

Abstract
Nav1.5 is the principal voltage-gated sodium channel expressed in heart, and is also expressed at lower abundance in embryonic dorsal root ganglia (DRG) with little or no expression reported postnatally. We report here the expression of Nav1.5 mRNA isoforms in adult mouse and rat DRG. The major isoform of mouse DRG is Nav1.5a, which encodes a protein with an IDII/III cytoplasmic loop reduced by 53 amino acids. Western blot analysis of adult mouse DRG membrane proteins confirmed the expression of Nav1.5 protein. The Na+ current produced by the Nav1.5a isoform has a voltage-dependent inactivation significantly shifted to more negative potentials (by ~5 mV) compared to the full-length Nav1.5 when expressed in the DRG neuroblastoma cell line ND7/23. These results imply that the alternatively spliced exon 18 of Nav1.5 plays a role in channel inactivation and that Nav1.5a is likely to make a significant contribution to adult DRG neuronal function.
Introduction
Mammalian voltage-gated sodium channels are critical determinants of electrical excitability in sensory neurons, underlie action potential generation and play a major role in nociception by controlling afferent impulse discharge (Devor, 2006; Lai et al., 2004; Wood et al., 2004). Nine sodium channel pore-forming α-subunits have been cloned, designated Nav1.1-Nav1.9, each of which contain four internally homologous transmembrane domains (I-IV) connected by the three interdomain cytoplasmic loops IDI/II, IDII/III and the smaller IDIII/IV. The α-subunits can be classified into two pharmacologically distinct groups on the basis of their sensitivity to the specific inhibitor tetrodotoxin (TTX): the TTX-resistant (TTX-R) channels Nav1.5, Nav1.8 and Nav1.9 are blocked by concentrations of TTX at least 200-fold greater than those that block TTX-sensitive (TTX-S) channels (Plummer and Meisler, 1999; Yu and Catterall, 2003). Each of the TTX-R channels have different electrophysiological properties: Nav1.5 produces a Na+ current with fast activation and inactivation (Renganathan et al., 2002), Nav1.8 produces a slowly activating and inactivating Na+ current (Akopian et al., 1999) and Nav1.9 produces a persistent Na+ current (Renganathan et al., 2002).
Expression of Nav1.8 is restricted to small-diameter sensory neurons in DRG, trigeminal ganglia and nodose ganglia (Akopian et al., 1996; Sangameswaran et al., 1996), whilst Nav1.9 is preferentially expressed in small-diameter DRG and trigeminal ganglia neurons (Dib-Hajj et al., 1998; Tate et al., 1998). Nav1.5 is the predominant cardiac subtype (Plummer and Meisler, 1999), with much lower levels also being expressed in brain, intestinal smooth muscle and embryonic skeletal muscle (Donahue et al., 2000; Gersdorff Korsgaard et al., 2001; Hartmann et al., 1999; Kallen et al., 1990; Ou et al., 2002). Two groups failed to demonstrate Nav1.5 mRNA expression in adult DRG (Akopian et al., 1999; Renganathan et al., 2002), though in vitro patch-clamp studies have detected a subset of rat small- and medium-diameter DRG neurons that have TTX-R voltage-gated Na+ currents with the properties of Nav1.5 (Renganathan et al., 2002; Rush et al., 1998; Scholz et al., 1998).
In addition to the full-length Nav1.5 cDNA sequence (Gellens et al., 1992; Rogart et al., 1989; Zimmer et al., 2002), a Nav1.5a/H1-2/hNbR1-2 (hereafter Nav1.5a) isoform with an in-frame deletion of exon 18 has been described in mouse, rat and a human neuroblastoma cell line, that encodes a protein with an IDII/III cytoplasmic loop reduced by 53 amino acids (Gersdorff Korsgaard et al., 2001; Ou et al., 2005; Zimmer et al., 2002). Other splice variants of Nav1.5 include a non-functional Nav1.5b (H1-3) isoform, with an in-frame deletion of exons 17 and 18 that removes 201 amino acids from the IDII/III cytoplasmic loop (Wang et al., 1996; Zimmer et al., 2002), and Nav1.5c with the introduction of a CAG trinucleotide into exon 18 that encodes an additional glutamine residue (Kerr et al., 2004). Studies of functional differences between the exon 18-deleted Nav1.5a and full-length Nav1.5 isoforms have produced apparently contradictory results. No differences between the mouse Nav1.5a and Nav1.5 channels were detected when expressed in the human embryonic kidney 293 (HEK293) cell line (Zimmer et al., 2002), whereas in the same cell line human Nav1.5a (hNbR1–2) showed differences in voltage dependence of activation and inactivation from a human full-length Nav1.5 isoform (hNbR1) (Ou et al., 2005). Potential differences when expressed in neuronal cells were not assessed.
In this study, we investigated the expression of Nav1.5 mRNA isoforms in the intact adult DRG and after axotomy, identifying Nav1.5a rather than the full-length Nav1.5 channel as the major isoform of adult mouse DRG, and demonstrate functional differences between the two isoforms when expressed in the DRG neuroblastoma cell line ND7/23 (previously validated as a suitable heterologous expression system for other tetrodotoxin-resistant sodium channels).
Results
Nav1.5 mRNA isoforms are expressed in adult mouse and rat DRG
Using reverse transcription PCR (RT-PCR), we show expression of Nav1.5 mRNA in adult mouse DRG for the first time (Fig. 1A). Although we have previously detected Nav1.5 mRNA in neonatal mouse DRG (Kerr et al., 2004), to the best of our knowledge, this is the first demonstration of its expression in the adult DRG of any species. The more abundant product of 358 basepairs (bp) was cloned and identified as the Nav1.5a isoform, which has an in-frame deletion of exon 18 (Gersdorff Korsgaard et al., 2001; Wang et al., 1996; Zimmer et al., 2002). The larger and much rarer product is of the expected size of 517/520 bp for the Nav1.5 (CAG-skipped) full-length isoform unresolved from the alternatively spliced Nav1.5c (CAG-inclusive) isoform (Kerr et al., 2004; Zimmer et al., 2002), with cloning and DNA sequencing identifying 18 cDNA clones as the Nav1.5 isoform and 2 as the Nav1.5c isoform. Therefore, in adult mouse DRG the apparent relative expression of isoforms is: Nav1.5a (Δ exon 18) ≫ Nav1.5 (CAG-skipped)>Nav1.5c (CAG-inclusive).
Fig. 1.

(A) Expression of Nav1.5 mRNA isoforms in adult and neonatal mouse dorsal root ganglia (DRG). A faint Nav1.5/Nav1.5c product of approximately 520 bp and a more abundant Nav1.5a (Δ exon 18) product of approximately 360 bp were detected in adult (lane 3) and neonatal DRG (lane 5) RNA that had been reverse transcribed (RT+), but not from either adult or neonatal DRG RNA that has not been reverse transcribed (RT-; lanes 2 and 4, respectively. Lane 1 was a 1 kb DNA ladder (Invitrogen). The neonatal sample was from postnatal day 2 (P2), and RT-PCR was for 35 cycles. (B) Expression of Nav1.5 mRNA isoforms in adult rat tissues. Nav1.5/Nav1.5c and Nav1.5a products of approximately 370 and 210 bp were detected in adult rat DRG (lane 3), trigeminal ganglia (lane 4) and heart (lane 5). Lane 2 was a water control, and lane 1 was a 1 kb DNA ladder. RT-PCR was for 35 cycles.
Nav1.5 cDNA was also cloned from adult rat DRG, with DNA sequencing identifying 46 clones as being the Nav1.5 isoform and 2 clones as the Nav1.5c isoform. Unlike mouse, the level of expression of Nav1.5a (Δ exon 18) was similar to Nav1.5/Nav1.5c (Fig. 1B), such that the apparent relative expression of isoforms in adult rat DRG is Nav1.5a≈Nav1.5>Nav1.5c. We have previously reported expression of Nav1.5/Nav1.5c mRNA in adult rat trigeminal ganglia (Kerr et al., 2004), but as the primer set used in those experiments could not amplify the Nav1.5a isoform, we also demonstrate here that the relative expression of Nav1.5/Nav1.5c to Nav1.5a in the rat trigeminal ganglia is similar to that in DRG (Fig. 1B). In contrast, adult cardiac expression of Nav1.5/Nav1.5c markedly exceeded Nav1.5a, as previously reported (Gersdorff Korsgaard et al., 2001).
The effect of axotomy on Nav1.5 and Nav1.8 mRNA expression in adult mice
In rats, peripheral nerve transection (axotomy) is known to result in a decrease in Nav1.8 mRNA levels in small-diameter lumbar DRG neurons (Dib-Hajj et al., 1996; Okuse et al., 1997). We therefore optimized real-time quantitative RT-PCR assays for mouse Nav1.5a, Nav1.5/Nav1.5c and Nav1.8/Nav1.8c mRNAs in order to assess the effects of peripheral axotomy on DRG expression of these channels. The specificity of each assay was demonstrated by testing against Nav1.5a, Nav1.5, Nav1.8 and Nav1.9 cDNA plasmids (data not shown). Expression levels were normalized to GAPDH mRNA, which has previously been shown to be unchanged following sciatic nerve lesion (Macdonald et al., 2001), and the suitability of GAPDH as a control was confirmed in the present study by the mean threshold cycle values (Ct) not being altered 1 week after axotomy (P=0.16824). Seven days after peripheral axotomy, the expression of Nav1.5a mRNA decreased by 48.1%, Nav1.5/Nav1.5c mRNA decreased by 46.4% and Nav1.8/Nav1.8c mRNA also decreased by 46.4% in mouse lumbar L4 and L5 DRG (Fig. 2).
Fig. 2.
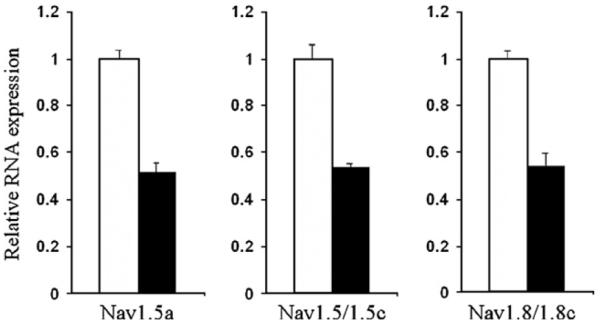
Expression of Nav1.5 and Nav1.8 mRNAs decrease in adult mouse DRG after axotomy. In quantitative RT-PCR assays the expression of Nav1.5a mRNA decreased to 0.519±0.027 of control (n=5; P<0.0005), Nav1.5/Nav1.5c mRNA decreased to 0.536±0.019 of control (n=6; P<0.0001) and Nav1.8/Nav1.8c mRNA decreased to 0.536±0.065 of control (n=6; P<0.0005) in pooled ipsilateral (axotomized) lumbar L4 and L5 DRG compared to contralateral (unaxotomized) controls, 7 days after axotomy. Data are shown as mean±S.E., in which expression after axotomy (filled boxes) was compared to contralateral controls of 1.00 relative units (unfilled boxes).
Expression of Nav1.5 protein in adult DRG
To address the potential expression of Nav1.5 protein in adult DRG, Western blots of detergent-solubilized membrane preparations from adult mouse lumbar DRG and heart were probed with an anti-Nav1.5 antibody that has previously been used to detect Nav1.5 protein in rat cardiac myocytes (Dhar et al., 2001). A single major band of >250 kDa was detected in both DRG and heart, with expression in DRG being much lower (Fig. 3; note 8-fold more DRG membrane protein used). This size was a little larger than previous reports from cardiac tissue (Cohen and Levitt, 1993; Dhar et al., 2001; Mohler et al., 2004), and the specificity of the band was confirmed by the absence of signal detected in heart membrane proteins when the antibody was pre-adsorbed to immunizing peptide (data not shown). The predicted difference in size between Nav1.5 and Nav1.5a isoforms of only 5.7 kDa (Wu et al., 2003) would not be expected to be detected under the conditions used.
Fig. 3.

Detection of Nav1.5 protein in DRG and heart membrane proteins isolated from adult mice. A Western blot of detergent-solubilized membrane proteins from heart (50 μg, lane 1) and DRG (400 μg, lane 3) was probed with an anti-Nav1.5 antibody, resulting in the detection in both tissues of a single major band with an apparent molecular weight (Mr)of >250 kDa. Lane 2 is blank. Positions of molecular weight markers are indicated in kDa.
Electrophysiological properties of Nav1.5 and Nav1.5a in DRG neuroblastoma cells
Although we have found Nav1.5a mRNA to be the major isoform expressed in mouse DRG, it is not known whether there are functional differences between the currents produced by this isoform and the full-length Nav1.5 isoform when expressed in neuronal cells. Therefore we made use of the ND7/23 DRG neuroblastoma cell line, which is known to produce endogenous TTX-S but not TTX-R Na+ currents, and has been used successfully to express Nav1.8 channels (Choi et al., 2004; John et al., 2004; Zhou et al., 2003). We found that in untransfected ND7/23 cells, the endogenous TTX-S INa was blocked completely by 30 nM TTX (INa at −12 mV: 12.13±1.8 pA/pF, TTX 0±0 pA/pF, n=7, P<0.001). The IC50 for TTX blockade of the endogenous Na+ current was between 1 and 3 nM, consistent with a previous report (Zhou et al., 2003).
ND7/23 cells were transiently transfected with Nav1.5 or Nav1.5a cDNA, using a vector that co-expresses EGFP, and whole-cell voltage-clamp recordings were made from the fluorescent, transfected cells. Endogenous Na+ current was blocked by 30 nM TTX added to the bath solution, a concentration that will have had negligible effect on Nav1.5 channel currents (Ou et al., 2005; Zimmer et al., 2002), and a low extracellular Na+ concentration (20 mM) was used in order to reduce the amplitude of Na+ currents (INa) and thereby to improve the control of voltage (Zimmer et al., 2002). Depolarizations positive to −72 mV elicited rapidly activating and inactivating currents in both Nav1.5- and Nav1.5a-transfected cells (Figs. 4A, B). Following correction of the current–voltage relation for the voltage-drop across the series resistance between the pipette electrode and the cell interior using Eq. (1) (Fig. 4B), there was no difference between the two isoforms in the half-maximal voltage of activation of the currents (Fig. 4C), although the slope factor of activation of the Nav1.5a currents was slightly greater than that of the full-length isoform, reflecting a less steep voltage-dependence of activation (Fig. 4D).
Fig. 4.

Voltage-gated inward currents through Nav1.5 and Nav1.5a channels. (A) Example of inward currents through Nav1.5 channels activated by depolarizing pulses to the command potentials (CP) indicated. Holding potential was −92 mV. Solid line superimposed on current trace at −47 mV shows an example of a fit to a decaying exponential (Eq. (4)). Inset shows voltage protocol. (Identical findings were observed using the Nav1.5a construct.) (B) Peak inward current–voltage relation for data shown in A. Open circles—uncorrected data; filled triangles—data corrected for voltage-drop across the series resistance (Eq. (1)). Solid lines represent a fit to a modified Boltzmann relation (Eq. (2)). In this representative example, V0.5,act for uncorrected data shown was −46.5 mVand for corrected data was −41.6 mV. (C) Mean (±S.E.M.) V0.5,act for Nav1.5 and Nav1.5a channel currents. (D) Mean (±S.E.M.) for slope factors (s). *, P<0.05.
The current traces provided evidence of marked differences in both voltage- and time-dependent inactivation of the currents. The time course of current inactivation was fitted with a single decaying exponential (e.g. Fig. 4A), the time constant of which was voltage-dependent (Fig. 5A). At moderately depolarizing command potentials (i.e. −52 mV to −42 mV), currents through the exon 18-deleted Nav1.5a isoform showed faster inactivation than currents through the full-length Nav1.5 channel (Fig. 5A). There were no differences between the isoforms in the rate of recovery from inactivation (Fig. 5B), whereas the steady-state inactivation of Nav1.5a was shifted negatively by ~5 mV in comparison to the full-length isoform (Fig. 5D).
Fig. 5.

Inactivation of Nav1.5 and Nav1.5a channel currents. (A) Voltage-dependence of time constant (τ; Eq. (4)) of inactivation for Nav1.5 (open circles) and Nav1.5a (filled triangles). ***, P<0.001; *, P<0.05. (B) Recovery from inactivation of Nav1.5 (open circles) and Nav1.5a (filled triangles) channel currents. Solid lines represent fits to double exponential relation (Eq. (5)). Fitted parameters were as follows: Nav1.5, A=0.96±0.05, τ1=5.04±0.24 ms, τ2=158.9±28.8 ms (n=28); Nav1.5a, A=0.97±0.04, τ1=4.52±0.22 ms, τ2=246.9±60.3 ms (n=28, n.s.). (C) Representative current traces obtained at a command potential of −22 mV following preconditioning potentials indicated using voltage protocol shown. (D) Left-hand panel shows mean (±S.E.M.) V0.5,inact for Nav1.5 and Nav1.5a channel currents. **, P<0.01. Right-hand panel shows mean (±S.E.M.) for slope factors (s).
Due to the overlap between the voltage-dependence of steady-state activation and inactivation, a so-called ‘window current’ (Herzog et al., 2001) would occur at membrane potentials from −80 mV to −40 mV (Fig. 6). The shift in the voltage-dependence of inactivation together with the lower slope of activation of Nav1.5a compared to the full-length isoform results in a shift of the membrane potential of peak window current from −62.7 mV (Nav1.5) to −66.4 mV (see inset, Fig. 6).
Fig. 6.
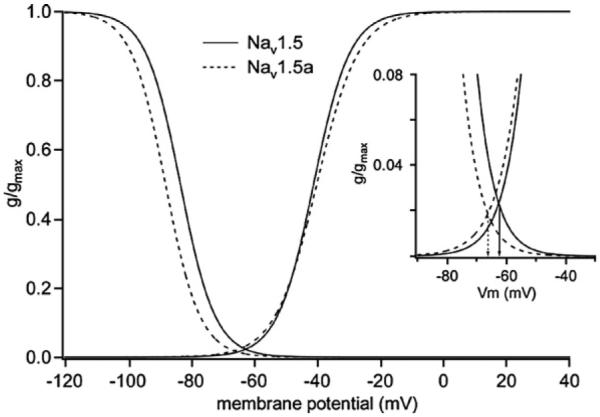
Voltage-dependence of activation and inactivation for Nav1.5 and Nav1.5a currents. The mean fitted parameters used are from Figs. 4C, D and 5D. Nav1.5: V0.5,act=−41.1 mV, sact=5.8 mV; V0.5,inact=−83.3 mV, sinact=−5.5 mV. Nav1.5a: V0.5,act=−40.2 mV, sact=6.6 mV; V0.5,inact=−88.1 mV, sinact=−5.5 mV. Inset shows region of overlap between activation and inactivation that would produce a window current. Arrows indicate voltage of peak window current for each isoform.
Discussion
In this study, the expression of Nav1.5 mRNA in adult mouse and rat DRG was detected by RT-PCR and confirmed by DNA sequencing, with Nav1.5 protein expression being detected by Western blot analysis. A previous study that did not detect Nav1.5 mRNA in adult mouse DRG may have been limited by the sensitivity of the RT-PCR, as rat sequence primers were used because the mouse cDNA had not been cloned at that time (Akopian et al., 1999) (four nucleotide differences in forward primer). Another study of Nav1.5 mRNA expression using a combined RT-PCR and restriction enzyme polymorphism assay in embryonic and neonatal rat DRG suggested that Nav1.5 levels decrease during development from embryonic day 15 (E15) to little or no detectable Nav1.5 in the DRG at birth or postnatal day 7 (P7) (Renganathan et al., 2002). That result may also have been influenced by the primer pair used, which amplifies both Nav1.5 (three substitutions and an insertion in reverse primer) and Nav1.6 (Black et al., 1998; Renganathan et al., 2002), and by Nav1.6 mRNA expression increasing substantially between E17 and P15 (Felts et al., 1997).
We have previously reported that the apparent ratio of Nav1.5/Nav1.5c to the Nav1.5a mRNA isoform varies widely between different mouse tissues (Kerr et al., 2004). Expression in the adult DRG is similar to neonatal DRG in which Nav1.5a ≫ Nav1.5/Nav1.5c (Fig. 1A), in contrast to similar levels of each isoform in neonatal heart (Haufe et al., 2005; Kerr et al., 2004) and Nav1.5/Nav1.5c ≫ Nav1.5a in adult heart (Gersdorff Korsgaard et al., 2001; Haufe et al., 2005; Kerr et al., 2004; Zimmer et al., 2002). These data indicate that in the adult mouse there is a marked tissue-specific bias in the pre-mRNA alternative splicing of exon 18, with retention in heart but preferential excision in DRG. Nav1.5a mRNA is also a major isoform expressed in adult rat DRG and trigeminal ganglia (Fig. 1B). A species-specific mechanism appears to control the alternative splicing that produces either the Nav1.5 (CAG-skipped) or Nav1.5c (CAG-inclusive) isoforms: the ratio of Nav1.5 to Nav1.5c isoforms is approximately 9:1 in adult mouse DRG and heart, 20:1 in adult rat DRG and trigeminal ganglia (see above; (Kerr et al., 2004)), and in humans the corresponding Q1077del and Q1077 isoforms have a ratio of 2:1 in heart (Makielski et al., 2003; Tan et al., 2005).
After peripheral axotomy, expression of Nav1.5a, Nav1.5/Nav1.5c and Nav1.8/Nav1.8c mRNAs each decreased by around 50% in mouse lumbar L4 and L5 DRG (Fig. 2), similar in extent to that previously described for Nav1.8 mRNA in rat (Dib-Hajj et al., 1996; Okuse et al., 1997). The down-regulation of Nav1.8 after axotomy has been attributed to a lack of peripheral target-derived signals, such as nerve growth factor (NGF) and glial cell line-derived neurotrophic factor (GDNF), as transection of the central axonal projections (dorsal rhizotomy) did not change Nav1.8 levels ((Sleeper et al., 2000); and references therein). Interestingly, whereas Nav1.5 mRNA levels decreased in the DRG after axotomy, it has previously been reported that expression within adult rat hind leg muscle increased from undetectable by at least 100-fold after axotomy (Kallen et al., 1990).
Our electrophysiological results demonstrate for the first time that there are functional differences between the exon 18-deleted Nav1.5a isoform and the corresponding full-length isoform when expressed in a neuronal cell line. The time constants of inactivation at moderate potentials (i.e. −42 to −52 mV) were faster (Fig. 5A), and the voltage-dependence of steady-state inactivation shifted ~5 mV negatively (Fig. 5D), in Nav1.5a compared to the full-length Nav1.5 channels. These results are in contrast to the findings of Zimmer and colleagues who, using precisely the same mouse Nav1.5 and Nav1.5a constructs, found no functional differences between the isoforms when heterologously expressed in HEK293 cells (Zimmer et al., 2002). The remarkable similarity in V0.5,act and V0.5,inact of the full-length isoform between this study (V0.5,act~−41 mV; V0.5,inact~−83 mV) and Zimmer et al. (V0.5act~−39 mV; V0.5,inact~−83 mV; Zimmer et al., 2002) suggests that it is the neuronal phenotype of the ND7/23 cells interacting with the Nav1.5a isoform, that is responsible for the differences reported here. In contrast, functional differences in both voltage-dependent activation and inactivation have been reported between human Nav1.5a (hNbR1–2) and a full-length isoform when expressed in HEK293 cells (Ou et al., 2005). However, of the human full-length Nav1.5 isoforms Q1077del and Q1077 that correspond to rodent Nav1.5 and Nav1.5c, respectively (Kerr et al., 2004; Makielski et al., 2003; Tan et al., 2005), the one used by Ou et al. (2005; hNbR1; AB158469) corresponds to human Q1077, so that this study was equivalent to comparing the rodent Nav1.5c and Nav1.5a isoforms. Recent evidence has demonstrated the importance of the glutamine at position 1077 to channel function: when the human Q1077del and Q1077 isoforms were each expressed in HEK293 cells in the presence of eight different common polymorphisms of SCN5A (Nav1.5), six showed distinct phenotypes dependent on the splice variant used (Makielski et al., 2003; Tan et al., 2005).
Our results implicate the 53 residues encoded by the alternatively spliced exon 18 in the control of channel inactivation. At least two possible mechanisms exist for the observed differences between the two isoforms, which are not mutually exclusive: (a) the deletion of the 53 amino acids may directly affect the biophysical properties of the channel and the resulting sodium current, and (b) the inclusion or exclusion of the 53 amino acids may alter a protein interaction site, permitting or preventing association with target proteins. Cytoplasmic loops are potential sites for interaction with intracellular proteins that modulate channel function (Malik-Hall et al., 2003; Plummer and Meisler, 1999), but the portion of the Nav1.5 IDII/III cytoplasmic loop encoded by exon 18 is C-terminal to the motif necessary for interaction with ankyrin-G (Lemaillet et al., 2003; Mohler et al., 2004) and has only one type of predicted protein motif (PROSITE, release 19.23): [S/T]-X-X-[D/E] putative phosphorylation sites for CK2 (formerly casein kinase 2) (Meggio and Pinna, 2003). In the mouse exon 18-encoded sequence there are four putative sites for phosphorylation by CK2, two of which are conserved between mouse, rat and human. However, in preliminary experiments, we found no difference between the mouse Nav1.5 and Nav1.5a isoforms in the effect on current inactivation of pre-treatment with the CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB; Sarno et al., 2001), suggesting that CK2 phosphorylation of these sites does not account for the functional differences between the isoforms (Kerr, Gao, James & Wynick, unpublished data). Nevertheless, it is clear from work on the human SCN5A (Nav1.5) common polymorphisms P1090L and S1103Y that the exon 18-encoded region can influence voltage-dependent gating (Splawski et al., 2002; Tan et al., 2005).
Although we have demonstrated the presence of Nav1.5 protein in adult DRG (Fig. 3), the functional significance of this expression is as yet unclear. In a study of small-diameter neurons from rat DRG, TTX-R voltage-gated Na+ currents with the properties of Nav1.5 were detected in 20% of neonatal and 3% of adult neurons (Renganathan et al., 2002; Rush et al., 1998), and similar currents have been detected in medium-diameter rat DRG neurons (Scholz et al., 1998). In those studies the range of half-maximal voltages of activation and of inactivation (V0.5,act~−42 mV to −31 mV; V0.5,inact~−113 mV to −66 mV) are comparable with those found in the present study (Renganathan et al., 2002; Rush et al., 1998; Scholz et al., 1998). As the resting membrane potential of the soma of adult DRG neurones in vivo has been reported to range from −69 mV to −41 mV (Fang et al., 2005), similar to in vitro values (Rush et al., 1998), one might predict that the majority of Nav1.5 and Nav1.5a channels in the ND7/23 cells would be in the inactivated state and therefore not available for activation. However, this overlooks the existence of a window current (Fig. 6) that, assuming the half-maximal voltages of activation and inactivation in the present study apply to adult DRG neurones in vivo, could be expected to contribute to the resting membrane potential and membrane excitability as suggested for the persistent TTX-R Na+ current (Herzog et al., 2001). Moreover, negative shifts in voltage-dependent inactivation that are thought to be due to dialysis of cell contents during whole-cell patch-clamp recordings (Kunze et al., 1985; Sakakibara et al., 1993) may result in the underestimation of the contribution of Nav1.5a channels to the window current and membrane excitability. Thus, the properties of Nav1.5a channels are consistent with their making a significant contribution to adult DRG neuronal function.
In summary, we have demonstrated the expression of Nav1.5 mRNA isoforms in adult mouse and rat DRG, with the predominant Nav1.5a isoform in adult mouse DRG decreasing following peripheral nerve injury. Expression of Nav1.5 protein in adult DRG was confirmed by Western blot analysis, and differences have been identified between the Nav1.5 and Nav1.5a isoforms in voltage-dependent inactivation of the currents when expressed in a neuronal cell line. Since these differences may be of considerable significance to DRG function, further investigation of the role of Nav1.5 channels in the adult DRG is now warranted, though this cannot currently be addressed in Nav1.5-knockout (Scn5a−/−) mice due to lethality around embryonic day 11.5 (Papadatos et al., 2002).
Experimental methods
Animals, surgery and tissue collection
All animals were fed standard chow and water ad libitum, and animal care procedures were carried out within UK Home Office protocols and guidelines. Neonatal (postnatal day 2) and adult (12-week-old) lumbar DRG were each pooled from 129/OlaHsd mice (Bristol University colony). For studies on peripheral nerve transection (axotomy), the right sciatic nerve of 10-12-week-old male 129/OlaHsd mice were transected at the mid-thigh level (Kerr et al., 2004), prior to killing 7 days later by cervical dislocation to obtain ipsilateral (axotomized) and contralateral (control) lumbar L4 and L5 DRG pools. Lumbar DRG, trigeminal ganglia and hearts were each pooled from two adult male Wistar rats (Bristol University colony). Tissues were frozen on dry ice and stored at −80 °C.
RNA extraction and reverse transcription
Total RNA isolation, DNase treatment and re-extraction, and reverse transcription (RT) reactions with random hexamers were as previously published (Kerr et al., 2004).
RT-PCR of Nav1.5 cDNA from mouse and rat tissues
Nav1.5 cDNA was amplified from adult male or neonatal 129/OlaHsd mouse DRG using 5 μl (100 ng total RNA equivalent) of RT reaction in a 50 μl RT-PCR with recombinant Taq DNA polymerase (Invitrogen) and the primers 5for1 (Kerr et al., 2004) and 5′-GTCTTGCGCAGTCTCCACCAGAC-3′ (nucleotides (nt) 3646-3624 of AJ271477 (Zimmer et al., 2002)). These amplify a portion of the IDII/III cytoplasmic loop coding region, spanning DNA corresponding to human SCN5A (Nav1.5) exons 17-20 (Wang et al., 1996). PCR conditions were: 94 °C for 2 min, and 35 cycles of 94 °C, 30 sec; 64 °C, 45 sec; 72 °C, 45 sec, with a final 72 °C for 10 min. Adult products were excised from a 3% agarose gel, purified (GenElute, Sigma-Aldrich) and cloned into pCRII-TOPO (Invitrogen) (Kerr et al., 2004). Mouse Nav1.5 cDNA sequences included the three previously reported nucleotide substitutions (Kerr et al., 2004), compared to AJ271477 (Zimmer et al., 2002).
Rat Nav1.5 cDNA was amplified from adult male tissues, as above, with primers 5′-ACACGATTCGAGGAGGACAAGCGA-3′ and 5′-CGGCGGTGTTGGTCATGTCAGCT-3′ that correspond, respectively, to nt 3282-3305 and 3648-3626 of M27902 (Rogart et al., 1989) and span DNA corresponding to human SCN5A exons 17-19 (Wang et al., 1996).
DNA sequencing was performed by the Department of Biochemistry, Oxford University.
Real-time quantitative RT-PCR assays of mouse Nav1.5 and Nav1.8 mRNAs
Real-time quantitative RT-PCR assays (reviewed in Giulietti et al., 2001) used primer and probe sets designed using default parameters of Primer Express software (Applied Biosystems). Probes had the 5′ fluorescent reporter dye FAM (6-carboxyfluorescein) and the 3′ quencher dye TAMRA (6-carboxy-tetramethyl-rhodamine), except for the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) control which had a 5′ VIC reporter dye.
Primers to detect the Nav1.5a isoform cDNA (Gersdorff Korsgaard et al., 2001; Wang et al., 1996; Zimmer et al., 2002) were based on a reverse primer spanning the exon 17/19 junction sequence: forward primer 5′-CATCGCAGTGGCTGAGTCA-3′, reverse primer 5′-GTAACTGTCCTCGGGAGTCTGT-3′ and TaqMan antisense probe 5′-CCTCCTCCGTGCCAAGGCTGTTCT-3′, that correspond, respectively, to nt 3200-3218, (3464-3447+3287-3284) and 3270-3247 of AJ271477 (Zimmer et al., 2002), except for the previously described nt 3449, 3448 and 3284 substitutions (underlined; (Kerr et al., 2004)). Primers to detect Nav1.5/Nav1.5c isoform cDNAs were for a product that crossed a 1.28 kb intron (anonymous genomic clone AC121922 nt 56143-57425/57428) analogous to human SCN5A intron 17 (Wang et al., 1996): forward primer 5′-CAGGAAGAGGATGAGGAGAACAG-3′, reverse primer 5′-CACCAGACACAACTTGGGATTC-3′ and TaqMan antisense probe 5′-TGTTTGCTGGACTCTTCCTCCTCCGTG-3′, that correspond, respectively, to nt 3231-3253, 3309-3288 and 3286-3260 of AJ271477 (Zimmer et al., 2002), except for the previously described nt 3284 substitution (underlined; (Kerr et al., 2004)). Primers to detect Nav1.8/Nav1.8c isoform cDNAs were for a product that crossed Scn10a intron 16 of approximately 1.4 kb (Kerr et al., 2004; Souslova et al., 1997): forward primer 5′-GACCTTGATGAGCTCGAGGAA-3′, reverse primer 5′-GGTGATCTTCACACTTTTGGACTTG-3′ and internal TaqMan probe 5′-TCCTGGCAGGAAGAGAGCCCCAA-3′, that correspond, respectively, to nt 3051-3071, 3165-3141 and 3096-3118 of Y09108 (Souslova et al., 1997), except for the previously described nt 3159 substitution (underlined; Table I of Kerr et al., 2004). Endogenous control GAPDH cDNA primers were: forward primer 5′-GCAGTGGCAAAGTGGAGATTG-3′, reverse primer 5′-CTGGAACATGTAGACCATGTAGTTGA-3′ and TaqMan probe 5′-CCATCAACGACCCCTTCATTGAC-3′, that correspond, respectively, to nt 111-131, 184-159 and 135-157 of M32599 (Sabath et al., 1990).
Replicate 25 μl PCRs included 12.5 μl TaqMan Universal PCR Master Mix (Applied Biosystems), water (Sigma-Aldrich), primers, probe and template, and used standard cycling conditions (95 °C for 10 min, followed by 50 cycles of 95 °C, 15 sec and 60 °C, 1 min) in an ABI PRISM 7900 Sequence Detection System (Applied Biosystems). Primer and probe concentrations were optimized against corresponding cDNA plasmids, and single products of the expected size were amplified from DRG, as detected by gel electrophoresis on 4% agarose gels (data not shown). For each RNA sample, duplicate incubations with reverse transcriptase (RT+) and a single incubation without enzyme (RT−; control for RT-dependence) were carried out at 1 μg total RNA per 50 μl (Kerr et al., 2004), from each of which triplicate PCRs were set up. Sodium channel and GAPDH templates were amplified in separate wells of the same 96 well plate, all threshold cycle (Ct) values were determined at the 0.2 default threshold, and relative expression levels were determined by the comparative Ct method (Giulietti et al., 2001). Results are presented as mean±S.E. of log transformed data, with statistical significance judged by one-tail Student t-test (Proudnikov et al., 2003).
Preparation of membrane proteins and Western blotting
Adult male 129/OlaHsd mouse lumbar DRG (pooled 10- and 15-week-old) and hearts (15-week-old) were frozen on dry ice and stored at −80 °C. Ice-cold homogenization buffer (Mohler et al., 2004), including protease inhibitor cocktail (Sigma-Aldrich; P8340) but without EGTA, was added to DRG, vortexed briefly and homogenized using an ice-cold Dounce homogenizer. Hearts were ground into a fine powder by mortar and pestle prior to the addition of homogenization buffer. Homogenates were centrifuged at 1000×g for 10 min at 4 °C, to pellet large membranes and nuclei (Mohler et al., 2004), the supernatant was saved and the pellet resuspended (Hartshorne and Catterall, 1984; Nishiwaki et al., 1998) in homogenization buffer, and centrifuged as above. The combined supernatants were centrifuged at 100,000×g for 1 h at 4 °C. The pellets were solubilized in 75 μl (DRG) or 400 μl (heart) of ice-cold homogenization buffer (Mohler et al., 2004), without sucrose or EGTA, containing 1.5% (v/v) Triton X-100 (VWR International) and 1% (w/v) sodium deoxycholate (Sigma-Aldrich), and incubated at 4 °C with rotation for 1 h. Insoluble material was removed by centrifugation at 20,000×g for 1 h (Nishiwaki et al., 1998) at 4 °C, and the supernatant protein concentration was determined by Pierce BCA assay kit (Perbio Science UK).
Protein samples and molecular weight markers (Precision Plus Protein Standards, All Blue; Bio-Rad Laboratories) were boiled in SDS-PAGE sample buffer containing 5% β-mercaptoethanol, fractionated on 4% stacking and 6% resolving acrylamide (37.5:1 acrylamide: bisacrylamide; Sigma-Aldrich) gels, and transferred onto nitrocellulose (Hybond-C Extra, GE Healthcare). Membranes were blocked overnight in 0.5% Blocker (1×TBS [20 mM Tris-HCl (pH 7.6), 137 mM NaCl] with diluted 10% Western Blocking Reagent [Roche]) and probed for 1 h with anti-Nav1.5 antibody (Alomone Labs; ASC-005) at a dilution of 1:200 in 10 ml of 0.5% Blocker, each at 4 °C. Subsequent stages at room temperature were: washing for 2×10 min in TBST (1×TBS with 0.1% (v/v) Tween 20 [VWR International]); re-blocking for 2×10 min in 0.5% Blocker; incubation for 45 min with HRP-conjugated goat anti-rabbit secondary antibody (Cell Signaling Technology; 7727) at a dilution of 1:2000 in 10 ml of 0.5% Blocker; and washing for 4×10 min in TBST. Antibody was detected using Super-Signal West Pico Chemiluminescent Substrate (Pierce) and Hyperfilm ECL (GE Healthcare).
Pre-incubation of anti-Nav1.5 antibody with control peptide followed manufacturerTs instructions (Alomone Labs).
Cell culture and transfection
The ND7/23 DRG neuroblastoma cell line (Wood et al., 1990) obtained from ECACC was cultured as previously reported (Roobol et al., 1995). For electrophysiological experiments, cells were plated at low density (4 or 6×104 cells) onto coverslips in 40×11 mm petri dishes (Helena Bioscience). 24 h later cells in 1.5 ml antibiotic-free medium were transfected with 500 μl lipid–DNA complex, containing 4 μg endotoxin-free maxiprep DNA (Qiagen) and 25 μl Optifect (Invitrogen), following manufacturer’s recommendations (Invitrogen). Plasmids of mouse Nav1.5 (mH1) or Nav1.5a (mH1-2) in the vector pTSV40Gnew were kindly provided by Thomas Zimmer (University of Jena, Germany) (Zimmer et al., 2002), and were verified by DNA sequencing. In these constructs the sodium channel is expressed from the SV40 promoter, and enhanced green fluorescent protein (EGFP) from the CMV promoter.
Electrophysiological recordings
All whole-cell patch-clamp recordings were carried out between 24 h and 32 h after transfection. The coverslips were mounted in a perfusion chamber on the stage of an inverted microscope (Olympus CK-40, Olympus UK Ltd). Patch-pipettes were pulled (Narishige PP-830) from borosilicate glass (Corning 7052, A-M Systems) and polished with a microforge (Narishige MF-83). The tip resistance of the pipette was 1.0-1.5 MΩ when filled with pipette solution, modified from Choi et al. (2004), which contained (in mM): 140 CsF, 1 EGTA, 10 NaCl, 10 HEPES, 5 glucose, pH 7.25 (CsOH). The bath solution for recordings, from Zimmer et al. (2002), contained (in mM): 120 CsCl, 20 NaCl, 0.1 CaCl2, 10 HEPES, 1 MgCl2, 10 glucose, 0.00003 tetrodotoxin (TTX), pH 7.35 (CsOH). Liquid junction potentials were compensated electronically with the pipette in the bath prior to formation of the giga-seal. It was estimated that there would be a junction potential of ~−8mV(−8.0±0.3 mV, n=3) in the whole-cell configuration, for which all data has been corrected. Whole-cell currents were passed through a 5 kHz lowpass Bessel filter and recorded at 20 kHz to the hard disk of an Apple Macintosh G3 computer using an Axopatch 200B patch-clamp amplifier (Axon Instruments Inc) and Pulse software (HEKA Elektronik GmbH). The experiments were conducted at room temperature (20-22 °C). The currents were elicited by 25 ms test potentials from −82 to +18 mV in 5 mV increment from a holding potential of −92 mV at 2 ms interval. Currents were normalized to the whole-cell capacitance as a measure of cell size. Test potentials were corrected for the voltage drop error across the series resistance (2.15±0.16 MΩ, n=71) using the following equation:
| (1) |
where Vm is the corrected test potential, Vcom is the uncorrected command test potential, Ipk is the peak inward current and Rs is the series resistance, according to Marty and Neher (1995). For example, the corrected test potential at a command potential of −32 mV was −26.75±0.79 mV (n=71). The current–voltage (I-V) relationship for INa was constructed and the data were fitted with the modified Boltzmann equation (Yuill et al., 2000):
| (2) |
where INa is the current density at the corrected test potential of Vm, Gmax is the maximal Na conductance, Vrev is the reversal potential, V0.5,act is the membrane potential of half-maximal current activation, and s is the slope factor.
Steady-state inactivation was determined using a double-pulse protocol consisting of 500 ms conditioning pulses to command potentials of between −122 and −22 mV, followed by a constant test pulse of 25 ms to −22 mV (2 s pulse interval). The amplitude of peak INa during the test pulses was normalized to the maximum peak current and plotted as function of the conditioning potential. Data were fitted with a Boltzmann equation:
| (3) |
where V is the test potential, V0.5,inact is the potential of half-maximal inactivation, and s is the slope factor.
The time course of INa decline during depolarizing pulses to command potentials from −52 mV to +18 mV was fitted with a single decaying exponential, as follows:
| (4) |
where I(t) is the current at time t, A is the maximum current amplitude at t=0, t is the time following depolarization, τ is the time constant and C is the non-inactivating component of the current.
Recovery from inactivation was evaluated by using a double-pulse protocol from a holding potential of −112 mV. A 25 ms control pulse (Icon) to −22 mV was followed after a variable delay by a 25 ms test pulse (Itest) to −22 mV. The time course of the recovery from inactivation was obtained by plotting the ratio, Itest/Icon, against the delay period (Δt); this was fitted with the following double exponential function:
| (5) |
where A is the fraction of the total current described by a fast time constant (τ1) and τ2 represents a slow time constant.
Electrophysiology data analysis
Data were analyzed using PulseFit (HEKA Elektronic GmbH) and IgorPro Vs3.16B (Wavemetrics Inc). Statistical comparisons of data were made using one-way analysis of variance (ANOVA) with, where appropriate, Student–Newman–Keuls multiple comparisons post hoc test. P<0.05 was accepted as being significant (n.s. denotes lack of statistical significance). Statistical analyses were carried out using GraphPad 4.02 (GraphPad Software Inc.).
Drugs
TTX (Sigma-Aldrich) was prepared as a stock solution of 3 mM in deionized water and stored at −20 °C. The final concentration of 30 nM was freshly prepared by series dilution with extracellular solution on the days of experiments.
References
- Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Black JA, Dib-Hajj S, Cohen S, Hinson AW, Waxman SG. Glial cells have heart: rH1 Na+ channel mRNA and protein in spinal cord astrocytes. Glia. 1998;23:200–208. [PubMed] [Google Scholar]
- Choi JS, Tyrrell L, Waxman SG, Dib-Hajj SD. Functional role of the C-terminus of voltage-gated sodium channel Na(v)1.8. FEBS Lett. 2004;572:256–260. doi: 10.1016/j.febslet.2004.07.047. [DOI] [PubMed] [Google Scholar]
- Cohen SA, Levitt LK. Partial characterization of the rH1 sodium channel protein from rat heart using subtype-specific antibodies. Circ. Res. 1993;73:735–742. doi: 10.1161/01.res.73.4.735. [DOI] [PubMed] [Google Scholar]
- Devor M. Sodium channels and mechanisms of neuropathic pain. J. Pain. 2006;7:S3–S12. doi: 10.1016/j.jpain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Dhar MJ, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, Brosius FC, Kass RS, Isom LL. Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation. 2001;103:1303–1310. doi: 10.1161/01.cir.103.9.1303. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj S, Black JA, Felts P, Waxman SG. Down-regulation of transcripts for Na channel alpha-SNS in spinal sensory neurons following axotomy. Proc. Natl. Acad. Sci. U. S. A. 1996;93:14950–14954. doi: 10.1073/pnas.93.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc. Natl. Acad. Sci. U. S. A. 1998;95:8963–8968. doi: 10.1073/pnas.95.15.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue LM, Coates PW, Lee VH, Ippensen DC, Arze SE, Poduslo SE. The cardiac sodium channel mRNA is expressed in the developing and adult rat and human brain. Brain Res. 2000;887:335–343. doi: 10.1016/s0006-8993(00)03033-x. [DOI] [PubMed] [Google Scholar]
- Fang X, McMullan S, Lawson SN, Djouhri L. Electrophysiological differences between nociceptive and non-nociceptive dorsal root ganglion neurones in the rat in vivo. J. Physiol. 2005;565:927–943. doi: 10.1113/jphysiol.2005.086199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts PA, Yokoyama S, Dib-Hajj S, Black JA, Waxman SG. Sodium channel alpha-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res. Mol. Brain Res. 1997;45:71–82. doi: 10.1016/s0169-328x(96)00241-0. [DOI] [PubMed] [Google Scholar]
- Gellens ME, George AL, Jr., Chen LQ, Chahine M, Horn R, Barchi RL, Kallen RG. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. U. S. A. 1992;89:554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersdorff Korsgaard MP, Christophersen P, Ahring PK, Olesen SP. Identification of a novel voltage-gated Na+ channel rNa(v)1.5a in the rat hippocampal progenitor stem cell line HiB5. Pflugers Arch. 2001;443:18–30. doi: 10.1007/s004240100625. [DOI] [PubMed] [Google Scholar]
- Giulietti A, Overbergh L, Valckx D, Decallonne B, Bouillon R, Mathieu C. An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods. 2001;25:386–401. doi: 10.1006/meth.2001.1261. [DOI] [PubMed] [Google Scholar]
- Hartmann HA, Colom LV, Sutherland ML, Noebels JL. Selective localization of cardiac SCN5A sodium channels in limbic regions of rat brain. Nat. Neurosci. 1999;2:593–595. doi: 10.1038/10147. [DOI] [PubMed] [Google Scholar]
- Hartshorne RP, Catterall WA. The sodium channel from rat brain. Purification and subunit composition. J. Biol. Chem. 1984;259:1667–1675. [PubMed] [Google Scholar]
- Haufe V, Camacho JA, Dumaine R, Gunther B, Bollensdorff C, von Banchet GS, Benndorf K, Zimmer T. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J. Physiol. 2005;564:683–696. doi: 10.1113/jphysiol.2004.079681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RI, Cummins TR, Waxman SG. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J. Neurophysiol. 2001;86:1351–1364. doi: 10.1152/jn.2001.86.3.1351. [DOI] [PubMed] [Google Scholar]
- John VH, Main MJ, Powell AJ, Gladwell ZM, Hick C, Sidhu HS, Clare JJ, Tate S, Trezise DJ. Heterologous expression and functional analysis of rat Nav1.8 (SNS) voltage-gated sodium channels in the dorsal root ganglion neuroblastoma cell line ND7-23. Neuropharmacology. 2004;46:425–438. doi: 10.1016/j.neuropharm.2003.09.018. [DOI] [PubMed] [Google Scholar]
- Kallen RG, Sheng ZH, Yang J, Chen LQ, Rogart RB, Barchi RL. Primary structure and expression of a sodium channel characteristic of denervated and immature rat skeletal muscle. Neuron. 1990;4:233–242. doi: 10.1016/0896-6273(90)90098-z. [DOI] [PubMed] [Google Scholar]
- Kerr NC, Holmes FE, Wynick D. Novel isoforms of the sodium channels Nav1.8 and Nav1.5 are produced by a conserved mechanism in mouse and rat. J. Biol. Chem. 2004;279:24826–24833. doi: 10.1074/jbc.M401281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunze DL, Lacerda AE, Wilson DL, Brown AM. Cardiac Na currents and the inactivating, reopening, and waiting properties of single cardiac Na channels. J. Gen. Physiol. 1985;86:691–719. doi: 10.1085/jgp.86.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Porreca F, Hunter JC, Gold MS, Lai J, Porreca F, Hunter JC, Gold MS. Voltage-gated sodium channels and hyperalgesia. [Review] [184 refs] Annu. Rev. Pharmacol. Toxicol. 2004;44:371–397. doi: 10.1146/annurev.pharmtox.44.101802.121627. [DOI] [PubMed] [Google Scholar]
- Lemaillet G, Walker B, Lambert S. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J. Biol. Chem. 2003;278:27333–27339. doi: 10.1074/jbc.M303327200. [DOI] [PubMed] [Google Scholar]
- Macdonald R, Bingham S, Bond BC, Parsons AA, Philpott KL. Determination of changes in mRNA expression in a rat model of neuropathic pain by Taqman quantitative RT-PCR. Brain Res. Mol. Brain Res. 2001;90:48–56. doi: 10.1016/s0169-328x(01)00086-9. [DOI] [PubMed] [Google Scholar]
- Makielski JC, Ye B, Valdivia CR, Pagel MD, Pu J, Tester DJ, Ackerman MJ. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ. Res. 2003;93:821–828. doi: 10.1161/01.RES.0000096652.14509.96. [DOI] [PubMed] [Google Scholar]
- Malik-Hall M, Poon WY, Baker MD, Wood JN, Okuse K. Sensory neuron proteins interact with the intracellular domains of sodium channel NaV1.8. Brain Res. Mol. Brain Res. 2003;110:298–304. doi: 10.1016/s0169-328x(02)00661-7. [DOI] [PubMed] [Google Scholar]
- Marty A, Neher E. Tight-seal whole-cell recording. In: Sakmann B, Neher E, editors. Single Channel Recording. 2nd ed. Plenum; New York: 1995. pp. 31–52. [Google Scholar]
- Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- Mohler PJ, Rivolta I, Napolitano C, Lemaillet G, Lambert S, Priori SG, Bennett V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiwaki T, Maeda N, Noda M. Characterization and developmental regulation of proteoglycan-type protein tyrosine phosphatase zeta/RPTPbeta isoforms. J. Biochem. (Tokyo) 1998;123:458–467. doi: 10.1093/oxfordjournals.jbchem.a021959. [DOI] [PubMed] [Google Scholar]
- Okuse K, Chaplan SR, McMahon SB, Luo ZD, Calcutt NA, Scott BP, Akopian AN, Wood JN. Regulation of expression of the sensory neuron-specific sodium channel SNS in inflammatory and neuropathic pain. Mol. Cell. Neurosci. 1997;10:196–207. doi: 10.1006/mcne.1997.0657. [DOI] [PubMed] [Google Scholar]
- Ou Y, Gibbons SJ, Miller SM, Strege PR, Rich A, Distad MA, Ackerman MJ, Rae JL, Szurszewski JH, Farrugia G. SCN5A is expressed in human jejunal circular smooth muscle cells. Neurogastroenterol. Motil. 2002;14:477–486. doi: 10.1046/j.1365-2982.2002.00348.x. [DOI] [PubMed] [Google Scholar]
- Ou SW, Kameyama A, Hao LY, Horiuchi M, Minobe E, Wang WY, Makita N, Kameyama M. Tetrodotoxin-resistant Na+ channels in human neuroblastoma cells are encoded by new variants of Nav1.5/SCN5A. Eur. J. Neurosci. 2005;22:793–801. doi: 10.1111/j.1460-9568.2005.04280.x. [DOI] [PubMed] [Google Scholar]
- Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. U. S. A. 2002;99:6210–6215. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer NW, Meisler MH. Evolution and diversity of mammalian sodium channel genes. Genomics. 1999;57:323–331. doi: 10.1006/geno.1998.5735. [DOI] [PubMed] [Google Scholar]
- Proudnikov D, Yuferov V, LaForge KS, Ho A, Jeanne KM. Quantification of multiple mRNA levels in rat brain regions using real time optical PCR. Brain Res. Mol. Brain Res. 2003;112:182–185. doi: 10.1016/s0169-328x(03)00056-1. [DOI] [PubMed] [Google Scholar]
- Renganathan M, Dib-Hajj S, Waxman SG. Na(v)1.5 underlies the ‘third TTX-R sodium current’ in rat small DRG neurons. Brain Res. Mol. Brain Res. 2002;106:70–82. doi: 10.1016/s0169-328x(02)00411-4. [DOI] [PubMed] [Google Scholar]
- Rogart RB, Cribbs LL, Muglia LK, Kephart DD, Kaiser MW. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc. Natl. Acad. Sci. U. S. A. 1989;86:8170–8174. doi: 10.1073/pnas.86.20.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roobol A, Holmes FE, Hayes NV, Baines AJ, Carden MJ. Cytoplasmic chaperonin complexes enter neurites developing in vitro and differ in subunit composition within single cells. J. Cell Sci. 1995;108:1477–1488. doi: 10.1242/jcs.108.4.1477. [DOI] [PubMed] [Google Scholar]
- Rush AM, Brau ME, Elliott AA, Elliott JR. Electrophysiological properties of sodium current subtypes in small cells from adult rat dorsal root ganglia. J. Physiol. 1998;511:771–789. doi: 10.1111/j.1469-7793.1998.771bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath DE, Broome HE, Prystowsky MB. Glyceraldehyde-3-phosphate dehydrogenase mRNA is a major interleukin 2-induced transcript in a cloned T-helper lymphocyte. Gene. 1990;91:185–191. doi: 10.1016/0378-1119(90)90087-8. [DOI] [PubMed] [Google Scholar]
- Sakakibara Y, Furukawa T, Singer DH, Jia H, Backer CL, Arentzen CE, Wasserstrom JA. Sodium current in isolated human ventricular myocytes. Am. J. Physiol. 1993;265:H1301–H1309. doi: 10.1152/ajpheart.1993.265.4.H1301. [DOI] [PubMed] [Google Scholar]
- Sangameswaran L, Delgado SG, Fish LM, Koch BD, Jakeman LB, Stewart GR, Sze P, Hunter JC, Eglen RM, Herman RC. Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J. Biol. Chem. 1996;271:5953–5956. doi: 10.1074/jbc.271.11.5953. [DOI] [PubMed] [Google Scholar]
- Sarno S, Reddy H, Meggio F, Ruzzene M, Davies SP, Donella-Deana A, Shugar D, Pinna LA. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (‘casein kinase-2’) FEBS Lett. 2001;496:44–48. doi: 10.1016/s0014-5793(01)02404-8. [DOI] [PubMed] [Google Scholar]
- Scholz A, Appel N, Vogel W. Two types of TTX-resistant and one TTX-sensitive Na+ channel in rat dorsal root ganglion neurons and their blockade by halothane. Eur. J. Neurosci. 1998;10:2547–2556. [PubMed] [Google Scholar]
- Sleeper AA, Cummins TR, Dib-Hajj SD, Hormuzdiar W, Tyrrell L, Waxman SG, Black JA. Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J. Neurosci. 2000;20:7279–7289. doi: 10.1523/JNEUROSCI.20-19-07279.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souslova VA, Fox M, Wood JN, Akopian AN. Cloning and characterization of a mouse sensory neuron tetrodotoxin-resistant voltage-gated sodium channel gene, Scn10a. Genomics. 1997;41:201–209. doi: 10.1006/geno.1997.4669. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, Beggs AH, Cappuccio FP, Sagnella GA, Kass RS, Keating MT. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297:1333–1336. doi: 10.1126/science.1073569. [DOI] [PubMed] [Google Scholar]
- Tan BH, Valdivia CR, Rok BA, Ye B, Ruwaldt KM, Tester DJ, Ackerman MJ, Makielski JC. Common human SCN5A polymorphisms have altered electrophysiology when expressed in Q1077 splice variants. Heart Rhythm. 2005;2:741–747. doi: 10.1016/j.hrthm.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Tate S, Benn S, Hick C, Trezise D, John V, Mannion RJ, Costigan M, Plumpton C, Grose D, Gladwell Z, Kendall G, Dale K, Bountra C, Woolf CJ. Two sodium channels contribute to the TTX-R sodium current in primary sensory neurons. Nat. Neurosci. 1998;1:653–655. doi: 10.1038/3652. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li Z, Shen J, Keating MT. Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics. 1996;34:9–16. doi: 10.1006/geno.1996.0236. [DOI] [PubMed] [Google Scholar]
- Wood JN, Bevan SJ, Coote PR, Dunn PM, Harmar A, Hogan P, Latchman DS, Morrison C, Rougon G, Theveniau M. Novel cell lines display properties of nociceptive sensory neurons. Proc. Biol. Sci. 1990;241:187–194. doi: 10.1098/rspb.1990.0084. [DOI] [PubMed] [Google Scholar]
- Wood JN, Boorman JP, Okuse K, Baker MD, Wood JN, Boorman JP, Okuse K, Baker MD. Voltage-gated sodium channels and pain pathways. [Review] [95 refs] J. Neurobiol. 2004;61:55–71. doi: 10.1002/neu.20094. [DOI] [PubMed] [Google Scholar]
- Wu CH, Yeh LS, Huang H, Arminski L, Castro-Alvear J, Chen Y, Hu Z, Kourtesis P, Ledley RS, Suzek BE, Vinayaka CR, Zhang J, Barker WC. The protein information resource. Nucleic Acids Res. 2003;31:345–347. doi: 10.1093/nar/gkg040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol. 2003;4:207. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuill KH, Convery MK, Dooley PC, Doggrell SA, Hancox JC. Effects of BDF 9198 on action potentials and ionic currents from guineapig isolated ventricular myocytes. Br. J. Pharmacol. 2000;130:1753–1766. doi: 10.1038/sj.bjp.0703476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Dong XW, Crona J, Maguire M, Priestley T. Vinpocetine is a potent blocker of rat NaV1.8 tetrodotoxin-resistant sodium channels. J. Pharmacol. Exp. Ther. 2003;306:498–504. doi: 10.1124/jpet.103.051086. [DOI] [PubMed] [Google Scholar]
- Zimmer T, Bollensdorff C, Haufe V, Birch-Hirschfeld E, Benndorf K. Mouse heart Na+ channels: primary structure and function of two isoforms and alternatively spliced variants. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H1007–H1017. doi: 10.1152/ajpheart.00644.2001. [DOI] [PubMed] [Google Scholar]