

Abstract
Controlling the loading of Rad51 onto DNA is important for governing when and how homologous recombination is used. Here we use a combination of genetic assays and indirect immunofluorescence to show that the F-box DNA helicase (Fbh1) functions in direct opposition to the Rad52 orthologue Rad22 to curb Rad51 loading onto DNA in fission yeast. Surprisingly, this activity is unnecessary for limiting spontaneous direct-repeat recombination. Instead it appears to play an important role in preventing recombination when replication forks are blocked and/or broken. When overexpressed, Fbh1 specifically reduces replication fork block-induced recombination, as well as the number of Rad51 nuclear foci that are induced by replicative stress. These abilities are dependent on its DNA helicase/translocase activity, suggesting that Fbh1 exerts its control on recombination by acting as a Rad51 disruptase. In accord with this, overexpression of Fbh1 also suppresses the high levels of recombinant formation and Rad51 accumulation at a site-specific replication fork barrier in a strain lacking the Rad51 disruptase Srs2. Similarly overexpression of Srs2 suppresses replication fork block-induced gene conversion events in an fbh1Δ mutant, although an inability to suppress deletion events suggests that Fbh1 has a distinct functionality, which is not readily substituted by Srs2.
Homologous recombination (HR) is often described as a double-edged sword: it can maintain genome stability by promoting DNA repair, while its injudicious action can disturb genome stability by causing gross chromosome rearrangement (GCR) or loss of heterozygosity (LOH). Both GCR and LOH are potential precursors of diseases such as cancer, and consequently there is need to control when and how HR is used.
A key step in most HR is the loading of the Rad51 recombinase onto single-stranded DNA (ssDNA), which forms a nucleoprotein filament (nucleofilament) that catalyzes the pairing of homologous DNAs and subsequent strand invasion (32). This is a critical point at which recombination can be regulated through the removal of the Rad51 filament (60). Early removal can prevent strand invasion altogether, freeing the DNA for alternative processing. Later removal may limit unnecessary filament growth, free the 3′-OH of the invading strand to prime DNA synthesis, and ultimately enable ejection of the invading strand, which is important for the repair of double-strand breaks (DSBs) by synthesis-dependent strand annealing (SDSA). SDSA avoids the formation of Holliday junctions that can be resolved into reciprocal exchange products (crossovers), which may result in GCR or LOH if the recombination is ectopic or allelic, respectively.
One enzyme that appears to be able to control Rad51 in the aforementioned manner is the yeast superfamily 1 DNA helicase Srs2 (42). In Saccharomyces cerevisiae, Srs2 is recruited to stalled replication forks by the SUMOylation of PCNA, and there it appears to block Rad51-dependent HR in favor of Rad6- and Rad18-dependent postreplication repair (1, 2, 35, 50, 53, 58). In vitro Srs2 can strip Rad51 from ssDNA via its DNA translocase activity (31, 62) and therefore probably controls HR at stalled replication forks by acting as a Rad51 disruptase. In accord with this, chromatin immunoprecipitation analysis has shown that Rad51 is enriched at or near replication forks in an srs2 mutant (50). Srs2 also plays an important role in crossover avoidance during DSB repair, where it is thought to promote SDSA by both disrupting Rad51 nucleofilaments and dissociating displacement (D) loops (20, 27).
Srs2 is conserved in the fission yeast Schizosaccharomyces pombe (19, 43, 63) and has a close relative in bacteria called UvrD, which can similarly control HR by disrupting RecA nucleofilaments (61). However, an obvious homologue in mammals has not been detected. Recently, two mammalian members of the RecQ DNA helicase family, BLM and RECQL5, were shown to disrupt Rad51 nucleofilaments in vitro (11, 25), although in the case of BLM, this activity appears to be relatively weak (5, 55). Nevertheless these data have led to speculation that both BLM and RECQL5 might perform a function similar to that of Srs2 in vivo (6). Certainly mutational inactivation of either helicase results in elevated levels of HR and genome instability, with an associated increased rate of cancer (23, 25). However, BLM and RECQL5 are not the only potential Rad51 disruptases in mammals; a relative of Srs2 and UvrD called FBH1 was recently implicated in this role by genetic studies of its orthologue in S. pombe and by its ability to partially compensate for the loss of Srs2 in S. cerevisiae, which, unlike S. pombe, lacks an FBH1 orthologue (15). FBH1 is so named because of an F box near its N terminus—a feature that makes it unique among DNA helicases (28). The F box is important for its interaction with SKP1 and therefore the formation of an E3 ubiquitin ligase SCF (SKP1-Cul1-F-box protein) complex (29). The targets of this complex are currently unknown. In S. pombe, mutations within Fbh1's F-box block interaction with Skp1 and prevent Fbh1 from localizing to the nucleus and forming damage-induced foci therein (57). Fbh1's role in constraining Rad51 activity in S. pombe is evidenced by the increase in spontaneous Rad51 foci and accumulation of UV irradiation-induced Rad51-dependent recombination intermediates in an fbh1Δ mutant (47). Moreover, loss of both Fbh1 and Srs2 in S. pombe results in a synergistic reduction in cell viability, and like Srs2, Fbh1 is essential for viability in the absence of the S. pombe RecQ family DNA helicase Rqh1, which processes recombination intermediates (47, 48). In both cases the synthetic interaction is suppressed by deleting rad51, suggesting that Fbh1 works in parallel with Srs2 and Rqh1 to prevent the formation of toxic recombination intermediates. In yeast, Rad51-mediated recombination is dependent on Rad52 (Rad22 in S. pombe), which is believed to promote the nucleation of Rad51 onto DNA that is coated with the ssDNA binding protein replication protein A (RPA) (18, 32). Intriguingly, the genotoxin sensitivity and recombination deficiency of a rad22 mutant are suppressed in a Rad51-dependent manner by deleting fbh1 (48). This suggests that Fbh1 and Rad22 act in opposing ways to modulate the assembly of the Rad51 nucleofilament. Although current data indicate a role for Fbh1 in controlling HR, the only evidence so far that Fbh1 limits recombinant formation is in chicken DT40 cells, for which a modest increase in sister chromatid exchange has been noted when FBH1 is deleted (30).
Here we present in vivo evidence suggesting that Fbh1 does indeed act as a Rad51 disruptase, which is dependent on its DNA helicase/translocase activity. We confirm predictions that this activity works in opposition to Rad22 for the loading of Rad51 onto DNA and show that Fbh1's modulation of Rad51 activity, while not essential for limiting spontaneous direct-repeat recombination, is critical for preventing recombination at blocked replication forks. Finally, we highlight similarities and differences between Fbh1 and Srs2, based on their mutant phenotypes and relative abilities to suppress recombination when overexpressed. Overall our data affirm that Fbh1 is one of the principal modulators of Rad51 activity in fission yeast and therefore may play a similar role in vertebrates.
MATERIALS AND METHODS
S. pombe strains and plasmids.
S. pombe strains are listed in Table 1. The lacO array-containing strains were made by integration of derivatives of pMW700 and pMW701 (pMW734 and pMW735) (3) (see below), which had been linearized with BlpI, at ade6-M375 in FO1236. Colony PCR was used to determine that the linear plasmid had integrated at the correct site. pMW734 and pMW735 were constructed by subcloning an ∼1.7-kb HincII lacO array fragment from pLAU43 (34) into the PvuII site downstream of his3+ in pMW701 and pMW700, respectively. pREP41-fbh1 (pMW637) and pREP41-fbh1D485N (pMW658) have been described (48). pREP41-2myc-fbh1 (pMW650) and pREP41-2myc-fbh1D485N (pMW837) are derivatives of pREP41MHN (16). pREP2 is a derivative of pREP1 containing the wild-type nmt promoter and a ura4+ marker instead of LEU2 (44). pREP2-fbh1 (pMW867) was made by subcloning the fbh1 gene from pMW637 immediately downstream of the nmt promoter in pREP2. pREP41-srs2 (pIJ9) contains the cDNA of srs2 cloned into the NdeI and BamHI sites in pREP41. pREP41-srs2D238N (pIJ12) is derived from pIJ9 using a QuikChange site-directed mutagenesis kit (Stratagene). The pREP81-LacI-ECFP plasmid will be described elsewhere. In all cases the correct plasmid sequences were confirmed by nucleotide sequencing.
TABLE 1.
Schizosaccharomyces pombe strains
| Strain | Genotype | Source or reference |
|---|---|---|
| MCW1088 | h+rad51Δ::arg3+ura4-D18 leu1-32 his3-D1 arg3-D4 | Lab strain |
| MCW1221 | h+ura4-D18 leu1-32 his3-D1 arg3-D4 | Lab strain |
| MCW1230 | h+rad54Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 | Lab strain |
| MCW1262 | h−ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 1/ade6-L469 | 3 |
| MCW1285 | h+rad22Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 | 48 |
| MCW1433 | h−ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 2/ade6-L469 | 3 |
| MCW1443 | h−rqh1Δ::kanMX6 ura4-D18 his3-D1 leu1-32 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 1/ade6-L469 | 3 |
| MCW1447 | h−rqh1Δ::kanMX6 ura4-D18 his3-D1 leu1-32 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 2/ade6-L469 | 3 |
| MCW1490 | h+fbh1Δ::kanMX6 ura4-D18 leu1-32 his3-D1 arg3-D4 | 48 |
| MCW1553 | h+fbh1Δ::kanMX6 rad22Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 | 48 |
| MCW1647 | h+fbh1Δ::kanMX6 rad54Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 | This study |
| MCW1687 | h+rad22Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 1/ade6-L469 | 3 |
| MCW1688 | h+rad22Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 2/ade6-L469 | 3 |
| MCW1691 | h+rad51Δ::arg3+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 1/ade6-L469 | 3 |
| MCW1692 | h+rad51Δ::arg3+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 2/ade6-L469 | 3 |
| MCW1759 | h+fbh1D485N::kanMX6 ura4-D18 leu1-32 his3-D1 arg3-D4 | This study |
| MCW2349 | h+fbh1Δ::kanMX6 ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/lacOx40/his3+/RTS1 site A orientation 1/ade6-L469 | This study |
| MCW2351 | h+fbh1Δ::kanMX6 ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/lacOx40/his3+/RTS1 site A orientation 2/ade6-L469 | This study |
| MCW2366 | h+srs2Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/lacOx40/his3+/RTS1 site A orientation 1/ade6-L469 | This study |
| MCW2368 | h+srs2Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/lacOx40/his3+/RTS1 site A orientation 2/ade6-L469 | This study |
| MCW2406 | h+rqh1Δ::kanMX6 ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/lacOx40/his3+/RTS1 site A orientation 1/ade6-L469 | This study |
| MCW2408 | h+rqh1Δ::kanMX6 ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/lacOx40/his3+/RTS1 site A orientation 2/ade6-L469 | This study |
| MCW2648 | h+fbh1D485N::KanMX6ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 1/ade6-L469 | This study |
| MCW2873 | h+fbh1D485N::KanMX6ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 2/ade6-L469 | This study |
| FO1236 | h−ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 | Lab strain |
| FO1748 | h+srs2Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 1/ade6-L469 | This study |
| FO1750 | h+srs2Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 2/ade6-L469 | This study |
| FO1752 | h+rad22+::YFP-kanMX6 ura4-D18 leu1-32 his3-D1 | This studya |
| FO1776 | h+rad11+::GFP-kanMX6 ura4-D18 leu1-32 his3-D1 arg3-D4 | This studyb |
| FO1791 | h−ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/lacOx40/his3+/RTS1 site A orientation 1/ade6-L469 | This study |
| FO1792 | h−ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/lacOx40/his3+/RTS1 site A orientation 2/ade6-L469 | This study |
| FO1814 | h+fbh1Δ::KanMX6ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 1/ade6-L469 | This study |
| FO1816 | h+fbh1Δ::KanMX6ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 2/ade6-L469 | This study |
| FO1831 | h+fbh1Δ::arg3+rad22Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 1/ade6-L469 | This study |
| FO1833 | h+fbh1Δ::arg3+rad22Δ::ura4+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 2/ade6-L469 | This study |
| FO1860 | h+fbh1Δ::KanMX6rad51Δ::arg3+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 1/ade6-L469 | This study |
| FO1862 | h+fbh1Δ::KanMX6rad51Δ::arg3+ura4-D18 leu1-32 his3-D1 arg3-D4 ade6-M375 int::pUC8/his3+/RTS1 site A orientation 2/ade6-L469 | This study |
Derivative of SP220 (45).
Derivative of a rad11+::GFP-kanMX6 strain kindly provided by M. G. Ferreira and J. P. Cooper (CRUK, London, United Kingdom).
Media and genetic methods.
The media and genetic methods used standard protocols (46). The complete and minimal media were yeast extract with supplements (YES) and Edinburgh minimal medium plus 3.7 mg/ml sodium glutamate (EMMG) and appropriate amino acids (0.2475 mg/ml), respectively. Ade+ recombinants were selected on YES lacking adenine and supplemented with 200 mg/liter guanine to prevent uptake of residual adenine. Strains containing plasmids were grown in EMMG lacking leucine/uracil to maintain selection for the plasmid. Thiamine (4 μM final concentration) was included where appropriate to repress expression from the nmt promoter.
Spot assays.
Exponentially growing cells from liquid cultures were harvested, washed, and resuspended in water at a density of 1 × 107 to 103 cells/ml. Aliquots (10 μl) of the cell suspensions were spotted onto EMMG plates lacking leucine and containing methyl methanesulfonate (MMS), hydroxurea (HU), or camptothecin (CPT) as indicated. For UV, plates were irradiated using a Stratalinker (Stratagene). Plates were photographed after 5 days of growth at 30°C, unless stated otherwise.
Recombination assays.
Direct-repeat recombination was assayed by measuring the frequency of Ade+ recombinants, as described previously (3), except for strains containing plasmids, which were grown on EMMG lacking leucine and thiamine for 4 days at 30°C prior to selecting for Ade+ recombinants. Recombinant frequencies represent the mean value from at least 15 colonies for each strain, and for strains containing plasmids, at least three independent transformants were tested. Two sample t tests were used to determine the statistical significance of differences in recombinant frequencies between strains. For P values, see Tables S1 to S3 in the supplemental material.
DNA preparation and 2D gels.
Chromosomal DNA was purified from logarithmically growing cultures, as described previously (26), and enriched for replication intermediates on benzoylated naphtholyated DEAE-cellulose. Two-dimensional (2D) gels were run according to the method of Brewer and Fangman (8), using 0.4% and 1% agarose for the first and second dimensions, respectively. Gels were Southern blotted onto Amersham Hybond-N+ membrane and probed with 32P-labeled probe A (3). Blots were analyzed by phosphorimaging using a Fuji FLA3000 and Image Gauge software.
Microscopic preparation, immunostaining, and detection.
To 10 ml of asynchronously cycling yeast cultures at a density of ∼1 × 107 cells/ml sodium azide was added at a final concentration of 0.1%. The cells were harvested by centrifugation, washed once with sterile demineralized water, resuspended in a spheroplasting solution containing a cocktail of lysing enzymes, and incubated at 30°C for 27 min, as described previously (39). The spheroplasts were spread onto slides after being treated with detergent and fixative (4, 40).
The slides were washed three times in 1× phosphate-buffered saline containing 0.05% Triton X-100 for 15 min each. The following primary antibodies, diluted in 1× PBS as indicated, were applied under a coverslip at room temperature for ∼18 h: 1:50 rabbit polyclonal anti-human Rad51 (Santa Cruz Biotechnology Inc., Santa Cruz, CA), 1:100 mouse monoclonal anti-c-Myc (Sigma, St. Louis, MO), and 1:2,000 chicken polyclonal anti-green fluorescent protein (GFP) (Millipore Corp., Billerica, MA). The preparations were washed three times in 1× phosphate-buffered saline plus 0.05% Triton X-100 for 15 min each, and secondary antibodies (1:1,000 goat anti-rabbit immunoglobulin G [IgG] conjugated to Cy3 [GE Healthcare UK Ltd., Little Chalfont, United Kingdom], 1:1,000 goat anti-mouse IgG conjugated to Cy3 [GE Healthcare UK Ltd.], 1:200 goat anti-rabbit IgG conjugated to fluorescein isothiocyanate [Sigma], and 1:400 goat anti-chicken IgY conjugated to FITC [Novus Biologicals Inc., Littleton, CO]) were applied under a coverslip at room temperature for at least 4 h. Finally, the slides were washed again as described above and mounted in Vectashield (Vector Laboratories, Burlingame, CA) containing 1 μg/ml DAPI (4′,6- diamidino-2-phenylindole) to stain the DNA.
Immunostained slides were analyzed using a 100×/1.30 UPlanFl objective of an Olympus BX50 epifluorescence microscope equipped with the appropriate filter sets to detect blue, green, and red fluorescence. Black and white pictures of the single fluorescence channels were acquired with a cooled charge-coupled-device camera (Princeton Instruments Inc., NJ) controlled by Metamorph software (version 6.1r6; Molecular Devices, Sunnyvale, CA). Single-channel pictures were assigned pseudocolors and merged using Adobe Photoshop 7.0 (Adobe Systems Inc., San Jose, CA).
RESULTS
Fbh1 and Rad22 play opposing roles in modulating the loading of Rad51 onto DNA.
We previously identified Fbh1's link to recombination through its genetic interaction with Rad22 (48). Deletion of fbh1 suppresses the severe DNA repair and recombination deficiencies of a rad22Δ mutant, and in return the deletion of rad22 suppresses the poor growth and genotoxin hypersensitivity of an fbh1Δ mutant. Based on this genetic observation, we hypothesized that Fbh1 and Rad22 play opposing roles in modulating the loading of Rad51 onto DNA, with Fbh1 curbing loading and Rad22 promoting it. To test this model, we analyzed Rad51 loading onto DNA by immunocytochemistry in four different genetic backgrounds (wild type, rad22Δ, fbh1Δ, and rad22Δ fbh1Δ) (Fig. 1). Initially we looked at cells undergoing logarithmic growth. Here spread nuclei from wild-type or rad22Δ mutant cells exhibited few if any Rad51 foci (Fig. 1A and B). In contrast, >80% of fbh1Δ nuclei under the same conditions exhibit at least one Rad51 focus, and approximately 40% show more than five, which is consistent with previous data (Fig. 1B) (47). Importantly, this elevated level of spontaneous Rad51 foci is suppressed by deleting rad22, suggesting that they are a consequence of Rad51 loading at sites of ssDNA (Fig. 1B).
FIG. 1.

The effect of fbh1Δ and rad22Δ on Rad51 foci. (A) Examples of spread nuclei from cells grown in YES with or without CPT (10 μM)/MMS (0.03%) for 4 to 5 h. Nuclei are stained with anti-Rad51 (red) and DAPI (blue). (B) Quantification of Rad51 foci in wild-type (MCW1221), rad22Δ (MCW1285), fbh1Δ (MCW1490), and rad22Δ fbh1Δ (MCW1553) strains. Each value represents the analysis of 100 nuclei from two independent experiments. The number on top of each bar is the mean number of Rad51 foci per nucleus.
We next analyzed Rad51 focus formation in cells exposed to agents MMS and CPT, which can cause replication fork blockage and/or breakage (Fig. 1). Exposure to either agent results in multiple Rad51 foci in the majority of wild-type nuclei, and similar levels of foci are observed in nuclei from fbh1Δ cells under these conditions (Fig. 1A and B). In contrast, rad22Δ nuclei exhibit relatively few Rad51 foci following CPT or MMS exposure, with less than 10% showing greater than five foci (Fig. 1A and B). This reduction in Rad51 foci is suppressed by the deletion of fbh1 (Fig. 1A and B). Taken together, these data support our hypothesis that Fbh1 and Rad22 play opposing roles in modulating the loading of Rad51 onto DNA. Without Fbh1, Rad22 promotes excessive loading of Rad51 onto DNA, at least under normal growth conditions, whereas without Rad22, Fbh1 promotes its excessive removal. In the absence of both Rad22 and Fbh1, the balance is restored and Rad51 can successfully load onto DNA in response to DNA damage.
Fbh1 limits recombination at blocked replication forks.
An fbh1Δ mutant exhibits defects in chromosome segregation due seemingly to excessive Rad51 activity, either creating DNA junctions between chromatids and/or physically impeding the replication/repair/segregation machinery (48). Interestingly this heightened Rad51 activity does not result in a detectable increase in spontaneous or UV-induced conversion-type recombinants, as measured by a direct-repeat recombination assay (48). This may be because it leads to cell death rather than to a viable, and therefore detectable, recombinant. Alternatively the sites where Rad51 spontaneously accumulates in an fbh1Δ mutant may be chromosomal regions that are especially susceptible to “DNA damage”; for example, they might be regions that are difficult to replicate and/or prone to DNA breakage (13, 36, 56).
Recently we showed that replication fork blockage by a polar site-specific barrier (RTS1) positioned between a direct repeat of ade6− heteroalleles results in a very strong stimulation in Rad51-dependent Ade+ recombinant formation (Fig. 2A) (3). This stimulation is seen only with one orientation of the barrier due to a strong bias in the direction in which the ade6 locus is replicated. To see if Fbh1 limits replication block-induced recombination we deleted fbh1 in the barrier-containing strains and measured the frequency of recombinant formation. Both wild-type and fbh1Δ strains exhibit similar levels of recombinants when the RTS1 barrier is orientated such that forks traversing ade6 are not impeded (Fig. 2B). Consistent with our previous data, reversing the orientation of RTS1 blocks replication and results in a massive stimulation of recombination (Fig. 2A and C) (3). However, this stimulation in the fbh1Δ strain is even greater (by ∼10-fold; P = <0.0001) than that in the wild type (Fig. 2C; see also Table S1 in the supplemental material).
FIG. 2.

Spontaneous and RTS1-induced direct-repeat recombination. (A) Recombination substrate integrated at the ade6 locus on chromosome 3. The panels on the right are 2D gel analyses of the replication intermediates in the EcoNI fragment and show an uninterrupted arc of replication intermediates with RTS1 in orientation 1 (top) and an accumulation of replication intermediates at RTS1 in orientation 2 (bottom). (B and C) Ade+ recombinant frequencies. The data for rqh1Δ are from Sun et al. (59). (D) 2D gel analysis of the replication intermediates in the EcoNI fragment shown in panel A. In each strain, RTS1 is in orientation 2. The schematic describes the relevant features of these gels, including the RFB. The bar chart shows a quantification of the RFB signal in each strain. Each value is the mean of three experiments, and the error bars are the standard deviations. asc., ascending.
The presence of a His+ marker between the ade6− repeats enables two types of Ade+ recombinants to be distinguished: those that retain the marker (conversion types) and those that lose it by deletion of the region between the repeats (deletion types). Fork blockage at RTS1 typically results in equal proportions of conversion and deletion types; however, in the fbh1Δ strain the majority of induced recombinants are deletion types (Fig. 2C; see also Table S1 in the supplemental material). As deletion types can arise from replication fork breakage (3), we analyzed DNA in this region by gel electrophoresis and Southern blotting. However, we detected no obvious increase in DSBs in the fbh1Δ strain (data not shown). Moreover, a comparison of replication intermediates in the region surrounding RTS1 by native 2D gel electrophoresis reveals no difference between wild-type and fbh1Δ strains in the amounts of blocked forks that accumulate (Fig. 2D). These data suggest that the elevated recombination in the fbh1Δ strain is not a consequence of decreased fork stability or altered barrier activity.
The observation that Fbh1 restricts Rad51 focus formation under normal growth conditions (Fig. 1) (47) suggests that it might control recombination at the RTS1 barrier by curbing Rad51 binding to ssDNA at the blocked fork. If this is true, then the elevated recombination in an fbh1Δ mutant should be dependent on Rad51. It was important to test this because the formation of deletion types does not always depend on Rad51. In fact, loss of Rad51 results in a marked increase in spontaneous deletion types (Fig. 2B; see also Table S1 in the supplemental material) (3, 18). However, contrary to its requirement for spontaneous recombination, the elevated level of deletion types in an fbh1Δ single mutant is entirely dependent on Rad51 (Fig. 2C; see also Table S1 in the supplemental material). Furthermore, this dependence is not a consequence of reduced barrier activity, because the rad51Δ and fbh1Δ rad51Δ strains accumulate amounts of replication intermediates at RTS1 similar to that of the wild-type strain (Fig. 2D). Instead these data indicate that Fbh1 controls Rad51-dependent recombination at blocked replication forks, possibly by curbing its loading onto DNA.
As shown in Fig. 1, Rad22 is needed for the normal and excessive loading of Rad51 onto DNA in the presence and absence of Fbh1, respectively. If these cytological data accurately reflect what happens at the RTS1 barrier, then Rad22 should be needed for both wild-type levels and fbh1Δ mutant levels of recombination. The data in Fig. 2C (see also Table S1 in the supplemental material) show that this is indeed the case, with replication fork block-induced recombination being effectively lost in a rad22Δ single mutant and reduced to below wild-type levels in a rad22Δ fbh1Δ double mutant.
A requirement for helicase/translocase activity in controlling recombination.
To see if Fbh1's helicase/translocase activity is necessary for controlling recombination at blocked replication forks, we used a strain carrying a mutation that results in a D485N change in helicase motif II. The equivalent mutation in other DNA helicases impairs ATP hydrolysis and DNA unwinding (51). The fbh1D485N strain is comparable to an fbh1Δ strain for both elevated levels of spontaneous Rad51 foci and replication fork block-induced recombination (Fig. 2C; see also Table S1 in the supplemental material; also data not shown). These data indicate that Fbh1's helicase/translocase activity is necessary for its role in controlling Rad51 activity.
Comparing the effect of fbh1Δ, srs2Δ, and rqh1Δ on spontaneous and RTS1-induced direct-repeat recombination.
We have previously shown that both the rqh1Δ and srs2Δ mutants, unlike fbh1Δ, exhibit a heightened frequency of spontaneous direct-repeat recombination compared to the wild type (3, 17, 19, 59) (Fig. 2B; see also Table S1 in the supplemental material). We have also shown that Rqh1, like Fbh1, limits Rad51-dependent recombination at the RTS1 replication fork barrier (RFB) (3, 59). In fact, a comparison of these data reveals similar increases in the overall frequency of recombinants in both mutants, albeit the frequency of deletion types is slightly higher in the rqh1Δ mutant and the frequency of conversion types is slightly lower (Fig. 2C; see also Table S1 in the supplemental material). An srs2Δ mutant also exhibits an increase in RTS1-induced recombinant formation, albeit the overall frequency of Ade+ recombinants is slightly less than with either fbh1Δ or rqh1Δ (Fig. 2; see also Table S1 in the supplemental material). Importantly, the proportion of recombinants that are conversion types is much higher in an srs2Δ mutant (∼66%) than in either the fbh1Δ (∼24%) or rqh1Δ (∼13%) mutant. The potential significance of this is discussed later.
Accumulation of Rad51 at the RTS1 RFB in an srs2Δ mutant.
In S. cerevisiae, Srs2 limits Rad51 accumulation at stalled replication forks presumably by disrupting the Rad51 nucleofilament (31, 50, 62). To see whether Srs2 and/or Fbh1 can similarly restrict Rad51 accumulation at the RTS1 RFB in S. pombe, we introduced a short array of lac operator (lacO) sequences downstream of RTS1 so that we could visualize the RTS1-containing region with an enhanced cyan fluorescent protein (ECFP)-tagged version of the lac repressor protein LacI (Fig. 3A). In both Escherichia coli and S. pombe, such lacO-LacI arrays can impede DNA replication (52, 54; our unpublished data). However, by using low levels of LacI expression, this problem is avoided, and the lacO-LacI array can be used as an “inert cytological marker” for RTS1 to assess its colocalization with Rad51 by immunostaining of spread nuclei (Fig. 3B and data not shown). We used this system to compare wild-type, fbh1Δ, srs2Δ, and rqh1Δ strains, with RTS1 in orientation 1 and 2, for the percentage of Rad51 foci that colocalize with RTS1 (lacO-LacI). Initially our aim was to use synchronized populations of cells for this analysis so that we could see whether Rad51 colocalized with RTS1 during S phase. However, we were unable to obtain synchronized populations of fbh1Δ mutant cells using standard approaches (data not shown). We therefore resorted to using asynchronously growing cells. Surprisingly the orientation of RTS1 made little difference to the mean number of Rad51 foci in either wild-type or mutant strains (Fig. 3C). This contrasts with data using a related system in which RTS1 elements flank a ura4 marker and thereby prevent its replication (33). Here a general increase in the number of cells with Rad51 foci was observed with asynchronous populations when the RTS1 RFBs were activated by expression of the barrier protein Rtf1 (33). We are uncertain why we do not see a similar effect, but it could relate to the fact that in our system the presence of only one RTS1 element means that converging replication forks can merge. The data in Fig. 3C also show no significant difference in the mean numbers of Rad51 foci in wild-type and fbh1Δ cells, which contrasts with the heightened levels of spontaneous foci in fbh1Δ cells observed earlier (47) (Fig. 1). This difference appears to be due to the conditions used to culture fbh1Δ cells; in rich media they exhibit increased numbers of spontaneous Rad51 foci, whereas in minimal media they do not and growth and viability are improved, although increased RTS1-induced recombination is still observed (data not shown). A possible explanation for the improvement in fbh1Δ growth/viability in minimal media is discussed later. Even though an fbh1Δ mutant exhibits a big increase in Rad51-dependent recombination when RTS1 is in orientation 2, a significant increase in the percentage of Rad51 foci that colocalize with RTS1 in orientation 2 compared to orientation 1 is not seen (Fig. 3D). The same is true for both wild-type and rqh1Δ strains (Fig. 3D). In contrast, the srs2Δ mutant shows a significant increase in the number of Rad51 foci that colocalize with RTS1 in orientation 2, which is consistent with the idea that Srs2 limits Rad51 accumulation at blocked replication forks (50).
FIG. 3.

Colocalization of Rad51 foci at the RTS1 RFB. (A) Schematic of the recombination substrate plus the lacO array integrated at the ade6 locus on chromosome 3. Asterisks indicate point mutations in the ade6 alleles. (B) Representative spread nuclei from srs2Δ mutant cells containing RTS1 (orientation 2), the adjacent lacO array, and pREP81-LacI-ECFP. The cells were grown to logarithmic phase in EMMG supplemented with thiamine. Spreads were stained with antibodies against Rad51 (red) and the ECFP tag on LacI (green). The DNA is stained with DAPI (blue). Two merged images are shown; the one on the left is an example in which foci are not colocalizing, and the one on the right is one in which they are. (C) Mean number of Rad51 foci per cell in strains FO1791, FO1792, MCW2349, MCW2351, MCW2366, MCW2368, MCW2406, and MCW2408. (D) The percentage of Rad51 foci shown in panel C that colocalize with the lacO array. The data shown in panels C and D are mean values from three independent experiments in which a total of 150 spread nuclei (50 per experiment) were assessed for each strain. The error bars are the standard deviations. The one-tailed Fisher exact test was used to obtain the P values for the indicated comparisons. wt, wild type.
Overexpression of Fbh1 sensitizes wild-type cells to genotoxins and limits RTS1-induced recombination.
The failure to detect an accumulation of Rad51 at the RTS1 RFB in an fbh1Δ mutant is not consistent with the idea that Fbh1, like Srs2, limits recombination by acting as a Rad51 disruptase. However, the sensitivity of our analysis may have been compromised by the inability to synchronize fbh1Δ cells, especially if Rad51 accumulation at RTS1 occurs only when forks are blocked in S phase. We therefore looked for other evidence that Fbh1 might control recombination by acting as a Rad51 disruptase. We reasoned that if Fbh1 acts in this way, then simply overexpressing it should result in the displacement of Rad51 from DNA and thereby produce a phenocopy of a rad51Δ mutant. As rad51Δ mutants are hypersensitive to a range of genotoxins, we first analyzed the effect of Fbh1 overexpression on the sensitivity of a wild-type strain to UV, HU, MMS, and CPT. The fbh1 gene was overexpressed from the thiamine-repressible nmt promoter in plasmid pREP41. In the presence of thiamine, the nmt promoter is repressed, and under these conditions the pREP41-fbh1 plasmid has little or no effect on the genotoxin sensitivity of the wild-type strain (data not shown). However, in the absence of thiamine, the expression of fbh1 is switched on and results in a marked increase in hypersensitivity to UV, HU, MMS, and CPT, compared to the same strain carrying the empty pREP41 plasmid (Fig. 4A). In contrast, overexpression of Fbh1 in a rad51Δ mutant does not dramatically increase sensitivity, indicating that the sensitization of the wild type is a consequence of Fbh1 perturbing Rad51 activity (Fig. 4B). To see if Fbh1's helicase/translocase activity is responsible for this sensitization, we also analyzed the effect of overexpressing fbh1D485N. Intriguingly, Fbh1D485N increases sensitivity even more than wild-type Fbh1 and is especially toxic in a rad51Δ mutant (Fig. 4A and B). Toxicity is also seen with an fbh1Δ mutant, indicating that it is not simply mediated by poisoning endogenous wild-type Fbh1 (data not shown). When overexpressed, Fbh1D485N forms multiple foci on chromatin spreads, even when cells are not challenged with genotoxins (data not shown). We therefore suspect that it causes toxicity by failing to turn over at sites at which Fbh1 normally acts and consequently impedes other repair processes. A failure to turn over could also act to titrate its interaction partner Skp1 (57), potentially resulting in changes in global ubiquitin ligase activity, which could have pleiotropic effects, in terms of sensitizing cells to genotoxins.
FIG. 4.

The effect of Fbh1 overexpression on repair and recombination. (A and B) Spot assays showing the sensitization to genotoxins of wild-type (MCW1221) and rad51Δ strains (MCW1088) by Fbh1 or Fbh1D485N overexpression. (C) Ade+ recombinant frequencies in a wild-type strain (MCW1262) containing the recombination substrate shown in Fig. 2A (RTS1 orientation 1) and carrying plasmids pREP41, pREP41-fbh1, and pREP41-fbh1D485N. (D) Same representation shown in panel C, except that the strain (MCW1433) contains RTS1 in orientation 2. Error bars are the standard deviations for mean values. Note that the recombination data shown here and in subsequent figures are derived from strains grown on minimal media (EMMG), whereas the data shown in Fig. 2B and C are from strains grown on YES. In most cases we observed a general reduction in the frequency of recombinants when equivalent strains were grown on EMMG compared to YES. (E) 2D gel analysis and quantification of the RFB in strain MCW1433 carrying the indicated plasmids. See the Fig. 2D legend for further details.
To extend this analysis further we examined the effect of Fbh1 overexpression on both spontaneous and RTS1-induced direct-repeat recombination (Fig. 4C and D). As mentioned earlier, the deletion of rad51 results in a loss of spontaneous conversion-type recombinants together with a marked increase in deletion types, whereas for RTS1-induced recombination both types of recombinants are reduced (Fig. 2B and C). If overexpressing Fbh1 indiscriminately removes Rad51 from DNA, then it should produce a phenocopy of rad51Δ for both spontaneous and RTS1-induced recombination; however, pREP41-fbh1 has no effect on the frequency of spontaneous recombinants but does significantly reduce RTS1-induced recombinants (Fig. 4C and D; see also Table S2 in the supplemental material). This reduction is not due to a loss of RTS1 barrier activity, as judged by the analysis of replication intermediates by 2D gel electrophoresis (Fig. 4E). Instead it is likely to be a consequence of Fbh1 limiting Rad51 activity. Intriguingly, the reduction in deletion-type recombinants (∼4-fold) caused by Fbh1 overexpression is similar to that obtained by the deletion of rad51 (∼2.5-fold), whereas for conversion types, the reduction (∼7-fold) is somewhat less (rad51Δ reduces RTS1-induced conversion types by ∼100-fold). These data suggest that Fbh1 is more effective at blocking the formation of Rad51-dependent deletion-type recombinants than it is at blocking conversion-type recombinants.
Like wild-type Fbh1, the overexpression of Fbh1D485N has no significant effect on the frequency of spontaneous conversion- or deletion-type recombinants, whereas it has a dramatic impact on the frequency of RTS1-induced recombinants (Fig. 4C and D; see also Table S2 in the supplemental material). However, unlike wild-type Fbh1, it causes an increase in recombinants rather than a decrease. Importantly there is a greater increase in deletion-type recombinants than conversion types (6-fold versus 2.3-fold), and this is similar to the effect of fbh1 deletion and suggests that Fbh1D485N overexpression impedes the activity of the endogenous wild-type protein. Consistent with this, Fbh1D485N overexpression has no effect on the frequency of RTS1-induced recombinants in an fbh1Δ strain (data not shown). Taken together, these data indicate that Fbh1 retains its specificity for blocked replication forks, even when overexpressed, and suggest that its helicase/translocase activity is necessary for limiting Rad51 activity at these sites.
Fbh1 overexpression reduces DNA damage-induced Rad51 foci without affecting Rad11 or Rad22 foci.
To more directly test whether Fbh1 limits the accumulation of Rad51 on DNA, we analyzed the effect of its overexpression on the formation of CPT-induced Rad51 foci in wild-type cells (Fig. 5A and B). Control cells containing pREP41 showed a typical induction of Rad51 foci following CPT treatment, albeit the number of induced foci are less than that shown in Fig. 1, because of the longer cell generation time in minimal media. In contrast, the mean number of Rad51 foci following CPT treatment is reduced by approximately threefold when Fbh1 is overexpressed from the pREP41-fbh1 plasmid. This reduction is statistically significant (P < 6 × 10−9) (Fig. 5B). Moreover, the number of nuclei containing more than five Rad51 foci is reduced by >16-fold. Again this reduction is significant (P < 0.05). With the same cells, we also measured the induction of RPA foci using an antibody directed against a GFP tag on its Rad11 subunit (Fig. 5A and B). Similar to those of Rad51, Rad11-GFP foci are induced by CPT treatment. However, unlike with Rad51 foci, the mean number of Rad11-GFP foci following CPT treatment remains largely unaffected by Fbh1 overexpression, indicating that the early steps in DNA damage processing are unperturbed by Fbh1 (Fig. 5B). We then repeated the experiment using a strain that expresses Rad22 fused to yellow fluorescent protein (YFP) from the endogenous rad22 promoter. Rad22-YFP foci are induced by CPT treatment and, like Rad11-GFP foci, are unaffected by the overexpression of Fbh1 (Fig. 5A and B). This shows that Fbh1 does not inhibit the loading of Rad22 onto DNA, which immediately precedes Rad51 loading. Finally we examined the effect of Fbh1D485N overexpression on the formation of Rad51 foci. As with fbh1Δ and fbh1D485N mutant strains, high levels of Rad51 foci were present, even in the absence of CPT treatment, providing further evidence that Fbh1D485N overexpression might impede the function of the endogenous wild-type Fbh1 (see Fig. S1 in the supplemental material).
FIG. 5.

The effect of Fbh1 overexpression on Rad51, Rad11-GFP, and Rad22-YFP focus formation. (A) Representative spread nuclei from wild-type cells carrying pREP41 or pREP41-fbh1 grown for 5 h in EMMG containing 10 μM CPT. Spreads were stained with antibodies against Rad51 or the tags on Rad11 and Rad22, as indicated. DAPI-stained DNA (blue). (B) Quantification of data shown in panel A. Each value represents the analysis of at least 100 nuclei from at least three different transformants. Rad51 and Rad11 foci were assessed with the same strain (FO1776) and cultures, whereas Rad22 was assessed with strain FO1752. Numbers on top of each bar are the mean number of foci per nucleus and are compared by t testing, where indicated. Note that neither strain FO1752 (containing rad22-YFP) nor FO1776 (containing rad11-GFP) shows any overt growth defects or hypersensitivity to genotoxins, indicating that the tagged forms of Rad22 and Rad11 are functional (data not shown).
The data above suggest that Fbh1 specifically acts on Rad51 to curb its loading onto DNA. To provide further evidence for this, we looked to see whether Fbh1 and Rad51 foci colocalize following CPT treatment. To detect Fbh1 by immunostaining we overexpressed it from the pREP41 nmt promoter with a double myc tag (2myc) on its N terminus and used a monoclonal anti-c-Myc antibody. 2myc-Fbh1 fully complements the genotoxin hypersensitivities of an fbh1Δ mutant (data not shown) and when overexpressed reduces the number of Rad51 foci that are induced by CPT (Fig. 6A), indicating that it is fully functional. However, only 7% of 2myc-Fbh1 foci and 27% of Rad51 foci colocalize following CPT treatment (Fig. 6B). Presumably the high levels of 2myc-Fbh1 result in the rapid removal of Rad51 from DNA, which in turn results in the apparent lack of colocalization. Consistent with this idea ∼66% of Rad11-GFP foci and ∼63% of Rad22-YFP foci colocalize with 2myc-Fbh1 under these conditions, indicating that Fbh1 is recruited to sites where Rad51 would be expected to act (Fig. 6C and D). In apparent contradiction to the data above, Fbh1 tagged at its N terminus with YFP, when overexpressed from the pREP41 nmt promoter, forms CPT-induced foci that colocalize mostly (>90%) with CFP-tagged Rad51 foci (48). However, unlike 2myc-Fbh1, YFP-Fbh1 complements only partially the genotoxin hypersensitivities of an fbh1Δ mutant and therefore probably exhibits strong colocalization with Rad51 because it is impaired for its removal (data not shown). Altogether, these data are consistent with Fbh1 targeting specifically Rad51 to curb its loading onto DNA.
FIG. 6.
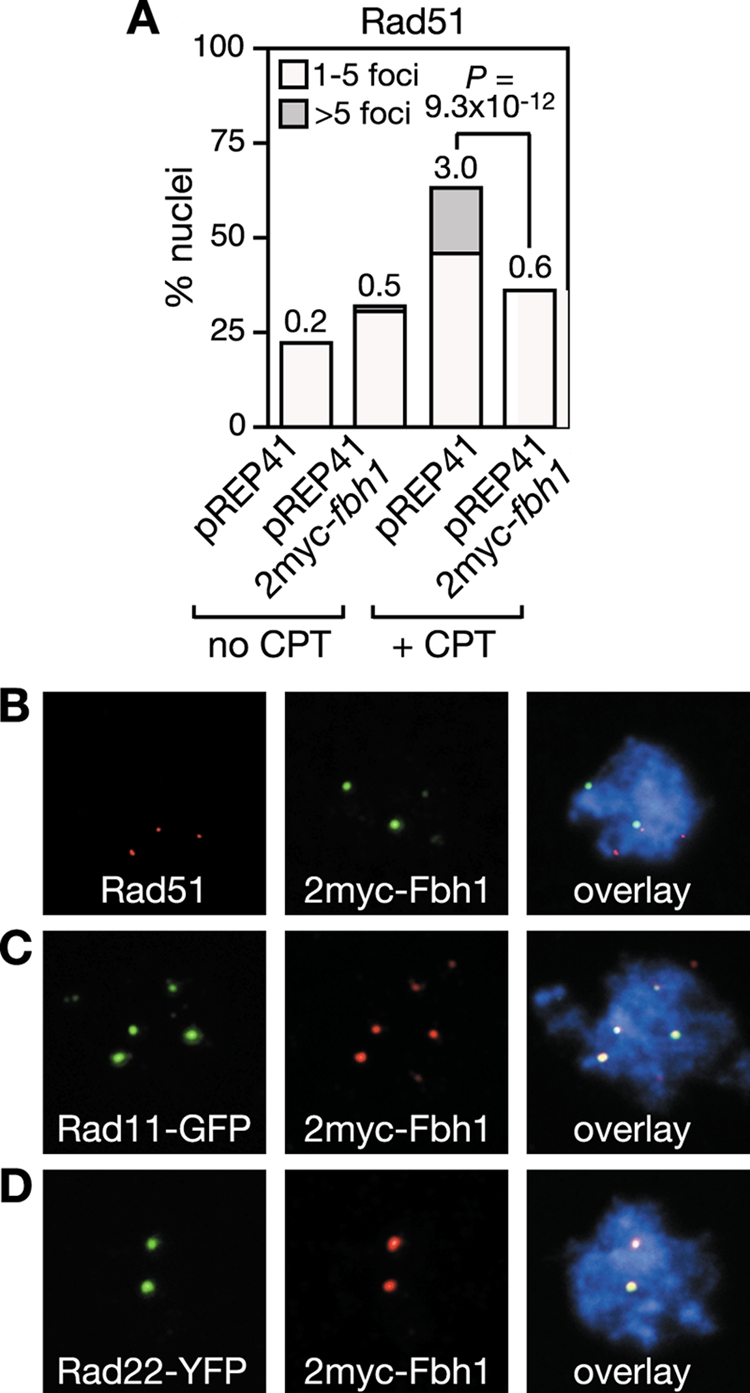
Fbh1 colocalizes with Rad11 and Rad22 in nuclear foci. (A) Effect of 2myc-Fbh1 overexpression on Rad51 focus formation in strain FO1776. Culturing and quantification are the same as those shown in Fig. 5. Colocalization of 2myc-Fbh1 foci with Rad51 (B), Rad11 (C), and Rad22 (D) foci following growth in CPT, as shown in panel A. Out of 59 Rad51 foci from 100 nuclei, 16 colocalized with 2myc-Fbh1. In contrast, 87 out of 131 Rad11 foci from 48 nuclei and 59 out of 94 Rad22 foci from 50 nuclei colocalized with 2myc-Fbh1.
Fbh1 overexpression limits recombination and Rad51 accumulation at RTS1 in an srs2Δ mutant.
To gain further evidence that Fbh1 can limit recombination at RTS1 by acting as a Rad51 disruptase, we determined what effect its overexpression would have on the increased levels of spontaneous and RTS1-induced recombination in an srs2Δ mutant (Fig. 7A and B; see also Table S2 in the supplemental material). In the case of spontaneous recombinants, Fbh1 overexpression from the pREP41 nmt promoter has no effect on the frequency of deletion types but does significantly reduce conversion types (Fig. 7A; see also Table S2 in the supplemental material). In contrast, both types of RTS1-induced recombinants are reduced (Fig. 7B; see also Table S2 in the supplemental material). This last effect correlates with a reduction in Rad51 foci that colocalize with the RTS1 RFB (Fig. 7C). Together these data suggest that Fbh1 can substitute for Srs2 in limiting recombination at blocked replication forks, most probably by acting as a Rad51 disruptase.
FIG. 7.

The effect of Fbh1 overexpression on the spontaneous and RTS1-induced hyperrecombination in an srs2Δ mutant. (A) Ade+ recombinant frequencies in an srs2Δ mutant strain (FO1748) containing the recombination substrate shown in Fig. 2A (RTS1 orientation 1) and carrying plasmid pREP41 or pREP41-fbh1, as indicated. (B) Same analysis as shown in panel A, except that the strain (FO1750) contains RTS1 in orientation 2. Error bars are the standard deviations for the mean values. (C) The effect of Fbh1 overexpression on the percentage of Rad51 foci that colocalize with lacO-LacI foci in strain MCW2368. Note that in this experiment, Fbh1 is expressed from the wild-type nmt promoter in pREP2. This produces approximately the same level of Fbh1 overexpression in the presence of thiamine (needed to limit expression of LacI-ECFP from the pREP81 nmt promoter) as the pREP41 nmt promoter does in its absence. The data are mean values from three independent experiments in which a total of 150 spread nuclei (50 per experiment) were assessed for each strain. The error bars are the standard deviations, and the P value was obtained using a one-tailed Fisher exact test.
Srs2 overexpression limits RTS1-induced recombination in both wild-type and fbh1Δ mutant strains.
If Fbh1 is a Rad51 disruptase, then overexpression of Srs2 should provide a good substitute for it. Initially we looked to see whether the genotoxin sensitivities of an fbh1Δ mutant could be suppressed. However, a general toxic effect of Srs2 overexpression masked any potential benefits that it might have here (data not shown). We next analyzed its effect on RTS1-induced recombination in both wild-type and fbh1Δ strains (Fig. 8A and B; see also Table S3 in the supplemental material). Here Srs2 overexpression reduces the frequency of conversion types by more than eightfold in the wild type and more than threefold in the fbh1Δ mutant, while having relatively little or no effect on the frequency of deletion types. This reduction appears to depend mainly on Srs2's helicase/translocase activity because overexpression of Srs2 carrying a D238N mutation in helicase motif II, which should prevent ATP hydrolysis (51), has little or no effect on recombinant frequencies. Altogether these data are consistent with the idea that recombination at the RTS1 RFB can stem from the accumulation of Rad51 at the blocked fork in wild-type and fbh1Δ cells. However, the relative inability of Srs2 to limit deletion types highlights a difference from Fbh1.
FIG. 8.

The effect of Srs2 overexpression on RTS1-induced recombination in wild-type and fbh1Δ strains. (A) Ade+ recombinant frequencies in a wild-type strain (MCW1433) containing the recombination substrate shown in Fig. 2A (RTS1 orientation 2) and carrying plasmids pREP41, pREP41-srs2, and pREP41-srs2D238N, as indicated. (B) Same analysis as shown in panel A, except the strain (FO1816) is an fbh1Δ mutant. Error bars are the standard deviations for mean values.
DISCUSSION
Here we have provided further evidence that Fbh1 controls HR by preventing or limiting Rad51 nucleofilament assembly and in so doing counters the pronucleofilament assembly function of Rad22. We suspect that the balance between Fbh1 and Rad22 is critical to achieve appropriately formed Rad51 nucleofilaments, which are active yet not predisposed to aberrant behavior. Exactly how Fbh1 controls Rad51 activity is still not certain, although it is clear from the data presented here and elsewhere that its helicase/translocase activity is required (48, 57). Fbh1 could act as part of a larger complex that controls Rad51 nucleofilament formation possibly through promoting the ubiquitination of target proteins (57). Here its translocase activity might be used solely to localize the complex to sites where it is needed (e.g., blocked replication forks). However, based on its similarity to Srs2, we suspect that Fbh1 can use its translocase activity to displace Rad51 presynaptic filaments as it moves along the DNA. Indeed this idea provides a simple explanation for how overexpressing just Fbh1 can reduce RTS1-induced recombination, the number of Rad51 foci that form in response to CPT, and the heightened colocalization of Rad51 with RTS1 in an srs2Δ mutant. However, it is worth noting that both Srs2 and RECQL5, which disrupt Rad51 nucleofilaments, also physically interact with Rad51 (25, 31), and currently we have no direct evidence that Fbh1 shares this property.
The induction of Fbh1 nuclear foci in response to various genotoxins suggests that it routinely functions to modulate Rad51 nucleofilament formation in response to DNA damage (47, 48). However, it does not appear to act whenever/wherever Rad51 mediates HR because, unlike an srs2Δ mutant (19), an fbh1Δ mutant does not display an increase in spontaneous direct-repeat recombination at the ade6 locus. In fact, the only situation where we have seen an effect of fbh1Δ on spontaneous recombination is with a rad22Δ mutant (48), and it could be argued that this is a pathological case and Fbh1 is acting only because of an abnormality in Rad51 nucleofilament formation. Although Fbh1 is not required to limit spontaneous recombination at the ade6 locus, it does appear to limit Rad51 activity elsewhere, because fbh1Δ mutant cells growing in rich media display abnormally high levels of Rad51 nuclear foci (47) (Fig. 1). Our favored explanation for this is that Fbh1 may act only where there is a free duplex DNA end, produced either by double-strand breakage or replication fork regression. Certainly, Fbh1 forms foci in response to genotoxins, such as CPT, that cause fork breakage, as well as meiotic DSBs generated by the Spo11 orthologue Rec12 (N. Sun and M. C. Whitby, unpublished data). Under normal growth conditions, DSBs and fork regression may be restricted mainly to fragile genomic sites, which are difficult to replicate (13, 36). In contrast, spontaneous recombination at the ade6 locus may be initiated mainly from single-strand gaps rather than DSBs (37).
An fbh1Δ mutant has improved growth and viability when grown in minimal media (EMMG) compared to growth in rich media (YES). This difference correlates with a reduction in the levels of spontaneous Rad51 nuclear foci (e.g., compare the data shown in Fig. 1 and 3D). If Fbh1 functions predominantly at fragile sites, then the improved growth/viability of an fbh1Δ mutant in minimal media suggests that these sites manifest their fragility to Fbh1 mainly under optimal growth conditions. Why this might be is unclear; however, one possibility is that under slower growth conditions (i.e., in EMMG) there is a greater induction of the induced core environmental stress response genes (7, 12, 14, 41), which could limit the amount of spontaneous DNA damage (e.g., antioxidants limiting the levels of reactive oxygen species and therefore in turn the amount of oxidative damage that the DNA suffers). The faster growth in YES would therefore be accompanied by a greater amount of spontaneous DNA damage. Such damage might provoke a greater recombinational response when coincident with fragile sites. Indeed the frequency of recombination induced by the RTS1 barrier when cells are grown on YES is always greater than that on EMMG (e.g., compare the wild-type recombinant frequencies for RTS1 orientation 2 shown in Fig. 2C, for which cells were grown on YES, and those shown in Fig. 4D, for which cells were grown on EMMG). It is also possible that an increase in reactive oxygen species in YES could directly damage components of the replisome, leading to impaired replication fork progression (22), and since replisome impairment can induce DNA breakage at fragile sites (21, 36), the need for Fbh1 at these sites might be greater when cells are grown in YES rather than EMMG.
Consistent with the idea that Fbh1 acts at sites that are difficult to replicate, it strongly suppresses Rad51-dependent recombinant formation when replication forks are blocked at the programmed RFB RTS1 (Fig. 2). However, our inability to detect an increase in Rad51 accumulation at RTS1 in an fbh1Δ mutant means that we cannot be certain whether the increase in recombinants is a consequence of Fbh1 failing to prevent Rad51 nucleofilament formation at the blocked fork. Indeed, if this is its function why is it not redundant with Srs2? Here one could argue that there might be limiting amounts of both proteins. Consistent with this, overexpression of Fbh1 suppresses the hyper-recombination and increase in Rad51 foci that colocalize with RTS1 in an srs2Δ mutant (Fig. 7). However, the proportions of RTS1-induced conversion- and deletion-type recombinants in the srs2Δ and fbh1Δ mutants (Fig. 2) are different, and this is mirrored by the relative propensities of the overexpressed proteins to suppress the different types of recombinants (Fig. 7 and 8). These data suggest that, while there might be some overlap in function, Fbh1 and Srs2 have distinct roles in limiting Rad51 activity at RFBs. The absence of Fbh1 in S. cerevisiae suggests that Srs2 would perform both roles in this organism.
It is interesting to speculate what the distinct roles of Fbh1 and Srs2 might be. However, the likelihood that these factors play different roles in different situations generates a level of complexity that makes the derivation of an all-encompassing model difficult. The discussion that follows therefore provides possibilities that fit with at least some of the data. One idea is that Fbh1 and Srs2 function primarily on different Rad51 nucleofilaments with different propensities to give rise to conversion- and deletion-type recombinants. For example, Srs2 might work on Rad51 nucleofilaments that form at single-stranded gaps, whereas Fbh1 could work on those that form on ssDNA tails (arising as a consequence of fork regression and/or breakage). As alluded to above, this could explain why Srs2, and not Fbh1, is required to limit spontaneous direct-repeat recombination at ade6. If fork regression/breakage at RTS1 is relatively infrequent, this could also explain why we do not see Rad51 accumulation at RTS1 in an fbh1Δ mutant.
Another idea is that Fbh1 and Srs2 might function at different stages of the recombination reaction. For example, Fbh1 might act at an early stage to limit Rad51 nucleofilament formation (i.e., during presynapsis), whereas Srs2 might act later, after strand invasion (i.e., during postsynapsis), combining both D-loop dissociation and Rad51 nucleofilament disruption in a single reaction (20). We have incorporated this idea into a hypothetical model for RTS1-induced recombinant formation (Fig. 9). Here Fml1 promotes regression of the replication fork blocked at RTS1, generating a double-stranded DNA end (Fig. 9, step 2) (59). This end is then resected to generate an ssDNA tail with a 3′-OH terminus, onto which Rad51 can load (Fig. 9, step 3). As discussed previously the purpose of this activity at a programmed RFB is unclear but may reflect an attempt to promote a recombination-dependent replication restart (3, 59). The loading of Rad51 onto the ssDNA is controlled by the opposing activities of Rad22 and Fbh1, as discussed above (Fig. 9, step 4). Once loaded, Rad51 can presumably promote Ade+ recombinant formation by catalyzing strand invasion from ade6-M375 into ade6-L469 (Fig. 9, step 5). In wild-type cells, approximately 50% of the Ade+ recombinants derive from Rad51-dependent gene conversion of ade6-M375, and approximately 30% from Rad51-dependent deletion events (3, 59) (see also Table S1 in the supplemental material). The remaining 20% are mostly Rad51-independent deletions (3, 59) (see also Table S1 in the supplemental material). As proposed previously, we imagine that the RTS1-induced Rad51-dependent Ade+ recombinants stem from alternative processing of the D loop that is formed by strand invasion (3, 59). DNA synthesis primed from the invading 3′ end results in the copying of the wild-type ade6 sequence (Fig. 9, step 7a). Subsequent unwinding of the D loop by helicases, such as Rqh1 and/or Fml1, results in heteroduplex formation at one of the copies of ade6-M375, which may ultimately result in its conversion to ade+ (Fig. 9, step 8a to 9a). Alternatively the D loop and blocked fork are cleaved by a nuclease, such as Mus81-Eme1 (49, 64), and thereby give rise to an Ade+ deletion type (Fig. 9, steps 7b to 9b). We think that Fbh1's role in limiting Rad51 nucleofilament formation (Fig. 9, step 4) might result in shorter D loops that are more susceptible to disruption by Srs2, which thereby aborts recombination (Fig. 9, steps 5a to 6a). In S. cerevisiae, Srs2 is thought to translocate in a 3′-to-5′ direction along the RPA-coated displaced strand and upon reaching the four-way DNA junction switches to the Rad51-coated heteroduplex, where it unwinds the D loop in a reaction that is stimulated by Rad51 (20) (Fig. 9, insert). It is also thought that Rad54 translocation on duplex DNA displaces Rad51 from the 3′ end of the invading strand to enable priming of DNA synthesis (38) (Fig. 9, insert). We imagine that if DNA synthesis is primed before Srs2 reaches the end of the Rad51 nucleofilament, then D-loop disassembly will be avoided. In this way the balance between Rad54 and Srs2 activities would determine whether the D loop gives rise to a recombinant or not (Fig. 9, steps 6a and b). However, even if DNA synthesis is primed, the shorter D loops that result from the action of first Fbh1 and then Srs2 might be predisposed to give rise to conversion types rather than deletion types. This idea is based on the notion that DNA unwinding is the default mode for processing a D loop and that cleavage occurs only if the D loop persists. Therefore, the D loops formed by the longer Rad51 nucleofilaments in an fbh1Δ mutant are not only more resistant to dissociation by Srs2, resulting in an overall increase in recombinant formation, but may also persist longer, allowing a greater chance for cleavage and thereby deletion-type formation (Fig. 9, steps 5b, 6c, and 7b to 9b). In contrast, the D loops formed by the shorter Rad51 nucleofilaments in an srs2Δ mutant, while persisting long enough for DNA synthesis to be primed, may nevertheless be more susceptible to being unwound and thereby give rise to more conversion types than deletion types (Fig. 9, steps 5a, 6b, and 7a to 9a). Overexpression of Srs2 may preferentially reduce conversion types in both wild-type and fbh1Δ strains (Fig. 8) because these recombinants tend to derive from the D loops formed by the shorter Rad51 nucleofilaments and are therefore susceptible to dissociation by Srs2.
FIG. 9.

Hypothetical model for direct-repeat recombination induced by replication fork blockage at RTS1. Where there is a bifurcation of pathways, arrow sizes indicate relative pathway bias. See Discussion for further details. This figure is adapted from references 3 and 59.
Some support for this speculative model comes from genetic interaction data. For example, the reciprocal suppression of the fbh1Δ and rad22Δ single mutant phenotypes in a fbh1Δ rad22Δ double mutant is consistent with the idea that Fbh1 acts during presynapsis to modulate Rad51 nucleofilament assembly (48). In contrast, an srs2Δ rad22Δ double mutant exhibits exactly the same poor growth and genotoxin sensitivity as a rad22Δ single mutant (data not shown). This is consistent with a postsynaptic role for Srs2, which is needed only if Rad22 is present.
Another genetic interaction that distinguishes fbh1 from srs2 is with rad54. Specifically an srs2Δ rad54Δ double mutant is inviable (19, 43), whereas a fbh1Δ rad54Δ double mutant is viable and, in fact, grows slightly better than an fbh1Δ single mutant (see Fig. S2 in the supplemental material). The viability of an srs2Δ rad54Δ mutant is restored by deleting Rad51, which is consistent with the idea that Srs2 and Rad54 function in alternative pathways for processing Rad51-dependent recombination intermediates (19, 43). Rad54 is a member of the Swi2/Snf2 family of DNA-dependent ATPases and is believed to fulfill multiple roles in HR. These roles include ATP-independent promotion of Rad51 nucleofilament formation/stability, as well as ATP-dependent processes, such as chromatin remodeling, promotion of Rad51-mediated strand invasion, D-loop dissociation, Holliday junction branch migration, and removal of Rad51 from duplex DNA (9, 10, 24). The last function is thought to promote efficient turnover of Rad51 after strand invasion and ensure accessibility of the invading 3′ end for priming DNA synthesis (24, 38). Exactly which of these functions is critical in the absence of Srs2 is not certain, although in terms of our model it is reasonable to think that Rad54 provides an alternative to Srs2 for processing the D loop. If Srs2 plays an important role in D-loop processing, the slightly improved growth of an fbh1Δ rad54Δ double mutant compared to an fbh1Δ single mutant suggests that Fbh1 does not. However, one might predict that Rad54's activity would become more critical in the absence of Fbh1 due to a greater need to process D loops formed by long Rad51 nucleofilaments. Perhaps the absence of Rad54's early role in promoting Rad51 nucleofilament formation/stability suppresses some of the heightened Rad51 activity in an fbh1Δ mutant, thereby explaining why a fbh1Δ rad54Δ double mutant exhibits improved growth rather than reduced viability.
In conclusion, we have provided further evidence suggesting that Fbh1 and Srs2 are Rad51 disruptases and discussed how such seemingly equivalent activities could be utilized in distinct ways within the cell. This may be relevant for understanding how FBH1 is deployed in humans, where BLM and RECQL5 are also proposed to act as Rad51 disruptases (11, 25). From our data we would predict that, while there might be an overlap in activity, these different Rad51 disruptases will have distinct functions, and clarifying what these are is an important goal for future research. Finally, as defects in both BLM and RECQL5 result in cancer predisposition (23, 25), it would not be surprising if the same were true of FBH1.
Supplementary Material
Acknowledgments
We thank J. Cooper and G. Baldacci for strains. We also thank H. Jahari for constructing plasmids pIJ9 and pIJ12.
This work was supported by a Wellcome Trust Senior Research Fellowship awarded to M.C.W. A.L. was supported in part by an Erwin Schrödinger Fellowship from the Austrian Science Fund. S.S. was supported by a Skaggs-Oxford scholarship.
Footnotes
Published ahead of print on 22 June 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Aboussekhra, A., R. Chanet, Z. Zgaga, C. Cassier-Chauvat, M. Heude, and F. Fabre. 1989. RADH, a gene of Saccharomyces cerevisiae encoding a putative DNA helicase involved in DNA repair. Characteristics of radH mutants and sequence of the gene. Nucleic Acids Res. 177211-7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguilera, A., and H. L. Klein. 1988. Genetic control of intrachromosomal recombination in Saccharomyces cerevisiae. I. Isolation and genetic characterization of hyper-recombination mutations. Genetics 119779-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahn, J. S., F. Osman, and M. C. Whitby. 2005. Replication fork blockage by RTS1 at an ectopic site promotes recombination in fission yeast. EMBO J. 242011-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahler, J., T. Wyler, J. Loidl, and J. Kohli. 1993. Unusual nuclear structures in meiotic prophase of fission yeast: a cytological analysis. J. Cell Biol. 121241-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barber, L. J., J. L. Youds, J. D. Ward, M. J. McIlwraith, N. J. O'Neil, M. I. Petalcorin, J. S. Martin, S. J. Collis, S. B. Cantor, M. Auclair, H. Tissenbaum, S. C. West, A. M. Rose, and S. J. Boulton. 2008. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 135261-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Branzei, D., and M. Foiani. 2007. RecQ helicases queuing with Srs2 to disrupt Rad51 filaments and suppress recombination. Genes Dev. 213019-3026. [DOI] [PubMed] [Google Scholar]
- 7.Brauer, M. J., C. Huttenhower, E. M. Airoldi, R. Rosenstein, J. C. Matese, D. Gresham, V. M. Boer, O. G. Troyanskaya, and D. Botstein. 2008. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol. Biol. Cell 19352-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brewer, B. J., and W. L. Fangman. 1987. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51463-471. [DOI] [PubMed] [Google Scholar]
- 9.Bugreev, D. V., F. Hanaoka, and A. V. Mazin. 2007. Rad54 dissociates homologous recombination intermediates by branch migration. Nat. Struct. Mol. Biol. 14746-753. [DOI] [PubMed] [Google Scholar]
- 10.Bugreev, D. V., O. M. Mazina, and A. V. Mazin. 2006. Rad54 protein promotes branch migration of Holliday junctions. Nature 442590-593. [DOI] [PubMed] [Google Scholar]
- 11.Bugreev, D. V., X. Yu, E. H. Egelman, and A. V. Mazin. 2007. Novel pro- and anti-recombination activities of the Bloom's syndrome helicase. Genes Dev. 213085-3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castrillo, J. I., L. A. Zeef, D. C. Hoyle, N. Zhang, A. Hayes, D. C. Gardner, M. J. Cornell, J. Petty, L. Hakes, L. Wardleworth, B. Rash, M. Brown, W. B. Dunn, D. Broadhurst, K. O'Donoghue, S. S. Hester, T. P. Dunkley, S. R. Hart, N. Swainston, P. Li, S. J. Gaskell, N. W. Paton, K. S. Lilley, D. B. Kell, and S. G. Oliver. 2007. Growth control of the eukaryote cell: a systems biology study in yeast. J. Biol. 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cha, R. S., and N. Kleckner. 2002. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297602-606. [DOI] [PubMed] [Google Scholar]
- 14.Chen, D., W. M. Toone, J. Mata, R. Lyne, G. Burns, K. Kivinen, A. Brazma, N. Jones, and J. Bahler. 2003. Global transcriptional responses of fission yeast to environmental stress. Mol. Biol. Cell 14214-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiolo, I., M. Saponaro, A. Baryshnikova, J. H. Kim, Y. S. Seo, and G. Liberi. 2007. The human F-Box DNA helicase FBH1 faces Saccharomyces cerevisiae Srs2 and postreplication repair pathway roles. Mol. Cell Biol. 277439-7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Craven, R. A., D. J. Griffiths, K. S. Sheldrick, R. E. Randall, I. M. Hagan, and A. M. Carr. 1998. Vectors for the expression of tagged proteins in Schizosaccharomyces pombe. Gene 22159-68. [DOI] [PubMed] [Google Scholar]
- 17.Doe, C. L., J. Dixon, F. Osman, and M. C. Whitby. 2000. Partial suppression of the fission yeast rqh1− phenotype by expression of a bacterial Holliday junction resolvase. EMBO J. 192751-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doe, C. L., F. Osman, J. Dixon, and M. C. Whitby. 2004. DNA repair by a Rad22-Mus81-dependent pathway that is independent of Rhp51. Nucleic Acids Res. 325570-5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doe, C. L., and M. C. Whitby. 2004. The involvement of Srs2 in post-replication repair and homologous recombination in fission yeast. Nucleic Acids Res. 321480-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dupaigne, P., C. Le Breton, F. Fabre, S. Gangloff, E. Le Cam, and X. Veaute. 2008. The Srs2 helicase activity is stimulated by Rad51 filaments on dsDNA: implications for crossover incidence during mitotic recombination. Mol. Cell 29243-254. [DOI] [PubMed] [Google Scholar]
- 21.Durkin, S. G., and T. W. Glover. 2007. Chromosome fragile sites. Annu. Rev. Genet. 41169-192. [DOI] [PubMed] [Google Scholar]
- 22.Girard, P. M., M. Pozzebon, F. Delacote, T. Douki, V. Smirnova, and E. Sage. 2008. Inhibition of S-phase progression triggered by UVA-induced ROS does not require a functional DNA damage checkpoint response in mammalian cells. DNA Repair (Amsterdam) 71500-1516. [DOI] [PubMed] [Google Scholar]
- 23.Hanada, K., and I. D. Hickson. 2007. Molecular genetics of RecQ helicase disorders. Cell. Mol. Life Sci. 642306-2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heyer, W. D., X. Li, M. Rolfsmeier, and X. P. Zhang. 2006. Rad54: the Swiss Army knife of homologous recombination? Nucleic Acids Res. 344115-4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu, Y., S. Raynard, M. G. Sehorn, X. Lu, W. Bussen, L. Zheng, J. M. Stark, E. L. Barnes, P. Chi, P. Janscak, M. Jasin, H. Vogel, P. Sung, and G. Luo. 2007. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 213073-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huberman, J. A., L. D. Spotila, K. A. Nawotka, S. M. el-Assouli, and L. R. Davis. 1987. The in vivo replication origin of the yeast 2 μm plasmid. Cell 51473-481. [DOI] [PubMed] [Google Scholar]
- 27.Ira, G., A. Malkova, G. Liberi, M. Foiani, and J. E. Haber. 2003. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115401-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim, J., J. H. Kim, S. H. Lee, D. H. Kim, H. Y. Kang, S. H. Bae, Z. Q. Pan, and Y. S. Seo. 2002. The novel human DNA helicase hFBH1 is an F-box protein. J. Biol. Chem. 27724530-24537. [DOI] [PubMed] [Google Scholar]
- 29.Kim, J. H., J. Kim, D. H. Kim, G. H. Ryu, S. H. Bae, and Y. S. Seo. 2004. SCFhFBH1 can act as helicase and E3 ubiquitin ligase. Nucleic Acids Res. 322287-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kohzaki, M., A. Hatanaka, E. Sonoda, M. Yamazoe, K. Kikuchi, N. Vu Trung, D. Szuts, J. E. Sale, H. Shinagawa, M. Watanabe, and S. Takeda. 2007. Cooperative roles of vertebrate Fbh1 and Blm DNA helicases in avoidance of crossovers during recombination initiated by replication fork collapse. Mol. Cell. Biol. 272812-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krejci, L., S. Van Komen, Y. Li, J. Villemain, M. S. Reddy, H. Klein, T. Ellenberger, and P. Sung. 2003. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423305-309. [DOI] [PubMed] [Google Scholar]
- 32.Krogh, B. O., and L. S. Symington. 2004. Recombination proteins in yeast. Annu. Rev. Genet. 38233-271. [DOI] [PubMed] [Google Scholar]
- 33.Lambert, S., A. Watson, D. M. Sheedy, B. Martin, and A. M. Carr. 2005. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 121689-702. [DOI] [PubMed] [Google Scholar]
- 34.Lau, I. F., S. R. Filipe, B. Soballe, O. A. Okstad, F. X. Barre, and D. J. Sherratt. 2003. Spatial and temporal organization of replicating Escherichia coli chromosomes. Mol. Microbiol. 49731-743. [DOI] [PubMed] [Google Scholar]
- 35.Lawrence, C. W., and R. B. Christensen. 1979. Metabolic suppressors of trimethoprim and ultraviolet light sensitivities of Saccharomyces cerevisiae rad6 mutants. J. Bacteriol. 139866-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lemoine, F. J., N. P. Degtyareva, K. Lobachev, and T. D. Petes. 2005. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120587-598. [DOI] [PubMed] [Google Scholar]
- 37.Lettier, G., Q. Feng, A. A. de Mayolo, N. Erdeniz, R. J. Reid, M. Lisby, U. H. Mortensen, and R. Rothstein. 2006. The role of DNA double-strand breaks in spontaneous homologous recombination in S. cerevisiae. PLoS Genet. 2e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li, X., and W. D. Heyer. 2009. RAD54 controls access to the invading 3′-OH end after RAD51-mediated DNA strand invasion in homologous recombination in Saccharomyces cerevisiae. Nucleic Acids Res. 37638-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lorenz, A., A. Estreicher, J. Kohli, and J. Loidl. 2006. Meiotic recombination proteins localize to linear elements in Schizosaccharomyces pombe. Chromosoma 115330-340. [DOI] [PubMed] [Google Scholar]
- 40.Lorenz, A., J. L. Wells, D. W. Pryce, M. Novatchkova, F. Eisenhaber, R. J. McFarlane, and J. Loidl. 2004. S. pombe meiotic linear elements contain proteins related to synaptonemal complex components. J. Cell Sci. 1173343-3351. [DOI] [PubMed] [Google Scholar]
- 41.Lu, C., M. J. Brauer, and D. Botstein. 2009. Slow growth induces heat-shock resistance in normal and respiratory-deficient yeast. Mol. Biol. Cell 20891-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macris, M. A., and P. Sung. 2005. Multifaceted role of the Saccharomyces cerevisiae Srs2 helicase in homologous recombination regulation. Biochem. Soc. Trans. 331447-1450. [DOI] [PubMed] [Google Scholar]
- 43.Maftahi, M., J. C. Hope, L. Delgado-Cruzata, C. S. Han, and G. A. Freyer. 2002. The severe slow growth of Δsrs2 Δrqh1 in Schizosaccharomyces pombe is suppressed by loss of recombination and checkpoint genes. Nucleic Acids Res. 304781-4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maundrell, K. 1993. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 123127-130. [DOI] [PubMed] [Google Scholar]
- 45.Meister, P., M. Poidevin, S. Francesconi, I. Tratner, P. Zarzov, and G. Baldacci. 2003. Nuclear factories for signalling and repairing DNA double strand breaks in living fission yeast. Nucleic Acids Res. 315064-5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreno, S., A. Klar, and P. Nurse. 1991. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194795-823. [DOI] [PubMed] [Google Scholar]
- 47.Morishita, T., F. Furukawa, C. Sakaguchi, T. Toda, A. M. Carr, H. Iwasaki, and H. Shinagawa. 2005. Role of the Schizosaccharomyces pombe F-Box DNA helicase in processing recombination intermediates. Mol. Cell. Biol. 258074-8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osman, F., J. Dixon, A. R. Barr, and M. C. Whitby. 2005. The F-Box DNA helicase Fbh1 prevents Rhp51-dependent recombination without mediator proteins. Mol. Cell. Biol. 258084-8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Osman, F., J. Dixon, C. L. Doe, and M. C. Whitby. 2003. Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis. Mol. Cell 12761-774. [DOI] [PubMed] [Google Scholar]
- 50.Papouli, E., S. Chen, A. A. Davies, D. Huttner, L. Krejci, P. Sung, and H. D. Ulrich. 2005. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 19123-133. [DOI] [PubMed] [Google Scholar]
- 51.Pause, A., and N. Sonenberg. 1992. Mutational analysis of a DEAD box RNA helicase: the mammalian translation initiation factor eIF-4A. EMBO J. 112643-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Payne, B. T., I. C. van Knippenberg, H. Bell, S. R. Filipe, D. J. Sherratt, and P. McGlynn. 2006. Replication fork blockage by transcription factor-DNA complexes in Escherichia coli. Nucleic Acids Res. 345194-5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pfander, B., G. L. Moldovan, M. Sacher, C. Hoege, and S. Jentsch. 2005. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436428-433. [DOI] [PubMed] [Google Scholar]
- 54.Possoz, C., S. R. Filipe, I. Grainge, and D. J. Sherratt. 2006. Tracking of controlled Escherichia coli replication fork stalling and restart at repressor-bound DNA in vivo. EMBO J. 252596-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prakash, R., D. Satory, E. Dray, A. Papusha, J. Scheller, W. Kramer, L. Krejci, H. Klein, J. E. Haber, P. Sung, and G. Ira. 2009. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2367-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raveendranathan, M., S. Chattopadhyay, Y. T. Bolon, J. Haworth, D. J. Clarke, and A. K. Bielinsky. 2006. Genome-wide replication profiles of S-phase checkpoint mutants reveal fragile sites in yeast. EMBO J. 253627-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sakaguchi, C., T. Morishita, H. Shinagawa, and T. Hishida. 2008. Essential and distinct roles of the F-box and helicase domains of Fbh1 in DNA damage repair. BMC Mol. Biol. 927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schiestl, R. H., S. Prakash, and L. Prakash. 1990. The SRS2 suppressor of rad6 mutations of Saccharomyces cerevisiae acts by channeling DNA lesions into the RAD52 DNA repair pathway. Genetics 124817-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun, W., S. Nandi, F. Osman, J. S. Ahn, J. Jakovleska, A. Lorenz, and M. C. Whitby. 2008. The fission yeast FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during double-strand break repair. Mol. Cell 32118-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Symington, L. S., and W. D. Heyer. 2006. Some disassembly required: role of DNA translocases in the disruption of recombination intermediates and dead-end complexes. Genes Dev. 202479-2486. [DOI] [PubMed] [Google Scholar]
- 61.Veaute, X., S. Delmas, M. Selva, J. Jeusset, E. Le Cam, I. Matic, F. Fabre, and M. A. Petit. 2005. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 24180-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Veaute, X., J. Jeusset, C. Soustelle, S. C. Kowalczykowski, E. Le Cam, and F. Fabre. 2003. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423309-312. [DOI] [PubMed] [Google Scholar]
- 63.Wang, S. W., A. Goodwin, I. D. Hickson, and C. J. Norbury. 2001. Involvement of Schizosaccharomyces pombe Srs2 in cellular responses to DNA damage. Nucleic Acids Res. 292963-2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whitby, M. C., F. Osman, and J. Dixon. 2003. Cleavage of model replication forks by fission yeast Mus81-Eme1 and budding yeast Mus81-Mms4. J. Biol. Chem. 2786928-6935. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.