

SUMMARY
Acute myelogenous leukemia (AML) is organized as a cellular hierarchy initiated and maintained by a subset of self-renewing leukemia stem cells (LSC). We hypothesized that increased CD47 expression on human AML LSC contributes to pathogenesis by inhibiting their phagocytosis through the interaction of CD47 with an inhibitory receptor on phagocytes. We found that CD47 was more highly expressed on AML LSC than their normal counterparts, and that increased CD47 expression predicted worse overall survival in 3 independent cohorts of adult AML patients. Furthermore, blocking monoclonal antibodies directed against CD47 preferentially enabled phagocytosis of AML LSC and inhibited their engraftment in vivo. Finally, treatment of human AML LSC-engrafted mice with anti-CD47 antibody depleted AML and targeted AML LSC. In summary, increased CD47 expression is an independent poor prognostic factor that can be targeted on human AML stem cells with blocking monoclonal antibodies capable of enabling phagocytosis of LSC.
INTRODUCTION
According to the cancer stem cell model, tumors are organized as a cellular hierarchy maintained by a small pool of self-renewing cancer stem cells which must be eliminated in order to eradicate the tumor (Jordan et al., 2006; Reya et al., 2001). For the development of cancer stem cell-targeted therapies, it is necessary to identify molecules and pathways that are preferentially expressed in these cancer stem cells and that are critical for pathogenesis.
To date, human acute myeloid leukemia (AML) stem cells (LSC) are the most well studied cancer stem cell population (Wang and Dick, 2005). AML is an aggressive malignancy with five year overall survival between 30–40%, and much lower for those over age 65 (Estey and Dohner, 2006; Lowenberg et al., 1999). Cytogenetic abnormalities are prognostic in adults with AML, however, up to 50% have a normal karyotype (Byrd et al., 2002; Grimwade et al., 1998). In these patients, the presence of specific molecular mutations can provide prognostic information, particularly internal tandem duplications within the fms-related tyrosine kinase 3 gene (FLT3-ITD) (Mrozek et al., 2007; Schlenk et al., 2008).
In published reports assaying a variety of subtypes of AML, LSC were found to be negative for expression of lineage markers (Lin−), positive for expression of CD34, and negative for expression of CD38 (Bonnet and Dick, 1997; Wang and Dick, 2005). We have recently shown that the Lin−CD34+CD38−CD90− fraction of human cord blood contains a non-HSC multipotent progenitor (MPP), and have hypothesized that this MPP is the cell of origin for human AML (Majeti et al., 2007). Consistent with this hypothesis, we have shown that pre-leukemic mutations occur in a clonal HSC population, eventually leading to the development of LSC at the MPP stage in AML or the granulocyte-macrophage progenitor (GMP) stage in myeloid blast crisis CML (Jamieson et al., 2004; Miyamoto et al., 2000; Weissman, 2005).
We report here the identification of higher expression of CD47 on AML LSC compared to their normal counterparts, HSC and MPP, a finding corroborated by microarray gene expression analysis (Majeti et al., 2009). CD47 is a widely expressed transmembrane protein (Brown and Frazier, 2001). CD47 serves as the ligand for signal regulatory protein alpha (SIRPα), which is expressed on phagocytic cells including macrophages and dendritic cells, that when activated initiates a signal transduction cascade resulting in inhibition of phagocytosis (Barclay and Brown, 2006; Blazar et al., 2001; Okazawa et al., 2005; Oldenborg et al., 2001; Oldenborg et al., 2000). In our own studies, we have found that expression of mouse CD47 in a human AML cell line inhibits phagocytosis and facilitates engraftment in immunodeficient mice, and that CD47 expression on mouse HSC and progenitors increases upon mobilization and is required for engraftment upon transplantation (Jaiswal et al., companion manuscript). We hypothesize that increased expression of CD47 on human AML contributes to pathogenesis by inhibiting phagocytosis of these cells through the interaction of CD47 with SIRPα.
RESULTS
CD47 is More Highly Expressed on AML LSC Than Their Normal Counterparts and is Associated with the FLT3-ITD Mutation
In our investigation of several mouse models of myeloid leukemia, we identified increased expression of CD47 on mouse leukemia cells compared to normal bone marrow (Jaiswal et al., companion manuscript). This prompted investigation of CD47 expression on human AML LSC and their normal counterparts. Using flow cytometry, CD47 was more highly expressed on multiple specimens of AML LSC than normal bone marrow HSC and MPP (Figure 1). This increased expression extended to the bulk leukemia cells, which expressed CD47 similarly to the LSC-enriched fraction.
Figure 1. CD47 is More Highly Expressed on AML LSC Compared to Their Normal Counterparts.
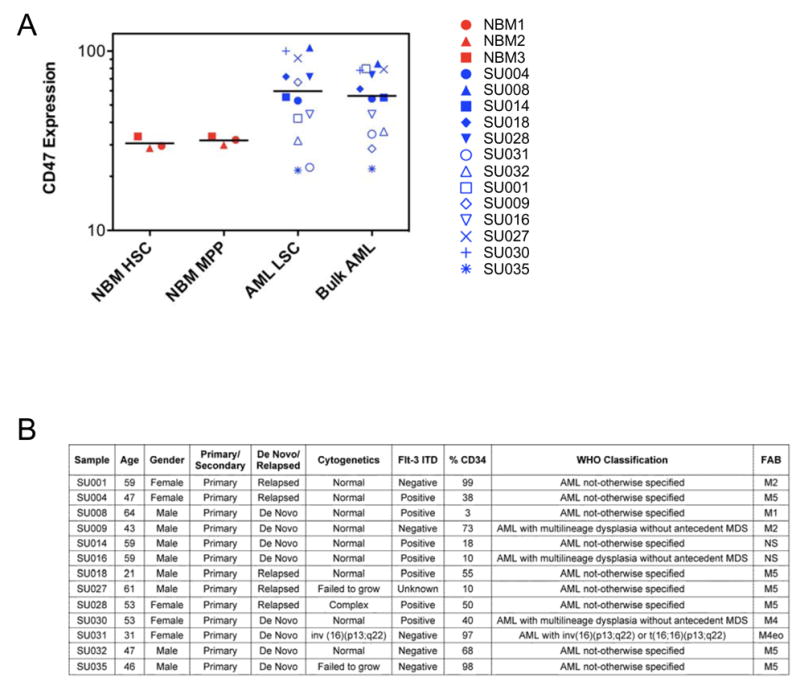
A. Relative CD47 expression on normal bone marrow HSC (Lin−CD34+CD38−CD90+) and MPP (Lin−CD34+CD38−CD90−CD45RA−), as well as LSC (Lin−CD34+CD38−CD90−) and bulk leukemia cells from human AML samples, was determined by flow cytometry. Mean fluorescence intensity was normalized for cell size and against lineage positive cells to account for analysis on different days. The same sample of normal bone marrow (red, n=3) or AML (blue, n=13) is indicated by the same symbol in the different populations. Normalized mean expression (and range) for each population was: HSC 30.6 (28.8–33.4), MPP 31.8 (30.0–33.4), LSC 59.8 (21.6–104.7), and bulk AML 56.3 (22.1–85.1). The differences between the mean expression of HSC with LSC (p=0.003), HSC with bulk leukemia (p=0.001), MPP with LSC (p=0.004), and MPP with bulk leukemia (p=0.002) were statistically significant using a 2-sided Student’s t-test. The difference between the mean expression of AML LSC compared to bulk AML was not statistically significant with p=0.50 using a paired 2-sided Student’s t-test.
B. Clinical and molecular characteristics of primary human AML samples manipulated in vitro and/or in vivo.
Examination of a subset of these samples indicated that CD47 surface expression correlated with CD47 mRNA expression (Supplemental Figure 1). To investigate CD47 expression across morphologic, cytogenetic, and molecular subgroups of AML, gene expression data from a previously described cohort of 285 adult patients were analyzed (Valk et al., 2004). No significant difference in CD47 expression among FAB (French-American-British) subtypes was found (Supplemental Figure 2A). In most cytogenetic subgroups, CD47 was expressed at similar levels, except for cases harboring t(8;21)(q22;q22), a favorable risk group which had a statistically significant lower CD47 expression (Supplemental Figure 2B). In molecularly characterized AML subgroups, no significant association was found between CD47 expression and mutations in the tyrosine kinase domain of FLT3 (FLT3-TKD), over-expression of EVI1, or mutations in CEBPA, NRAS, or KRAS. However, higher CD47 expression was strongly correlated with the presence of FLT3-ITD (p<0.001), which is observed in nearly one third of AML with normal karyotypes and is associated with worse overall survival (Mrozek et al., 2007; Schlenk et al., 2008). This finding was separately confirmed in two independent datasets of 214 and 137 AML patients (Supplemental Table 1) (Bullinger et al., 2008; Jongen-Lavrencic et al., 2008).
Identification and Separation of Normal HSC From Leukemia Cells in the Same Patient Based On Differential CD47 Expression
In the LSC-enriched Lin−CD34+CD38− fraction of specimen SU008, a rare population of CD47lo-expressing cells was detected, in addition to the majority CD47hi-expressing cells (Figure 2A). These populations were isolated by fluorescence-activated cell sorting (FACS) to >98% purity and either transplanted into newborn NOG mice or plated into complete methylcellulose. The CD47hi cells failed to engraft in vivo or form any colonies in vitro, as can be observed with some AML specimens (Ailles et al., 1997). However, the CD47lo cells engrafted with normal myelo-lymphoid hematopoiesis in vivo and formed numerous morphologically normal myeloid colonies in vitro (Figure 2B,C). This specimen harbored the FLT3-ITD mutation, which was detected in the bulk leukemia cells (Figure 2D). The purified CD47hi cells contained the FLT3-ITD mutation, and therefore, were part of the leukemic clone, while the CD47lo cells did not. Human cells isolated from mice engrafted with the CD47lo cells contained only wild type FLT3, indicating that the CD47lo cells contained normal hematopoietic progenitors.
Figure 2. Identification and Separation of Normal HSC From Leukemia Cells in the Same Patient Based On Differential CD47 Expression.
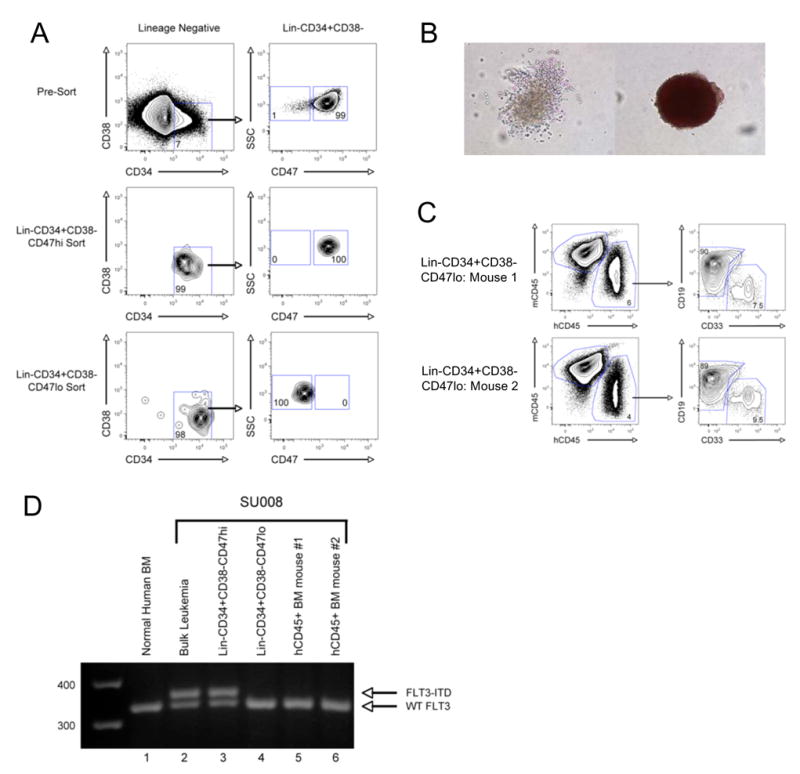
A. CD47 expression on the Lin−CD34+CD38− LSC-enriched fraction of specimen SU008 was determined by flow cytometry. CD47hi- and CD47lo-expressing cells were identified and purified using FACS. The left panels are gated on lineage negative cells, while the right panels are gated on Lin−CD34+CD38− cells.
B. Lin−CD34+CD38−CD47lo and Lin−CD34+CD38−CD47hi cells were plated into complete methylcellulose, capable of supporting the growth of all myeloid colonies. 14 days later, myeloid colony formation was determined by morphologic assessment. Representative CFU-G/M (left) and BFU-E (right) are presented.
C. Lin−CD34+CD38−CD47lo cells were transplanted into 2 newborn NOG mice. 12 weeks later, the mice were sacrificed and the bone marrow was analyzed for the presence of human CD45+CD33+ myeloid cells and human CD45+CD19+ lymphoid cells by flow cytometry.
D. Normal bone marrow HSC, bulk SU008 leukemia cells, Lin−CD34+CD38−CD47hi cells, Lin−CD34+CD38−CD47lo cells, or human CD45+ cells purified from the bone marrow of mice engrafted with Lin−CD34+CD38−CD47lo cells were assessed for the presence of the FLT3-ITD mutation by PCR. The wild type FLT3 and the FLT3-ITD products are indicated.
Increased CD47 Expression in Human AML is Associated with Poor Clinical Outcomes
We hypothesized that increased CD47 expression on human AML contributes to pathogenesis, and predicted that AML with higher expression of CD47 would be associated with worse clinical outcomes. Consistent with this hypothesis, analysis of a previously described group of 285 adult AML patients with diverse cytogenetic and molecular abnormalities (Valk et al., 2004) revealed that a dichotomous stratification of patients into low CD47 and high CD47 expression groups was associated with a significantly increased risk of death in the high expressing group (p=0.03, Supplemental Figure 3A–C). The association of overall survival with this dichotomous stratification of CD47 expression was validated in a second test cohort of 242 adult patients (Metzeler et al., 2008) with normal karyotypes (NK-AML) (p=0.01, Supplemental Figure 3A,D).
Applying this stratification to a distinct validation cohort of 137 adult patients with normal karyotypes (Bullinger et al., 2008), we confirmed the prognostic value of CD47 expression for both overall and event-free survival (Figure 3). Analysis of clinical characteristics of the low and high CD47 expression groups in this cross-validation cohort also identified statistically significant differences in white blood cell (WBC) count and FLT3-ITD status, and no differences in rates of complete remission and type of consolidative therapy including allogeneic transplantation (Supplemental Table 1). Kaplan-Meier analysis demonstrated that high CD47 expression at diagnosis was significantly associated with worse event-free and overall survival (Figure 3A,B). Patients in the low CD47 expression group had a median event-free survival of 17.1 months compared to 6.8 months in the high CD47 expression group, corresponding to a hazard ratio of 1.94 (95% confidence interval 1.30 to 3.77, p=0.004). For overall survival, patients in the low CD47 expression group had a median of 22.1 months compared to 9.1 months in the high CD47 expression group, corresponding to a hazard ratio of 2.02 (95% confidence interval 1.37 to 4.03, p=0.002). When CD47 expression was considered as a continuous variable, increased expression was also associated with a worse event-free (p=0.02) and overall survival (p=0.02).
Figure 3. Increased CD47 Expression in Human AML is Associated with Poor Clinical Outcomes.
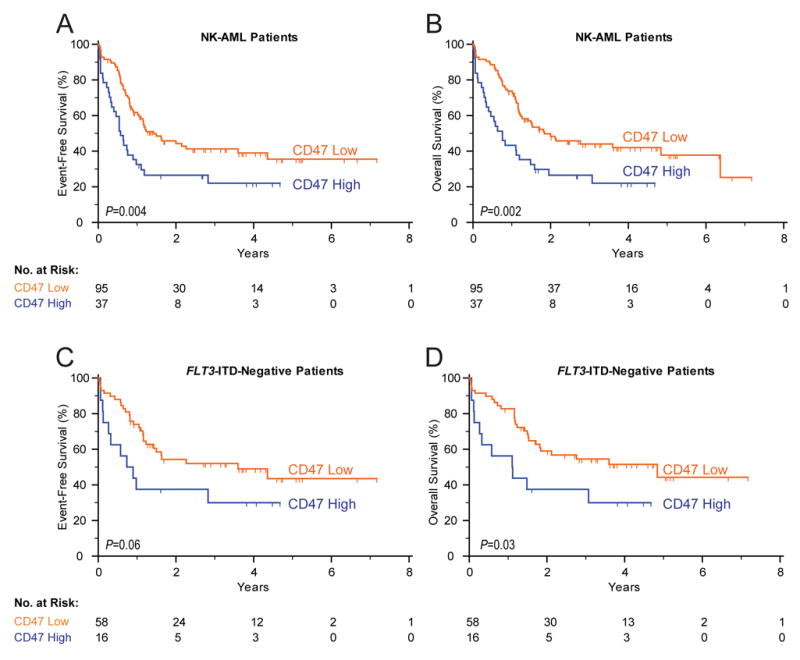
Event-free (A,C) and overall (B,D) survival of 132 AML patients with normal cytogenetics (A,B) and the subset of 74 patients without the FLT3-ITD mutation (C,D). Patients were stratified into low CD47 and high CD47 expression groups based on an optimal threshold (28% high, 72% low) determined by microarray analysis from an independent training data set. The significance measures are based on log-likelihood estimates of the p-value, when treating the model with CD47 expression as a binary classification.
Despite the association with FLT3-ITD (Supplemental Table 1), increased CD47 expression at diagnosis was significantly associated with worse event-free and overall survival in the subgroup of 74 patients without FLT3-ITD, when considered either as a binary classification (Figure 3C,D) or as a continuous variable (p=0.02 for both event-free and overall survival). In multivariable analysis considering age, FLT3-ITD status, and CD47 expression as a continuous variable, increased CD47 expression remained associated with worse event-free survival with a hazard ratio of 1.33 (95% confidence interval 1.03 to 1.73, p=0.03) and overall survival with a hazard ratio of 1.31 (95% confidence interval 1.00 to 1.71, p=0.05) (Supplemental Table 2).
Monoclonal Antibodies Directed Against Human CD47 Preferentially Enable Phagocytosis of AML LSC by Human Macrophages
We hypothesized that increased CD47 expression on human AML contributes to pathogenesis by inhibiting phagocytosis of leukemia cells, leading us to predict that disruption of the CD47-SIRPα interaction with a monoclonal antibody directed against CD47 will preferentially enable the phagocytosis of AML LSC. Several anti-human CD47 monoclonal antibodies have been generated including some capable of blocking the CD47-SIRPα interaction (B6H12.2 and BRIC126) and others unable to do so (2D3) (Subramanian et al., 2006). The ability of these antibodies to enable phagocytosis of AML LSC, or normal human bone marrow CD34+ cells, by human macrophages in vitro was tested. Incubation of AML LSC with human macrophages in the presence of IgG1 isotype control antibody or mouse anti-human CD45 IgG1 monoclonal antibody did not result in significant phagocytosis as determined by either immunofluorescence microscopy (Figure 4A) or flow cytometry (Supplemental Figure 5). However, addition of the blocking anti-CD47 antibodies B6H12.2 and BRIC126, but not the non-blocking anti-CD47 antibody 2D3, enabled phagocytosis of AML LSC (Figure 4A,C). No phagocytosis of normal CD34+ cells was observed with any of the antibodies (Figure 4C).
Figure 4. Monoclonal Antibodies Directed Against Human CD47 Preferentially Enable Phagocytosis of Human AML LSC by Human and Mouse Macrophages In Vitro.
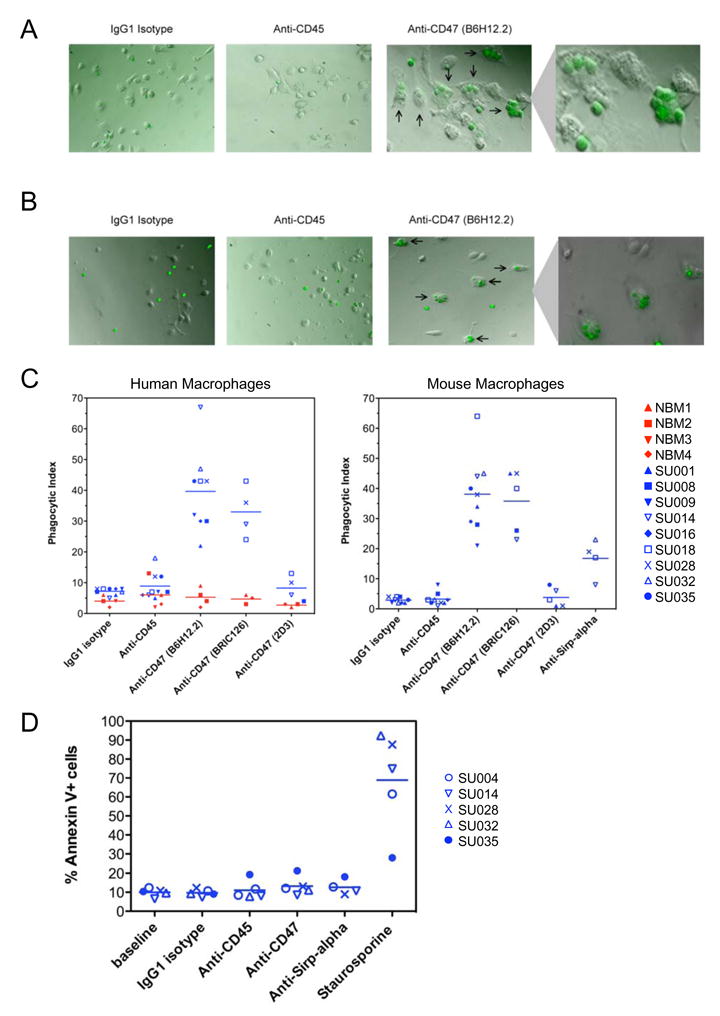
A,B. CFSE-labeled AML LSC were incubated with human peripheral blood-derived macrophages (A) or mouse bone marrow-derived macrophages (B) in the presence of IgG1 isotype control, anti-CD45 IgG1, or anti-CD47 (B6H12.2) IgG1 antibody. These cells were assessed by immunofluorescence microscopy for the presence of fluorescently-labeled LSC within the macrophages (indicated by arrows).
C. CFSE-labeled AML LSC or normal bone marrow CD34+ cells were incubated with human (left) or mouse (right) macrophages in the presence of the indicated antibodies and then assessed for phagocytosis by immunofluorescence microscopy. The phagocytic index was determined for each condition by calculating the number of ingested cells per 100 macrophages. For AML LSC, the differences between isotype or anti-CD45 antibody with blocking anti-CD47 antibody treatment (B6H12.2 and BRIC126) were statistically significant with p<0.001 for all pairwise comparisons with human and mouse macrophages. For human macrophages, the differences between AML LSC and normal CD34+ cells were statistically significant for B6H12.2 (p<0.001) and BRIC126 (p=0.002). For mouse macrophages, the difference between istotype control and anti-SIRPα antibody was statistically significant (p=0.02).
D. AML LSC were incubated in the presence of the indicated antibodies or the staurosporine positive control as described above, but in the absence of macrophages. At the end of the incubation, apoptotic cells were identified by Annexin V staining as determined by flow cytometry. No statistically significant increase in apoptosis was detected with any of the antibodies.
Monoclonal Antibodies Directed Against Human CD47 or Mouse SIRPα Enable Phagocytosis of AML LSC by Mouse Macrophages
The CD47-SIRPα interaction has been implicated as a critical regulator of xenotransplantation rejection in several cross species transplants; however, there are conflicting reports of the ability of CD47 from one species to bind and stimulate SIRPα of a different species (Ide et al., 2007; Subramanian et al., 2006; Takenaka et al., 2007). In order to directly assess the effect of inhibiting the interaction of human CD47 with mouse SIRPα, the in vitro phagocytosis assays described above were conducted with mouse macrophages. Incubation of AML LSC with mouse macrophages in the presence of IgG1 isotype control antibody or mouse anti-human CD45 IgG1 monoclonal antibody did not result in significant phagocytosis as determined by either immunofluorescence microscopy (Figure 4B) or flow cytometry (Supplemental Figure 5). However, addition of the blocking anti-CD47 antibodies B6H12.2 and BRIC126, but not the non-blocking 2D3, enabled phagocytosis of AML LSC (Figure 4B,C). The CD47-SIRPα interaction was alternatively disrupted by a monoclonal antibody directed against mouse SIRPα, which also enabled phagocytosis of AML LSC (Figure 4C).
A Monoclonal Antibody Directed Against CD47 Does Not Induce Apoptosis of AML LSC
Antibodies directed against CD47 have been shown to directly induce apoptosis of several malignant hematopoietic cell lines, as well as primary human chronic lymphocytic leukemia B cells only when immobilized or cross-linked (Kikuchi et al., 2005; Kikuchi et al., 2004; Mateo et al., 1999; Uno et al., 2007). These prior reports raise the alternative hypothesis that anti-CD47 antibodies induce apoptosis of AML LSC, which are then recognized by macrophages and phagocytosed. In order to assess the ability of the blocking anti-CD47 antibody B6H12.2 to directly induce apoptosis of primary human AML LSC, these cells were incubated in vitro with antibodies as described above, but in the absence of macrophages, and expression of Annexin V was determined by flow cytometry. No increase in Annexin V-positive apoptotic cells was detected with the anti-CD47 antibody compared to controls over the time period tested (Figure 4D). Even when plate-bound, the anti-CD47 antibody did not induce apoptosis of AML LSC (Supplemental Figure 7D). Furthermore, phagocytosis of AML LSC was detected as early as 15 minutes after incubation with blocking anti-CD47 antibody, while no apoptosis was detected at 2 hours (Supplemental Figure 7E). These results indicate that the blocking anti-CD47 antibody B6H12.2 does not directly induce apoptosis of human AML LSC.
A Monoclonal Antibody Directed Against Human CD47 Inhibits AML LSC Engraftment and Depletes AML In Vivo
The ability of the blocking anti-CD47 antibody B6H12.2 to target AML LSC in vivo was tested. First, a pre-coating strategy was utilized in which AML LSC were purified by FACS and incubated with IgG1 isotype control, anti-human CD45, or anti-human CD47 antibody. An aliquot of the cells was analyzed for coating by staining with a secondary antibody demonstrating that both anti-CD45 and anti-CD47 antibody bound the cells (Supplemental Figure 9A). The remaining cells were transplanted into newborn NOG mice that were analyzed for leukemic engraftment 13 weeks later. In all but one mouse, the isotype control and anti-CD45 antibody coated cells exhibited long-term leukemic engraftment; however, most mice transplanted with cells coated with anti-CD47 antibody had no detectable leukemia engraftment (Supplemental Figure 9B).
Next, a treatment strategy was utilized in which mice were first engrafted with human AML LSC and then administered daily intraperitoneal injections of 100 micrograms of either mouse IgG or anti-CD47 antibody for 14 days, with leukemic engraftment determined pre- and post-treatment. Analysis of the peripheral blood showed near complete elimination of circulating leukemia in mice treated with anti-CD47 antibody, often after a single dose, with no response in control mice (Figure 5A,B). Similarly, there was a significant reduction in leukemic engraftment in the bone marrow of mice treated with anti-CD47 antibody, while leukemic involvement increased in control IgG-treated mice (Figure 5C,D and Supplemental Figure 10A). Histologic analysis of the bone marrow identified monomorphic leukemic blasts in control IgG-treated mice (Figure 5E, panels 1,2) and cleared hypocellular areas in anti-CD47 antibody-treated mice (Figure 5E, panels 4,5). In the bone marrow of some anti-CD47 antibody-treated mice that contained residual leukemia, macrophages were detected containing phagocytosed pyknotic cells (Figure 5E, panels 3,6).
Figure 5. A Monoclonal Antibody Directed Against Human CD47 Depletes AML In Vivo.
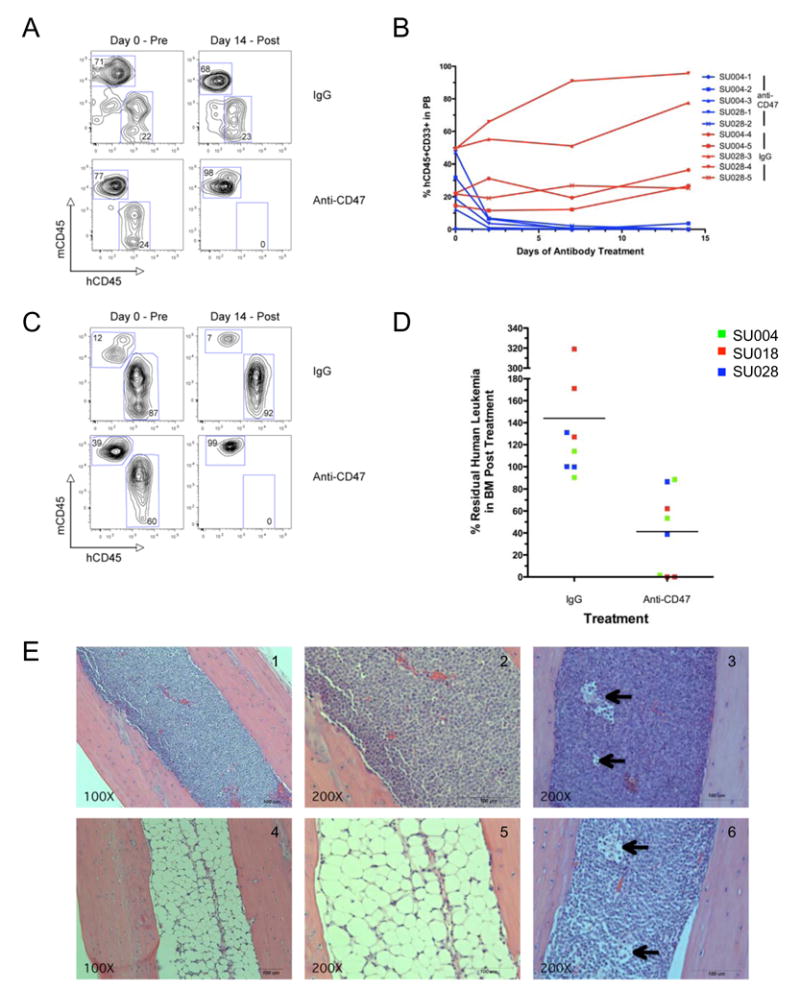
A–D. Newborn NOG mice were transplanted with AML LSC, and 8–12 weeks later, peripheral blood (C,D) and bone marrow (E,F) were analyzed for baseline engraftment prior to treatment with anti-CD47 (B6H12.2) or control IgG antibody (Day 0). Mice were treated with daily 100 microgram intraperitoneal injections for 14 days, at the end of which, they were sacrificed and peripheral blood and bone marrow were analyzed for the percentage of human CD45+CD33+ leukemia. (A) Pre- and post-treatment human leukemic chimerism in the peripheral blood from representative anti-CD47 antibody and control IgG-treated mice as determined by flow cytometry. (B) Summary of human leukemic chimerism in the peripheral blood assessed on multiple days during the course of treatment demonstrated elimination of leukemia in anti-CD47 antibody treated mice compared to control IgG treatment (p=0.007). (C) Pre- and post-treatment human leukemic chimerism in the bone marrow from representative anti-CD47 antibody or control IgG-treated mice as determined by flow cytometry. (D) Summary of human leukemic chimerism in the bone marrow on day 14 relative to day 0 demonstrated a dramatic reduction in leukemic burden in anti-CD47 antibody treated mice compared to control IgG treatment (p=0.006).
E. H&E sections of representative mouse bone marrow cavities from mice engrafted with SU004 AML LSC post-treatment with either control IgG (panels 1,2) or anti-CD47 antibody (panels 4,5). IgG-treated marrows were packed with monomorphic leukemic blasts, while anti-CD47-treated marrows were hypocellular, demonstrating elimination of the human leukemia. In some anti-CD47 antibody-treated mice that contained residual leukemia, macrophages were detected containing phagocytosed pyknotic cells (panels 3,6 arrows).
A Monoclonal Antibody Directed Against Mouse CD47 Enables Phagocytosis of Mouse AML and Does Not Deplete Normal HSC In Vivo
CD47 is expressed at low levels on most normal tissues, including HSC. In order to investigate the viability of targeting CD47 as a therapeutic strategy, we utilized a mouse model of AML and a blocking anti-mouse CD47 monoclonal antibody (MIAP301) (Oldenborg et al., 2001). A serially transplantable mouse model of AML was generated by transduction of 5-fluoruracil-treated wild type bone marrow with a retrovirus encoding HoxA9 and Meis1, as well as GFP (Lessard and Sauvageau, 2003). These mouse leukemia cells exhibited a 3–5 fold increase in CD47 surface expression compared to normal bone marrow (data not shown), similar to that observed with human AML. We first investigated the ability of the blocking anti-mouse CD47 monoclonal antibody to enable phagocytosis of the mouse leukemia cells and found that unlike an isotype-matched control, anti-mouse CD47 antibody enabled phagocytosis of GFP-positive leukemia cells by mouse macrophages in vitro (Figure 6A,B). Next, wild type mice were administered daily intraperitoneal injections of 200 micrograms of anti-mouse CD47 antibody for 14 days. This dose resulted in antibody coating of 100 percent of total bone marrow cells (data not shown). The mice appeared grossly normal and were sacrificed at the end of the treatment course. Analysis of the bone marrow showed no difference in overall cellularity (data not shown), percentage of Lin−Kit+Sca+ (KLS) cells (Figure 6C), or percentage of HSC (Figure 6D). Complete blood counts showed no evidence of anemia, but did indicate isolated neutropenia in the anti-CD47 antibody treated mice (Supplemental Table 3). Metabolic panels showed no serological evidence of hepatic or renal damage (Supplemental Table 4). Finally, in a pilot experiment, we found that treatment of mouse leukemia engrafted mice with the anti-mouse CD47 antibody resulted in a statistically significant increased survival compared to control IgG (Supplemental Figure 11). These results suggest that targeting CD47 with a blocking monoclonal antibody yields no unacceptable toxicity and is a viable therapeutic strategy.
Figure 6. A Monoclonal Antibody Directed Against Mouse CD47 Enables Phagocytosis of Mouse AML and Does Not Deplete Normal HSC In Vivo.
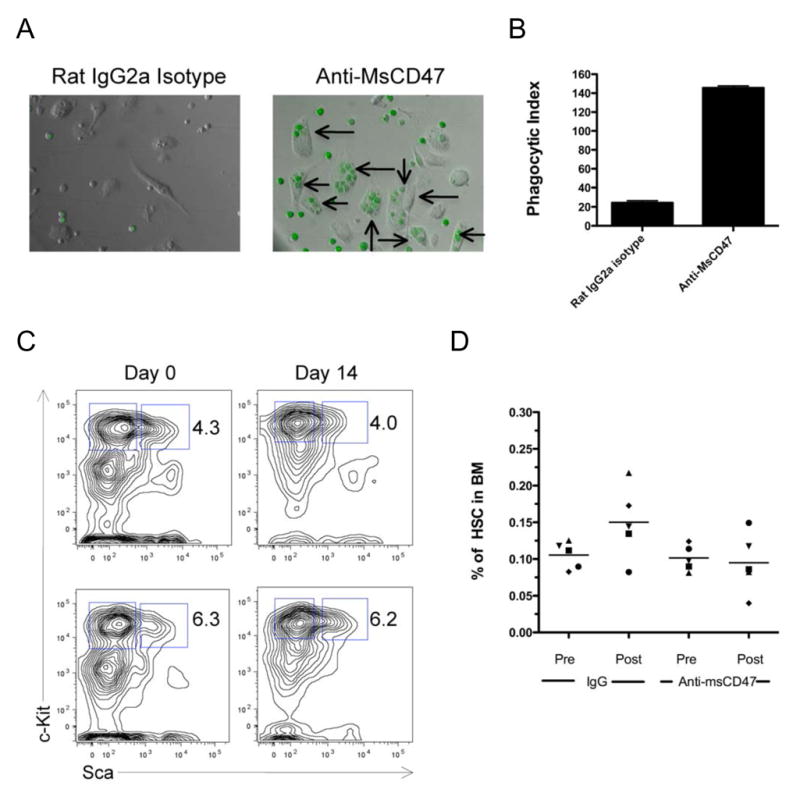
A. GFP+ mouse AML cells were incubated with mouse bone marrow-derived macrophages in vitro in the presence of 10 micrograms/ml of rat IgG2a isotype control or anti-mouse CD47 antibody for two hours. Phagocytosis of GFP+ leukemia cells was observed by fluorescence microscopy (arrows).
B. Quantitative analysis of phagocytosis was determined by calculating the phagocytic index in triplicate assays. Anti-MsCD47 antibody enabled a statistically significant increase in phagocytosis of mouse leukemia cells compared to isotype control (p<0.001). C,D. C57BL/6 wild type mice were treated for 14 days with daily 200 microgram intraperitoneal injections of either anti-msCD47 or rat IgG control antibody. Bone marrow from these mice was aspirated pre- and post-treatment and indicated no effect of either treatment on the frequency of Lin−Kit+Sca+ (KLS) cells (C) or Lin−Kit+Sca+Flk2−CD34− HSC (D) in the bone marrow. Representative flow cytometry plots are shown in panel C. No differences in the percentage of HSC pre and post-treatment were observed with either control IgG (p=0.09) or anti-msCD47 (p=0.81).
A Monoclonal Antibody Directed Against Human CD47 Enables Phagocytosis of AML In Vivo and Targets AML LSC
The in vivo mechanism of the anti-human CD47 antibody was investigated using two approaches to determine if the blocking B6H12.2 anti-CD47 antibody eliminates human AML in vivo by enabling phagocytosis of these cells. First, primary human AML LSC were transduced with a lentivirus expressing GFP and transplanted into NOG mice. Engrafted mice were treated with a single dose of mouse IgG or anti-CD47 antibody and 4 hours later bone marrow, spleen, and liver were examined for the presence of GFP-positive human leukemia cells within F4/80 positive mouse phagocytes by flow cytometry. Unlike IgG control-treated mice, human GFP+ AML cells were detected within phagocytes from all three tissues in anti-CD47-treated mice (Figure 7A,B). In the second experiment, mouse phagocytes were depleted in human AML LSC-engrafted mice prior to treatment with anti-CD47 antibody by administering liposomal clodronate, which accumulates in lysosomes resulting in death of phagocytes (Supplemental Figure 12A). Depletion of phagocytes inhibited the ability of anti-CD47 antibody to eliminate human AML from both the peripheral blood and bone marrow in vivo (Figure 7C).
Figure 7. A Monoclonal Antibody Directed Against Human CD47 Enables Phagocytosis of AML In Vivo and Targets LSC.
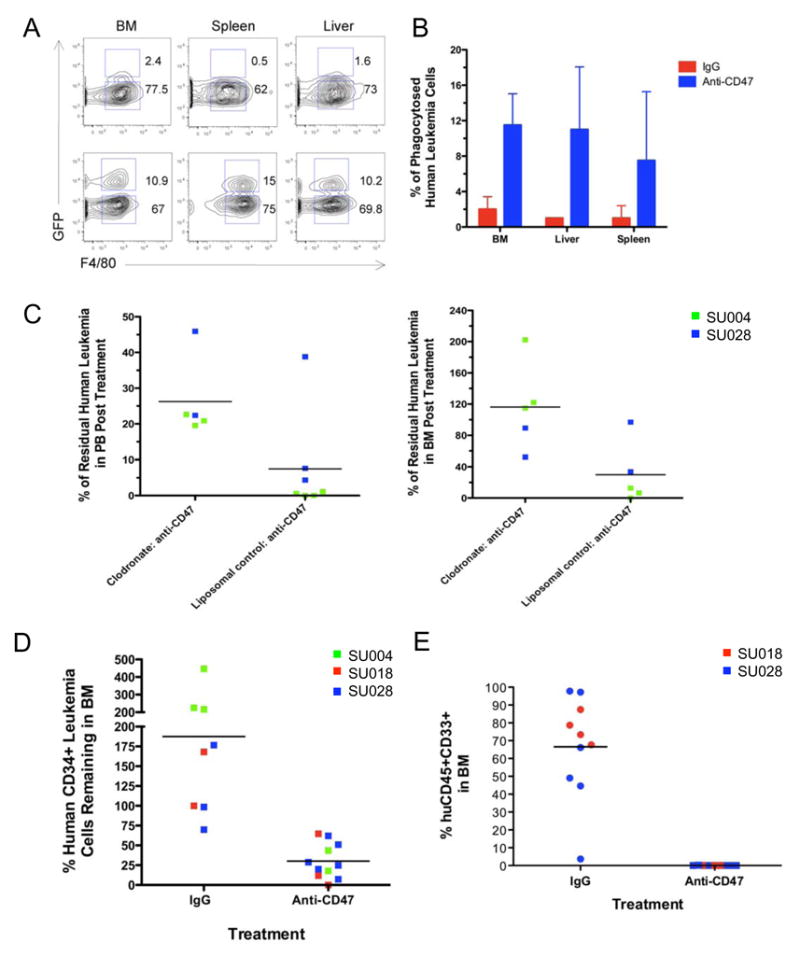
A,B. Flow cytometry plots (A) and quantitation (B) from NOG mice engrafted with lentivirally-transduced GFP-positive SU028 AML LSC, 4 hours after treatment with a single 100 microgram intraperitoneal dose of anti-CD47 antibody (B6H12.2) or control IgG (n=2 for each). Cell suspensions from bone marrow, spleen, and liver were stained for human CD45 and mouse F4/80, which recognizes phagocytes. All plots are gated on human CD45-negative cells. Double positive events represent GFP-positive leukemia cells within mouse phagocytes.
C. NOG mice engrafted with the indicated AML LSC, were treated with liposomal clodronate to deplete phagocytes, and administered daily intraperitoneal injections of anti-CD47 antibody for 14 days. The percentage of residual human leukemia cells in the peripheral blood (left) and bone marrow (right) was determined as described above. Depletion of phagocytes resulted in a statistically significant inhibition of the ability of anti-CD47 antibody to eliminate human AML from both the peripheral blood (p=0.03) and bone marrow (p=0.04). Clodronate treatment by itself had no effect on leukemic engraftment (Supplemental Figure 12 A,B).
D. The percentage of human CD34+ LSC-enriched human leukemia cells remaining in the bone marrow after treatment with either IgG control or anti-CD47 antibody was determined by flow cytometry. Treatment with anti-CD47 antibody resulted in a statistically significant decrease (p<0.001) compared to the control.
E. 500,000 whole bone marrow cells from IgG control (n=12) or anti-CD47 (n=9) antibody-treated mice were secondarily transplanted into NOG mice. 12 weeks later, secondary mice were sacrificed and analyzed for human leukemia engraftment in the peripheral blood (Supplemental Figure 13) and bone marrow as described above. Secondary mice transplanted from IgG treated mice engrafted human leukemia in the bone marrow, while secondary mice transplanted from anti-CD47 treated mice developed no engraftment (p<0.001). Statistical significance was determined using Fisher’s exact test.
Finally, in vivo targeting of AML LSC was investigated. First, the percentage of CD34+ LSC-enriched human leukemia cells present in the bone marrow after treatment was determined by flow cytometry. Treatment with anti-CD47 antibody resulted in a statistically significant decrease in the percentage of human CD34+ leukemia cells remaining in the bone marrow after treatment (Figure 7D and Supplemental Figure 10B). Next, targeting of AML LSC was functionally assessed by secondary transplantation of bone marrow from IgG control or anti-CD47 antibody-treated mice. Secondary mice transplanted from IgG treated mice engrafted human leukemia in the peripheral blood (Supplemental Figure 13) and bone marrow (Figure 7E). However, secondary mice transplanted from anti-CD47 treated mice developed no engraftment in the blood or marrow, which could be the result of in vivo antibody coating with anti-CD47. Regardless, the lack of secondary engraftment clearly indicates that treatment with anti-CD47 antibody targeted AML LSC in vivo.
DISCUSSION
We report here the identification of higher expression of CD47 on AML LSC compared to their normal counterparts and hypothesize that increased expression of CD47 on human AML contributes to pathogenesis by inhibiting phagocytosis of these cells through the interaction of CD47 with SIRPα (Supplemental Figure 14A). Consistent with this hypothesis, we demonstrate that increased expression of CD47 in human AML is associated with decreased overall survival. We also demonstrate that disruption of the CD47-SIRPα interaction with monoclonal antibodies directed against CD47 preferentially enables phagocytosis of AML LSC by macrophages, inhibits engraftment, and targets AML LSC in vivo. Together, these results establish the rationale for considering the use of an anti-CD47 monoclonal antibody as a novel therapy for human AML.
The enabling of phagocytosis by blocking monoclonal antibodies directed against CD47 is a novel mechanism of action for a therapeutic monoclonal antibody in the treatment of cancer. Currently approved antibody therapies are believed to act via stimulation of antibody-dependent cellular cytotoxicity (ADCC), disruption of critical receptor-ligand interactions, or through unknown mechanisms (Adams and Weiner, 2005). Blocking anti-CD47 monoclonal antibodies would treat human AML by enabling phagocytosis and elimination of AML LSC (Supplemental Figure 14B). In support of this mechanism of action in vivo, we show that treatment of human AML LSC-engrafted mice with anti-CD47 antibody results in rapid phagocytosis of AML cells (Figure 7A,B), and,, that depletion of phagocytes with clodronate abrogates this effect (Figure 7C).
As demonstrated here, CD47 contributes to pathogenesis by conferring a survival advantage to LSC and progeny blasts through evasion of phagocytosis by the innate immune system. Moreover, some dendritic cells express SIRPα (Braun et al., 2006; Latour et al., 2001; Sarfati et al., 2008; Seiffert et al., 1999), and we propose that increased CD47 expression on AML LSC also serves to prevent the activation of adaptive T cell immune responses.
AML LSC are enriched in the Lin−CD34+CD38− fraction, which in normal bone marrow contains HSC and MPP. The identification of cell surface molecules that can distinguish between leukemic and normal stem cells is essential for flow cytometry-based assessment of minimal residual disease (MRD) and for the development of prospective separation strategies for use in cellular therapies. Several candidate molecules have recently been identified, including CD123 (Jordan et al., 2000), CD44 (Jin et al., 2006), CD96 (Hosen et al., 2007), CLL-1 (van Rhenen et al., 2007), and now CD47. We demonstrate that not only is CD47 more highly expressed on AML LSC compared to normal HSC and MPP, but also that this differential expression can be used to separate normal HSC/MPP from leukemia cells. This is the first demonstration of the prospective separation of normal HSC from leukemia cells in the same patient sample, and offers the possibility of leukemia-depleted autologous HSC transplantation therapies.
Targeting of CD47 on AML LSC with Therapeutic Monoclonal Antibodies
Cell surface molecules preferentially expressed on AML LSC compared to their normal counterparts are candidates for targeting with therapeutic monoclonal antibodies. Thus far, several molecules have been targeted on AML including CD33 (Adams and Weiner, 2005), CD44 (Jin et al., 2006), CD123 (Lock et al., 2007), and now CD47. Here we report that a monoclonal antibody directed against CD47 targets AML LSC in vivo, as shown by direct reduction in the percentage of human CD34+ LSC-enriched leukemia cells in the bone marrow, and complete elimination of engraftment in secondary transplants (Figure 7D,E).
Targeting of leukemia cells and cell lines with anti-CD47 antibodies has previously been reported to directly induce apoptosis. Treatment of primary human B-CLL cells was shown to induce caspase-independent cell death (Mateo et al., 1999), while a different anti-CD47 antibody was shown to induce apoptosis of several hematopoietic cell lines (Kikuchi et al., 2005; Kikuchi et al., 2004; Uno et al., 2007). These reports raise the alternative hypothesis that anti-CD47 antibodies induce apoptosis of AML LSC, which are then recognized by macrophages and phagocytosed. However, several caveats must be considered when comparing these prior reports to our current study. First, the report on B-CLL involved a mature lymphocytic neoplasm, which is very biologically different from immature aggressive AML, and demonstrated apoptosis not with soluble antibody, but only with cross-linking of antibody, which can result in different effects. Second, the additional reports utilized cell lines and not primary leukemia cells, which are very biologically distinct regarding both proliferation and cell death. Ultimately, we feel that it is not possible to extrapolate the effect of anti-CD47 antibodies from these reports to primary human AML cells.
Several lines of evidence suggest that targeting of CD47 with a monoclonal antibody acts by disrupting the CD47-SIRPα interaction, thereby preventing a phagocytic inhibitory signal, rather than by acting through induction of apoptosis, ADCC, or other mechanisms. First, two blocking anti-CD47 antibodies enabled AML LSC phagocytosis, while one non-blocking antibody did not, even though all three bind the cells similarly (Figure 4C and Supplemental Figure 6). Second, an anti-mouse SIRPα antibody also enabled phagocytosis of human AML LSC by mouse macrophages, demonstrating phagocytosis without direct binding of antibody to AML LSC (Figure 4C). Third, in the case of the B6H12.2 antibody used for most of our experiments, no direct induction of apoptosis of primary AML LSC was detected when added either as a soluble antibody or as an immobilized plate-bound antibody (Figure 4D and Supplemental Figure 7D). Fourth, phagocytosis of AML LSC was detected as early as 15 minutes after addition of anti-CD47 antibody, while no apoptosis was detected at 2 hours (Figure 4D and Supplemental Figure 7E). In fact, only minimal Annexin V-positive staining was detected on Jurkat cells 2 hours after incubation with immobilized plate-bound anti-CD47 antibody (Supplemental Figure 7C). Fifth, if phagocytosis were occurring secondary to apoptosis, then depletion of phagocytes with clodronate should not inhibit the effect of the antibody, which would still directly kill the leukemia cells. However, clodronate did inhibit the ability of anti-CD47 antibody to deplete human AML (Figure 7C), indicating that enabling of phagocytosis is the most likely mechanism. Finally, the isotype-matched anti-CD45 antibody, which also binds LSC, failed to produce the same effects, making ADCC less likely (Figure 4). In fact, the B6H12.2 antibody is mouse isotype IgG1, which is less effective at engaging mouse Fc receptors than antibodies of isotype IgG2a or IgG2b (Nimmerjahn and Ravetch, 2007). For human clinical therapies, blocking CD47 on AML LSC with humanized monoclonal antibodies should promote LSC phagocytosis through a similar mechanism, as indicated by the human macrophage-mediated in vitro phagocytosis (Figure 4A,C).
Higher CD47 expression is detected on AML LSC; however, CD47 is expressed on normal tissues, including bone marrow HSC. We identified a preferential effect of anti-CD47 antibodies in enabling the phagocytosis of AML LSC compared to normal bone marrow CD34+ cells by human macrophages in vitro. In fact, no increased phagocytosis of normal CD34+ cells compared to isotype control was detected, suggesting that blocking CD47 with monoclonal antibodies is a viable therapeutic strategy for human AML. We speculate that this difference is due to the presence of as yet unknown stimuli for phagocytosis on AML LSC that are lacking on normal CD34+ cells. We have now administered a blocking anti-mouse CD47 antibody to normal mice on a schedule similar to the anti-human CD47 antibody, and find coating of all bone marrow cells with no reduction in HSC, no liver or kidney toxicity, and only isolated neutropenia in complete blood counts (Figure 6 and Supplemental Tables 3,4), suggesting that this antibody has no unacceptable toxicity and does not deplete normal mouse HSC.
The experimental evidence presented here provides the rationale for anti-CD47 monoclonal antibodies as monotherapy for AML. However, such antibodies may be equally, if not more effective as part of a combination strategy. The combination of a blocking anti-CD47 antibody with a second antibody able to bind an LSC-specific molecule (for example CD96) and engage Fc receptors on phagocytes may result in a synergistic stimulus for phagocytosis and specific elimination of AML LSC (Supplemental Figure 14C). Furthermore, combinations of monoclonal antibodies to AML LSC that include blocking anti-CD47 and human IgG1 antibodies directed against two other cell surface antigens will be more likely to eliminate leukemia cells with pre-existing epitope variants or antigen loss that are likely to recur in patients treated with a single antibody.
EXPERIMENTAL PROCEDURES
Human Samples
Normal human bone marrow mononuclear cells were purchased from AllCells Inc. (Emeryville, CA). Human AML samples (Figure 1B) were obtained from patients at the Stanford Medical Center with informed consent, according to an IRB-approved protocol (Stanford IRB# 76935 and 6453). Human CD34-positive cells were enriched with magnetic beads (Miltenyi Biotech).
Flow Cytometry Analysis and Cell Sorting
A panel of antibodies was used for analysis and sorting of AML LSC (Lin−CD34+CD38−CD90−, where lineage included CD3, CD19, and CD20), HSC (Lin−CD34+CD38−CD90+), and MPP (Lin−CD34+CD38−CD90−CD45RA−) as previously described (Majeti et al., 2007). Analysis of CD47 expression was performed with an anti-human CD47 PE antibody (clone B6H12, BD Biosciences, San Jose CA). For analysis of mouse bone marrow, the following antibodies were used: Sca1 PB, cKit Alexa 750, Flk2 PE, CD34 FITC, Lineage (CD3, CD4, CD5, CD8, B220, Mac1) PeCy5 (Ebiosciences. San Diego, CA).
Anti-Human and Mouse CD47 and Anti-Mouse SIRPα Antibodies
Monoclonal mouse anti-human CD47 antibodies included: BRIC126, IgG2b (Abcam, Cambridge, MA), 2D3, IgG1 (Ebiosciences), and B6H12.2, IgG1. Monoclonal rat anti-mouse CD47 antibody used was MIAP301, IgG2a. The B6H12.2 and MIAP301 hybridomas were obtained from the American Type Culture Collection (Rockville, MD). Antibody was either purified from hybridoma supernatant using protein G affinity chromatography according to standard procedures or obtained from BioXCell (Lebanon, NH). Monoclonal rat anti-mouse SIRPα, P84, IgG1 was purchased from BD Pharmingen. Isotype controls included mouse IgG1 and rat IgG2a antibodies (Ebiosciences).
In Vitro Phagocytosis Assays
Human AML LSC or normal bone marrow CD34+ cells were CFSE-labeled and incubated with either mouse or human macrophages in the presence of 7μg/ml IgG1 isotype control, anti-CD45 IgG1, anti-CD47 (clones B6H12.2, BRIC126, or 2D3), or anti-mouse SIRPα antibody for 2 hours. Mouse GFP-positive leukemia cells were incubated with mouse macrophages in the presence of 10 μg/ml of rat IgG2a isotype control or anti-mouse CD47 (MIAP301) for 2 hours. Cells were then analyzed by fluorescence microscopy to determine the phagocytic index (number of cells ingested per 100 macrophages). In some experiments, cells were then harvested and stained with either a mouse or human macrophage marker and phagocytosed cells were identified by flow cytometry as macrophage+CFSE+. Statistical analysis using Student’s t-test was performed with GraphPad Prism. See Supplemental Methods for detailed procedures.
In Vivo Antibody Treatment of Human AML LSC Engrafted Mice
1–2.5×105 FACS-purified LSC were transplanted into NOG pups. Eight to twelve weeks later, human AML engraftment (hCD45+CD33+ cells) was assessed in the peripheral blood and bone marrow by tail bleed and aspiration of the femur, respectively. Engrafted mice were then treated with daily intraperitoneal injections of 100 micrograms of anti-CD47 antibody or IgG control for 14 days. On day 15 mice were sacrificed and the peripheral blood and bone marrow were analyzed for AML.
In Vivo Human AML Phagocytosis Assay
A GFP encoding lentivirus was prepared from the pCDH-CMV construct (System Biosciences, Mountain View, CA) using standard techniques. AML LSC from sample SU028 were transduced overnight and transplanted into newborn NOG pups as described. Twelve weeks later human CD45+CD33+GFP+ leukemia engraftment was assessed in the peripheral blood, and GFP+ human leukemia-engrafted mice were injected intraperitoneally with a single 100 microgram dose of either anti-CD47 antibody (clone B6H12.2) or IgG control. Four hours later, mice were sacrificed and bone marrow, spleen, and liver were analyzed for the presence of GFP+ leukemia cells within F4/80 positive mouse phagocytes by flow cytometry. The presence of human CD45−GFP+mouse F4/80+ events identified mouse phagocytes with ingested human leukemia cells.
In Vivo Macrophage Depletion
Liposomal clodronate and control liposomes were prepared as described (Jaiswal et al., companion manuscript). Macrophages were depleted in AML LSC-engrafted NOG mice with the following treatment schedule: 200 microliters of either clodronate or liposomal control was injected intravenously via the retro-orbital sinus two days prior to treatment of these mice with anti-CD47 antibody for IgG control. 100μl of either clodronate or liposomal control was then injected in the same manner on days 2, 6, and 10 after initiation of daily antibody treatment. Mice were then sacrificed on day 14 to assess human leukemic engraftment as described.
Secondary Transplantation
AML LSC-engrafted mice treated with daily injections of either IgG control or anti-CD47 antibody were sacrificed at the end of 14 days of treatment. 5×105 whole bone marrow cells were transplanted into newborn NOG mice. Twelve weeks later peripheral blood and bone marrow was harvested and analyzed for human CD45+CD33+ leukemia engraftment as described.
AML Patients, Microarray Gene Expression Data, and Statistical Analysis
Gene expression and clinical data were analyzed for three previously described cohorts of adult AML patients: (1) a training dataset of 285 patients with diverse cytogenetic and molecular abnormalities described by Valk et al.(Valk et al., 2004), (2) a test dataset of 242 patients with normal karyotypes described by Metzeler et al.(Metzeler et al., 2008), and (3) a validation dataset of 137 patients with normal karyotypes described by Bullinger et al.(Bullinger et al., 2008) See Supplemental Methods for details of therapy. The clinical end points analyzed included overall and event-free survival, with events defined as the interval between study enrollment and removal from the study owing to a lack of complete remission, relapse, or death from any cause, with data censored for patients who did not have an event at the last follow-up visit. See Supplemental Methods for detailed procedures.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Libuse Jerabek and Theresa Storm for excellent lab management, Adriane Mosely for animal husbandry, Chrissy Muscat for antibody preparation, Chris Park for histology assistance, and Garry Nolan for support. The authors are grateful to Drs. Jonathan Pollack, Lars Bullinger, Hartmut Dohner, Peter J.M. Valk, Ruud Delwel, and Bob Lowenberg for sharing microarray data as well as the corresponding clinical covariates for AML patients, and to Rob Tibshirani for statistical advice. R.M. is supported by grants from the Walter and Idun Y. Berry Foundation, the Doctors Cancer Foundation, and the American Association for Cancer Research. R.M. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund. M.P.C. is supported by a Medical Student Training Fellowship from the Howard Hughes Medical Institute, an American Medical Association Seed Research Grant, and the Stanford Medical Scholars Research Program. W.W.P. is supported by the Stanford Medical Scientist Training Program and a NIH Interdisciplinary Regenerative Medicine Predoctoral Fellowship. S.J. is supported by the Stanford Medical Scientist Training Program. K.D.G. is supported by the National Science Foundation Graduate Research Fellowship and Stanford DARE Fellowship. I.L.W. is the Virginia and Daniel Ludwig Professor of Clinical Cancer at Stanford University. R.M., M.P.C., A.A.A., W.W.P., S.J., K.D.G., and N.v.R. have no conflicts of interest to disclose. I.L.W. is a co-founder and Director of Stem Cells Inc., and co-founded Cellerant, Inc., but holds no stock in the company. I.L.W. is an inventor of U.S. Patent Application 11/528,890 entitled “Methods for Diagnosing and Evaluating Treatment of Blood Disorders”. I.L.W., S.J., M.P.C., and R.M. have filed U.S. Patent Application Serial No. 12/321,215 entitled “Methods For Manipulating Phagocytosis Mediated by CD47”. This research is supported by National Institutes of Health grant R01CA86017 to I.L.W, and funding from the Smith Family Fund.
Footnotes
AUTHOR CONTRIBUTIONS
R.M., M.P.C., A.A.A., S.J., and I.L.W. designed the experiments and wrote the manuscript. R.M., M.P.C., A.A.A., and W.W.P. performed the experiments and analyzed data. K.D.G. generated and provided the mouse model of AML. N.v.R. prepared and provided reagents.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- Ailles LE, Gerhard B, Hogge DE. Detection and characterization of primitive malignant and normal progenitors in patients with acute myelogenous leukemia using long-term coculture with supportive feeder layers and cytokines. Blood. 1997;90:2555–2564. [PubMed] [Google Scholar]
- Barclay AN, Brown MH. The SIRP family of receptors and immune regulation. Nat Rev Immunol. 2006;6:457–464. doi: 10.1038/nri1859. [DOI] [PubMed] [Google Scholar]
- Blazar BR, Lindberg FP, Ingulli E, Panoskaltsis-Mortari A, Oldenborg PA, Iizuka K, Yokoyama WM, Taylor PA. CD47 (integrin-associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J Exp Med. 2001;194:541–549. doi: 10.1084/jem.194.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Braun D, Galibert L, Nakajima T, Saito H, Quang VV, Rubio M, Sarfati M. Semimature stage: a checkpoint in a dendritic cell maturation program that allows for functional reversion after signal-regulatory protein-alpha ligation and maturation signals. J Immunol. 2006;177:8550–8559. doi: 10.4049/jimmunol.177.12.8550. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11:130–135. doi: 10.1016/s0962-8924(00)01906-1. [DOI] [PubMed] [Google Scholar]
- Bullinger L, Dohner K, Kranz R, Stirner C, Frohling S, Scholl C, Kim YH, Schlenk RF, Tibshirani R, Dohner H, Pollack JR. An FLT3 gene-expression signature predicts clinical outcome in normal karyotype AML. Blood. 2008;111:4490–4495. doi: 10.1182/blood-2007-09-115055. [DOI] [PubMed] [Google Scholar]
- Byrd JC, Mrozek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, Pettenati MJ, Patil SR, Rao KW, Watson MS, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–4336. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, Rees J, Hann I, Stevens R, Burnett A, Goldstone A. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998;92:2322–2333. [PubMed] [Google Scholar]
- Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, Krensky AM, Weissman IL. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A. 2007;104:11008–11013. doi: 10.1073/pnas.0704271104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide K, Wang H, Tahara H, Liu J, Wang X, Asahara T, Sykes M, Yang YG, Ohdan H. Role for CD47-SIRPalpha signaling in xenograft rejection by macrophages. Proc Natl Acad Sci U S A. 2007;104:5062–5066. doi: 10.1073/pnas.0609661104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- Jongen-Lavrencic M, Sun SM, Dijkstra MK, Valk PJ, Lowenberg B. MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood. 2008;111:5078–5085. doi: 10.1182/blood-2008-01-133355. [DOI] [PubMed] [Google Scholar]
- Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, Meyerrose T, Rossi R, Grimes B, Rizzieri DA, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14:1777–1784. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, Uno S, Kinoshita Y, Yoshimura Y, Iida S, Wakahara Y, Tsuchiya M, Yamada-Okabe H, Fukushima N. Apoptosis inducing bivalent single-chain antibody fragments against CD47 showed antitumor potency for multiple myeloma. Leuk Res. 2005;29:445–450. doi: 10.1016/j.leukres.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, Uno S, Yoshimura Y, Otabe K, Iida S, Oheda M, Fukushima N, Tsuchiya M. A bivalent single-chain Fv fragment against CD47 induces apoptosis for leukemic cells. Biochem Biophys Res Commun. 2004;315:912–918. doi: 10.1016/j.bbrc.2004.01.128. [DOI] [PubMed] [Google Scholar]
- Latour S, Tanaka H, Demeure C, Mateo V, Rubio M, Brown EJ, Maliszewski C, Lindberg FP, Oldenborg A, Ullrich A, et al. Bidirectional negative regulation of human T and dendritic cells by CD47 and its cognate receptor signal-regulator protein-alpha: down-regulation of IL-12 responsiveness and inhibition of dendritic cell activation. J Immunol. 2001;167:2547–2554. doi: 10.4049/jimmunol.167.5.2547. [DOI] [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- Lock RB, Jin L, Lee EM, Ramshaw HS, Busfield S, Peoppl AG, Wilkinson L, Guthridge MA, Boyd A, Gearing D, et al. CD123 (IL-3 Receptor {alpha} Chain) Neutralization by a Monoclonal Antibody Selectively Eliminates Human Acute Myeloid Leukemic Stem Cells. ASH Annual Meeting Abstracts. 2007;110:161. [Google Scholar]
- Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- Majeti R, Becker MW, Tian Q, Lee TL, Yan X, Liu R, Chiang JH, Hood L, Clarke MF, Weissman IL. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0900089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1:635–645. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo V, Lagneaux L, Bron D, Biron G, Armant M, Delespesse G, Sarfati M. CD47 ligation induces caspase-independent cell death in chronic lymphocytic leukemia. Nat Med. 1999;5:1277–1284. doi: 10.1038/15233. [DOI] [PubMed] [Google Scholar]
- Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112:4193–4201. doi: 10.1182/blood-2008-02-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci U S A. 2000;97:7521–7526. doi: 10.1073/pnas.97.13.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109:431–448. doi: 10.1182/blood-2006-06-001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr Opin Immunol. 2007;19:239–245. doi: 10.1016/j.coi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y, Oldenborg PA, Ishikawa O, Matozaki T. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J Immunol. 2005;174:2004–2011. doi: 10.4049/jimmunol.174.4.2004. [DOI] [PubMed] [Google Scholar]
- Oldenborg PA, Gresham HD, Lindberg FP. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J Exp Med. 2001;193:855–862. doi: 10.1084/jem.193.7.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Sarfati M, Fortin G, Raymond M, Susin S. CD47 in the immune response: role of thrombospondin and SIRP-alpha reverse signaling. Curr Drug Targets. 2008;9:842–850. doi: 10.2174/138945008785909310. [DOI] [PubMed] [Google Scholar]
- Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, Habdank M, Spath D, Morgan M, Benner A, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- Seiffert M, Cant C, Chen Z, Rappold I, Brugger W, Kanz L, Brown EJ, Ullrich A, Buhring HJ. Human signal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood. 1999;94:3633–3643. [PubMed] [Google Scholar]
- Subramanian S, Parthasarathy R, Sen S, Boder ET, Discher DE. Species- and cell type-specific interactions between CD47 and human SIRPalpha. Blood. 2006;107:2548–2556. doi: 10.1182/blood-2005-04-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka K, Prasolava TK, Wang JC, Mortin-Toth SM, Khalouei S, Gan OI, Dick JE, Danska JS. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol. 2007;8:1313–1323. doi: 10.1038/ni1527. [DOI] [PubMed] [Google Scholar]
- Uno S, Kinoshita Y, Azuma Y, Tsunenari T, Yoshimura Y, Iida S, Kikuchi Y, Yamada-Okabe H, Fukushima N. Antitumor activity of a monoclonal antibody against CD47 in xenograft models of human leukemia. Oncol Rep. 2007;17:1189–1194. [PubMed] [Google Scholar]
- Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, Delwel R. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B, Stigter-van Walsum M, Zweegman S, Ossenkoppele GJ, Jan Schuurhuis G. The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110:2659–2666. doi: 10.1182/blood-2007-03-083048. [DOI] [PubMed] [Google Scholar]
- Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Weissman I. Stem cell research: paths to cancer therapies and regenerative medicine. Jama. 2005;294:1359–1366. doi: 10.1001/jama.294.11.1359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.