

Abstract
The REACH (realistic extension algorithm via covariance Hessian) coarse-grained biomolecular simulation method is a self-consistent multiscale approach directly mapping atomistic molecular dynamics simulation results onto a residue-scale model. Here, REACH is applied to calculate the dynamics of protein-protein interactions. The intra- and intermolecular fluctuations and the intermolecular vibrational densities of states derived from atomistic molecular dynamics are well reproduced by the REACH normal modes. The phonon dispersion relations derived from the REACH lattice dynamics model of crystalline ribonuclease A are also in satisfactory agreement with the corresponding all-atom results. The REACH model demonstrates that increasing dimer interaction strength decreases the translational and rotational intermolecular vibrational amplitudes, while their vibrational frequencies are relatively unaffected. A comparative study of functionally interacting biological dimers with crystal dimers, which are formed artificially via crystallization, reveals a relation between their static structures and the interprotein dynamics: i.e., the consequence of the extensive interfaces of biological dimers is reduction of the intermonomer translational and rotational amplitudes, but not the frequencies.
Introduction
Many cellular processes are governed by networks of directly-interacting protein molecules. On binding, the internal dynamics of interacting protein molecules can change, involving, in principle, either stiffening or softening of the binding partners. The accompanying thermodynamic change will contribute to the binding free energy, and understanding dynamical changes accompanying the formation of protein complexes is, therefore, an important goal in molecular biophysics (1–9). The development of reliable and fast methods for determining these dynamical changes will be necessary for the characterization of many complexes of potential interest using a structural bioinformatics approach (10–15).
For the characterization of large numbers of protein complexes, computational techniques have a considerable advantage over experiments in that they can be relatively fast while providing detailed information. One widely-used computational tool for calculating atomic-detail motions of proteins from three-dimensional structures is normal mode analysis (NMA) (16–18), in which the potential energy is approximated as harmonic, leading to the internal dynamics being represented as a superposition of independent vibrational modes. To reduce computational costs in NMA for large systems, a simplified, coarse-grained version of NMA, called the elastic network model (ENM), has been proposed (19–23). In this model, a protein molecule is represented by point-interaction residues connected by harmonic springs, enabling collective vibrational motions to be rapidly calculated. Although simple in form, ENM has been usefully applied to a variety of problems concerning protein dynamics and structural change (23–27). More extended coarse-grained force field models, that include, for example, local virtual bond stretching, angle bending, and dihedral and nonlocal interactions, have also been developed and applied to proteins (28–31).
In previous work we introduced a coarse-graining biomolecular simulation methodology, REACH (realistic extension algorithm via covariance Hessian), for deriving residue-scale ENM force constants directly from the variance-covariance fluctuation matrix calculated from atomic-detail molecular dynamics (MD) simulations (32,33). The residue-scale REACH method is a self-consistent direct mapping, requiring no iterative fitting and no input of experimental data. The REACH normal modes were shown to reproduce the amplitudes and frequencies of MD-derived fluctuations (32,33). REACH was also found to provide a force field transferable between proteins of different structural classes (33). Low-temperature coarse-grained MD simulations with the REACH force field were also found to successfully reproduce the corresponding atomistic MD results (33).
In this work, the REACH method is applied to crystallographically characterized protein complexes. Additional methodology is developed for calculating the required force constants in a crystalline environment, in which a protein molecule interacts with both the other molecules in the asymmetric unit (i.e., the molecules arranged in the same unit cell) and the image molecules in neighboring unit cells. As a test case, an MD trajectory of crystalline dimeric myoglobin is used to derive the REACH force constants. Analytical model functions are derived for the distance dependence of the force constants. The REACH NMA mean-square fluctuations are found to reproduce those of atomistic MD. The contributions of the intramolecular and intermolecular (translational and rotational) motions are decomposed. Furthermore, to estimate the relation between the binding strength and the intermolecular dynamics, the effect of varying the effective intermolecular force constant is examined.
In a further application, the harmonic REACH force field is applied to calculate the lattice dynamics of a protein crystal: ribonuclease A. The lattice dynamics originates from both protein-protein interactions and protein internal dynamics. The phonon dispersion relations of the crystal are calculated from the dynamical matrix in residue-scale coarse-grained model and the sound velocities for the acoustic modes calculated are compared with those derived from atomistic normal modes (34).
Because the REACH normal modes can be determined only by using the Cα coordinates of each residue together with the simple model functions of the distance dependence of the interaction force constants, the method is computationally inexpensive. Furthermore, the demonstrated transferability of the REACH force field allows the force constants derived from the atomistic MD simulation of dimeric myoglobin to be applied to other proteins (33). Therefore, REACH is applied in a bioinformatics-style study, analyzing a large number of Protein Data Bank (PDB) structures (35). The dynamics of functional dimers (here called biological dimers) are compared with proteins dimerized nonfunctionally by way of crystallization (here called crystal dimers). Previous studies have shown that the contact regions at the binding interfaces in the biological dimers are more extensive than the crystal dimers (36–39). The classification of protein-protein interfaces into crystal and biological origins has been achieved with >95% accuracy, based on structural bioinformatics studies using a support vector machine (40,41). The REACH methodology allows the dynamical consequences of the structural differences to be examined.
Theory and Methods
Dynamical simulations
Molecular dynamics simulations of crystalline dimeric myoglobin were performed. The simulation details are described in Kurkal-Siebert and Smith (42). Briefly, the simulation was performed at constant temperature (300 K) and pressure (1 atm) conditions (the NPT ensemble) using the monoclinic crystal (PDB: 1A6G (43) space group: P 21,). Additional MD simulation of tetrameric myoglobin (PDB: 1U7R (44)) with a different space group, P 212121, was also performed to examine the dependence of the REACH crystalline force field on the space group. Production runs were performed for 10 ns at 300 K. The atomic coordinates were saved every 50 fs for analysis.
REACH: calculation of force constants in a crystalline environment
The REACH coarse-grained method is described in detail in Moritsugu and Smith (32). Here is given a brief summary. The method uses only Cα atom coordinates; heteroatoms, such as ions and the heme in myoglobin, are not included. The potential energy in the elastic network model is written as (19)
| (1) |
where dij (d0ij) is the distance between the dynamical (equilibrium) positions of the Cα atoms in residues i and j, and kij is the force constant for the harmonic spring between i and j. Calculation of the Hessian (second-derivative) matrix from Eq. 1 leads to the relation
| (2) |
where Kij is the off-diagonal component of the Hessian associated with i and j. Making the harmonic approximation under the equilibrium condition at constant temperature, T, allows the Hessian matrix to be calculated from the variance-covariance matrix, C = (Cij) = (〈(ri – 〈ri〉)(rj − 〈rj〉)〉) as
| (3) |
where kB is the Boltzmann constant. The value kij is then derived by combining Eqs. 2 and 3 as
| (4) |
In this study, the 10-ns myoglobin MD trajectory was separated into ten 1-ns trajectories from each of which C was calculated. Subsequently, the ten C-matrices were averaged and the associated force constants derived using Eq. 4. To calculate the force constants for local interactions, i.e., the virtual 1-2 (between residues i and i+1), 1-3 (between residues i and i+2), and 1-4 (between residues i and i+3) terms, segments of 20 residues were fitted individually to calculate submatrices of C. This avoids the error associated with the best fit to the overall structure arising from the incorporation of external motions of the segments, which would result in errors in the pairwise covariances (32).
In a crystal environment the calculation of long-range interactions is not as straightforward as Eq. 2, since the potential energy (Eq. 1) includes not only the direct interaction between residue i and j in the same molecule but also the interactions with residues in other proteins in the neighboring cells. Here, to save computational cost, we assume traditional symmetry between unit cells, i.e., that the equilibrium symmetry in the crystal structure holds during protein fluctuation such that residue j in one unit cell moves identically to residues j′ (the residues corresponding to j in the other unit cells). Given this assumption, Kij can be described using the interaction energy of residue i with both residue j and all the possible residues j′, i.e., . Each Vij′ contributes to Kij as
However, we do not assume any further symmetry between the two myoglobin monomers, e.g., such that an atom in one monomer moves anticorrelated with the corresponding atom in the other monomer. Keeping the motion of the monomer anticorrelated with that of the other monomer yields only the fully symmetric vibrations, which are not sufficient to describe the full vibrational characteristics of the system. In this study, the full normal modes are calculated without freezing any degree of freedom and are used to describe vibrational amplitudes and frequencies as well as the sound velocity (see the descriptions below).
For simplifying the interaction calculation, analytical model functions were constructed of the distance-dependent nonlocal intramolecular and intermolecular interactions. As in the previous work (32), the two model functions are assumed to be single exponentials, i.e., kintra(r) = aintra exp(−bintrar) and kinter(r) = ainter exp(−binterr), with parameters aintra, bintra, ainter, and binter. The resulting force constant parameters determining k(r) are listed as follows:
| (5) |
Using the protein structure, r0 (the coordinates in the PDB file) and crystal information (i.e., the space group defining the arrangement of molecular positions in the asymmetric unit together with the unit cell parameters defining the image cell shape), the molecules in the primary and neighboring cells were constructed. The protein-protein interaction energy can then be calculated together with the local interactions, allowing derivation of the Hessian matrix elements using the four model parameters: KREACH = KREACH(aintra, bintra, ainter, binter). Only those nonlocal interactions shorter than 30 Å were used for calculating KREACH, as trial calculations showed that a longer cutoff length leads to negligible changes in the parameters derived (results not shown). The parameters were determined so as to minimize the following estimation function,
| (6) |
by analogy with the correspondence between 1/Kii and 〈x2i〉 = Cii.
In a rigorous calculation of crystal dynamics, an average should be performed over the wavevectors, q, in the dynamical matrix (see the Supporting Material). However, the inclusion of q ≠ 0 preludes the direct calculation of the force constant parameters using Eq. 6. However, it has been found that taking only q = 0, i.e., considering the protein-protein interactions between only the adjacent unit cells, leads to similar protein internal fluctuations as obtained by averaging the contributions over q (45). This finding is confirmed by the result that the REACH force field does not depend on the space group (see Results).
Calculation of mean-square fluctuation and vibrational density of states using normal modes
Using the potential energy (Eq. 1) together with the equilibrium structure and the force constant models derived, the REACH normal modes can be calculated, comprising the set of eigenvalues, {λi}, and eigenvectors, {v0,i}. The mass of each coarse-grained residue is defined as the sum of the atomic masses comprising the corresponding residue. In the crystal normal modes there are three modes with eigenvalues of zero (as compared with six for an isolated system), all of which correspond to translational motion. The other modes may contain protein rotational contributions.
For deriving the internal mean-square fluctuation of residue n, x2n, new eigenvectors, {vi}, were calculated by subtracting whole-molecule rotational motion from {v0,i}. The value x2n was calculated using the equilibrium condition at the temperature, T as
| (7) |
where vn,i is the displacement of residue n in mode i and mn the residue mass. To calculate the contribution to x2n from intermolecular motion, the {vi} in Eq. 7 were replaced by , in which the intramolecular contributions are subtracted. Further decomposition was performed of the intermolecular displacement into the translation and rotation between two monomers as
The vibrational density of states, g(ω), was calculated from the REACH normal modes. A previous study showed that the single protein all-atom MD-derived g(ω) in the low frequency range (<∼100 cm−1) is well reproduced by the REACH method (32). Using the normal-mode frequencies, {ωi }, g(ω) is calculated as
| (8) |
where σi is the variance of the Gaussian distribution of each vibrational mode, and can be considered to represent, for example, spectrometric instrumental resolution. The expression σi = 1 cm−1 was used for all modes in this study. The intermolecular contribution to g(ω), ginter(ω), was calculated using the ratio of intermolecular to overall motion, , as
| (9) |
The above procedure is also applicable to the translational and rotational components, gtra(ω) and grot(ω), by defining and and applying Eq. 9.
Bioinformatics-style study of biological and crystal dimers
Finally, the REACH method was applied to examine intermolecular dynamics of protein dimers in the PDB. For this purpose, 83 biological dimers and 46 crystal dimers were selected, as a subset of the database described in the literature (38,39), and are listed in Table S1 in the Supporting Material. The biological dimers are proteins in the PDB reported as also being dimers in solution based on biochemical or biophysical data such as analytical centrifugation. Pairs of the biological dimers were selected based on the criteria of having twofold symmetry and a large interface area. The crystal dimers were identified as PDB entries containing monomeric proteins making large crystal packing interfaces and including two or more identical chains in the asymmetric unit and/or a space group with at least one twofold rotation operation, but for which no evidence of dimerization in solution has been reported (39). Only homodimer proteins were examined to compare with the dimer myoglobin results. The PDB entries, with the coordinates of the homodimers being explicitly described herein, were used so as to determine the interfaces of two monomers.
For each of the PDB entries, the Cα coordinates were extracted and used to derive the equilibrium structures for calculating REACH normal modes. The force-constant model functions derived from the crystalline myoglobin dimer MD simulations were employed.
To evaluate the binding strength between the two monomers, the all-atom interaction energy and buried surface area were calculated using the CHARMM program (46) and CHARMM 22 all-atom potential function (47). For this purpose, hydrogen atoms were added to the atomic coordinates in the PDB file using the HBUILD module of CHARMM (46) and the structures were energy-minimized using 1000 steps of the steepest-descent method and 1000 steps of adopted-basis Newton-Raphson minimization (46) with the main-chain atoms fixed. The interaction energy between the two monomers was then calculated using the structures derived. For simplicity, minimizations were performed in vacuum using a distance-dependent dielectric constant, ɛ = 2r, to approximate solvent effects. The buried surface area, S, was calculated using the accessible surface areas of the dimer, S0, and two monomers, S1 and S2, as S = (S1 + S2 – S0)/2. The accessible surface areas were calculated from the energy-minimized structures using a water probe of 1.4 Å radius (48).
Results
Myoglobin dimer in crystal: a test case for calculating force constants
The REACH force constants for dimeric myoglobin were calculated from the corresponding MD trajectory of the protein in the crystalline state. Using the REACH normal modes calculated in combination with the force constants derived, the mean-square fluctuation and the vibrational density of states were calculated and decomposed into intra- and intermolecular (translational and rotational) components.
In Fig. 1 a, the REACH force constants, k, between the residue pairs are plotted as a function of the pairwise distance, r. The distributions of the virtual 1-2, 1-3, 1-4, and nonlocal and intramolecular interactions are very similar to those previously derived for monomeric and dimeric myoglobin in aqueous solution (32). The distribution of the intermolecular interaction force constants (Fig. 1 e) is rather scattered, due probably to the low correlation of the residue pair fluctuations, even at the nearest distance of r ∼7 Å. However, the averages over the range r = 7−10 Å are statistically meaningful, nonzero quantities.
Figure 1.
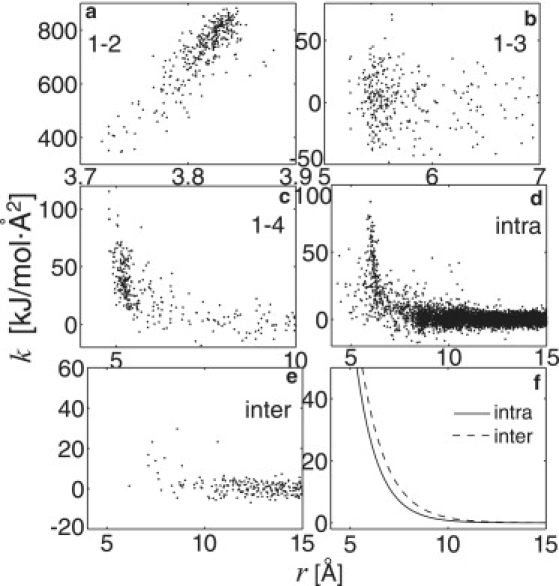
REACH force constants, k, of virtual (a) 1-2, (b) 1-3, and (c) 1-4 interactions in the crystalline myoglobin dimer are shown as a function of pairwise distance, r. (d and e) Off-diagonal elements of Hessian via nonlocal intramolecular and intermolecular pairs are shown. (f) Model functions of k derived by Eq. 6 (intra, dotted curve; and inter, dashed curve). See text for details.
Model functions of the distance dependence of the force constants were constructed. The model functions of the local interactions (k12, k13, k14) were assumed to be constant, as in the previous REACH method (32,33) and another coarse-grained MD study (28). The distance dependence of the model functions for both the intra- and intermolecular interactions was assumed to be a single exponential: the corresponding parameters were determined by minimizing Eq. 6. The resulting values and standard-deviation errors are listed in Table 1.
Table 1.
REACH force constants derived from MD trajectories
| k12 | k13 | k14 | aintra | bintra | ainter | binter | |
|---|---|---|---|---|---|---|---|
| Crystalline dimer | 716 ± 7.8 | 4.63 ± 1.7 | 28.6 ± 1.7 | 6140 ± 380 | 0.901 ± 0.072 | 3920 ± 710 | 0.777 ± 0.15 |
| Crystalline tetramer† | 723 ± 4.2 | 13.8 ± 1.3 | 29.6 ± 1.9 | 6000 ± 200 | 0.896 ± 0.062 | 2650 ± 270 | 0.750 ± 0.11 |
| Solvated monomer‡ | 735 ± 9.7 | 5.59 ± 2.0 | 31.9 ± 2.1 | 1980 ± 260 | 0.749 ± 0.031 | ||
| Solvated dimer‡ | 712 ± 24 | 6.92 ± 2.3 | 32.0 ± 2.4 | 2560 ± 530 | 0.800 ± 0.034 | 1630 ± 17 | 0.772 ± 0.17 |
Note that kintra(r) = aintra exp(−bintrar) and kinter(r) = ainter exp(−binterr). Units used are kJ/mol·Å2, except for b, which is Å−1.
Result from tetramic myoglobin with P 212121 space group.
Results in Moritsugu and Smith (32).
The virtual 1-2, 1-3, and 1-4 force constant parameters for crystal dimer myoglobin are similar to those found for the solvated monomer and dimer (32). However, the intra- and intermolecular force constants are slightly larger than the previous values obtained in solution, due possibly to the effects of the neighboring crystal molecules. At any given distance, the model function of the intermolecular force constant is found to be slightly larger value than for the intramolecular interaction (see Fig. 1 f). The distributions (results not shown) and model functions (in Table 1) of the REACH force constants from P 212121 myoglobin tetramer are similar to those from P 21 myoglobin, suggesting that the REACH force field does not depend on the space group.
Using the model parameter functions in Table 1, the REACH normal modes of the crystalline myoglobin dimer were calculated, including the protein-protein interactions between different unit cells, and the associated x2 and g(ω) were derived. Fig. 2 a1 shows a good correspondence between the REACH- and atomistic MD-derived x2, with a high correlation coefficient of 0.683. The magnitude of x2 from the REACH normal modes is slightly smaller than from the atomistic MD (the average is 0.413 Å2 from REACH and 0.512 Å2 from the all-atom MD) but is in satisfactory agreement given the simplicity of the REACH method. The 1-ns atomistic MD trajectory may include additional motions beyond the effective harmonic approximation employed in REACH.
Figure 2.
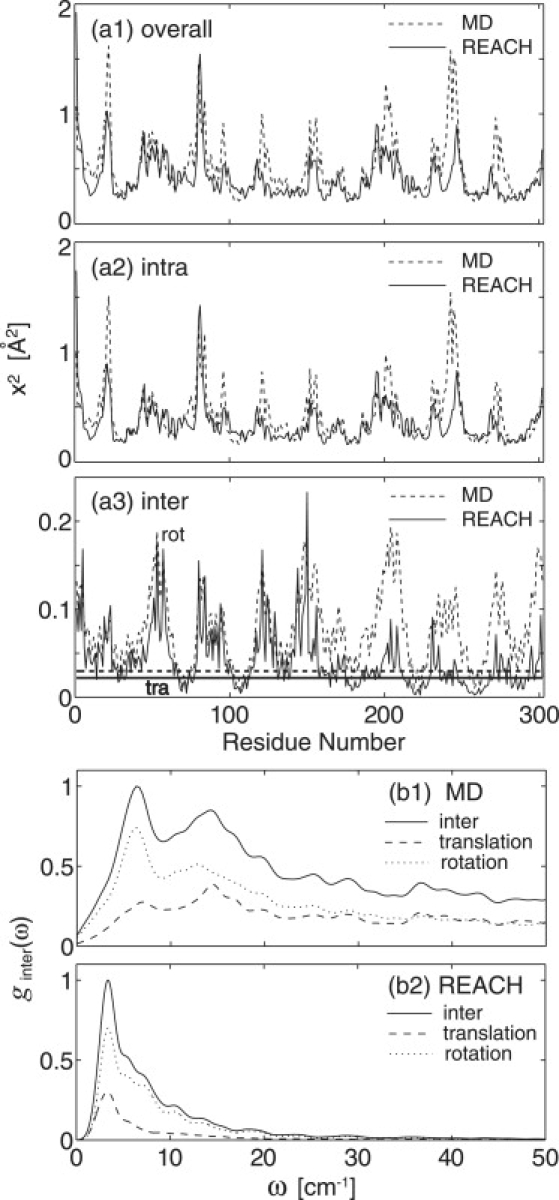
(a1–a3) Mean-square fluctuations of each residue in myoglobin dimer derived from MD (dashed curve) and REACH normal modes (solid curve): contributions of overall, intramolecular, and translational (thick line) and rotational (constant line) motion, respectively. (b1 and b2) Vibrational density of states for intermolecular (translation + rotation, solid curve), translational (dashed curve), and rotational (dotted curve) vibration from MD and REACH normal modes. To calculate the MD spectra, 216 MD trajectory frames separated by 50 fs (total length of ∼3.3 ns) were used for calculating the velocity autocorrelation function (VACF) and the VACF derived was Fourier-transformed.
The x2 values from the intra- and intermolecular (translational and rotational) motions were calculated separately using Eq. 7, together with the associated decomposed eigenvector components. The REACH intramolecular x2 values shown in Fig. 2 a2 are also in good agreement with the all-atom MD, as evidenced by both the high correlation coefficient (0.668) and the similar averages (0.354 Å2 from the REACH normal modes and 0.411 Å2 from the all-atom MD). An ENM force field with a one-parameter force constant (k = 3.2 kJ/mol) and a cutoff of 12 Å was found to derive similar x2 values, but the agreement is somewhat worse in the ratio of the amplitudes of the intermolecular to overall motion (11% for ENM, and 15% for REACH and 20% for MD). The stronger force constants for local interactions and decaying function for nonlocal interactions in REACH may balance these contributions better.
Decomposition of the intermolecular x2 values into the translational and rotational components is shown in Fig. 2 a3. The magnitudes are underestimated by REACH, both for the translation (0.0219 Å2 from the REACH and 0.0297 Å2 from the all-atom MD) and for the rotation (0.0413 Å2 from the REACH and 0.0768 Å2 from the all-atom MD). At 300 K, the rotational contribution is larger than that from translation. The rotational contribution correlates with the overall x2 values, indicating, as expected, that the residues far from the molecular center (i.e., near the surface) are more likely to undergo both larger internal fluctuation and rigid-body rotation than those in the protein core (49).
The vibrational densities of states for the intermonomer motions, ginter(ω), were calculated from both the MD trajectory and the REACH normal modes using Eqs. 8 and 9. Fig. 2 b shows that the peak amplitude of the translational spectrum is smaller than that from the rotation, a result consistent with the relative x2 values. A clear peak in the very low-frequency region of the spectrum, at ∼6.3 cm−1 (MD) and ∼3.4 cm−1 (REACH) is seen. The peak corresponds roughly to a ∼8 cm−1 peak seen in the experimental neutron scattering spectrum from plastocyanin at 100 K (50), which may therefore originate from interprotein interaction. The MD spectrum shifts slightly to higher frequency and, for ω > 10 cm−1, is more broadly distributed, due probably to the increased roughness of the potential energy surface at atomic resolution. The second peak at ∼15 cm−1 in the MD spectrum is not well reproduced by the present REACH normal modes, again due to the absence of high-frequency vibrations in the coarse-grained model. This peak, arising mainly from rotational contributions, may involve the interaction of explicit side chains of interfacial residues.
In conclusion, the residue-scale REACH method, with a simple potential energy function and force field, satisfactorily reproduces protein fluctuations and frequencies, associated with the overall motion and also the internal motion and intermonomer translation and rotation, although only the overall motion contribution in K was used for determining the intermolecular force constants in Eq. 6.
Lattice dynamics via crystalline REACH
The phonon dispersion relations, ω(q), and the associated acoustic-mode sound velocities of a crystalline protein, ribonuclease A, calculated using the REACH crystalline force constants derived, indicate a qualitative agreement with the atomistic result (34). Details of the calculation procedures and main results are described in the Supporting Material.
Binding strength and intermolecular translation and rotation in myoglobin dimer
To examine the relation between the monomer-monomer interaction strength and the intermonomer fluctuation the REACH normal-mode calculations were performed using model dimeric crystalline myoglobin systems, in which the monomer-monomer force constants are multiplied by a scaling factor, β, i.e., kinter,dimer → βkinter,dimer, and the intermonomer translational and rotational motions were evaluated as a function of β. An additional scaling factor, β0, multiplying uniformly all the protein-protein interactions in the crystal, was also introduced. β = 1 and β0 = 1 thus correspond to the unscaled crystalline myoglobin system. Variation of the scaling factor allows the significance of the relation between interaction strength and intermolecular dynamics to be uncovered. Further, the results derived from this model system aid in understanding the results of the comparison between the biological and crystal dimers (see the next section).
Fig. 3 a1 shows that x2 decreases with β, indicating that smaller residue fluctuations accompany stronger intermolecular interaction. This result holds for the overall motion and for both the intermolecular translational and rotational components. The ratio of the intermolecular to the overall x2 contributions, R = 〈x2inter〉/〈x2〉, shown in Fig. 3 a2, also decreases with increasing β. Thus, as β increases, the decrease in 〈x2inter〉 is larger than that in 〈x2〉; i.e., a strong intermolecular interaction preferentially reduces the intermolecular motion. The decrease of the translational x2 (and R) is larger than that of the rotation, i.e., the monomer-monomer translation is thus more strongly reduced by strong binding interaction than the rotation.
Figure 3.
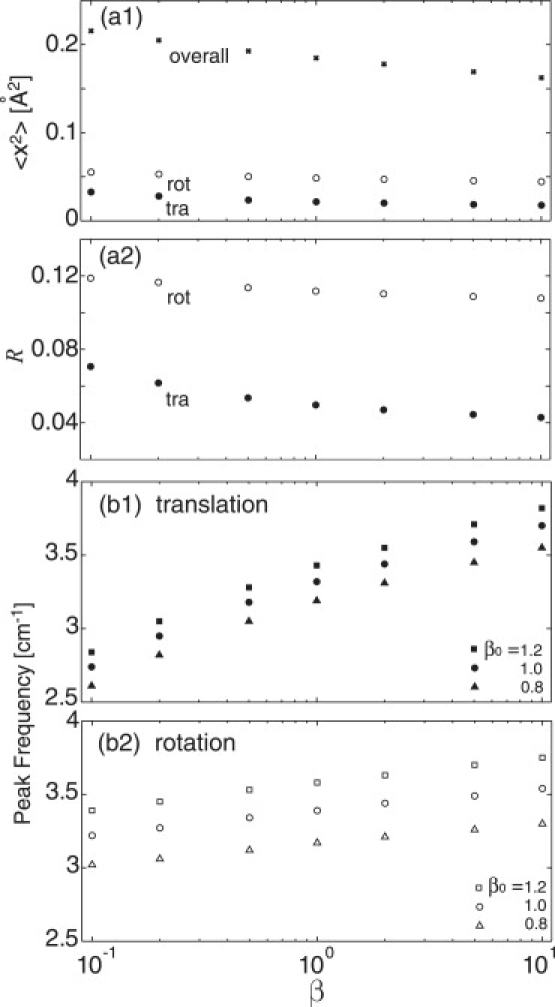
(a1) Average of REACH mean-square fluctuation over residues in myoglobin dimer from the overall (cross) as well as translational (solid circle) and rotational (open circle) motions are plotted as a function of monomer-monomer interaction scaling factor, β. (a2) Ratio of the translational (solid circle) and rotational (open circle) to overall contributions of mean-square fluctuation, R, as a function of β. (b1 and b2) Peak frequency of the vibrational density of states in translation and rotation as a function of β. Data for all the protein-protein interactions multiplied by β0 are plotted as crosses (β0 = 1.2), circles (β0 = 1.0), and triangles (β0 = 0.8). See text for details.
In Fig. 3 b, the frequency of the highest peak in the REACH intermonomer vibrational density of states is plotted as a function of β. The peak shifts to higher frequency with increasing β, consistent with the decrease of x2, i.e., strengthening the intermolecular interaction narrows the curvature of the associated potential and thus decreases the vibrational period. The spectral frequency shift is larger for translation than rotation: the shift in grot(ω) is only ∼±7% for a 10-fold increase/decrease of the intermonomer interaction, whereas for the translation it is ∼±15%.
Next, all the intermolecular interactions were multiplied by β0, i.e., not only between pairwise monomers (kinter,dimer → β0β kinter,dimer), but also between all the molecules in the asymmetric unit and image cell (kinter → β0kinter). Fig. 3 b shows that the intermolecular peak frequency increases much more with β0 than with β, indicating that the protein-protein vibration in the crystalline state is highly suppressed by the interactions with the other crystal molecules. For the rotation, the peak shift in grot(ω) resulting from a 20% increase of β0 corresponds to that obtained from a 100-fold increase of β, implying that the intermolecular rotation is mainly governed by the interaction with the surrounding protein molecules, rather than the pairwise dimer interaction.
Bioinformatics-style study of the biological and crystal dimers
Finally, the REACH method is now applied to analyze the dynamics of a large number of protein dimers in the PDB (see Table S1). For these calculations, the model force-constant functions derived from the crystalline myoglobin dimer were employed, thus making use of the demonstrated transferability of the REACH force field (33). Differences in the intermonomer dynamics between the biological and crystal dimers are determined, allowing relationships between the static and dynamical dimer structures to be characterized.
In Fig. 4 a is shown the number of intermonomer residue pairs at r < 12 Å, Npair, as a function of the number of residues of each dimer. For dimers of a similar size, Npair tends to be larger in biological dimers than in crystal dimers, indicating that biological dimers have more extensive monomer-monomer interfaces than crystal dimers. Fig. 4 b shows a high correlation between the dimer interaction energy, Eint, and the buried surface area, S. Eint in the all-atom force fields can thus, in principle, be derived from S, i.e., Eint [kcal/mol] ≈ 0.6 S [Å2]. The biological dimers have larger S and lower Eint, suggesting more stabilized static structures than the crystal dimers.
Figure 4.
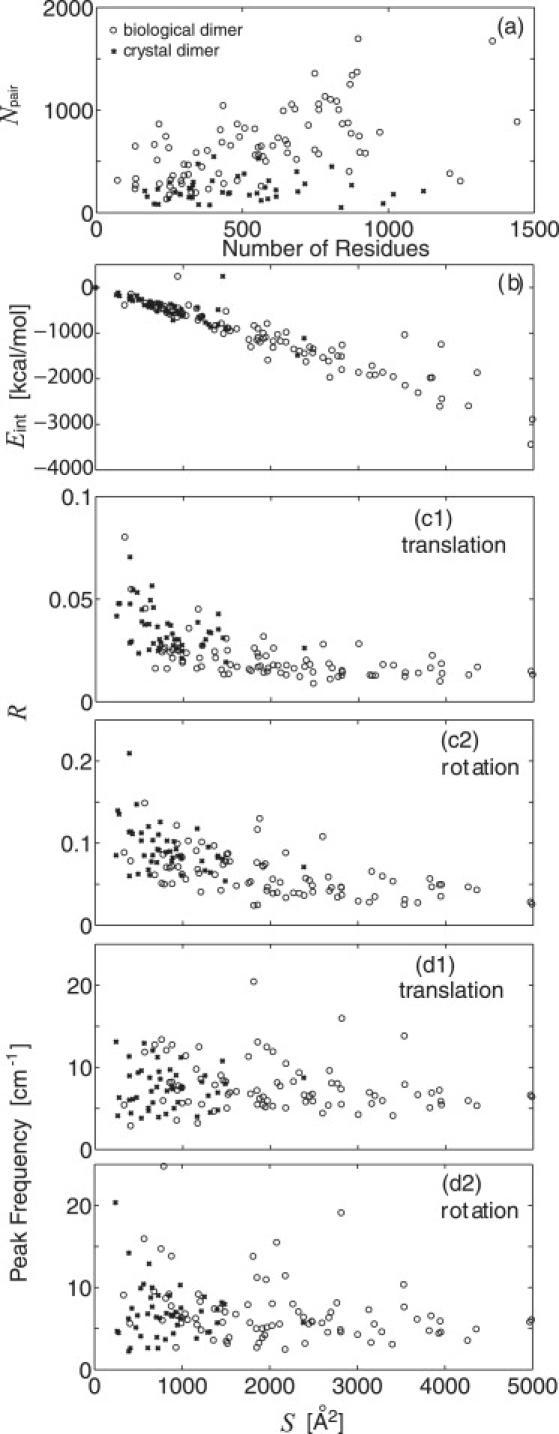
Bioinformatics-style study of the 83 biological (circles) and 46 crystal (crosses) dimers in Table S1. (a) Number of intermolecular atomic pairs at r < 12 Å, Npair, as a function of the number of residues. (b–d) Buried surface area (S) dependence of: (b) dimer interaction energy of the pairwise monomers, Eint; (c1 and c2) ratio of the intermolecular translation and rotation to the overall contribution of REACH mean-square fluctuation, R; and (d1 and d2) peak frequency of the vibrational density of states for intermolecular translation and rotation.
In Fig. 4 c, the ratio of the intermolecular (translational and rotational) to the overall mean-square fluctuation, R, is shown as a function of S. A static-dynamic relationship can clearly be seen: increasing S leads, on average, to a decrease of R, meaning that strong intermolecular binding via a large binding interface suppresses the intermonomer motion. The distributions of the biological and crystal dimers are different although not clearly demarcated: the biological dimers occupying larger S and smaller R, and the crystal dimers smaller S and larger R. The rotational x2 (Fig. 4 c2) is larger than the translational x2 (Fig. 4 c1) irrespective of S, i.e., of the interaction strength. In summary, the extensive monomer-monomer contacts arising in biological dimers lead to stable binding complexes that tend to suppress the associated intermonomer translational and rotational fluctuations.
Next, the frequencies of the intermonomer motions were analyzed via their vibrational spectra. For each dimer the vibrational densities of states for the intermolecular translation and rotation were calculated from the REACH normal modes using Eq. 9, and the frequencies of the highest amplitude were determined (see dimeric myoglobin spectra in Fig. 2 b2). Fig. 4 d reveals little dependence on S of the distribution of the peak frequencies. The average frequency of the rotational spectrum peak is 6.9 cm−1 for the biological and 6.7 cm−1 for crystal dimers, values smaller than for the translation (8.0 cm−1 for biological and 7.2 cm−1 for crystal dimers). However, these differences in the average peak frequencies are within the distribution widths, implying that the characteristic frequencies of the intermolecular motions are not strongly dependent on the dimer interaction strength. This result is in agreement with that derived from the model system study of dimeric myoglobin in the previous section, in which it was found that the peak frequency changes only ∼±7% for rotation and ∼±15% for translation by a 10-fold increase/decrease of the intermonomer interaction. The monomer-monomer vibrational frequency may therefore be determined by the interaction with the other molecules in the crystal rather than by the dimer interaction strength.
Concluding Remarks
In this article, protein dimer interaction dynamics has been analyzed using the REACH coarse-grained simulation methodology. The REACH method derives residue-scale force constants from an atomistic MD trajectory (in this case, of crystalline myoglobin). The REACH method is applied to a number of crystal and biological dimers in the PDB.
The REACH force constants, plotted as a function of pairwise distance, show distinct distributions depending on the local (virtual 1-2, 1-3, 1-4 bonds) or nonlocal (intramolecular and intermolecular) interaction considered. Model functions of the distance dependence of the local interactions in crystalline myoglobin are found to be similar to those derived for the solvated dimer (32). However, the nonlocal interaction strength in the crystal is slightly larger than in solution, due probably to crystal packing effects. Both the overall and the intra-/intermolecular (translational and rotational) contributions to x2 from the all-atom MD are well reproduced by the present REACH normal modes. The REACH method also adequately reproduces the vibrational densities of states for the intermolecular translation and rotation in the lowest frequency region. The underestimation of the vibrational frequencies may be improved by calibration of the mass of each residue, and this is planned for future work.
The coarse-grained model based only on Cα coordinate of each residue cannot distinguish between dimer interaction modes correctly. All-atom MD simulation is required to examine the bound structure including elements such as side-chain packing, water molecule rearrangement, and so on. However, the aim of this study is not to elucidate the structures of the dimers, but instead to examine collective, intermolecular dynamics of the dimer in the equilibrium-bound state. There is significant evidence from our previous work and other studies that global collective dynamics can be well reproduced with a coarse-grained model.
It is useful to summarize here the comparison between REACH and finer-grained, all-atom MD, and the more simplified force field of ENM:
-
1.
Coarse-graining leads to the absence of high-frequency modes relative to the all-atom MD. However, this absence is not essential, as high-frequency modes are low-amplitude and relatively unimportant in determining internal dynamics. Further, although it is found that the harmonic vibrations in our REACH coarse-grained model undergo shifts to low-frequency relative to the atomistic motions, the agreement between the REACH and atomistic vibrational amplitudes and frequencies is such that the REACH model provides useful information on the collective dimer interaction dynamics.
-
2.
The ENM force field (a step function with a cutoff length) is a simplification of that of REACH. The REACH force-field model functions and the associated model parameters are consequences from a direct mapping of atomistic MD force field, whereas the ENM model function is only an assumption. In this sense, it is considered that the usefulness of the ENM step-function-like force field is first validated by the result that the REACH force constants decay as a function of residue-pair distance.
Potential advantages of REACH over ENM are:
-
—
The REACH force field is transferable, whereas the unique parameter of ENM calculated by fitting internal fluctuation is sensitive to the system studied and, furthermore, to the cutoff length.
-
—
The (nonzero) decaying function of the REACH force constant avoids the case that some residues, e.g., close to N- or C termini, are isolated from the other residues beyond the ENM cutoff length, which leads to meaningless motions for these segments.
-
—
The intermolecular and intramolecular vibrational amplitudes are better balanced by using the REACH force field rather than ENM, in which the one-parameter force constant is determined by fitting only the overall fluctuation (as found in this study). This result is significant in the study of the interaction dynamics of protein complexes.
The relation between the binding strength and intermonomer dynamics in the myoglobin dimer was studied using REACH. The analysis of a hypothetical model system in which the intermonomer force constants are artificially multiplied by a factor, β, demonstrates that the dimer binding strength determines the amplitude of the intermonomer motion. The vibrational amplitude and frequency of the translational vibration depends more strongly on β than the amplitude and frequency of the rotation, suggesting that stability of protein complexes may be accompanied by suppression of protein-protein translation. In contrast, when all the intermolecular interactions in a crystal, including those between two adjacent monomers as well as those between the molecules in the asymmetric unit and in the image cell, are uniformly strengthened by β0, the intermonomer vibrations undergo a shift to high frequencies. Increasing β thus mimics the effect of strengthening specific interaction via increase of the binding interface area or via specific electrostatic interactions, whereas β0 mimics the magnification of the general interaction strength with the surrounding molecules in a multiprotein complex. The magnitude of the frequency shift affected by β0 is larger for rotation than that for translation, implying that rotation is more highly affected by surrounding molecules than translation.
The REACH coarse-grained method enables internal dynamics of large numbers of dimer systems (here, 129) to be analyzed. Most structural bioinformatics studies have been based on the comparative analysis of static structures. Here, we extend this into an analysis of interaction dynamics, which are of thermodynamic importance in complex association phenomena. To do this, the force constants derived from the dimeric myoglobin analysis were used, thus inviting the assumption that the myoglobin force field is transferable to all protein dimers. The REACH method enables a direct mapping of the atomistic MD onto the coarse-grained model without iterative steps and thus avoids system-dependence of the force constants (32,33). Indeed, previous work demonstrated strong similarity in the intramolecular force constants obtained from three different proteins with different structural folds (51), i.e., myoglobin (α-fold), plastocyanin (β-fold), and dihydrofolate reductase (α/β fold), providing evidence for generality of the REACH force field (33).
Using the REACH normal modes, the intermonomer dynamics of biological and crystal dimers were compared. The static structures (i.e., the number of contact pairs, the buried accessible surface, and the interaction energy between two monomers) indicate that the biological dimers have more extensive binding interfaces than the crystal dimers, as previously detailed (38,39). It is found here that the consequence of the extensive interface is suppression of the intermonomer translational and rotational amplitudes. However, in contrast to the amplitudes, the peak frequencies of intermonomer vibrations in biological and crystal dimers are similar.
Protein-protein binding processes can be separated into two phenomena: the diffusive approach of two proteins so as to form an encounter complex, then the consequent rearrangement of the interface atoms. Crystal dimers may be considered to mimic encounter complexes. This comparative study indicates that the formation of the interfacial interactions stabilizing the complex structure after encounter-complex formation will change the amplitude of the intermolecular vibrations, especially the translational component. Future work may be directed at examining these effects in detail using all-atom MD simulation and/or the experiments on comparative systems: for example, wild-type and variant complexes with mutated interfacial residues.
In addition, in future work, this REACH approach will be extended to enable coarse-grained MD simulation with anharmonic potential functions. Anharmonic effects on protein interaction dynamics have been recently shown to be significant (42). Furthermore, an extension of the method to anharmonic crystal, using an anharmonic REACH force field derived from a full crystal all-atom MD simulation, is planned. Indeed, in another study, a hydration-dependent transition in interprotein motion at ∼180 K simulated by atomistic MD of crystalline myoglobin indicates significant anharmonicity in crystal modes at physiological temperature (52).
Supporting Material
Two tables and one figure are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)01017-0.
Supporting Material
Acknowledgments
The authors thank Dr. L. Meinhold for useful discussions.
We acknowledge funds from the U.S. Department of Energy via a Laboratory-Directed Research and Development grant. K.M. acknowledges support by the MEXT Grand Challenge Program using Next-Generation Supercomputing.
References
- 1.Steinberg I.Z., Scheraga H.A. Entropy changes accompanying association reactions of proteins. J. Biol. Chem. 1963;238:172–181. [PubMed] [Google Scholar]
- 2.Page M.I., Jencks W.P. Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect. Proc. Natl. Acad. Sci. USA. 1971;68:1678–1683. doi: 10.1073/pnas.68.8.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sturtevant J.M. Heat capacity and entropy changes in processes involving proteins. Proc. Natl. Acad. Sci. USA. 1977;74:2236–2240. doi: 10.1073/pnas.74.6.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erickson H.P. Co-operativity in protein-protein association: the structure and stability of the actin filament. J. Mol. Biol. 1989;206:465–474. doi: 10.1016/0022-2836(89)90494-4. [DOI] [PubMed] [Google Scholar]
- 5.Finkelstein A.V., Janin J. The price of lost freedom: entropy of biomolecular complex formation. Protein Eng. 1989;3:1–3. doi: 10.1093/protein/3.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Tidor B., Karplus M. The contribution of vibrational entropy to molecular association. The dimerization of insulin. J. Mol. Biol. 1994;238:405–414. doi: 10.1006/jmbi.1994.1300. [DOI] [PubMed] [Google Scholar]
- 7.Fischer S., Verma C.S. Binding of buried structural water increases the flexibility of proteins. Proc. Natl. Acad. Sci. USA. 1999;96:9613–9615. doi: 10.1073/pnas.96.17.9613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer S., Smith J.C., Verma C.S. Dissecting the vibrational entropy change on protein/ligand binding: burial of a water molecule in bovine pancreatic trypsin inhibitor. J. Phys. Chem. B. 2001;105:8050–8055. [Google Scholar]
- 9.Balog E., Becker T., Oettl M., Lechner R., Daniel R. Direct determination of vibrational density of states change on ligand binding to a protein. Phys. Rev. Lett. 2004;93:028103. doi: 10.1103/PhysRevLett.93.028103. [DOI] [PubMed] [Google Scholar]
- 10.Baker D., Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 11.Park J., Lappe M., Teichmann S.A. Mapping protein family interactions: intramolecular and intermolecular protein family interaction repertoires in the PDB and yeast. J. Mol. Biol. 2001;307:929–938. doi: 10.1006/jmbi.2001.4526. [DOI] [PubMed] [Google Scholar]
- 12.Aloy P., Russell R.B. Potential artifacts in protein-interaction networks. FEBS Lett. 2002;530:253–254. doi: 10.1016/s0014-5793(02)03427-0. [DOI] [PubMed] [Google Scholar]
- 13.Edwards A., Kus B., Jansen R., Greenbaum D., Greenblatt J. Bridging structural biology and genomics: assessing protein-interaction data with known complexes. Trends Genet. 2002;18:529–536. doi: 10.1016/s0168-9525(02)02763-4. [DOI] [PubMed] [Google Scholar]
- 14.Sali A., Glaeser R., Earnest T., Baumeister W. From words to literature in structural proteomics. Nature. 2003;422:216–225. doi: 10.1038/nature01513. [DOI] [PubMed] [Google Scholar]
- 15.Russell R.B., Alber F., Aloy P., Davis F.P., Korkin D. A structural perspective on protein–protein interactions. Curr. Opin. Struct. Biol. 2004;14:313–324. doi: 10.1016/j.sbi.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Gō N., Noguti T., Nishikawa T. Dynamics of a small globular protein in terms of low-frequency vibrational modes. Proc. Natl. Acad. Sci. USA. 1983;80:3670–3696. doi: 10.1073/pnas.80.12.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brooks B.R., Karplus M. Harmonic dynamics of proteins: normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA. 1983;80:6571–6575. doi: 10.1073/pnas.80.21.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levitt M., Sander C., Stern P.S. Protein normal-mode dynamics: trypsin inhibitor, crambin, ribonuclease and lysozyme. J. Mol. Biol. 1985;181:423–447. doi: 10.1016/0022-2836(85)90230-x. [DOI] [PubMed] [Google Scholar]
- 19.Tirion M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 20.Bahar I., Atilgan A.R., Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 21.Haliloglu T., Bahar I., Erman B. Gaussian dynamics of folded proteins. Phys. Rev. Lett. 1998;79:3090–3093. [Google Scholar]
- 22.Bahar I., Atilgan A.R., Demirel M.C., Erman B. Vibrational dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 1998;80:2733–2736. [Google Scholar]
- 23.Tama F., Brooks C.L. Diversity and identity of mechanical properties of icosahedral viral capsids studied with elastic network normal mode analysis. J. Mol. Biol. 2005;345:299–314. doi: 10.1016/j.jmb.2004.10.054. [DOI] [PubMed] [Google Scholar]
- 24.Bahar I., Rader A.J. Coarse-grained normal mode analysis in structural biology. Curr. Opin. Struct. Biol. 2005;15:586–592. doi: 10.1016/j.sbi.2005.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kundu S., Jernigan R.L. Molecular mechanism of domain swapping in proteins: an analysis of slower motions. Biophys. J. 2004;86:3846–3854. doi: 10.1529/biophysj.103.034736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang L.W., Liu X., Jursa C.J., Holliman M., Rader A.J. iGNM: a database of protein functional motions based on Gaussian network model. Bioinformatics. 2005;21:2978–2987. doi: 10.1093/bioinformatics/bti469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tobi D., Bahar I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc. Natl. Acad. Sci. USA. 2005;102:18908–18913. doi: 10.1073/pnas.0507603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trylska J., Tozzini V., McCammon A. Exploring global motions and correlations in the ribosome. Biophys. J. 2005;89:1455–1463. doi: 10.1529/biophysj.104.058495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tozzini V., McCammon J.A. A coarse-grained model for the dynamics of flap opening in HIV-1 protease. Chem. Phys. Lett. 2005;413:123–128. [Google Scholar]
- 30.Chu J.W., Voth G.A. Coarse-grained modeling of actin filament derived from atomistic-scale solutions. Biophys. J. 2006;90:1572–1582. doi: 10.1529/biophysj.105.073924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou J., Thorpe I.F., Izvekov S., Voth G.A. Coarse-grained peptide modeling using a systematic multiscale approach. Biophys. J. 2007;92:4289–4303. doi: 10.1529/biophysj.106.094425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moritsugu K., Smith J.C. Coarse-grained biomolecular simulation with REACH: realistic extension algorithm via covariance Hessian. Biophys. J. 2007;93:3460–3469. doi: 10.1529/biophysj.107.111898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moritsugu K., Smith J.C. REACH coarse-grained biomolecular simulation: transferability between different protein structural classes. Biophys. J. 2008;95:1639–1648. doi: 10.1529/biophysj.108.131714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meinhold L., Merzel F., Smith J.C. Lattice dynamics of a protein crystal. Phys. Rev. Lett. 2007;99:138101. doi: 10.1103/PhysRevLett.99.138101. [DOI] [PubMed] [Google Scholar]
- 35.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lo Conte L., Chothia C., Janin J. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 37.Chakrabarti P., Janin J. Dissecting protein-protein recognition sites. Proteins. 2002;47:334–343. doi: 10.1002/prot.10085. [DOI] [PubMed] [Google Scholar]
- 38.Bahadur R.P., Chakrabarti P., Rodier F., Janin J. Dissecting subunit interfaces in homodimeric proteins. Proteins. 2003;53:708–719. doi: 10.1002/prot.10461. [DOI] [PubMed] [Google Scholar]
- 39.Bahadur R.P., Chakrabarti P., Rodier F., Janin J. A dissection of specific and non-specific protein-protein interfaces. J. Mol. Biol. 2004;336:943–955. doi: 10.1016/j.jmb.2003.12.073. [DOI] [PubMed] [Google Scholar]
- 40.Zhu H., Domingues F.S., Sommer I., Lengauer T. NOXclass: prediction of protein-protein interaction types. BMC Bioinformatics. 2006;7:27. doi: 10.1186/1471-2105-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Block P., Paern J., Hüllermeier E., Sanschagrin P., Sotriffer C.A. Physicochemical descriptors to discriminate protein-protein interactions in permanent and transient complexes selected by means of machine learning algorithms. Proteins. 2006;65:607–622. doi: 10.1002/prot.21104. [DOI] [PubMed] [Google Scholar]
- 42.Kurkal-Siebert V., Smith J.C. Low-temperature protein dynamics: a simulation analysis of interprotein vibrations and the boson peak at 150 K. J. Am. Chem. Soc. 2006;128:2356–2364. doi: 10.1021/ja055962q. [DOI] [PubMed] [Google Scholar]
- 43.Vojtechovsky J., Chu K., Berendzen J., Sweet R.M., Schlichting I. Crystal structures of myoglobin-ligand complexes at near-atomic resolution. Biophys. J. 1999;77:2153–2174. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kondrashov D.A., Zhang W., Aranda R., Stec B., Phillips G.N. Sampling of the native conformational ensemble of myoglobin via structures in different crystalline environments. Proteins. 2008;70:353–362. doi: 10.1002/prot.21499. [DOI] [PubMed] [Google Scholar]
- 45.Hinsen K. Structural flexibility in proteins; impact of the crystal environment. Struct. Bioinformatics. 2008;24:521–528. doi: 10.1093/bioinformatics/btm625. [DOI] [PubMed] [Google Scholar]
- 46.Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 47.MacKerell A.D., Bashford D., Bellott R.L., Dunbrack R.L., Evanseck J.D. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 48.Brunger A., Karplus M. Polar hydrogen positions in proteins: empirical energy placement and neutron diffraction comparison. Proteins. 1988;4:148–156. doi: 10.1002/prot.340040208. [DOI] [PubMed] [Google Scholar]
- 49.Kuriyan J., Weis W.I. Rigid protein motion as a model for crystallographic temperature factors. Proc. Natl. Acad. Sci. USA. 1991;88:2773–2777. doi: 10.1073/pnas.88.7.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bizzarri A.R., Paciaroni A., Arcangeli C., Cannistraro S. Low-frequency vibrational modes in proteins: a neutron scattering investigation. Eur. Biophys. J. 2001;30:443–449. doi: 10.1007/s002490100167. [DOI] [PubMed] [Google Scholar]
- 51.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 52.Kurkal-Siebert V., Agarwal R., Smith J.C. Hydration-dependent dynamical transition in protein: protein interactions at ≈240K. Phys. Rev. Lett. 2008;100:138102. doi: 10.1103/PhysRevLett.100.138102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.