

Abstract
Objective:
Succinic semialdehyde dehydrogenase (SSADH) deficiency is an autosomal recessive disorder of GABA metabolism characterized by elevated levels of GABA and gamma-hydroxybutyric acid. Clinical findings include intellectual impairment, hypotonia, hyporeflexia, hallucinations, autistic behaviors, and seizures. Autoradiographic labeling and slice electrophysiology studies in the murine model demonstrate use-dependent downregulation of GABA(A) receptors. We studied GABA(A) receptor activity in human SSADH deficiency utilizing [11C]-flumazenil (FMZ)-PET.
Methods:
FMZ binding was measured in 7 patients, 10 unaffected parents, and 8 healthy controls. Data analysis was performed using a reference region compartmental model, with time-activity curve from pons as the input function. Relative parametric binding potential (BPND) was derived, with MRI-based pixel by pixel partial volume correction, in regions of interest drawn on coregistered MRI.
Results:
In amygdala, hippocampus, cerebellar vermis, frontal, parietal, and occipital cortex, patients with SSADH deficiency had significant reductions in FMZ BPND compared to parents and controls. Mean cortical values were 6.96 ± 0.79 (controls), 6.89 ± 0.71 (parents), and 4.88 ± 0.77 (patients) (F ratio 16.1; p < 0.001). There were no differences between controls and parents in any cortical region.
Conclusions:
Succinic semialdehyde dehydrogenase (SSADH) deficient patients show widespread reduction in BZPR binding on [11C]-flumazenil-PET. Our results suggest that high endogenous brain GABA levels in SSADH deficiency downregulate GABA(A)-BZPR binding site availability. This finding suggests a potential mechanism for neurologic dysfunction in a serious neurodevelopmental disorder, and suggests that PET may be useful to translate studies in animal models to human disease.
GLOSSARY
- FMZ
= flumazenil;
- MRS
= magnetic resonance spectroscopy;
- PVC
= partial volume correction;
- ROI
= region of interest;
- SPGR
= spoiled gradient recall;
- SSADH
= succinic semialdehyde dehydrogenase.
Succinic semialdehyde dehydrogenase (SSADH) deficiency, also called 4-hydroxybutyric aciduria (McKusick 279180) and aldehyde dehydrogenase 5a1 (Aldh5a1), is an autosomal recessive disorder. About 350 patients are known, with about 85% under 18, making this the most prevalent pediatric neurotransmitter disorder.1 In the absence of SSADH, transamination of GABA to succinic semialdehyde is followed by its conversion to 4-hydroxybutryic acid (gamma-hydroxybutyric acid, or GHB), leading to CNS GABA and γ-hydroxy butyrate (GHB) accumulation.2 Major clinical manifestations include developmental delay, hypotonia, ataxia, and seizures. Hyperkinetic behavior, aggression, self-injurious behaviors, and hallucinations have also been described; EEG abnormalities include generalized and focal epileptiform discharges, photosensitivity, and background slowing.1
Neuroimaging findings in SSADH deficiency include T2 hyperintensities in multiple regions, most commonly the globus pallidus. A pattern of dentate-pallidal hyperintensity has been described.3 There are also areas of abnormal T2-weighted signal in cerebral white matter and brainstem. PET with [18F]-fluorodeoxyglucose ([18F]-FDG PET) showed decreased cerebellar glucose metabolism in patients with known cerebellar atrophy on structural MRI.1 Magnetic resonance spectroscopy (MRS) showed elevated occipital GABA in 5 affected subjects, but not in parents.4
A mouse model of SSADH deficiency showed high GABA and GHB levels in neural tissue, altered GABA binding in cerebral cortex, and reduced expression of GABAA and GABAB receptors.5,6 Of interest, while the GABAergic system is significantly altered in this mouse model, the GHBergic system appears not to be affected.7
PET with [11C]-flumazenil ([11C]-FMZ- PET) has been used to image the GABA-benzodiazepine receptor complex in man.8–10 [11C]-FMZ binds with high affinity and selectivity to the GABAA receptor α1, α2, α3, and α5 receptor subunits; in cerebellum, binding is mainly to the α1 subunit.11
In order to test the hypothesis that patients with SSADH deficiency would demonstrate reduced GABA receptor binding, building directly on studies in the murine knockout model, we used [11C]-FMZ-PET to study affected subjects, parents, and healthy volunteers.
METHODS
Subjects.
We studied 7 affected patients (4 male; age 15.7 ± 5.7 years), 10 parents (6 male; age 43.6 ± 5.8), and 8 healthy adult volunteer controls (4 male; 31.8 ± 10.2 years). All study participants provided informed consent. Healthy volunteers were screened with general physical and neurologic examination, and standard laboratory tests, by the National Institute of Neurological Disorders and Stroke Clinical Epilepsy Section. Age-matched pediatric controls were not recruited for ethical reasons related to radiation exposure.
Affected patients and family members were screened for inclusion by the Children’s National Medical Center Department of Neurology. Screening was performed by an experienced pediatric neurologist (P.L.P.) who has clinical expertise in this condition, and has followed the families personally. Family and social histories were reviewed carefully and paternity established by appropriate methods if deemed necessary. Inclusion criteria were clinical characteristics consistent with SSADH deficiency, persistent 4-hydroxybutyric aciduria (gamma-hydroxybutyric aciduria), and confirmed leukocyte extract SSADH enzyme deficiency and/or identification of a pathogenic mutation in DNA samples.12,13 All electrophysiologic and imaging studies were performed at the NIH Clinical Center.
MRI.
All subjects underwent MRI using a 1.5-T Signa scanner (GE Healthcare). T1-weighted spoiled gradient images (repetition time, 27 msec; minimum echo time; flip angle 20°) were acquired in coronal orientation. Standard coronal T2 and fluid-attenuated inversion recovery short tau inversion recovery and three-dimensional spoiled gradient recall (SPGR) images were obtained. All MRI scans were read by an experienced neuroradiologist who was unaware of PET results. In addition, magnetic resonance–based partial volume correction (PVC) and region of interest (ROI) coregistration were performed.14
PET scanning.
Patients fasted for at least 3 hours before each PET scan. As part of standard procedure, EEG leads were attached to the scalp for monitoring during scanning, and a thermoplastic facemask used for positioning.
PET scans were acquired using a GE Advance scanner with septa retracted (35 contiguous slices; 4.25 mm plane separation; reconstructed 3-dimensional spatial resolution 6–7 mm full-width at half-maximum). A transmission scan was acquired to correct for attenuation. Following transmission scanning, a target dose of 0.285 mCi/kg with a maximum of 20 mCi of high specific activity [11C]-FMZ was injected by IV bolus, and a 60-minute dynamic emission image of the brain acquired.
Subject motion correction during the PET acquisition was performed with mutual-information registration of each scan time-frame to a standard frame before attenuation correction.15 Based on the calculated motion, the transmission images were resliced and projected for final reconstruction and realignment.16
A summed PET image (0–10 minutes post injection) was registered to each subject’s MRI with a mutual information algorithm and all the PET images were resliced. Binding potential (BPND) images were created using the 2-step version of the simplified reference tissue model (SRTM2).17 The time-activity curve for the reference tissue was derived from the pons (drawn on the MRI), where the [11C]-FMZ binding is predominantly accounted for by free and nonspecifically bound radiotracer. This method also produces images of relative tracer delivery (R1), an index related to cerebral blood flow.
Anesthetic.
In order to avoid possible interference with [11C]-FMZ binding, we used an agent that does not affect GABA levels or GABA receptors. For patients who needed sedation during PET, we used dexmedetomidine (Precedex, Hospira, Lake Forest, IL), a highly selective α2-adrenoreceptor agonist that exerts its hypnotic effects at the locus coeruleus, and has minimal cardiovascular and respiratory effects.18,19 A bolus dose of 1 mg/kg was infused over 10 minutes and followed by an infusion of 0.7–2 mcg/kg/hour. The drug has no effect on release or extracellular levels of glutamate, aspartate, and GABA.20 The sedation was used in 5 of the 7 patients and in no controls.
Data analysis.
We used a simplified reference region method.17 This approach uses the time-activity curve from the pons, a region with little or no specific binding, as the input function and produces functional images of binding potential equivalent to
where fND is a measure of the fraction of free intracerebral concentration of tracer, Bavail is the maximum available (unoccupied) receptor concentration, and KD is the tracer dissociation equilibrium constant. Functional images were corrected for partial volume effects and gray-white matter ratios on a pixel by pixel basis.14 Operators blinded to subject status (affected, parent, or control) drew ROIs on coregistered three-dimensional SPGR MRI scans. ROIs were frontal and occipital cortex, amygdala, hippocampus, caudate nucleus, globus pallidus, thalamus, midbrain, dentate nucleus, and cerebellar vermis. The Duvernoy Brain Atlas was used to identify structures.21
Regional BPND and R1 were compared among patients, controls and parents, with one-way analysis of variance and post hoc Tukey test, using Systat (Systat Inc., Richmond, CA).
This study was approved by the institutional review boards of the NIH (National Institute of Neurological Disorders and Stroke) and Children’s National Medical Center.
RESULTS
Neurologic examination of parents and controls, and their structural MRI studies, were normal.
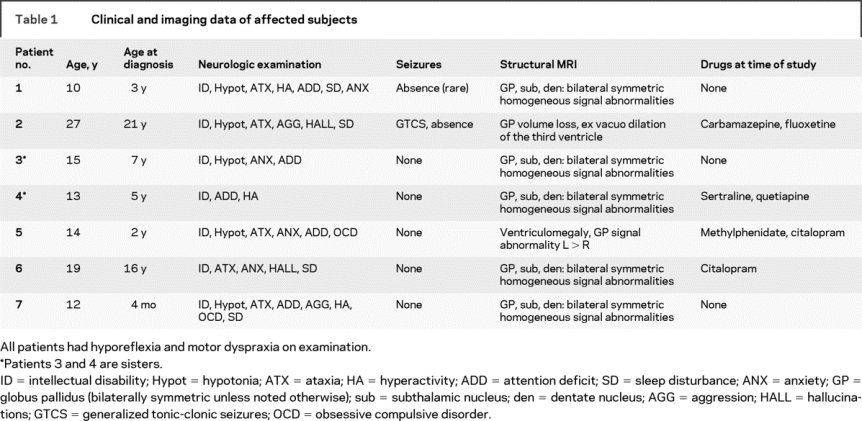
Affected subjects had a variety of neurologic findings (table 1). Two affected subjects had standard 21-channel EEG with hyperventilation and photic stimulation that was normal for age. Five had diffuse slowing.
Table 1 Clinical and imaging data of affected subjects
Structural MRI.
Five patients had a consistent pattern of bilaterally symmetric homogeneous signal abnormalities in the globus pallidus, subthalamic nuclei, and dentate nuclei (figure 1). Both the pars medialis and pars lateralis of the globus pallidus were involved. In the pars medialis, the pars interna and externa were not distinguishable. The signal abnormality appeared as hypointensity on T1- and hyperintensity on T2-weighted sequences, but was most clearly discernible using the short tau inversion recovery sequence, which combines T1 and T2 effects.

Figure 1 Coronal short tau inversion recovery sections from MRI in patient 1 showing bilateral symmetric homogeneous signal abnormalities in each globus pallidus pars lateralis (white arrowhead, A and B), pars medialis (black arrowheads, A), subthalamic nucleus (black arrows, B), and dentate nucleus (white arrows, C)
One patient had marked ventriculomegaly, and asymmetric involvement of the globus pallidus. In the globus pallidus, the signal abnormality was heterogeneous, with normal and abnormal portions. There was minimal abnormality present on the right, and a marked abnormality on the left, which was clearly expanded. Comparison to structural imaging done 5 years previously revealed the same degree of pallidal asymmetry and ventriculomegaly, and follow-up studies over 2 years showed completely stable findings.
In the oldest (27 years old) patient, the signal abnormality in the globus pallidus was subtle, but there was clear volume loss, with commensurate ex vacuo dilation of the third ventricle. Abnormalities of the STN and the dentate nuclei could not be identified.
No cortical or pontine abnormalities were identified on MRI.
PET.
There were statistically significant differences in 11C FMZ binding among patients, parents, and controls in all ROIs except midbrain; in the globus pallidus, a trend toward significance was found (table 2 and figure 2). Since no significant left-right difference in cortical BPND were found, left and right ROIs were averaged. Using a strict Bonferroni procedure to correct for 10 independent regional analyses of variance, significance is lost in dentate, and becomes marginal in caudate. Failure to find a significant group difference in the globus pallidus was due to particularly low values in one parent. Omitting this subject, the group difference was highly significant (F ratio 33.8). Overall cortical binding was significantly diminished in patients, with values about 70% of parents and controls (figure 3). There were no significant differences between the parent and control groups.
Table 2 Regional [11C]-flumazenil binding potential (BPND) data in patients, parents, and healthy controls
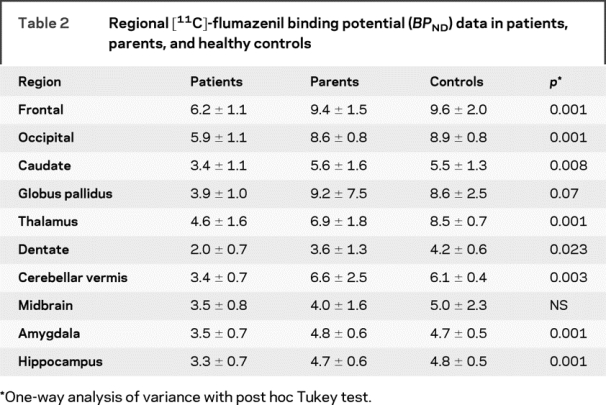

Figure 2 [11C]-flumazenil PET scans in an affected subject (A) and the subject’s parent (B) showing marked reduction of cortical binding potential in A
The color scale shows BPND.
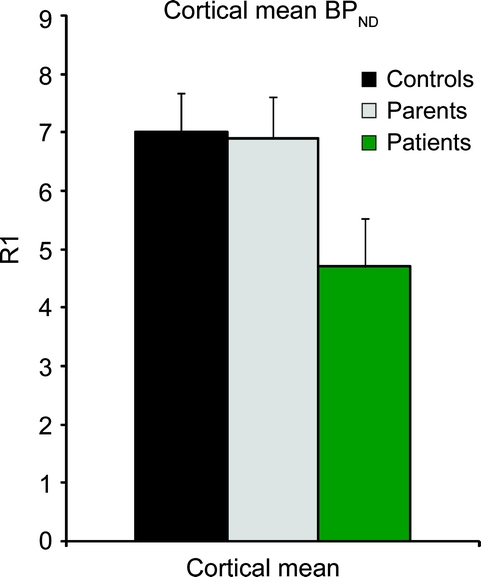
Figure 3 Global mean ± SD cortical binding in affected subjects, parents, and healthy volunteers
After analysis of variance, the post hoc Tukey procedure was used to determine intergroup differences. Setting the significance level at p < 0.05, after strict Bonferroni correction for multiple comparisons, there were significant control vs patient differences in frontal, occipital, thalamic, amygdala, and hippocampal regions; there were differences between parents and patients in frontal, occipital, amygdala, and hippocampal regions. There were no significant control vs parent differences either before or after correction.
In order to ensure that systematic differences in the pons time-activity curves did not affect the results, we compared the average pons activity counts, corrected for dose administered and patient weight among the 3 groups. There were no significant intergroup differences (F ratio 0.568; p > 0.50). We also calculated regional R1 tracer delivery values and found no difference among the groups in any region.
Two patients had scans without anesthesia. Both had cortical values below parents and controls; one was outside the mean plus 2 standard deviations (mean cortical values for controls, parents, and patients were 6.9 ± 0.7, 7.0 ± 0.7, and 4.7 ± 0.8, respectively). Values for these 2 patients were 5.0 and 6.3. Anesthesia did not affect tracer delivery R1.
Only one patient was on an antiepileptic drug at the time of the study; 4 (including both who had scans without anesthesia) were taking a selective serotonin reuptake inhibitor (table 1). Although FMZ binding tended to be higher in the patients on selective serotonin reuptake inhibitors, the differences were not significant.
We did not detect any relation of FMZ binding to age among the patients, who had a relatively restricted range, except for one outlier. Among parents and controls, age had no relation to binding.
DISCUSSION
We found that patients with SSADH deficiency have reduced benzodiazepine receptor binding measured with 11C-FMZ-PET. Parents, clinically unaffected, showed no binding reduction. This finding parallels results in SSADH null mice and suggests a potential mechanism for neurologic dysfunction in a serious neurodevelopmental disorder.
The subcortical reductions we found could have been related in part to structural abnormalities, because the PVC procedure is less accurate for subcortical structures.14 However, the patients had no cortical abnormalities on MRI, and PVC is reliable in these regions. The cortical binding reductions are unlikely to be due to partial volume effect.
Anesthesia and age were unlikely to have affected our results. Dexmedetomidine has been shown to have no interaction with GABA-benzodiazepine receptors.22 Of the 2 patients who did not receive anesthesia, BPND values were still below the parent and control range. Moreover, tracer delivery, measured by R1, did not differ across the groups. We were unable to study healthy child volunteers. However, previous studies have shown that [11C]-FMZ binding is high in young children, decreasing with age, but not reaching adult cortical values until age 20.23 The children studied (aged 2–17 years, mean 9.4 ± SD 4.1 years) had partial epilepsy and used only drugs that do not increase brain GABA. They were compared to adults (29 ± 7 years) with partial epilepsy matched with the children for the same anticonvulsant medications, as well as normal adults (40 ± 9 years) with no history of neurologic or psychiatric disorders. There was no difference between the adult normal and epilepsy groups. Thus, children without metabolic disease would be expected to have higher binding than adults.
Other PET studies have shown that 11C-FMZ-PET can detect alterations in GABA receptor binding. Prolonged vigabatrin treatment is associated with decreased binding in children with epilepsy.10 Since vigabatrin increases synaptic GABA availability by inhibiting enzymatic degradation, this study supports the concept of receptor downregulation of GABA receptors due to increased GABA levels, potentially exacerbated by elevations of other metabolites (e.g., GHB). Alternatively, reduced binding could be due to altered receptor properties rather than number. In a familial generalized epilepsy syndrome, patients with the GABRG2 (R43Q) mutation had global binding reductions of about 25%, particularly in anterior cortical regions.24 However, significant alterations of GABAAR β2 subunit levels and functional GABAAR activity have been demonstrated in murine SSADH-deficient hippocampal preparations.6
Several lines of evidence suggest a relationship between altered GABA receptor binding and the phenotype in SSADH deficiency. Evidence from the murine SSADH knockout suggests that GABA is significantly increased during embryonic development.25 During early development, and most likely in the early postnatal period, GABA is excitatory rather than inhibitory.26,27 The SSADH null animals have markedly smaller brains, with cerebellum particularly affected.28 The murine neurologic phenotype is characterized by ataxia and an epileptic transition from absence seizures to ultimately fatal convulsive status epilepticus by 3 weeks of age.29
GHB also may contribute to downregulation of presynaptic and postsynaptic GABA receptor expression, perhaps leading to the seizures and hyperactive behavior in SSADH deficiency. At the nonphysiologic concentrations (∼200–1000 μM) in SSADH deficiency, GHB acts as a weak GABABR agonist.30 This may lead to inhibition of GABAB interneurons, disinhibition of glutamatergic neurons, and pathologic hyperexcitability. Other intermediates accumulating in the mouse and human disorders may further heighten GABAergic effects. The GABA-analogues guanidinobutyrate, homocarnosine, succinic semialdehyde, and 4,5-dihydroxyhexanoic acid (DHHA) accumulate both in patient physiologic fluids and knockout mouse CNS.4,25,31–34 These species may exert previously undefined GABA receptor binding effects, and it has been shown that DHHA binds the high-affinity GHB receptor, albeit with low to moderate affinity.
A number of studies have shown reduced FMZ binding in patients with partial seizures, closely correlated with EEG distribution of epileptiform discharges.9,35 In some cases, reduced FMZ binding has been found in patients with normal MRI, as well as after partial volume correction in patients with mesial temporal sclerosis.9,36 The current study lays the groundwork for future clinical interventions in SSADH-deficient patients through identification of an important therapeutic endpoint. Along those lines, the nonphysiologic amino acid taurine has shown benefit in a single SSADH-deficient case.37 Among its many reported roles, taurine may activate GABA receptors.38 Other orally bioactive GABA receptor antagonists, such as SGS-742, can now also be considered in SSADH-deficient patients with monitoring of FMZ binding.39
DISCLOSURE
Supported by the National Institutes of Health, National Institute of Neurological Diseases and Stroke, Division of Intramural Research; National Institutes of Health NS40270 (Dr. Gibson); Pediatric Neurotransmitter Disease Association (Dr. Pearl); and Delman Family Fund for Pediatric Neurology Research (Dr. Pearl). Dr. Pearl receives research support from the Pediatric Neurotransmitter Disease Association and the Delman Family Fund for Pediatric Neurology Research. Dr. Gibson serves as Editor for the Journal of Inherited Metabolic Disease; receives royalties from Physician’s Guide to the Laboratory Diagnosis of Metabolic Diseases [1996, Springer, Berlin] and Laboratory Guide to the Methods in Biochemical Genetics [2008, Springer, Berlin]; and receives research support from the NIH [NS 40270] and the Pediatric Neurotransmitter Disease Association. Dr. Quezado receives research support from the NIH [NIH Clinical Center Intramural Research]. I. Dustin reports no disclosures. J. Taylor reports no disclosures. Dr. Trzcinski reports no disclosures. Dr. Schreiber reports no disclosures. K. Forester reports no disclosures. P. Reeves-Tyer reports no disclosures. Dr. Liew reports no disclosures. Dr. Shamim reports no disclosures. Dr. Herscovitch is employed full-time by the NIH and supervises all PET imaging for clinical research at NIH, has received funding for travel from the Canadian Foundation for Innovation, the Society of Nuclear Medicine, and the Crozer-Chester Medical Center, and has received honoraria from the Canadian Foundation for Innovation and the Crozer-Chester Medical Center. Dr. Carson serves as Editor for the Journal of Nuclear Medicine and the Journal of Cerebral Blood Flow and Metabolism; has received honoraria for lectures from Columbia University, the Connecticut PET Society, and the Japanese Society of Nuclear Medicine; and receives research support from Siemens Medical Systems, Pfizer Inc., GlaxoSmithKline Inc., Lilly Inc., NINDS [5R01NS058360], and Yale University. Dr. Butman is employed by the NIH and holds stock in Medtronic. Dr. Jakobs reports no disclosures. Dr. Theodore serves as Co-Editor-in-Chief of Epilepsy Research; serves on the editorial boards of Neurotherapeutics and Lancet Neurology; receives honoraria from Elsevier for editing epilepsy research; conducts research based in large part on PET scanning; owns stock in General Electric; and receives research support from NINDS [PI-Intramural Research Program].
Address correspondence and reprint requests to Dr. Phillip L. Pearl, Department of Neurology, Children’s National Medical Center, 111 Michigan Avenue, NW, Washington, DC 20010-2970 ppearl@cnmc.org
Disclosure: Author disclosures are provided at the end of the article.
Received December 18, 2008. Accepted in final form April 22, 2009.
REFERENCES
- 1.Pearl PL, Acosta MT, Gibson KM, et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 2003;60:1413–1417. [DOI] [PubMed] [Google Scholar]
- 2.Gibson KM, Hoffman GF, Hodson AK, et al. 4-Hydroxybutyric acid and the clinical phenotype of succinic semialdehyde dehydrogenase deficiency, an inborn error of GABA metabolism. Neuropediatrics 1998;29:14–22. [DOI] [PubMed] [Google Scholar]
- 3.Pearl PL, Gibson KM, Cortez MA, et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis 2009;32:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ethofer T, Seeger U, Klose U, et al. Proton MR spectroscopy in succinic semialdehyde dehydrogenase deficiency. Neurology 2004;62:1016–1018. [DOI] [PubMed] [Google Scholar]
- 5.Buzzi A, Wu Y, Frantsevaa MV, et al. Succinic semialdehyde dehydrogenase deficiency: GABAB receptor-mediated function. Brain Res 2006;1090:15–22. [DOI] [PubMed] [Google Scholar]
- 6.Wu Y, Buzzi A, Frantseva M, et al. Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABAA receptor-mediated mechanisms. Ann Neurol 2006;59:42–52. [DOI] [PubMed] [Google Scholar]
- 7.Mehta AK, Gould GG, Gupta M, et al. Succinate semialdehyde dehydrogenase deficiency does not down-regulate gamma-hydroxybutyric acid binding sites in the mouse brain. Mol Genet Metab 2006;88:86–89. [DOI] [PubMed] [Google Scholar]
- 8.Koepp MJ, Hand KS, Labbé C, et al. In vivo [11C]flumazenil-PET correlates with ex vivo [3H]flumazenil autoradiography in hippocampal sclerosis. Ann Neurol 1998;43:618–626. [DOI] [PubMed] [Google Scholar]
- 9.Muzik O, da Silva EA, Juhasz C, et al. Intracranial EEG versus flumazenil and glucose PET in children with extratemporal lobe epilepsy. Neurology 2000;5:171–179. [DOI] [PubMed] [Google Scholar]
- 10.Juhasz C, Muzik O, Chugani DC, et al. Prolonged vigabatrin treatment modifies developmental changes of GABA(A)-receptor binding in young children with epilepsy. Epilepsia 2001;42:1320–1326. [DOI] [PubMed] [Google Scholar]
- 11.Atack JR, Smith AJ, Emms F, McKernan RM. Regional differences in the inhibition of mouse in vivo [3H]Ro 15-1788 binding reflect selectivity for a1 versus a2 and a3 subunit-containing GABA A receptors. Neuropsychopharmacology 1999;20:255–262. [DOI] [PubMed] [Google Scholar]
- 12.Gibson KM, Aramaki S, Sweetman L, et al. Stable isotope dilution analysis of 4-hydroxybutyric acid: an accurate method for quantification in physiological fluids and the prenatal diagnosis of 4-hydroxybutyric aciduria. Biomed Environ Mass Spectrom 1990;19:89–93. [DOI] [PubMed] [Google Scholar]
- 13.Gibson KM, Lee CF, Chambliss KL, et al. 4-Hydroxybutyric aciduria: application of a fluorometric assay to the determination of succinic semialdehyde dehydrogenase activity in extracts of cultured human lymphoblasts. Clin Chim Acta 1991;196:219–222. [DOI] [PubMed] [Google Scholar]
- 14.Giovacchini G, Toczek MT, Bonwetsch R, et al. 5-HT1a receptors are reduced in temporal lobe epilepsy after partial volume correction. J Nucl Med 2005;46:1128–1135. [PMC free article] [PubMed] [Google Scholar]
- 15.Andersson JL, Vagnhammar BE, Schneider H. Accurate attenuation correction despite movement during PET imaging. J Nucl Med 1995;36:670–678. [PubMed] [Google Scholar]
- 16.Smith AM, Bruckbauer T, Wiendhard K, Pietrzyk U, Byars LG. Spatial transformation during 3D reconstruction in positron emission tomography. Eur J Nucl Med 1997;24:1413–1417. [DOI] [PubMed] [Google Scholar]
- 17.Wu Y, Carson RE. Noise reduction in the simplified reference tissue model for neuroreceptor functional imaging. J Cereb Blood Flow Metab 2002;22:1440–1452. [DOI] [PubMed] [Google Scholar]
- 18.Correa-Sales C, Rabin BC, Maze M. A hypnotic response to dexmedetomidine, an alpha 2 agonist, is mediated in the locus coeruleus in rats. Anesthesiology 1992;76:948–952. [DOI] [PubMed] [Google Scholar]
- 19.Maze M, Scarfini C, Cavaliere F. New agents for sedation in the intensive care unit. Crit Care Clin 2001;17:881–897. [DOI] [PubMed] [Google Scholar]
- 20.Valtonen P, Haapalinna A, Riekkinen P Sr, Halonen T. Effect of alpha 2-adrenergic drugs dexmedetomidine and atipamezole on extracellular amino acid levels in vivo. Eur J Pharmacol 1995;285:239–246. [DOI] [PubMed] [Google Scholar]
- 21.Duvernoy HM. The Human Brain: Surface, Three-Dimensional Sectional Anatomy and MRI. New York: Springer-Verlag; 1991. [Google Scholar]
- 22.Salonen M, Reid K, Maza M. Synergistic interaction between α2-adrenergic agonists and benzodiazepines in rats. Anesthesiology 1992;76:1004–1011. [DOI] [PubMed] [Google Scholar]
- 23.Chugani DC, Muzik O, Juhasz C, et al. Postnatal maturation of human GABA A receptors measured with positron emission tomography. Ann Neurol 2001;49:618–626. [PubMed] [Google Scholar]
- 24.Fedi M, Berkovic SF, Marini C, et al. A GABA A receptor mutation causing generalized epilepsy reduces benzodiazepine receptor binding. Neuroimage 2006;32:995–1000. [DOI] [PubMed] [Google Scholar]
- 25.Jansen EE, Struys E, Jakobs C, et al. Neurotransmitter alterations in embryonic succinate semialdehyde dehydrogenase (SSADH) deficiency suggest a heightened excitatory state during development. BMC Dev Biol Epub 2008 Nov 28;8:112. [DOI] [PMC free article] [PubMed]
- 26.Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 2001;105:521–532. [DOI] [PubMed] [Google Scholar]
- 27.Delpy A, Allain AE, Meyrand P, Branchereau P. NKCC1 cotransporter inactivation underlies embryonic development of chloride-mediated inhibition in mouse spinal motoneuron. J Physiol 2008;586:1059–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acosta MT, Munasinghe J, Theodore WH, et al. Neuroimaging in a murine model of inherited succinic semialdehyde dehydrogenase (SSADH) deficiency Ann Neurol 2005;58(suppl 9):S96–S96.
- 29.Cortez MA, Wu Y, Gibson KM, Snead OC. Absence seizures in succinic semialdehyde dehydrogenase-deficient mice: a model of juvenile absence epilepsy. Pharmacol Biochem Behav 2004;79:547–553. [DOI] [PubMed] [Google Scholar]
- 30.Lingenhoehl K, Brom R, Heid J, et al. Gamma-hydroxybutyrate is a weak agonist at recombinant GABA(B) receptors. Neuropharmacology 1999;38:1667–1673. [DOI] [PubMed] [Google Scholar]
- 31.Jansen EE, Verhoeven NM, Jakobs C, et al. Increased guanidino species in murine and human succinate semialdehyde dehydrogenase (SSADH) deficiency. Biochim Biophys Acta 2006;1762:494–498. [DOI] [PubMed] [Google Scholar]
- 32.Jansen EE, Gibson KM, Shigematsu Y, et al. A novel, quantitative assay for homocarnosine in cerebrospinal fluid using stable-isotope dilution liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2006;830:196–200. [DOI] [PubMed] [Google Scholar]
- 33.Gupta M, Polinsky M, Senephansiri H, et al. Seizure evolution and amino acid imbalances in murine succinate semialdehyde dehydrogenase (SSADH) deficiency. Neurobiol Dis 2004;16:556–562. [DOI] [PubMed] [Google Scholar]
- 34.Struys EA, Jansen EE, Gibson KM, Jakobs C. Determination of the GABA analogue succinic semialdehyde in urine and cerebrospinal fluid by dinitrophenylhydrazine derivatization and liquid chromatography-tandem mass spectrometry: application to SSADH deficiency. J Inherit Metab Dis 2005;28:913–920. [DOI] [PubMed] [Google Scholar]
- 35.Lamusuo S, Pitkanen A, Jutila L, et al. [11 C]Flumazenil binding in the medial temporal lobe in patients with temporal lobe epilepsy: correlation with hippocampal MR volumetry, T2 relaxometry, and neuropathology. Neurology 2000;54:2252–2260. [DOI] [PubMed] [Google Scholar]
- 36.Koepp MJ, Hammers A, Labbé C, et al. [11C]Flumazenil PET in patients with refractory temporal lobe epilepsy and normal MRI. Neurology 2000;54:332–339. [DOI] [PubMed] [Google Scholar]
- 37.Saronwala A, Tournay A, Gargus JJ. Taurine treatment of succinate semialdehyde dehydrogenase (SSADH) deficiency reverses MRI-documented globus lesions and clinical syndrome. Annual Meeting of the American College of Medical Genetics Proceedings (p 103); March 12–16, 1999; Phoenix, AZ.
- 38.Louzada PR, Lima AC, Mendonca-Silva DL, et al. Taurine prevents the neurotoxicity of beta-amyloid and glutamate receptor agonists: activation of GABA receptors and possible implications for Alzheimer’s disease and other neurological disorders. FASEB J 2004;18:511–518. [DOI] [PubMed] [Google Scholar]
- 39.Sunyer B, Patil S, Frischer C, et al. Strain-dependent effects of SGS742 in the mouse. Behav Brain Res 2007;181:64–75. [DOI] [PubMed] [Google Scholar]