

Abstract
The M2 protein of influenza virus A is a proton-selective ion channel activated by pH. Structure determination by solid-state and solution NMR and X-ray crystallography has contributed significantly to our understanding, but channel activation may involve conformations not captured by these studies. Indeed, solid-state NMR data demonstrate that the M2 protein possesses significant conformational heterogeneity. Here, we report molecular dynamics (MD) simulations of the M2 transmembrane domain (TMD) in the absence and presence of the antiviral drug amantadine. The ensembles of MD conformations for both apo and bound forms reproduced the NMR data well. The TMD helix was found to kink around Gly-34, where water molecules penetrated deeply into the backbone. The amantadine-bound form exhibited a single peak ≈10° in the distribution of helix-kink angle, but the apo form exhibited 2 peaks, ≈0° and 40°. Conformations of the apo form with small and large kink angles had narrow and wide pores, respectively, around the primary gate formed by His-37 and Trp-41. We propose a structural model for channel activation, in which the small-kink conformations dominate before proton uptake by His-37 from the exterior, and proton uptake makes the large-kink conformations more favorable, thereby priming His-37 for proton release to the interior.
Keywords: solid-state NMR, MD simulations, helix kink, histidine tetrad, amantadine
The M2 protein of influenza virus A is a proton-selective ion channel activated by pH. Its function, proton conductance, is essential for effective replication of the virus. The transmembrane domain (TMD) of this homotetrameric protein contains a single helix (residues Ser-22 to Leu-46) from each subunit. Within the TMD, residues His-37 and Trp-41 constitute the primary gate that is critical for proton conductance and selectivity and pH activation (1–3). On the N-terminal (i.e., virus exterior) side, disulfide bonds involving Cys-17 and Cys-19 provide stabilization (4). On the C-terminal side, an amphiphilic helix may also be involved in pH activation (5). The antiviral drug amantadine and its derivatives inhibit the channel function by putatively binding to the TMD (6–8). The structures of the M2 TMD in the apo form and the amantadine-bound form were first determined by solid-state NMR (9, 10). In addition, extensive experimental (1, 2, 11–16) and computational (17–20) studies have been performed to model the structure of the M2 TMD and understand the molecular mechanisms of conductance and selectivity of the proton channel. Recently, 2 new structures were solved by X-ray crystallography (21) and solution NMR spectroscopy (22). Those 2 studies have generated controversies, especially about the mechanism of drug inhibition. Additional structural results have recently been published that add to the controversies surrounding inhibitor binding (23, 24) and the closed- to open-state conformational transition (25).
Channel activation may involve conformations not captured by the X-ray and NMR structures. Indeed, solid-state NMR (9, 23, 26–28) and other studies have shown that the M2 TMD possesses significant conformational heterogeneity. For example, there is evidence that the transmembrane helix can be kinked around Gly-34, and the kink angle can be changed by the binding of amantadine (9, 26, 27). In the X-ray structure of the apo form, 1 helix of the M2 TMD exhibits a 15° kink around G34, whereas the other 3 helices appear straight (21). In the recent IR study a 129° change in the rotation about the helical axis is proposed between the low- and high-pH forms of the M2 TMD (25).
To explore conformations potentially involved in channel activation, we performed molecular dynamics (MD) simulations of the M2 TMD in explicit membrane bilayer and solvent, in both the apo form and the amantadine-bound form. Previously, our simulation study (19) led to the prediction of a secondary gate formed by Val-27, which was also shown by the X-ray (21) and solution NMR (22) structures and provides explanations for the low conductance of the M2 protein and some of the amantadine-resistant mutations. Based on our simulation results and other experimental observations, we propose a structural model for the pH activation of the M2 proton channel by injecting conformational details into the proton relay mechanism of Pinto et al. (14). In this model, both the unprotonated state and the protonated state are comprised of heterogeneous ensembles of conformations, with an increased population of large helix-kink angles around Gly-34 for the protonated state.
Results
Validation of the Ensembles of MD Conformations by Polarization Inversion Spin Exchange at Magic Angle (PISEMA).
The ensemble of conformations generated by the MD simulations can be directly tested by solid-state NMR spectroscopy. Specifically, we calculated PISEMA resonances on the conformations sampled from MD trajectories of the apo and amantadine-bound forms. These are compared in Fig. 1 against experimental data (9, 15). The high pH conditions of the experiments were modeled in the MD simulations (see Methods). Overall, calculated results for 15N anisotropic chemical shift and 15N-1H dipolar coupling reproduced the experimental PISEMA data well (see also Fig. S1). Particularly, the locations of the polar-index-slant-angle (PISA) wheels and pattern of resonances indicating the tilt of and rotation about the helical axis showed good agreement between calculation and experiment in both the apo and amantadine-bound forms. The agreement indicates that the tilt angles and overall conformations of the M2 helices were well reproduced in the MD trajectories.
Fig. 1.

Comparison between experimental and MD-derived PISEMA resonances of the M2 TMD. For the apo form, the experimental data are taken from ref. 15. Values for Ser-31, Gly-34, and His-37 were not available. For the amantadine-bound form, the experimental data are taken from ref. 9. Values for Ser-31 and His-37 were not available. Typical differences between experimental and MD resonances amount to <5° errors in peptide-plane orientations. The helix tilt angle obtained from the MD results as a whole agrees with the experimental counterpart to within ≈2°; the helix rotation angle agrees to within ≈10°.
The results shown in Fig. 1 were obtained by averaging over the 4 helices of the M2 TMD and over 2 independent MD trajectories each for the apo form and amantadine-bound form. The variations among the 8 individual results for each residue are presented below.
Helix Kinking and Conformational Heterogeneity.
In Fig. 1, both calculation and experiment show the PISA wheels for the N-terminal half (Leu-26–Ile-35) of the TMD helix and the C-terminal half (Leu-36–Leu-43) are centered at approximately the same location for the apo form but at different locations for the amantadine-bound form. This result was interpreted as indicating a straight helix for the apo form and a kinked helix, around Gly-34, for the amantadine-bound form (9). We calculated the angle, θ, between the 2 halves of the TMD helix in our simulations. As shown in Fig. 2, the distribution of θ for the amantadine-bound form exhibited a single peak ≈10°, indicating a kinked TMD helix.* However, the kink angle in the apo form exhibited wide variability. Its distribution consisted of 2 populations separated at θ = 24.5° (or cosθ = 0.91). The major population, accounting for 65% of the apo conformations, peaked at θ = 0°. The remaining 35% of the conformations had a relatively flat distribution between θ = 25° and θ = 50°.
Fig. 2.

Normalized distributions of the cosine of helix-kink angle for apo and amantadine-bound M2 TMD. For the apo form, dot and hash patterns under the curve indicate the small- and large-kink populations, respectively (separated at cosθ = 0.91).
Our simulations thus showed that, even though PISA wheels for the N-terminal and C-terminal halves were centered at approximately the same location, the TMD helix in the apo form actually could exhibit a wide range of kink angles. Helix kinking in the apo form has been observed by X-ray crystallography (21), EPR (5), and solid-state NMR (23). A very recent MD study of the apo form with various protontation states of His-37 also showed large kink angles (20).
The wider distribution of helix-kink angles in the simulations of the apo form indicates that it exhibited significantly greater conformational heterogeneity than the amantadine-bound form. Strong evidence for greater conformational heterogeneity in the apo form has been presented by broadened resonances in solid-state NMR spectra as observed in multiple studies (see Fig. S1) (9, 23, 26–28). To make comparison with this experimental observation, in Fig. 3 we display individual results for 15N anisotropic chemical shift and 15N-1H dipolar coupling, collected from the 4 helices of the apo or amantadine-bound form in 2 independent MD trajectories. Results for all 5 Leu residues (at positions 26, 36, 38, 40, and 43) are shown for illustration. The individual results for each residue in the amantadine-bound form clustered relatively tightly, whereas those in the apo form spread widely. Although we recognize that the time scales (tens of nanoseconds) sampled in the MD simulations are much shorter than the time scales (0.1–1 ms) sampled by solid-state NMR, the results in Fig. 3 appear to qualitatively mimic the narrow resonances of the amantadine-bound form and the broadened resonances of the apo form observed in the solid-state NMR experiments.
Fig. 3.

PISEMA resonances of 5 Leu residues (at positions 26, 36, 38, 40, and 43) calculated from the MD trajectories. There are 8 data points for each residue: the M2 TMD contains helices that are identical in sequence, and 2 independent MD trajectories were generated.
Water Penetration Around Gly-34.
Given the backbone flexibility of a Gly residue and its propensity to kink transmembrane helices (29–31), it was not surprising that helix kinking was observed in our simulations around Gly-34, the only Gly residue in the M2 TMD. The small size of the Gly side chain may allow for water access and hydrogen bonding with the backbone in the vicinity of the kink, thereby stabilizing it. We indeed found water hydrogen bonding with the backbone carbonyl and amide (Fig. 4). Moreover, some water molecules served simultaneously as donors and acceptors (SDA) while hydrogen bonding with the helix backbone around Gly-34. The occurrence of SDA water molecules was higher in the large-kink population of the apo form than in the small-kink population: 14% of helix conformations in the large-kink population contained at least 1 SDA water molecule, whereas the corresponding number in the small-kink population was only 1.4%. In comparison, SDA water molecules occurred in 7% of the conformations in the amantadine-bound form. It appears that water-backbone hydrogen bonding around G34 stabilized the kinked helices, and that involving SDA water, in particular, stabilized the large-kink conformations.
Fig. 4.

Water-backbone hydrogen bonding around Gly-34 in the absence (Left) and presence (Right) of amantadine. For clarity, only 2 of the 4 helices are shown, with Gly-34 presented as ball and stick. Typically each water molecule participated in a single water–Gly-34 hydrogen bond; sometimes a water molecule was found to bridge between helices. One water molecule, present in the apo form and shown as yellow spheres, simultaneously served as an acceptor to a backbone amide of Gly-34 and as a donor to a carbonyl of a neighboring residue in the same helix. Such water molecules are referred to as SDA.
Asp-44–Arg-45 Salt Bridges.
Salt bridges of Asp-44 and Arg-45, either from the same helix or 2 different helices of the M2 TMD, were observed in the X-ray structure (21). Based on the X-ray structure, Stouffer et al. (21) built models for the open and closed states of the M2 TMD, in which 4 Asp-44–Arg-45 salt bridges are formed intrahelically and interhelically, respectively. Intrahelical and interhelical Asp-44–Arg-45 salt bridges were also observed in our simulations. Interestingly, the partitions of the salt bridges into intrahelical and interhelical types were strikingly different for the apo and amantadine-bound forms (Fig. S2). In the apo form, the intrahelical type dominated over the interhelical type, with their number averaging 2.1 and 1.2, respectively, per snapshot along the MD trajectories. In the amantadine-bound form, the trend was reversed, with the intrahelical and interhelical types averaging 1.0, and 2.7, respectively. The increase in interhelical salt bridges in the amantadine-bound form might increase the stability of the tetramer and contribute to conformational homogeneity.
Side-Chain Conformations of Trp-41.
The implication of Trp-41 in channel gating (2, 32–34) prompted us to take a closer look at the side-chain conformations of this residue sampled in the MD simulations. Hu et al. (9) reported the PISEMA spectrum for the sidechain Nε1 of Trp-41 in the amantadine-bound form at high pH. The 15N anisotropic chemical shift was observed at 124.6 ppm and the 15N-1H dipolar coupling was observed at 0.0 kHz. From our MD simulations, the Trp-41 side-chain 15N anisotropic chemical shift was calculated to be 121.9 ppm, and the 15N-1H dipolar coupling was 0.2 kHz, both showing good agreement with the experimental results.
Witter et al. (34) reported static and magic-angle spinning (MAS) 19F NMR spectra of 6F-Trp-41 in apo M2 TMD at both high and low pH (8.0 and 5.3, respectively). The rotationally averaged chemical shift spans of the static 19F spectra were found to increase upon lowering pH: 10–20 ppm at pH = 8.0 vs. 20–30 ppm at pH = 5.3. As noted above (Fig. 2), in our simulations the TMD helices in the apo form exhibited 2 populations of kink angles (separated at θ = 24.5°). We calculated the chemical-shift spans from 19F chemical shift anisotropy (see SI Appendix) for the 2 populations separately. In principle F-F dipolar couplings may also contribute to the chemical-shift spans of the static spectra, but the experimental results of Witter et al. (34) indicate only relatively minor dipolar contributions. The calculated spans were 5 ppm for the small-kink population and 20 ppm for the large-kink population. These results are consistent with the observed trend upon changing pH, hinting that the relative population of the large-kink conformations increases at low pH.
The change in 19F chemical-shift span was a direct consequence of different preferences of the Trp-41 indole orientation, relative to the membrane normal, in the small- and large-kink populations (see SI Appendix and Fig. S3). The average angle between the indole normal and the membrane normal increased from 66° in the small-kink population to 80° in the large-kink population.† Interestingly, if the indole is viewed as a gate, then the gate becomes open in the latter orientation (see Figs. S4 and S5). Witter et al. (34) reported a decrease in F-F dipolar coupling upon lowering pH, which would be consistent with an increase in the large-kink population. However, 19F CODEX spectra of 5F-Trp-41 acquired under MAS by Luo et al. (35) do not support a significant change in F-F dipolar coupling with pH.
Dynamics of Amantadine Within the Binding Site.
Helix conformations with large-kink angles were not observed in the simulations of the amantadine-bound form; so apparently large-kink angles were incompatible with amantadine binding. The data presented above show that the occurrence of SDA water molecules in the amantadine-bound form is less prevalent than in the large-kink conformations of the apo form. This observation suggests that amantadine binding perhaps interferes with the interactions of SDA water molecules with the Gly-34 backbone, which probably are a stabilization factor for the large-kink conformations.
In the simulations, amantadine was observed to interact closely (< 3.5 Å between heavy atoms) on average with 2 of the 4 helices of the TMD at any given moment; the identities of these 2 helices changed frequently, indicating positional disorder. In addition, amantadine exhibited orientational disorder. Overall M2-bound amantadine was found to favor the upside down orientation in our previous report (19); that orientation appears to be the most compatible with X-ray diffraction data (21). We calculated here the order parameter of the amantadine Cα-N vector; a low value, ≈0.4, indicates that the Cα-N vector sampled a wide range of orientations (with a correlation time ≈300 ps for the internal motion).
Discussion
Conformational Heterogeneity of Apo M2 TMD.
A main finding of the present simulation study is that apo M2 TMD, in contrast to the amantadine-bound form, exhibits significant conformational heterogeneity. This finding is in agreement with solid-state NMR studies (9, 23, 26–28). Membrane proteins appear to be less stabilized compared with soluble proteins, and the resulting conformational flexibility may be necessary for protein function (26, 27, 36). For channel proteins in particular, which undergo conformational transitions upon activation, the existence of multiple low-energy conformational states is a requirement.
Our simulations further identified variation in helix-kink angles around G34 as a major source for the conformational heterogeneity of the M2 TMD. Importantly, the heterogeneity is suppressed by the binding of amantadine. Helix kinking, in particular around Gly residues, has been found to play an essential role in the activation of various ion channels (29–31). It seems to be a convenient way to affect significant changes in channel pore size. Our simulations show that water molecules can deeply penetrate into and form hydrogen bonds with the backbone around Gly, thereby stabilizing kinked helices.
Conformational Ensemble of Apo Form at Low pH.
The conformational heterogeneity of apo M2 TMD is exacerbated by low pH, as indicated by increased broadening in the solid-state NMR spectra of Li et al. (27). As a result, structural studies under low pH conditions, although essential for understanding pH activation, have been challenging. The available structures of the M2 TMD have been solved at high or neutral pH (10, 21) or in the presence of amantadine or its analog (9, 21, 22). Li et al. (27) indicated that overall the helix tilt and rotation seem to change little upon lowering pH. Other solid-state NMR (26, 34), EPR (5), and IR spectroscopy (25) have also provided structural information for apo M2 TMD at low pH. (The last study proposed a pH-dependent conformational change different from what is presented below; further comment is found in SI Appendix.) Khurana et al. (20) have modeled the low pH condition by protonating His-37 within multiple helices of the tetramer in MD simulations. However, simply changing the His-37 protonation may not adequately capture the chemistry involving the His-37 tetrad (tautomers, hydrogen bonding, etc.) (1). In addition, no appropriate starting structures are available because existing apo structures are not determined at low pH. For these reasons we refrained from carrying out “low pH” simulations.
The effect of pH on conformation can be considered as shifting the relative populations of the different conformations sampled by apo M2 TMD. In particular, we hypothesize that the small-kink population is likely dominant at any pH, but the large-kink population becomes more favored by lowering pH. The resulting increase in conformational heterogeneity is consistent with the data of Li et al. (27). In the large-kink conformations, the distances between helices in the tetramer remain the same in the N-terminal half but increase significantly in the C-terminal half (see Fig. S5). In line with our hypothesis, disulfide cross-linking experiments at pH 7.4 and 5.2 showed that, upon lowering pH, the probabilities for cross-linking remain the same in the N-terminal half but decrease significantly toward the C-terminal (37). As further support, the difference in static 19F chemical shift span of Trp-41 calculated on the 2 populations was found to be consistent with the observed trend upon lowering pH (34). Importantly, in the population that putatively becomes favored at low pH, the Trp-41 indole rings are oriented to open the channel pore. The shift in population by the binding of a ligand, a proton in the present case, is similar to what is suggested for a ligand-gated ion channel (38).
Structural Model for Channel Activation.
Based on MD simulations, Khurana et al. (20) proposed a transporter-like mechanism for proton conductance, in which the M2 TMD cycles between outside open/inside closed and outside closed/inside open conformations and His-37 is protonated/deprotonated during each cycle. Here, we put forward a more conventional structural model, by injecting conformational details into the proton relay mechanism proposed by Pinto et al. (14).
The proton relay mechanism posits that, under a proton gradient, a proton enters the pore from outside and binds with His-37; subsequently, the proton is released to the inside. In our model, illustrated in Fig. 5, the states before and after the release of the conducting proton from His-37 are comprised of 2 distinct conformational ensembles. Both states may have 2 of the 4 His-37 residues already protonated (1), but for convenience we refer to the 2 states as unprotonated and protonated, respectively. In the protonated state the population with large helix-kink angles is increased. The accompanying motions of the backbones and Trp-41 side chains open the pore, allowing the proton to be released to the interior. Upon proton release the population shifts toward the small-kink conformations, but a proton uptake from the exterior shifts the population back toward the large-kink conformations, and the cycle of proton release to the interior and proton uptake from the exterior repeats itself.
Fig. 5.

Structural model for channel activation by pH. Vertical arrows indicate equilibration between small- and large-kink conformations; the lengths of these arrows indicate the magnitudes of tendencies. Horizontal arrows indicate proton release or uptake. The curly brackets signify that the small- and large-kink conformations are in fast exchange on the time scales of proton release and uptake. Note that in the protonated state it is the minority population of large-kink conformations that is most likely to release the proton.
To gain further insight into the differences between the small-kink conformations in the unprotonated state and the large-kink conformations in the protonated state, we built a representative structure for each state by choosing a helix with either small kink (8.6°) or large kink (26.0°) and then generating a tetramer by enforcing 4-fold symmetry; the rms displacement between the 2 structures is 3.8 Å (Fig. S4). In the N-terminal half, the channel pores of the 2 structures are similar in size, both featuring a constriction at Val-27 (19, 21, 22). In contrast, in the C-terminal half, the pore is narrow in the unprotonated state but wide in the protonated state (Fig. S5).
Our model bears some resemblance to but differs both in the overall mechanism of conductance and structural details from that proposed by Khurana et al. (20). In our model, His-37 can bind a proton coming from either the exterior or interior, but the exterior is favored by concentration or other electrochemical gradients. Similarly, the proton can be released to either the interior or exterior, but the interior is favored because the large-kink conformations prime the channel pore in that way; if the rate constants for proton release to the interior and exterior are kin−0 and kex−0, respectively, then kin−0 > kex−0. In terms of structural details, our deprotonated state is dominated by small-kink conformations, with His-37 accessible to protons from both the exterior and interior. In fact, the rate constant, kin+0, for proton binding from the interior should be higher than the counterpart, kex+0, from the exterior, because kex−0/kex+0 = kin−0/kin+0 and kin−0 > kex−0 (see SI Appendix). In contrast, proton binding from the interior is effectively shut off in the model of Khurana et al. In the protonated state, we specifically propose that the proton is most likely released from the minor population of large-kink conformations.
Low and Asymmetric Conductance, Saturation at Low pH, and Rectification.
We can capture the above mechanism of proton conductance in a kinetic model, which is able to rationalize important observations on the M2 channel (39–41). According to this kinetic model (see Fig. S6 and SI Appendix), protons flow inward when the exterior pH, pHex, is lower than the interior pH, pHin. The inward proton flux reaches a maximum of kin−0 at very low pHex. With kin−0 = 102 s−1, kex−0 = 10 s−1, kin+0 = 108 M−1·s−1, kex+0 = 107 M−1·s−1 [the binding and unbinding rate constants for either the exterior or interior are constrained by a pKa = 6 for His-37 (1)], the inward flux is predicted to be 45 protons per s at pHex = 5 and pHin = 7, which is in the range of observed low conductance (39, 40). If the flux dependence on pHex is fitted to a 1-site binding function (see Fig. S7), as done by Chizhmakov et al. (41), the apparent dissociation constant is 12.1 μM, in good agreement with the value of 8.3 μM of Chizhmakov et al. (note that these values are ≈10-fold higher than expected from the His-37 pKa).
When the pH gradient across the membrane is reversed (i.e., pHin < pHex), the kinetic model predicts an outward proton flux, but with a smaller magnitude (see Fig. S7). For example, at pHin = 5 and pHex = 7, the outward flux is 9 protons per s, ≈5-fold lower than the corresponding inward flux. Such asymmetry has been observed (41), and in our model is attributed to the fact that kex−0 < kin−0. The predicted voltage-proton flux curve (see Fig. S8) also exhibits the observed inward rectification behavior.
The proton binding rate constants illustrated here are lower than that observed (≈1010 M−1·s−1) for proton binding to an imidazole in bulk water (42); the lower values are to be expected for binding to a buried His-37 (see SI Appendix), with Val-27 serving as a “dynamic” gate (43) along the pore on the N-terminal side (19) while the large-kink population, which may allow for efficient proton access from the C-terminal side, exists putatively only as a minority. Calculations of the binding rate constants based on the structure and dynamics (43) of the M2 protein remain to be carried out.
Conclusion
In this study we explored the conformational heterogeneity of the M2 TMD by MD simulations. The ensembles of MD conformations of both apo and amantadine-bound forms reproduced experimental PISEMA data well. A major source of the conformational heterogeneity of the apo form was the variation in helix-kink angles around G34. Conformations with small- and large-kink angles reproduced 19F NMR data on 6F-Trp-41 obtained at high and low pH, respectively. We further propose a structural model for channel activation by pH, in which the protein upon proton release is dominated by small-kink conformations and upon proton uptake large-kink conformations become more favorable.
Methods
MD Simulations.
The MD trajectories analyzed here were the same ones reported in a previous study (19), which focused on the secondary gate formed by Val-27. Starting from solid-state NMR structures of the M2 TMD (9, 10) fully hydrated DMPC bilayers, 2 independent trajectories each were accumulated for the apo form (both totaling 15 ns) and the amantadine-bound form (one totaling 15 ns and the other 22 ns). The simulations were designed to model the high-pH conditions under which the solid-state NMR experiments (9, 15) were carried out. Other details of the MD simulation protocol can be found in our previous report (19).
Calculation of PISEMA Resonances from MD Simulation.
Backbone 15N anisotropic chemical shift, δ, and 15N-1H dipolar coupling, ν, for either the apo form or amantadine-bound form of the M2 TMD were calculated from the 2 MD trajectories according to ref. 44:
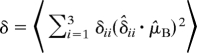 |
In these equations, δ11 = 57.3 ppm, δ22 = 81.2 ppm, and δ33 = 227.8 ppm are the principal values of the chemical shift tensor of 15N; δ̂ii denotes unit vectors along the 3 principal axes, shown in Fig. S3; μ̂B is a unit vector along the external magnetic field, which is parallel to the membrane normal; ν′ = 10.735 kHz; μ̂NH is a unit vector along the backbone NH bond; and P2(x) = (3x2 − 1)/2 (15). Average was first taken over 10,000 conformations sampled from the last 10 ns of each trajectory. When indicated, results from the 4 helices of the M2 TMD and the 2 independent trajectories were further averaged. All 15N chemical shifts are referenced to liquid ammonia.
The PISEMA resonance for the side-chain Nε1 of Trp-41 was calculated similarly, using the following parameters: δ11 = 61 ppm, δ22 = 129.6 ppm, and δ33 = 180.8 ppm, with the principal axes shown in Fig. S3; and ν′ = 10.17 kHz (45). Calculation of the rotationally averaged 19F chemical-shift span for 6F-Trp-41 is described in SI Appendix (see also Fig. S3).
Kink Angles of Helices.
To calculate the kink angle for each helix, a regular helix was built as reference by using average backbone φ and ψ angles from the MD trajectories. This reference helix was superimposed twice to each helix in every snapshot along the MD trajectories, once to match with the N-terminal half and once with the C-terminal half (separated at Gly-34). The kink angle was calculated as the angle between the helical axes of the 2 different superpositions.
Supplementary Material
Acknowledgments.
We thank Dr. Riqiang Fu for discussion on 19F NMR and Dr. William F. DeGrado for commenting on an earlier version of the article. This work is supported in part by National Institutes of Health Grant AI023007.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0906553106/DCSupplemental.
In the X-ray structure of the amantadine-bound form, the M2 TMD helices are straight, perhaps a reflection of the G34A mutation in that structure (21).
Note that the same indole orientation relative to the membrane normal can be obtained by different combinations of the orientation of the Trp41 peptide group (as dictated by the helix tilt and rotation) and the χ1 and χ2 angles of the side chain [e.g., Witter et al. (34) were able to fit their 19F NMR data at low and high pH by searching for χ1 and χ2 angles while fixing the backbone to the solid-state NMR structure (10)]. In contrast to the indole orientation, analysis of the individual angles, such as χ1, in our MD trajectories did not reveal informative contrast between the small- and large-kink populations.
References
- 1.Hu J, et al. Histidines, heart of the hydrogen ion channel from influenza A virus: Toward an understanding of conductance and proton selectivity. Proc Natl Acad Sci USA. 2006;103:6865–6870. doi: 10.1073/pnas.0601944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang Y, Zaitseva F, Lamb RA, Pinto LH. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J Biol Chem. 2002;277:39880–39886. doi: 10.1074/jbc.M206582200. [DOI] [PubMed] [Google Scholar]
- 3.Venkataraman P, Lamb RA, Pinto LH. Chemical rescue of histidine selectivity filter mutants of the M2 ion channel of influenza A virus. J Biol Chem. 2005;280:21463–21472. doi: 10.1074/jbc.M412406200. [DOI] [PubMed] [Google Scholar]
- 4.Holsinger LJ, Lamb RA. Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology. 1991;183:32–43. doi: 10.1016/0042-6822(91)90115-r. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen PA, et al. pH-induced conformational change of the influenza M2 protein C-terminal domain. Biochemistry. 2008;47:9934–9936. doi: 10.1021/bi801315m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinto LH, Lamb RA. The M2 proton channels of influenza A and B viruses. J Biol Chem. 2006;281:8997–9000. doi: 10.1074/jbc.R500020200. [DOI] [PubMed] [Google Scholar]
- 7.Jing X, et al. Functional studies indicate amantadine binds to the pore of the influenza A virus M2 proton-selective ion channel. Proc Natl Acad Sci USA. 2008;105:10967–10972. doi: 10.1073/pnas.0804958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stouffer AL, et al. The interplay of functional tuning, drug resistance, and thermodynamic stability in the evolution of the M2 proton channel from the influenza A virus. Structure (London) 2008;16:1067–1076. doi: 10.1016/j.str.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu J, et al. Backbone structure of the amantadine-blocked transmembrane domain M2 proton channel from influenza A virus. Biophys J. 2007;92:4335–4343. doi: 10.1529/biophysj.106.090183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimura K, Kim S, Zhang L, Cross TA. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid-state NMR. Biochemistry. 2002;41:13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 11.Sugrue RJ, Hay AJ. Structural characteristics of the M2 protein of influenza A viruses: Evidence that it forms a tetrameric channel. Virology. 1991;180:617–624. doi: 10.1016/0042-6822(91)90075-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinto LH, Holsinger LJ, Lamb RA. Influenza-virus M2 protein has ion channel activity. Cell. 1992;69:517–528. doi: 10.1016/0092-8674(92)90452-i. [DOI] [PubMed] [Google Scholar]
- 13.Wang C, Lamb RA, Pinto LH. Activation of the M2 ion channel of influenza virus: A role for the transmembrane domain histidine residue. Biophys J. 1995;69:1363–1371. doi: 10.1016/S0006-3495(95)80003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinto LH, et al. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc Natl Acad Sci USA. 1997;94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H+ channel. Protein Sci. 2001;10:2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stouffer AL, Nanda V, Lear JD, DeGrado WF. Sequence determinants of a transmembrane proton channel: An inverse relationship between stability and function. J Mol Biol. 2005;347:169–179. doi: 10.1016/j.jmb.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 17.Sansom MS, Kerr ID. Influenza virus M2 protein: A molecular modeling study of the ion channel. Protein Eng. 1993;6:65–74. doi: 10.1093/protein/6.1.65. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Wu Y, Voth GA. Proton transport behavior through the influenza A M2 channel: Insights from molecular simulation. Biophys J. 2007;93:3470–3479. doi: 10.1529/biophysj.107.105742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yi M, Cross TA, Zhou H-X. A secondary gate as a mechanism for inhibition of the M2 proton channel by amantadine. J Phys Chem B. 2008;112:7977–7979. doi: 10.1021/jp800171m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khurana E, et al. Molecular dynamics calculations suggest a conduction mechanism for the M2 proton channel from influenza A virus. Proc Natl Acad Sci USA. 2009;106:1069–1074. doi: 10.1073/pnas.0811720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stouffer AL, et al. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–599. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cady SD, Mishanina TV, Hong M. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: The role of Ser-31 in amantadine binding. J Mol Biol. 2009;385:1127–1141. doi: 10.1016/j.jmb.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pielak RM, Schnell JR, Chou JJ. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc Natl Acad Sci USA. 2009;106:7379–7384. doi: 10.1073/pnas.0902548106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manor J, et al. Gating mechanism of the influenza A M2 channel revealed by 1D and 2D IR spectroscopies. Structure (London) 2009;17:247–254. doi: 10.1016/j.str.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu J, Fu R, Cross TA. The chemical and dynamical influence of the antiviral drug amantadine on the M2 proton channel transmembrane domain. Biophys J. 2007;93:276–283. doi: 10.1529/biophysj.106.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C, Qin H, Gao FP, Cross TA. Solid-state NMR characterization of conformational plasticity within the transmembrane domain of the influenza A M2 proton channel. Biochim Biophys Acta. 2007;1768:3162–3170. doi: 10.1016/j.bbamem.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cady SD, Hong M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proc Natl Acad Sci USA. 2008;105:1483–1488. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sukharev S, Betanzos M, Chiang CS, Guy HR. The gating mechanism of the large mechanosensitive channel MscL. Nature. 2001;409:720–724. doi: 10.1038/35055559. [DOI] [PubMed] [Google Scholar]
- 30.Jiang Y, et al. The open pore conformation of potassium channels. Nature. 2002;417:523–526. doi: 10.1038/417523a. [DOI] [PubMed] [Google Scholar]
- 31.Gandhi CS, Rees DC. Opening the molecular floodgates. Science. 2008;321:1166–1167. doi: 10.1126/science.1162963. [DOI] [PubMed] [Google Scholar]
- 32.Okada A, Miura T, Takeuchi H. Protonation of histidine and histidine-tryptophan interaction in the activation of the M2 ion channel from influenza A virus. Biochemistry. 2001;40:6053–6060. doi: 10.1021/bi0028441. [DOI] [PubMed] [Google Scholar]
- 33.Czabotar PE, Martin SR, Hay AJ. Studies of structural changes in the M2 proton channel of influenza A virus by tryptophan fluorescence. Virus Res. 2004;99:57–61. doi: 10.1016/j.virusres.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Witter R, et al. Solid-state 19F NMR spectroscopy reveals that Trp41 participates in the gating mechanism of the M2 proton channel of influenza A virus. J Am Chem Soc. 2008;130:918–924. doi: 10.1021/ja0754305. [DOI] [PubMed] [Google Scholar]
- 35.Luo W, Mani R, Hong M. Side-chain conformation of the M2 transmembrane peptide proton channel of influenza A virus from 19F solid-state NMR. J Phys Chem B. 2007;111:10825–10832. doi: 10.1021/jp073823k. [DOI] [PubMed] [Google Scholar]
- 36.Bowie JU. Stabilizing membrane proteins. Curr Opin Struct Biol. 2001;11:397–402. doi: 10.1016/s0959-440x(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 37.Bauer CM, Pinto LH, Cross TA, Lamb RA. The influenza virus M2 ion channel protein: Probing the structure of the transmembrane domain in intact cells by using engineered disulfide cross-linking. Virology. 1999;254:196–209. doi: 10.1006/viro.1998.9552. [DOI] [PubMed] [Google Scholar]
- 38.Yi M, Tjong H, Zhou H-X. Spontaneous conformational change and toxin binding in α7 acetylcholine receptor: Insight into channel activation and inhibition. Proc Natl Acad Sci USA. 2008;105:8280–8285. doi: 10.1073/pnas.0710530105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mould JA, et al. Mechanism for proton conduction of the M2 ion channel of influenza A virus. J Biol Chem. 2000;275:8592–8599. doi: 10.1074/jbc.275.12.8592. [DOI] [PubMed] [Google Scholar]
- 40.Lin TI, Schroeder C. Definitive assignment of proton selectivity and attoampere unitary current to the M2 ion channel protein of influenza A virus. J Virol. 2001;75:3647–3656. doi: 10.1128/JVI.75.8.3647-3656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chizhmakov IV, et al. Differences in conductance of M2 proton channels of two influenza viruses at low and high pH. J Physiol (London) 2003;546:427–438. doi: 10.1113/jphysiol.2002.028910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eigen M, Hammes GG. Elementary steps in enzyme reactions (as studied by relaxation spectrometry) Adv Enzymol Relat Areas Mol Biol. 1963;25:1–38. doi: 10.1002/9780470122709.ch1. [DOI] [PubMed] [Google Scholar]
- 43.Zhou H-X, Wlodek ST, McCammon JA. Conformation gating as a mechanism for enzyme specificity. Proc Natl Acad Sci USA. 1998;95:9280–9283. doi: 10.1073/pnas.95.16.9280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Im W, Brooks CL., 3rd De novo folding of membrane proteins: An exploration of the structure and NMR properties of the fd coat protein. J Mol Biol. 2004;337:513–519. doi: 10.1016/j.jmb.2004.01.045. [DOI] [PubMed] [Google Scholar]
- 45.Ramamoorthy A, Wu CH, Opella SJ. Magnitudes and orientations of the principal elements of the 1H chemical shift, 1H-15N dipolar coupling, and 15N chemical shift interaction tensors in 15Nε1-tryptophan and 15Nπ-histidine side chains determined by three-dimensional solid-state NMR spectroscopy of polycrystalline samples. J Am Chem Soc. 1997;119:10479–10486. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.