalized to an internal S12 control performed in the same run. Data were analyzed by two-tailed, paired Student's t test (four p107+/+ and four p107−/− embryos from a minimum of two litters). *, P < 0.05. Bars represent standard errors.
Since we have previously reported that p107 deficiency results in an increase in the neural progenitor population (60, 61), we verified that the increase in FGF2 expression is not due simply to a reflection of this increased progenitor population. To address this issue, we cultured neural progenitor cells from control and p107-deficient embryos in the same concentrations of FGF2 and performed qRT-PCR. While neural precursors are grown in an excess of FGF in vitro (25 ng/ml), this approach provides the opportunity to precisely quantify cell numbers. A highly reproducible and significant increase in the levels of FGF2 transcript was observed with p107 deficiency compared to control cultures with the same number of progenitor cells (Fig. 1B). Thus, the increase in FGF2 that we observe with p107 deficiency is not a consequence of the increased cell population but, rather, is intrinsic to the progenitors themselves.
We have demonstrated that Rb, in addition to p107, is involved in regulating the pool of neural progenitors. In contrast to p107, however, Rb is required for terminal mitosis, as Rb deficiency resulted in ectopic proliferation of neural progenitors (18). Thus, having observed an increase in FGF2 expression in the absence of p107, we next asked whether this increase is unique to p107 or if it is shared among Rb family members. We examined the levels of FGF2 transcript at E14.5 in conditional telencephalon-specific Rb mutants and, similar to results with p107 deficiency, we observed an overall increase in the levels of FGF2 in the absence of Rb. In contrast to p107, however, this increase is not confined to the VZ, but rather is observed throughout the VZ and ectopically within the cortical plate (Fig. 1). These results demonstrate that both Rb family members are required for the correct regulation of FGF2 in the developing brain but that p107 is uniquely required only at the VZ.
p107 regulates FGF2 promoter activity.
The elevated levels of FGF2 transcript we observed for both p107- and Rb-deficient progenitors led us to question whether Rb family members regulate FGF2 gene expression through a direct or an indirect transcriptional mechanism. Both p107 and Rb interact primarily with E2F transcription factors to regulate transcription at the G1/S cell cycle checkpoint by two distinct mechanisms, first, by repressing transcription through interaction with repressive E2Fs, such as E2F4, and second, by inhibiting transcriptional activation through inhibition of the activating E2F1, E2F2, and E2F3 (15, 24, 55). As emerging data have suggested that Rb family members are capable of regulating transcription of genes with roles beyond the G1/S transition (17, 37; reviewed in reference 38), we examined the FGF2 promoter for potential E2F binding sites. The FGF2 promoter region has been previously characterized (19, 40, 44); however, E2F consensus sites have not yet been identified. Using Mulan/rVista analysis, we identified three putative E2F binding sites at bp −110, +17, and +148, which conform to the classical E2F consensus sequence of TTTSSCGC within the FGF2 promoter region and exon 1. To ask whether these E2F consensus sites were functional, a 1.2-kb region from the FGF2 promoter containing these sites was ligated into a pGL3B luciferase reporter construct (Fig. 2A). To first determine if the promoter is E2F responsive, the FGF2 reporter construct was cotransfected with expression vectors for E2F3, an activating E2F, or E2F4, which is believed to function as a repressor. While E2F4 had no observable effect on FGF2 promoter activity, E2F3 induced a ninefold activation, suggesting that E2F3 may be an activator of FGF2 transcription (Fig. 2B). Next, to determine whether Rb and p107 are each capable of repressing E2F3-mediated FGF2 promoter activation, Rb or p107 expression vectors were cotransfected in this reporter assay. In these experiments, p107 was able to repress the E2F3-mediated FGF2 promoter activation in a dose-dependent manner (Fig. 2C). This response appears to be unique to p107, as cotransfection of Rb did not result in a similar repression (not shown). These results demonstrate that p107 can regulate FGF2 promoter activity by repressing E2F-mediated transcriptional activation. Furthermore, these results not only identify a mechanism by which FGF2 levels become deregulated in vivo but also point toward a novel means by which p107 plays a role in regulating the progenitor population, independent of its direct role in regulating the cell cycle machinery.
FIG. 2.

p107 and E2F3 regulate FGF2 promoter activity. (A) Schematic of the FGF2 promoter and locations of E2F binding sites relative to the translation start site. A 1.2-kb region of the FGF2 promoter was inserted into the pGL3B vector. (B) Luciferase promoter assays in HEK-293A cells of the FGF2 promoter reveal that the addition of an activating E2F, E2F3, induces a ninefold induction of FGF2 promoter activity. In contrast, the coexpression of a repressive E2F, E2F4, did not affect FGF2 promoter activity. (C) E2F3 activation of the FGF2 promoter is repressed by coexpression of p107 in a concentration-dependent manner. *, P < 0.001.
p107 regulates E2F3-mediated activation of FGF2 transcription in a physiologically relevant context.
Having demonstrated that E2F3 and p107 coordinately regulate the activity of the FGF2 promoter in vitro, we next sought to determine if this functional interaction between p107 and E2F3 is preserved in a physiologically relevant context. First, we examined whether any of the E2F sites we identified within the promoter and first exon of the FGF2 gene are conserved in humans. Sequence conservation was observed between mouse and human genomes within the first exon (Fig. 3A). Next, we performed ChIP assays to determine whether E2F3 and p107 are capable of binding regions encompassing the E2F recognition sites within the FGF2 gene promoter and exon 1 in primary neural progenitor cultures. Chromatin was immunoprecipitated with antibodies to either E2F3 or p107, followed by PCR with primers designed around the bp −110 consensus site and the two sites within exon 1 (Fig. 3A, arrows). ChIP with both primer sets revealed the presence of both E2F3 and p107 in these regions, but no binding for either protein was detected with the negative control at bp −3,611 (Fig. 3B). Importantly, ChIP with E2f3 and p107 antibodies in Ef23−/− and p107−/− cells, respectively, resulted in PCR amplification signals below background levels, highlighting the specificity of the E2f3 and p107 antibodies used in these experiments. These results demonstrate E2F3 and p107 binding in the FGF2 gene 5′ regulatory region in the context of native chromatin. Further, the presence of both E2F3 and p107 within this region suggests that a functional interaction between p107/E2F3 could mediate FGF2 growth factor gene regulation in a physiological context.
FIG. 3.

p107 and E2F3 bind the FGF-2 promoter in a physiologically relevant context. (A) Schematic of E2F sites identified within the murine FGF2 gene regulatory region, and conservation of identified E2F sites with human sequences. Chromosomal positions of the aligned regions of the mouse (mm9) and human (hg18) genomes are indicated numerically, adjacent to the sequence shown. Regions highlighted in gray and black are E2F consensus sites in each genome corresponding to the mm9 bp +17 and +148 sites, respectively (GenBank accession numbers NM_008006, murine Fgf2; NM_002006, human Fgf2). The E2F consensus site identified at +148 bp in mouse chromosome 3 is conserved in sequence and position in human chromosome 4; the +17 mouse consensus site is mirrored by an E2F binding sequence in the human, located just 11 bp downstream of the site in the aligned mouse sequence. The arrows indicate the relative locations of the PCR primers used to examine potential E2F3/p107 binding by ChIP. (B) ChIP was performed to determine whether p107 and E2F3 were capable of binding the E2F recognition sites identified in vivo. ChIP assays were performed with embryonic neurospheres derived from E14.5 wild-type (WT), p107 null, and E2F3 null embryos. Chromatin was immunoprecipitated with antibodies to either E2F3 or p107, followed by PCR for the E2F consensus site and nonspecific region upstream. Immunoprecipitation with antibodies to E2F3 and p107 revealed that both factors are recruited to the region containing the conserved E2F site in the wild type, but not in E2f3 or p107 null cells, while no binding is displayed in a region without any nearby E2F binding sites. (C) For EMSA, total protein was extracted from primary proliferating neural precursors. Total protein extracts were incubated alone or in the presence of E2F or pocket protein antibodies with a double-stranded 32P-labeled E2F consensus probe. Antibodies used for the supershift are indicated above the corresponding lane. For wild-type cells, the presence of Rb/E2F and p107/E2F complexes is noted by arrowheads, which label the loss of DNA binding complexes with the addition of specific antibodies. Supershifting with E2F3 results in a notable decrease in the p107/E2F band (indicated by arrowheads), demonstrating the presence of E2F3 in the p107/E2F complex. cc, cold competition.
Our detection of p107 and E2F3 at the FGF2 promoter and the ability of p107 and E2F3 to regulate FGF2 promoter activity led us to ask if E2F3 and p107 are present within the same protein complex in proliferating neural progenitor cells. EMSA were performed on protein extracts from embryonic neural progenitors, and the composition of the complexes was identified using antibodies specific to Rb and E2F family members. In wild-type tissue, both free E2Fs and Rb family bound E2Fs are observed, indicating that members of this pathway are active in the context of neural progenitor proliferation (Fig. 3C). Indeed, in these cells, both Rb activity and p107 activity are readily identified and are thus present in complexes with E2F (Fig. 3C). The addition of E2F3 antibodies resulted in a supershift of the complex containing free E2F3 as well as a noticeable decrease in p107 binding activity. These results suggest that in this cellular context, E2F3 and p107 may be acting within the same complex. We also note that antibody shift with E2F4 results in a similar decrease in p107 activity, demonstrating the presence of E2F4 in the p107/E2F complex (Fig. 3C). Thus, these results suggest that E2F3 is among the E2Fs found in the p107/E2F complex. Together, these results suggest that a functional relationship exists whereby the cell cycle regulatory p107/E2F pathway is capable of modulating transcription of the FGF2 growth factor in neural progenitor cultures.
E2F3 mediates FGF2 gene expression in vivo.
Having observed a functional interaction between p107 and E2F3 in the context of FGF2 gene regulation in vitro, we next asked if such a relationship between E2F3 and FGF2 is preserved in vivo. To address this question, we asked if FGF2 gene expression is perturbed in embryos lacking E2F3. Since E2F3 is capable of activating FGF2 gene transcription, we asked if the absence of E2F3 results in a reduction of FGF2 expression in vivo. In situ hybridization for FGF2 was performed with control and E2F3−/− embryonic brains at E14.5 (Fig. 4). While control brains exhibited a moderate level of FGF2 transcript in the VZ, in the absence of E2F3, the levels of FGF2 within the VZ were reduced relative to those of the control (Fig. 4). This reduction of FGF2 in E2F3-deficient brains is in agreement with our in vitro results demonstrating that E2F3 activates transcription at the FGF2 promoter in the embryonic brain.
FIG. 4.

E2F3 mediates FGF2 gene expression in vivo. In situ hybridization was performed with E14.5 coronal brain sections by using riboprobes for FGF2. At E14.5, a reduced level of FGF2 expression was observed in the absence of E2F3. This decrease is apparent specifically within the VZ. Shown here are representative images from experiments performed in triplicate. Scale bar = 250 μm.
p107 mediates FGF2 responsiveness in neural progenitors during development.
The above data show that p107 mediates FGF2 expression and is present at the FGF2 promoter. Next, we asked if, in addition to influencing the levels of FGF2, p107 loss might affect the responsiveness of neural precursors to this growth factor. Cortical progenitor explants were cultured in the presence of FGF2 or EGF, a trophic factor known to promote progenitor cell survival and differentiation of a more mature population of progenitors relative to FGF (4, 27, 32). Progenitors were cultured from both E14.5 (at the initiation of EGF responsiveness) and E16.5 (fully established EGF and FGF2 responsiveness), and a single pulse of BrdU was added prior to fixation to label cells in S phase (Fig. 5). In the presence of FGF2, p107-deficient progenitors exhibited a significant increase in the number of BrdU-incorporated cells (Fig. 5). No difference in proliferation levels was observed in the presence of EGF between p107 and the control (data not shown). Similar results were obtained at E16.5, further supporting our findings that p107-deficient cells exhibit increased responsiveness to FGF2 (Fig. 5). These results demonstrate that throughout development, p107 mediates the mitogenic response of neural progenitors to FGF2.
FIG. 5.

p107-deficient progenitors exhibit enhanced responsiveness to FGF2 during development. (A) E14.5 and E16.5 cortical progenitors were cultured in defined media (DMEM-F12 with N2) with trophic factor FGF2, or no trophic factor (Control), for 22 h. BrdU was added to the cultures to label cells in S phase, and cultures were fixed 6 h later. BrdU immunohistochemistry and Hoechst nuclei staining of cortical progenitor cultures from wild-type and p107 null mice. (B) The rate of proliferation was assessed according to the number of BrdU-labeled cells divided by the total number of cells counted (Hoechst-stained nuclei) in E14.5 (five p107+/+ and five p107−/− embryos from a minimum of two litters) and E16.5 cultures (three wild type and three p107−/− embryos). The bars represent the change in the rate of proliferation for cultures without and with growth factor. The p107-deficient progenitors consistently exhibited a greater increase in BrdU-labeled cells relative to controls with FGF2 treatment. Significance was determined using a two-tailed t test. *, P < 0.05. Bar = 500 μm.
Increased responsiveness of p107 progenitors to FGF2 occurs in vivo.
Our data demonstrating that p107-deficient progenitors exhibit an increased proliferative response upon the addition of FGF2 lends itself to two possible interpretations. One explanation is that the in vivo microenvironment where progenitors reside retains the increased FGF2 that is present in p107 deficiency, creating an enhanced mitogenic environment relative to the control. Alternatively, a cell autonomous explanation for this observation is also possible. Neural progenitors are known to proliferate in a dose-responsive manner to increasing concentrations of FGF2, whereby a minimum threshold of FGF2 is required prior to the exponential proliferative response that occurs upon further addition of trophic factor (56). Thus, we hypothesize that p107-deficient neural progenitors, as a result of their elevated levels of endogenous FGF2, could exhibit a lower threshold requirement for exponential proliferation in response to exogenous FGF2. To distinguish between these two possibilities, we cultured progenitor cells derived from neurospheres along an FGF dose response curve. While normally used as an assay to retrospectively identify the number of neural precursor cells, the neurosphere assay relies on the principle that once a precursor divides, it will give rise to progenitors that will continue to proliferate and populate the clonally derived, observable neurosphere. Thus, the formation of neurospheres along an FGF dose response curve would represent a measurement of the progenitor cells' ability to respond to various concentrations of growth factor. Indeed, we show that culturing progenitors along increasing concentrations of growth factor does result in a corresponding increase in the number of neurospheres in both wild-type and p107-deficient cultures (Fig. 6). While the minimum FGF2 concentration at which wild-type progenitor cultures first exhibit neurospheres is between 0.6 ng and 2 ng, in p107 deficiency, neurospheres are first observed at significantly lower concentrations (0.2 ng). Further, the increase in the number of neurospheres observed between FGF2 concentration points is exponential and increased significantly relative to the control at these low to medium concentrations (0.2 ng to 2 ng) (Fig. 6). These results demonstrate that p107 deficiency results in a lower threshold concentration of FGF2 required to induce progenitor proliferation and suggest that p107-mediated neural progenitor responsiveness to FGF-2 occurs as a result of the inherent ability of progenitor cells to respond to low threshold levels of FGF2.
FIG. 6.
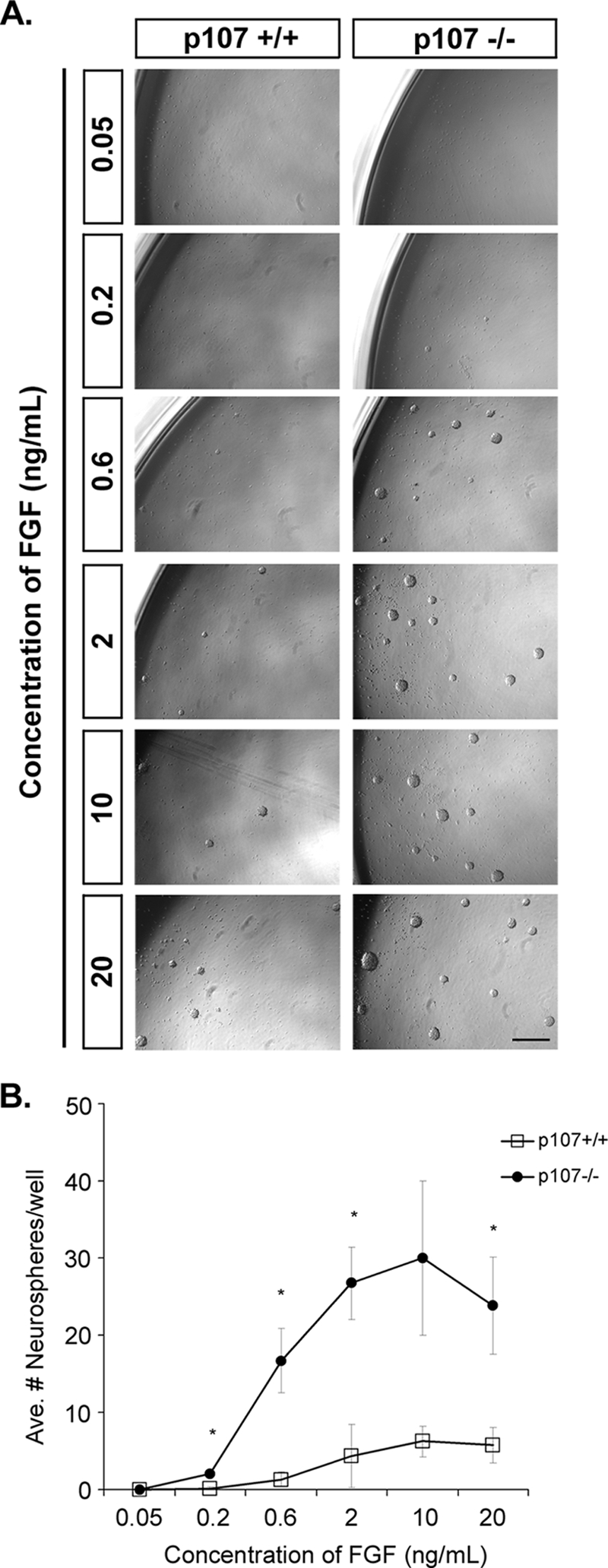
Increased responsiveness of p107-deficient progenitors to FGF2. E14.5 neural progenitors were cultured under neurosphere-promoting conditions with increasing concentrations of FGF2 (A), and the average numbers (Ave. #) of neurospheres from six replicate wells were counted after 7 days (B). p107 deficiency results in an increased number of spheres at each concentration, and the appearance of spheres is observed at lower FGF2 concentrations (0.2 ng/ml) relative to the control (0.6 ng/ml). Bars represent means ± standard errors of the means (SEM) (three control and four p107−/− embryos from a minimum of two litters). Significance was determined using paired two-tailed t tests. *, P < 0.05. Bar = 500 μm.
E2F3 positively regulates progenitor proliferation and FGF2 responsiveness.
Having demonstrated that E2F3 modulates FGF2 expression both in vitro and in vivo within the population of proliferating progenitor cells, we next sought to determine if E2F3 also affects FGF2 responsiveness, complementarily to p107. To address this question, we first examined the role of E2F3 in neural progenitor proliferation. While we have recently reported that E2F3 deficiency does result in decreased progenitor proliferation in the adult (37), no data exists as to the role of E2F3 in mediating the pool of embryonic precursors. Thus, we examined progenitor proliferation in embryos deficient for E2F3 using the M phase marker PH3 to label mitotic cells lining the lateral ventricles (Fig. 7A and B). E2F3-deficient embryos exhibited approximately 25% fewer PH3-labeled cells lining the ventricle than their wild type littermates (Fig. 7A and B). As this decrease in proliferation could be the result of either a genuine decrease in proliferation or an increase in cell death, we examined cell death in the absence of E2F3 during embryonic development. Sections from control and E2F3−/− embryos were subjected to TUNEL and AC-3 staining, and labeled cells in the dorsal telencephalon, including the VZ, were quantified. While a low level of cell death was observed for each of the control and E2F3-deficient embryos, we failed to detect a difference in the numbers of dying cells by the two methods (Fig. 7C). Thus, these data demonstrate that a smaller pool of proliferating neural progenitors is present in the absence of E2F3. Further, the observed reduction in progenitor proliferation supports our model involving a functional interaction between p107 and E2F3 to mediate FGF responsiveness in this cell population in vivo.
FIG. 7.

E2F3 positively regulates progenitor proliferation. Sections from E15.5 E2F3-deficient embryos and their corresponding wild-type littermates were labeled with an antibody to PH3 to label cells in the M phase of the cell cycle (A). PH3-labeled cells lining one lateral ventricle were counted every fifth section between anatomical landmarks, and the total number of PH3-labeled cells counted was expressed as the mean ± SEM. (B). Significantly fewer PH3-labeled cells were observed for E2F3−/− embryos than for wild-type littermates (B) (four embryos per genotype). Cell death was assessed by examining both TUNEL and AC-3 immunohistochemistry combined with Hoechst nuclear staining at E13.5 (C). Labeled cells were quantified throughout the telencephalic hemisphere from eight matched sections per embryo. No difference in the levels of cell death between control and E2F3-deficient embryos was detected (C) (three embryos per genotype). Significance was determined using a two-tailed t test. *, P < 0.05. Bar = 250 μm. ge, ganglionic eminence.
E2F3 mediates neural progenitor responsiveness to FGF2.
While the reduction in FGF2 levels and reduction of proliferating progenitors in the absence of E2F3 suggests that E2F3 functionally interacts with p107 to mediate FGF2 responsiveness, the data do not address the extent to which reduced FGF2 expression may contribute to the reduced proliferation of E2F3-deficient progenitors. To address this question, we performed analogous embryonic progenitor cultures in the presence or absence of FGF2 with E2F3-deficient embryos to examine their proliferative response (Fig. 8). One possibility is that in the absence of E2F3, the addition of exogenous FGF2 should result in reduced responsiveness as a result of the subthreshold endogenous levels of FGF2. Indeed, while the addition of FGF2 to the culture medium resulted in a 60% increase in proliferation among progenitor cells from control embryos, the addition of FGF2 to E2F3−/− progenitor cultures resulted in no appreciable increase in progenitor proliferation (control, 60.6 ± 14.4%; E2F3−/−, 5.0 ± 5.0%) (Fig. 8A and B). This lack of response upon growth factor addition prompted us to examine cell death under both conditions. A modest level of cell death was observed in the absence of FGF2 (Fig. 8C). We observed no difference, however, in the levels of cell death between E2F3−/− and control embryos (E2F3−/−, 3.1 ± 1.1%; control, 2.9 ± 1.8%). In the presence of growth factor, we observed a slight reduction in the levels of cell death for both control and mutant progenitor cultures; however, again, no difference in the levels of cell death was observed between control and E2F3−/− embryos (control, 1.32 ± 0.4%; E2F3−/−, 0.5 ± 0.4%) (Fig. 8C). Taken together, these data demonstrate that p107 and E2F3 functionally interact, in a physiologically relevant context, to mediate neural progenitor proliferation by regulating FGF2 responsiveness. Further, these data establish a unique mechanism preserved in vivo whereby a cell cycle regulatory pathway mediates proliferation by regulating growth factor responsiveness in vivo.
FIG. 8.

E2F3 mediates neural progenitor responsiveness to FGF2. E14.5 cortical progenitors were cultured in the absence or presence of FGF2 for 22 h. BrdU was added to the cultures to label cells in S phase, and cultures were fixed 6 h later. (A) BrdU immunohistochemistry (red) and Hoechst nuclei staining (blue) of cortical progenitor cultures from wild-type and E2F3-deficient mice in the absence or presence of FGF-2. The rate of proliferation was assessed according to the number of BrdU-labeled cells over total number of cells (Hoechst stained nuclei). (B) Bars represent the change in rate of proliferation for cultures without and with growth factor. While wild-type cultures exhibit a 60% increase in proliferation in response to FGF2 addition, E2F3-deficient progenitors exhibit no appreciable increase (three wild-type and four E2F3−/− embryos from a minimum of two litters). (C) Cell death in proliferating clumps in culture was examined by using Hoechst morphology to identify dying cells. In both the absence and presence of added FGF-2, no difference in cell death was observed between control and E2F3−/− embryos (three wild-type and four E2F3−/− embryos from a minimum of two litters). Bars represent means ± SEM. Significance was determined using a two-tailed t test. *, P < 0.05. Bar = 500 μm.
DISCUSSION
In this study, we asked whether members of the Rb/E2F pathway play a role in the regulation of growth factor responsiveness in the developing brain. The results of our studies support a number of conclusions. First, we show that Rb family proteins and their E2F transcription factor targets are required for the correct regulation of FGF2 expression in the developing brain. Second, we show that E2F3 and p107 are each mediators of FGF responsiveness in neural progenitor cells. Finally, we demonstrate that the FGF2 promoter contains functional E2F consensus sites occupied by E2F3 and p107 in the context of native chromatin. FGF2 gene transcription is controlled by p107 through direct repression of E2F3-mediated transcriptional activation. These results support a model whereby p107 functionally interacts with E2F3 to regulate the pool of FGF-responsive neural progenitors by directly regulating FGF2 gene expression in the developing brain. Furthermore, by demonstrating a novel role for p107/E2F in regulating the transcription of a growth factor that plays a key role in regulating the neural precursor population, we broaden the scope of cell cycle gene function beyond the regulation of the cell cycle machinery.
p107 regulates FGF2-mediated proliferation of neural progenitors.
We have previously demonstrated that p107 and E2F3 are each regulators of the pool of neural progenitors. First, we have shown that absence of E2F3 results in a smaller pool of neural progenitors in the adult (37), while absence of p107 leads to an enhanced pool of neural precursor cells in the embryo and adult, with a greater capacity for self-renewal (60). While the increased pool of precursors in p107 deficiency is, in part, the result of increased signaling by the Notch-Hes pathway (60, 61), what sustains this increased population through adulthood remains unknown. The trophic factors FGF2 and EGF have been previously shown to play a crucial role in regulating the neural precursor population. FGF2 is essential for maintaining the population of early neural precursor cells and for promoting progenitor cell proliferation and survival (21, 27). At early developmental stages (E8.5) neural precursor cells are FGF2 responsive; however, as development proceeds, EGF-responsive precursors develop such that by E14.5, both FGF2- and EGF-responsive cells are present (56). Due to the importance of these tropic factors in regulating the neural precursor population, we asked whether the expression of these growth factors or their receptors may be perturbed when Rb family proteins are absent. While no differences could be detected in the regulation of growth factor receptors (FGFR1, FGFR2), an aberrant increase in FGF2 transcript levels was seen in brains lacking p107 and pRb. We then questioned how such an increase in FGF2 ligand may impact the neural precursor population in the embryonic brain. To understand what sustains the expanded neural progenitor population found in animals lacking p107, we asked whether deregulation of FGF2 gene expression could modify progenitor responsiveness to FGF2 (a key trophic factor required for neural precursor regulation) and, if so, whether this may underlie the expanded neural progenitor populations found in animals lacking p107. Here, we demonstrate that p107-deficient progenitors exhibit increased proliferation specifically in response to FGF2 both in vitro and in vivo. No differences were found in responses to EGF alone between p107-deficient progenitors and wild-type controls throughout the course of development. Rather, our data demonstrating that p107-deficient progenitors exhibit increased proliferation in the presence of FGF2 alone support a model whereby p107 regulates a pool of progenitors that respond particularly to FGF2.
The question as to how an increase in FGF2 ligand as seen in p107 deficiency may affect progenitor responsiveness in vitro and in vivo was addressed in our studies. Previously, it has been shown that a minimal threshold of ligand is required to generate a proliferative response in progenitor cells (56). It has been previously shown that cell density has a major impact on progenitor expansion due to the release of endogenous growth factors into the cellular environment. To address this possibility in p107-deficient animals, we questioned whether the increased levels of endogenous FGF2 may result in increased progenitor responsiveness at lower concentrations of growth factor. Indeed our experiments presented here reveal that p107-deficient progenitor cells express increased levels of FGF2 and thus require a lower threshold of FGF ligand to induce neurosphere expansion mediated through progenitor proliferation. Given that the numbers of neural precursor cells are controlled by limiting amounts of growth factor, our data suggest that increased autocrine production of FGF2 in the p107 mutant may therefore explain the mechanism by which these progenitors could be maintained. Our results demonstrating that p107-deficient cells require a significantly lower threshold of FGF2 ligand to sustain an expanded precursor population are consistent with this interpretation.
Our studies demonstrating that E2F3 regulates the neural precursor pool during development establish the importance of E2F3 as a key regulator of the progenitor population from development through to adulthood. We show that this regulation appears as a result of changes in the proliferative capacity of progenitor cells themselves, as cell death among embryonic progenitors lacking E2F3 is unaffected. Consistent with the idea that E2F3 is a positive regulator of progenitor proliferation, we observe that E2F3 is also involved in mediating FGF2 responsiveness, as E2F3-deficient progenitors exhibit decreased proliferation in response to exogenous FGF2. We hypothesize that this is the result of the reduced levels of FGF2 observed in vivo, which could be the result of an increased threshold for progenitor proliferation in response to exogenous FGF2. Taken together, these studies establish the unique rolls played by multiple components of the Rb/E2F cell cycle regulatory pathway in mediating the pool of FGF-responsive neural progenitors, and they demonstrate novel context-dependent functions for these genes.
p107 regulates FGF2 promoter activity through an E2F-mediated mechanism in vivo.
We have shown that p107 and E2F3 are each capable of regulating the pool of FGF2-responsive neural progenitors, supporting a reciprocal interaction between these proteins in vivo. While E2F3 is traditionally a target for regulation by pRb, several lines of evidence support a functional interaction between E2F3 and p107. First, our previous studies have revealed that any Rb protein expressed in neural precursor cells is in a hyperphosphorylated state and is therefore inactive (5, 16, 52). Furthermore, Rb deficiency affects neither the population size of neural precursor cells nor their self-renewal capacity. Instead, Rb deficiency results in a defect in terminal mitosis following commitment to a neuronal fate. Second, p107 that is expressed only in the VZ plays an essential role in regulating the size of the neural precursor population and its self-renewal capacity (60, 61), implicating p107 and not Rb as the key regulator of E2F activity in this cell population. E2F3 is highly expressed in neural precursor cells, where, as we show, it activates FGF2 transcription. Our promoter analyses reveal that E2F3-mediated activation is repressed by p107, while Rb was ineffective. This could occur by an indirect interaction in which E2F3 activates transcription and p107 becomes recruited to the site through interactions with its traditional target E2F4 or, alternatively, p107 may directly bind to E2F3 to repress its activity. Since we have not been able to detect a direct interaction between p107 and E2F3 in neural progenitor cells, it is most likely that p107 is recruited to the 5′ regulatory region through another E2F.
Other E2Fs are likely found within the p107/E2F complex and could be contributing to the regulation of FGF2. Indeed, several sources of in vitro data from four different groups have shown E2F-mediated regulation of FGF ligands and their receptors in either microarray or ChIP-on-chip studies (33, 41, 67, 68). Further, deregulated expression of FGF2 itself was observed by a microarray analysis of cell lines with induced E2F1 expression (68). Finally, E2F1 was observed to be enriched at the promoters of a number of members of the FGF receptor and ligand superfamily, including FGF2 (67). Collectively, these data highlight a role for E2F-mediated regulation of members of the FGF gene family and support the hypothesis that multiple members of the E2F family are involved. Nevertheless, our biological observations regarding the specific role of E2F3 in mediating neural precursor proliferation and FGF2 responsiveness support our interpretation that E2F3 plays a significant role, acting in concert with p107 to mediate expression of FGF2.
The deregulated FGF2 expression we observe with Rb deficiency in fact further strengthens our interpretation of the importance of E2F3 in mediating FGF2 expression in vivo. Our previous studies have shown that significant deregulation of E2F3 occurs in the absence of Rb (5, 37). Further, we and others have shown that many of the neurodevelopmental defects, including both cell cycle and independent functions, observed with Rb deficiency occur as the result of deregulated E2F3 expression (8, 37). Thus, here we interpret the deregulated FGF2 expression that occurs in Rb deficiency as a consequence of the deregulated E2F3 activity in these cells. Together, these data suggest that p107 and E2F3 act coordinately to regulate the population of FGF2-responsive neural progenitors by controlling the transcription of the growth factor FGF2.
The E2F3 locus is particular in that two distinct transcripts are expressed, full-length E2F3a and N-terminal-truncated E2F3b transcribed from an intronic promoter within the E2F3 locus (22, 30). Biochemical studies have shown that in fibroblasts, E2F3a expression is cell cycle regulated and is similar to that of E2F1 (22, 30). E2F3b, however, is expressed equivalently in quiescent and proliferating cells and is a specific partner for Rb in quiescent cells and, thus, may have an role opposing that of E2F3a (22, 30). While these studies may suggest that E2F3a regulates genes related to cell division and E2F3b regulates gene expression outside of cell division, in vivo studies have shown that the roles of E2F3a and E2F3b are likely to be more complex in their regulation of pocket protein activity in different physiological contexts (9, 57). For example, mice deficient for individual species of E2F3 have been shown to have both unique and overlapping roles in mediating Rb-dependent activities. E2F3a was shown to mediate Rb activity in the placenta and nervous system, while both E2F3a and E2F3b are each capable of mediating Rb activity in lens fiber cells (9). Additionally, E2F3a was uniquely shown to mediate the differentiation of starburst amacrine cells in the retina (7), thus demonstrating that the activator E2F3a is capable of performing classical E2F repressor functions—in this case, mediating cellular differentiation. Thus, given the context-dependent nature of E2F activity, determining which E2F3 isoform is involved in mediating FGF2 expression remains a relevant goal.
Due to their action as tumor suppressors and their critical role in the regulation of cell division, the mechanisms by which Rb family proteins function is of great interest not only in the field of development but also in the study of carcinogenesis. Traditionally, the Rb family proteins are known to control cell growth by their well-established role in regulating the cell cycle machinery and G1/S phase transition. More recent studies using microarray analyses (3, 12, 25, 33, 41, 43, 68), however, have suggested new targets that extend beyond the regulation of the cell cycle machinery, implicating novel functions for the Rb/E2F pathway in the control of differentiation. The physiological context in which Rb family proteins may regulate these targets, however, remains unknown.
In the present study, we identify a new mechanism by which the Rb/E2F family controls cell division, which extends beyond the regulation of the cell cycle machinery. Further, these data run contrary to the traditional dogma that the relationship between growth factors and the cell cycle machinery is unidirectional and point to the existence of the inverse relationship. Here, we show that Rb and E2F also regulate cell division by controlling the autocrine production of the growth factor FGF2. FGF2, expressed in neural precursor cells, is a potent ligand for the family of FGF receptors that controls the size of the neural progenitor population in the developing brain (45, 58). Our results reveal that p107 and E2F3 coordinately regulate FGF2 growth factor responsiveness in neural precursor cells. By increasing the cellular production of FGF2, we show that neural progenitor cells respond to significantly lower threshold levels of growth factor. Our studies therefore demonstrate the importance of the p107/E2F3 pathway in controlling the levels of the growth factor FGF2 in the developing brain. These findings have major implications for the mechanisms by which the Rb family proteins function in regulating the neuronal precursor population and may also provide important insights regarding their role in the context of tumor development.
In conclusion, we identify a distinct mechanism by which p107 regulates the proliferative response in neural progenitor cells to FGF2 through repression of E2F3-mediated transcriptional activation of the growth factor FGF2. This represents a new mechanism of action, whereby the Rb/E2F cell cycle regulatory pathway mediates proliferation in a manner distinct from its role in regulating expression of classical targets known to regulate the cell cycle machinery.
Acknowledgments
We thank Alex Blais for assistance with the ChIP protocols and reviews of the E2F sequence data. Additionally, the invaluable technical assistance of Dominique Yelle, Linda Jui, and Eric Ouellet is gratefully acknowledged.
This work was supported by operating grants from the Canadian Institutes of Health Research to R.S.S. J.L.V. is a recipient of a Heart & Stroke Foundation of Canada fellowship. K.A.M. and L.M.J. are supported by Canada Graduate Doctoral Research Awards from the Canadian Institutes of Health Research.
Footnotes
Published ahead of print on 29 June 2009.
REFERENCES
- 1.Balciunaite, E., A. Spektor, N. H. Lents, H. Cam, H. te Riele, A. Scime, M. A. Rudnicki, R. Young, and B. D. Dynlacht. 2005. Pocket protein complexes are recruited to distinct targets in quiescent and proliferating cells. Mol. Cell. Biol. 258166-8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bieda, M., X. Xu, M. A. Singer, R. Green, and P. J. Farnham. 2006. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 16595-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Black, E. P., T. Hallstrom, H. K. Dressman, M. West, and J. R. Nevins. 2005. Distinctions in the specificity of E2F function revealed by gene expression signatures. Proc. Natl. Acad. Sci. USA 10215948-15953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burrows, R. C., D. Wancio, P. Levitt, and L. Lillien. 1997. Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron 19251-267. [DOI] [PubMed] [Google Scholar]
- 5.Callaghan, D. A., L. Dong, S. M. Callaghan, Y. X. Hou, L. Dagnino, and R. S. Slack. 1999. Neural precursor cells differentiating in the absence of Rb exhibit delayed terminal mitosis and deregulated E2F 1 and 3 activity. Dev. Biol. 207257-270. [DOI] [PubMed] [Google Scholar]
- 6.Cam, H., E. Balciunaite, A. Blais, A. Spektor, R. C. Scarpulla, R. Young, Y. Kluger, and B. D. Dynlacht. 2004. A common set of gene regulatory networks links metabolism and growth inhibition. Mol. Cell 16399-411. [DOI] [PubMed] [Google Scholar]
- 7.Chen, D., I. Livne-bar, J. L. Vanderluit, R. S. Slack, M. Agochiya, and R. Bremner. 2004. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell 5539-551. [DOI] [PubMed] [Google Scholar]
- 8.Chen, D., R. Opavsky, M. Pacal, N. Tanimoto, P. Wenzel, M. W. Seeliger, G. Leone, and R. Bremner. 2007. Rb-mediated neuronal differentiation through cell-cycle-independent regulation of E2f3a. PLoS Biol. 5e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chong, J. L., S. Y. Tsai, N. Sharma, R. Opavsky, R. Price, L. Wu, S. A. Fernandez, and G. Leone. 2009. E2f3a and E2f3b contribute to the control of cell proliferation and mouse development. Mol. Cell. Biol. 29414-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cobrinik, D., M. H. Lee, G. Hannon, G. Mulligan, R. T. Bronson, N. Dyson, E. Harlow, D. Beach, R. A. Weinberg, and T. Jacks. 1996. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 101633-1644. [DOI] [PubMed] [Google Scholar]
- 11.Cooper-Kuhn, C. M., M. Vroemen, J. Brown, H. Ye, M. A. Thompson, J. Winkler, and H. G. Kuhn. 2002. Impaired adult neurogenesis in mice lacking the transcription factor E2F1. Mol. Cell. Neurosci. 21312-323. [DOI] [PubMed] [Google Scholar]
- 12.Dimova, D. K., O. Stevaux, M. V. Frolov, and N. J. Dyson. 2003. Cell cycle-dependent and cell cycle-independent control of transcription by the Drosophila E2F/RB pathway. Genes Dev. 172308-2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dono, R. 2003. Fibroblast growth factors as regulators of central nervous system development and function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284R867-R881. [DOI] [PubMed] [Google Scholar]
- 14.Dono, R., G. Texido, R. Dussel, H. Ehmke, and R. Zeller. 1998. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 174213-4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dyson, N. 1998. The regulation of E2F by pRb-family proteins. Genes Dev. 122245-2262. [DOI] [PubMed] [Google Scholar]
- 16.Ferguson, K. L., S. M. Callaghan, M. J. O'Hare, D. S. Park, and R. S. Slack. 2000. The Rb-CDK4/6 signaling pathway is critical in neural precursor cell cycle regulation. J. Biol. Chem. 27533593-33600. [DOI] [PubMed] [Google Scholar]
- 17.Ferguson, K. L., K. A. McClellan, J. L. Vanderluit, W. C. McIntosh, C. Schuurmans, F. Polleux, and R. S. Slack. 2005. A cell-autonomous requirement for the cell cycle regulatory protein, Rb, in neuronal migration. EMBO J. 244381-4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferguson, K. L., J. L. Vanderluit, J. M. Hebert, W. C. McIntosh, E. Tibbo, J. G. MacLaurin, D. S. Park, V. A. Wallace, M. Vooijs, S. K. McConnell, and R. S. Slack. 2002. Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J. 213337-3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Florkiewicz, R. Z., and A. Sommer. 1989. Human basic fibroblast growth factor gene encodes four polypeptides: three initiate translation from non-AUG codons. Proc. Natl. Acad. Sci. USA 863978-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fortin, A., J. G. MacLaurin, N. Arbour, S. P. Cregan, N. Kushwaha, S. M. Callaghan, D. S. Park, P. R. Albert, and R. S. Slack. 2004. The proapoptotic gene SIVA is a direct transcriptional target for the tumor suppressors p53 and E2F1. J. Biol. Chem. 27928706-28714. [DOI] [PubMed] [Google Scholar]
- 21.Gritti, A., E. A. Parati, L. Cova, P. Frolichsthal, R. Galli, E. Wanke, L. Faravelli, D. J. Morassutti, R. Roisen, D. D. Nickel, and A. L. Vescovi. 1996. Multipotential stem cells from the adult mouse brain proliferate and self-renew in response to basic fibroblast growth factor. J. Neurosci. 161091-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He, Y., M. K. Armanious, M. J. Thomas, and W. D. Cress. 2000. Identification of E2F-3B, an alternative form of E2F-3 lacking a conserved N-terminal region. Oncogene 193422-3433. [DOI] [PubMed] [Google Scholar]
- 23.Hébert, J. M., and S. K. McConnell. 2000. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev. Biol. 222296-306. [DOI] [PubMed] [Google Scholar]
- 24.Hurford, R. K., D. Cobrinik, M. H. Lee, and N. Dyson. 1997. pRb and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 111447-1463. [DOI] [PubMed] [Google Scholar]
- 25.Ishida, S., E. Huang, H. Zuzan, R. Spang, G. Leone, M. West, and J. R. Nevins. 2001. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol. Cell. Biol. 214684-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin, V. X., A. Rabinovich, S. L. Squazzo, R. Green, and P. J. Farnham. 2006. A computational genomics approach to identify cis-regulatory modules from chromatin immunoprecipitation microarray data: a case study using E2F1. Genome Res. 161585-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kilpatrick, T. J., and P. F. Bartlett. 1995. Cloned multipotential precursors from the mouse cerebrum require FGF-2, whereas glial restricted precursors are stimulated with either FGF-2 or EGF. J. Neurosci. 153653-3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LeCouter, J. E., B. Kablar, W. R. Hardy, C. Ying, L. A. Megeney, L. L. May, and M. A. Rudnicki. 1998. Strain-dependent myeloid hyperplasia, growth deficiency, and accelerated cell cycle in mice lacking the Rb-related p107 gene. Mol. Cell. Biol. 187455-7465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee, M. H., B. O. Williams, G. Mulligan, S. Mukai, R. T. Bronson, N. Dyson, E. Harlow, and T. Jacks. 1996. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 101621-1632. [DOI] [PubMed] [Google Scholar]
- 30.Leone, G., F. Nuckolls, S. Ishida, M. Adams, R. Sears, L. Jakoi, A. Miron, and J. R. Nevins. 2000. Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol. Cell. Biol. 203626-3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leone, G., R. Sears, E. Huang, R. Rempel, F. Nuckolls, C. H. Park, P. Giangrande, L. Wu, H. I. Saavedra, S. J. Field, M. A. Thompson, H. Yang, Y. Fujiwara, M. E. Greenberg, S. Orkin, C. Smith, and J. R. Nevins. 2001. Myc requires distinct E2F activities to induce S phase and apoptosis. Mol. Cell 8105-113. [DOI] [PubMed] [Google Scholar]
- 32.Lillien, L., and H. Raphael. 2000. BMP and FGF regulate the development of EGF-responsive neural progenitor cells. Development 1274993-5005. [DOI] [PubMed] [Google Scholar]
- 33.Ma, Y., R. Croxton, R. L. Moorer, Jr., and W. D. Cress. 2002. Identification of novel E2F1-regulated genes by microarray. Arch. Biochem. Biophys. 399:212-224. [DOI] [PubMed] [Google Scholar]
- 34.MacPherson, D., J. Sage, D. Crowley, A. Trumpp, R. T. Bronson, and T. Jacks. 2003. Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol. Cell. Biol. 231044-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marino, S., D. Hoogervoorst, S. Brandner, and A. Berns. 2003. Rb and p107 are required for normal cerebellar development and granule cell survival but not for Purkinje cell persistence. Development 1303359-3368. [DOI] [PubMed] [Google Scholar]
- 36.Marino, S., M. Vooijs, H. van d er Gulden, J. Jonkers, and A. Berns. 2000. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 14994-1004. [PMC free article] [PubMed] [Google Scholar]
- 37.McClellan, K. A., V. A. Ruzhynsky, D. N. Douda, J. L. Vanderluit, K. L. Ferguson, D. Chen, R. Bremner, D. S. Park, G. Leone, and R. S. Slack. 2007. Unique requirement for Rb/E2F3 in neuronal migration: evidence for cell cycle-independent functions. Mol. Cell. Biol. 274825-4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McClellan, K. A., and R. S. Slack. 2006. Novel functions for cell cycle genes in nervous system development. Cell Cycle 51506-1513. [DOI] [PubMed] [Google Scholar]
- 39.McClellan, K. A., and R. S. Slack. 2007. Specific in vivo roles for E2Fs in differentiation and development. Cell Cycle 62917-2927. [DOI] [PubMed] [Google Scholar]
- 40.Moffett, J., E. Kratz, J. Myers, E. K. Stachowiak, R. Z. Florkiewicz, and M. K. Stachowiak. 1998. Transcriptional regulation of fibroblast growth factor-2 expression in human astrocytes: implications for cell plasticity. Mol. Biol. Cell 92269-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Müller, H., A. P. Bracken, R. Vernell, M. C. Moroni, F. Christians, E. Grassilli, E. Prosperini, E. Vigo, J. D. Oliner, and K. Helin. 2001. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 15267-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohkubo, Y., A. O. Uchida, D. Shin, J. Partanen, and F. M. Vaccarino. 2004. Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J. Neurosci. 246057-6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Polager, S., Y. Kalma, E. Berkovich, and D. Ginsberg. 2002. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene 21437-446. [DOI] [PubMed] [Google Scholar]
- 44.Prats, H., M. Kaghad, A. C. Prats, M. Klagsbrun, J. M. Lelias, P. Liauzun, P. Chalon, J. P. Tauber, F. Amalric, J. A. Smith, et al. 1989. High molecular mass forms of basic fibroblast growth factor are initiated by alternative CUG codons. Proc. Natl. Acad. Sci. USA 861836-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raballo, R., J. Rhee, R. Lyn-Cook, J. F. Leckman, M. L. Schwartz, and F. M. Vaccarino. 2000. Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J. Neurosci. 205012-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rabinovich, A., V. X. Jin, R. Rabinovich, X. Xu, and P. J. Farnham. 2008. E2F in vivo binding specificity: comparison of consensus versus nonconsensus binding sites. Genome Res. 181763-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren, B., H. Cam, Y. Takahashi, T. Volkert, J. Terragni, R. A. Young, and B. D. Dynlacht. 2002. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev. 16245-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reuss, B., and O. von Bohlen und Halbach. 2003. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 313:139-157. [DOI] [PubMed] [Google Scholar]
- 49.Reynolds, B. A., W. Tetzlaff, and S. Weiss. 1992. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 124565-4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruzhynsky, V. A., K. A. McClellan, J. L. Vanderluit, Y. Jeong, M. Furimsky, D. S. Park, D. J. Epstein, V. A. Wallace, and R. S. Slack. 2007. Cell cycle regulator E2F4 is essential for the development of the ventral telencephalon. J. Neurosci. 275926-5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scime, A., G. Grenier, M. S. Huh, M. A. Gillespie, L. Bevilacqua, M. E. Harper, and M. A. Rudnicki. 2005. Rb and p107 regulate preadipocyte differentiation into white versus brown fat through repression of PGC-1α. Cell Metab. 2283-295. [DOI] [PubMed] [Google Scholar]
- 52.Slack, R. S., P. A. Hamel, T. S. Bladon, R. M. Gill, and M. W. McBurney. 1993. Regulated expression of the retinoblastoma gene in differentiating embryonal carcinoma cells. Oncogene 81585-1591. [PubMed] [Google Scholar]
- 53.Storring, J. M., A. Charest, P. Cheng, and P. R. Albert. 1999. TATA-driven transcriptional initiation and regulation of the rat serotonin 5-HT1A receptor gene. J. Neurochem. 722238-2247. [DOI] [PubMed] [Google Scholar]
- 54.Templeton, D., and R. A. Weinberg. 1991. Nonfunctional mutants of the Rb protein are characterized by defects in phosphorylation, viral oncoprotein association and nucleal tethering. Proc. Natl. Acad. Sci. USA 883033-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trimarchi, J. M., and J. A. Lees. 2002. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 311-20. [DOI] [PubMed] [Google Scholar]
- 56.Tropepe, V., M. Sibilia, B. G. Ciruna, J. Rossant, E. F. Wagner, and D. van der Kooy. 1999. Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev. Biol. 208166-188. [DOI] [PubMed] [Google Scholar]
- 57.Tsai, S. Y., R. Opavsky, N. Sharma, L. Wu, S. Naidu, E. Nolan, E. Feria-Arias, C. Timmers, J. Opavska, A. de Bruin, J. L. Chong, P. Trikha, S. A. Fernandez, P. Stromberg, T. J. Rosol, and G. Leone. 2008. Mouse development with a single E2F activator. Nature 4541137-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vaccarino, F. M., M. L. Schwartz, R. Raballo, J. Nilsen, J. Rhee, M. Zhou, T. Doetschman, J. D. Coffin, J. J. Wyland, and Y. T. Hung. 1999. Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat. Neurosci. 2246-253. [DOI] [PubMed] [Google Scholar]
- 59.van den Heuvel, S., and N. J. Dyson. 2008. Conserved functions of the pRB and E2F families. Nat. Rev. Mol. Cell Biol. 9713-724. [DOI] [PubMed] [Google Scholar]
- 60.Vanderluit, J. L., K. L. Ferguson, V. Nikoletopoulou, M. Parker, V. Ruzhynsky, T. Alexson, S. M. McNamara, D. S. Park, M. Rudnicki, and R. S. Slack. 2004. p107 regulates neural precursor cells in the mammalian brain. J. Cell Biol. 166853-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vanderluit, J. L., C. A. Wylie, K. A. McClellan, N. Ghanem, A. Fortin, S. Callaghan, J. G. Maclaurin, D. S. Park, and R. S. Slack. 2007. The Retinoblastoma family member p107 regulates the rate of progenitor commitment to a neuronal fate. J. Cell Biol. 178129-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vooijs, M., M. van der Valk, H. te Riele, and A. Berns. 1998. Flp-mediated tissue-specific inactivation of the retinoblastoma tumor suppressor gene in the mouse. Oncogene 17:1-12. [DOI] [PubMed] [Google Scholar]
- 63.Wallace, V. A., and M. C. Raff. 1999. A role for Sonic hedgehog in axon-to-astrocyte signalling in the rodent optic nerve. Development 1262901-2909. [DOI] [PubMed] [Google Scholar]
- 64.Weinmann, A. S., S. M. Bartley, T. Zhang, M. Q. Zhang, and P. J. Farnham. 2001. Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol. Cell. Biol. 216820-6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weinmann, A. S., P. S. Yan, M. J. Oberley, T. H. Huang, and P. J. Farnham. 2002. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev. 16235-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wells, J., K. E. Boyd, C. J. Fry, S. M. Bartley, and P. J. Farnham. 2000. Target gene specificity of E2F and pocket protein family members in living cells. Mol. Cell. Biol. 205797-5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu, X., M. Bieda, V. X. Jin, A. Rabinovich, M. J. Oberley, R. Green, and P. J. Farnham. 2007. A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res. 171550-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Young, A. P., R. Nagarajan, and G. D. Longmore. 2003. Mechanisms of transcriptional regulation by Rb-E2F segregate by biological pathway. Oncogene 227209-7217. [DOI] [PubMed] [Google Scholar]