

Abstract
Patterns of polymorphism and divergence in Drosophila protein-coding genes suggest that a considerable fraction of amino acid differences between species can be attributed to positive selection and that genes with sex-biased expression, that is, those expressed predominantly in one sex, have especially high rates of adaptive evolution. Previous studies, however, have been restricted to autosomal sex-biased genes and, thus, do not provide a complete picture of the evolutionary forces acting on sex-biased genes across the genome. To determine the effects of X-linkage on sex-biased gene evolution, we surveyed DNA sequence polymorphism and divergence in 45 X-linked genes, including 17 with male-biased expression, 13 with female-biased expression, and 15 with equal expression in the 2 sexes. Using both single- and multilocus tests for selection, we found evidence for adaptive evolution in both groups of sex-biased genes. The signal of adaptive evolution was particularly strong for X-linked male-biased genes. A comparison with data from 91 autosomal genes revealed a “fast-X” effect, in which the rate of adaptive evolution was greater for X-linked than for autosomal genes. This effect was strongest for male-biased genes but could be seen in the other groups as well. A genome-wide analysis of coding sequence divergence that accounted for sex-biased expression also uncovered a fast-X effect for male-biased and unbiased genes, suggesting that recessive beneficial mutations play an important role in adaptation.
Keywords: positive selection, gene expression, sex linkage, fast-X effect, Drosophila
Introduction
Genes that differ in expression between males and females, known as sex-biased genes, can provide insight into a number of important issues in genome evolution. This is because they may be subject to differing selective constraints depending on the sex in which they are expressed or they may experience conflicting selective pressures in males and females (reviewed by Ellegren and Parsch 2007). In Drosophila, male-biased genes, especially those expressed in reproductive tissues, show consistently high levels of adaptive protein evolution (Pröschel et al. 2006), as well as elevated levels of amino acid divergence between species (Zhang et al. 2004; Haerty et al. 2007; Zhang et al. 2007; but see Metta et al. 2006 and Dorus et al. 2006 for exceptions). Female-biased genes show a signal of adaptive evolution that is weaker and less consistent than that of male-biased genes, but stronger than that of genes expressed equally in the 2 sexes (unbiased genes; Pröschel et al. 2006). Previous studies that have used combined polymorphism and divergence data to infer the type and strength of selection acting on sex-biased genes have thus far been limited to autosomal genes. Because males and females differ in ploidy of the X chromosome, a similar analysis of X-linked sex-biased genes may prove valuable for uncovering evolutionary differences between the X chromosome and the autosomes (reviewed by Vicoso and Charlesworth 2006).
Microarray studies of Drosophila species have revealed an unequal distribution of sex-biased genes between the X chromosome and autosomes, with male-biased genes greatly underrepresented and female-biased genes slightly overrepresented on the X (Parisi et al. 2003; Ranz et al. 2003; Sturgill et al. 2007). Because theory predicts that X-linked genes may respond differently to sexual antagonism, depending on the dominance of the antagonistic effects (Rice 1984; Charlesworth et al. 1987), these observations have led some authors to propose that the uneven distribution of sex-biased genes may be the result of such genetic conflict (Parisi et al. 2003; Ranz et al. 2003; Connallon and Knowles 2005; but see Rogers et al. 2003 and Vicoso and Charlesworth 2006). The evolution of genes residing on the X chromosome may also be influenced by the so-called “fast-X” effect, which leads to a higher rate of adaptive substitution at X-linked loci if new beneficial mutations are, on average, recessive (Charlesworth et al. 1987; Orr and Betancourt 2001). This effect may be especially relevant to male-biased genes, as they show high rates of adaptive evolution and are primarily subject to selection in the heterogametic sex (Vicoso and Charlesworth 2006; Ellegren and Parsch 2007).
A number of recent studies have provided evidence for the faster evolution of X-linked genes in mammals (Torgerson and Singh 2003; Wang and Zhang 2004; Khaitovich et al. 2005; Lu and Wu 2005; Nielsen et al. 2005; Torgerson and Singh 2006; Baines and Harr 2007) and Z-linked genes in birds (Mank et al. 2007). However, similar studies in Drosophila have produced mixed results. These studies adopted several different strategies to test for a fast-X effect, including 1) comparing large numbers of X-linked and autosomal genes (Betancourt et al. 2002; Richards et al. 2005; Musters et al. 2006; Connallon 2007), 2) comparing X-linked and autosomal duplicate genes (Thornton and Long 2002, 2005), and 3) comparing orthologs that differ in chromosomal location between lineages due to translocation (Counterman et al. 2004; Thornton et al. 2006). Although some of the above studies found evidence for a fast-X effect (Thornton and Long 2002, 2005; Counterman et al. 2004; Musters et al. 2006), 2 recent and extensive analyses found no evidence for faster-X evolution (Thornton et al. 2006; Connallon 2007). However, these studies either lacked polymorphism data necessary for estimating the rate of adaptive evolution (Thornton et al. 2006) or had limited data for sex-biased genes (Connallon 2007). A genome-wide analysis of polymorphism and divergence in Drosophila simulans revealed a general increase in the evolutionary rate of X-linked loci (including noncoding sequences), although this effect could not be attributed to an increased rate of adaptive evolution (Begun et al. 2007). Comparative analysis of 12 Drosophila genome sequences using codon-based substitution models uncovered a marginally significant excess of positively selected genes on the X chromosome (Drosophila 12 Genomes Consortium 2007), although this was not consistent over different lineages of the Drosophila phylogeny (Singh et al. 2007).
Here we analyze polymorphism and divergence in a set of 45 X-linked genes that were specifically chosen on the basis of their relative expression level in the 2 sexes. This includes 17 male-biased genes, 13 female-biased genes, and 15 unbiased genes. Genes and population samples were selected to be directly comparable to previously published data from 91 autosomal sex-biased genes (Pröschel et al. 2006). Overall, we detect a significant signal of positive selection in the X-linked sex-biased genes, with the strongest and most consistent signal in the male-biased genes. This matches the pattern seen for autosomal genes. Additionally, we find evidence for increased adaptive evolution of X-linked genes, which is consistent with a fast-X effect. Comparative genomic analysis of Drosophila melanogaster, D. simulans, and Drosophila yakuba also reveals a strong fast-X effect for male-biased genes and a weak, but significant, fast-X effect for unbiased genes. These results suggest the frequent occurrence of recessive beneficial mutations.
Materials and Methods
Gene Selection
Sex-biased and unbiased genes were chosen following the criteria of Pröschel et al. (2006), with the additional requirement that all genes be located on the X chromosome. Briefly, the combined expression data of 3 independent microarray experiments performed on D. melanogaster (Parisi et al. 2003; Ranz et al. 2003; Gibson et al. 2004) was used to define a high-quality consensus set of sex-biased genes (Gnad and Parsch 2006). Male-biased genes were required to have an average male/female expression ratio of at least 2 (mean = 6.0), whereas female-biased genes were required to have a male/female expression ratio <0.5 (mean = 0.36). Unbiased genes were required to have a male/female expression ratio between 0.5 and 2.0 but were generally selected to have a ratio very near 1.0 (mean = 1.01). A complete list of genes is given in supplementary table S1 (Supplemental Material online).
Because one of the microarray data sets used for gene selection compared male and female reproductive tissues (Parisi et al. 2003), we expect many of our genes to be expressed in the gonads. Using the tissue-specific expression data of FlyAtlas (Chintapalli et al. 2007), we find that 16 of the 17 X-linked male-biased genes have enriched expression in testis. The remaining gene (CG1503) shows enrichment in accessory gland, as well as in other tissues (brain, crop, midgut). On average, the degree of testis enrichment for the 17 genes was 11-fold in comparison to the whole fly. Similarly, we found that all 33 of the autosomal male-biased genes from Pröschel et al. (2006) were expressed in testis, with an average enrichment of 10-fold. For the X-linked female-biased genes, 12 out of 13 showed enriched expression in ovary (average = 1.9-fold), whereas the remaining gene (CG3004) showed enrichment in tubule, midgut, and hindgut. All 28 of the autosomal female-biased genes from Pröschel et al. (2006) showed ovary enrichment (average = 2.2-fold). In general, the male-biased genes showed greater gonad enrichment and greater sex bias in their expression than the female-biased genes, which is consistent with patterns reported for the whole genome (Gibson et al. 2004; Parisi et al. 2004).
In order to minimize the effects of other factors known to influence rates of evolution, such as coding sequence length, intron length, or recombination rate (Comeron and Kreitman 2002; Comeron and Guthrie 2005; Presgraves 2005; Zhang and Parsch 2005; Haddrill et al. 2007), genes were selected to fall within a relatively narrow length distribution, have similar intron/exon structures, and experience similar levels of recombination. The average gene lengths in base pairs (standard deviations) for male-, female-, and unbiased genes were 896 (204, n = 17), 968 (259, n = 13), and 837 (232, n = 15), respectively. The average recombination rates (standard deviations), in terms of the measure R (Hey and Kliman 2002) were 3.36 (1.37), 3.34 (1.24), and 2.96 (1.41) for male-, female-, and unbiased genes, respectively.
Aside from the male/female expression ratio, no other functional information was used in gene selection. The vast majority (89%) were unnamed genes known only by their annotation numbers, and only 24% were associated with a Gene Ontology molecular function. None of the genes was previously known to have undergone adaptive evolution, and none belonged to a functional group known to experience frequent adaptive evolution, such as accessory protein genes or immune response genes (Swanson et al. 2001; Schlenke and Begun 2003). Furthermore, interspecific divergence was not considered during gene selection. Thus, we expect that our gene set is a random sample of X-linked genes of each sex-biased expression class.
Fly Strains, Polymerase Chain Reaction, and Sequencing
For the polymorphism survey, we used 12 highly inbred D. melanogaster strains from Lake Kariba, Zimbabwe. These same strains were used in previous genome-wide polymorphism surveys (Glinka et al. 2003; Ometto et al. 2005; Pröschel et al. 2006), which allows us to directly compare results between studies. The D. melanogaster genome (release 4.0; http://www.flybase.org) was used to design polymerase chain reaction (PCR) primers flanking the coding sequence of each target gene. A complete list of the PCR primers, as well as the cycling conditions used for each gene, is provided in supplementary table S2 (Supplemental Material online). When possible, the same primers were used to amplify the orthologous gene from a highly inbred strain of D. simulans from Chapel Hill, NC (Meiklejohn et al. 2004). Following PCR, the amplified products were purified with ExoSAP-IT (USB, Cleveland, OH) and sequenced from both strands using BigDye chemistry and a 3730 automated sequencer (Applied Biosystems, Foster City, CA). The PCR primers were used as sequencing primers. When necessary, additional internal sequencing primers were used (see supplementary table S2, Supplemental Material online). For some genes, we were unable to get successful PCR or DNA sequence from all 12 D. melanogaster strains. The average number of strains sequenced per gene was 11 (see supplementary table S1, Supplemental Material online). For 29 genes, we were unable to obtain a PCR product from D. simulans. In these cases, we used the sequence from the D. simulans genome project (Washington University School of Medicine Genome Sequencing Center) downloaded from the UCSC Genome Browser (http://genome-test.cse.ucsc.edu/). For all genes, the D. yakuba sequence was downloaded from the above source. All new DNA sequences have been submitted to the GenBank/EMBL databases under the accession numbers AM998825–AM999334.
Analysis of Polymorphism and Divergence
Basic polymorphism and divergence statistics were calculated using DnaSP 4 (Rozas et al. 2003). For McDonald-Kreitman (MK) table data, we used the number of segregating mutations (instead of the number of segregating sites) because some genes had sites with 3 segregating variants. In these cases, the frequency of each mutation was considered separately for calculation of Tajima's D and the identification of singleton polymorphisms. For divergence, we included only sites showing a fixed difference between D. melanogaster and D. simulans. Multilocus Tajima's D tests were performed using the HKA program, which was kindly provided by J. Hey. To calculate the fraction of positively selected amino acid substitutions, α, with the method of Bierne and Eyre-Walker (2004), we used the DoFE program, which was kindly provided by A. Eyre-Walker. The method of Bustamante et al. (2002) for estimating the posterior distribution of the selection parameter, γ, was implemented through the MKPRF web server (http://cbsuapps.tc.cornell.edu/mkprf.aspx).
To estimate the fitness effects of the complete mutational distribution for each gene or group of genes, we modified the method of Sawyer et al. (2003, 2007) to include a third, outgroup species (D. yakuba). The population pedigree of the 3 species was parameterized by 2 time parameters, t1 for the distance from the present to the common ancestor of D. melanogaster and D. simulans and t2 for the distance between that common ancestor and the common ancestor of the 3 species. The times t1 and t2 are measured in generations scaled by the within-species haploid effective population size, which was assumed to be the same for the 3 species. In particular, the scaled evolutionary distance in the pedigree between the present and the root is t1 + t2 and the distance between the common ancestor of D. melanogaster and D. simulans, and D. yakuba is 2t2 + t1.
An extended 2 × 3 MK table (also known as a DPRS table) was calculated for each of the 45 loci, with rows of the tables corresponding to nonsynonymous (R) and synonymous (S) codon positions, respectively. The first column entries are the numbers of codon positions that are polymorphic within the D. melanogaster sequences, with adjustments for codon positions that show evidence for more than 1 mutation since the common ancestor of the 3 species. Specifically, a codon position with K > 2 segregating codons was counted as K − 1 > 1 polymorphisms instead of 1. Codon positions with missing data in any sequence were disregarded in the analysis.
The second column in the DPRS table is the number of “fixed difference” codon positions between D. simulans and D. melanogaster at that locus, which was computed as the number of codon positions at which the single D. simulans codon differs from all segregating codons in the D. melanogaster sample at that codon position. Similarly, the third DPRS column is the number of codon positions at which the D. yakuba codon differs from the D. melanogaster and D. simulans samples combined. In particular, the first 2 columns are determined entirely by the D. melanogaster and D. simulans sequences, although the counts are influenced by the need for a common alignment with the D. yakuba sequence.
The third-column DPRS counts are the result of mutations in a path of length 2t2 + t1 in the population pedigree that does not overlap with the pedigree of D. melanogaster and D. simulans. It follows from the basic model assumptions (Sawyer and Hartl 1992) that the third-column counts are independent of the first 2 columns, and from this that the DPRS counts are independent Poisson random variables with means given by model parameters. The 45 2 × 3 DPRS tables derived from the locus alignments were analyzed using a Markov Chain Monte Carlo method similar to that of Sawyer et al. (2007) for 2 × 2 DPRS tables. While the scaled synonymous and nonsynonymous mutation rates θs and θr for each locus are assumed constant on the 3-species pedigree, the only additional parameter is the second divergence time t2. As in previous models (Bustamante et al. 2002; Sawyer et al. 2003, 2007), the ratios θr/θs are not constant across loci because nonsynonymous mutations that result in strongly deleterious protein products are effectively censored in the diffusion time scale, which results in smaller estimated values of θr.
See Templeton (1996) for other examples of the use of MK-like tables of size larger than 2 × 2.
Comparative Genomic Analysis
Genome-wide comparisons of nonsynonymous/synonymous substitution rates (dN/dS) of male-, female-, and unbiased genes were performed using the high-quality 2-fold sex-biased gene set and dN/dS values downloaded from the Sebida database (Gnad and Parsch 2006; http://www.sebida.de). Genes located on chromosome 4 and those not mapped to chromosomal locations were excluded from the analysis. In addition, we required that the alignment of each gene between D. melanogaster and D. simulans (or D. yakuba) span at least 80% of the codons in the D. melanogaster protein-coding sequence. The higher quality of the D. yakuba sequence assembly (Drosophila 12 Genomes Consortium 2007) resulted in a greater number of aligned genes between D. melanogaster and D. yakuba than between D. melanogaster and D. simulans. Repeating the above analysis using a sex-biased gene set defined by a statistical cutoff (false discovery rate of 10%; Gnad and Parsch 2006) instead of a fold-change cutoff produced nearly identical results, which are not shown. Similarly, varying the percentage of aligned codons required between species from 50% to 100% had negligible effect on the results (not shown).
Results
MK Tests
In total, we collected DNA sequence polymorphism and divergence data for 45 X-linked protein-coding genes, including 17 with male-biased, 13 with female-biased, and 15 with unbiased expression. To evaluate the type of selection operating on individual genes, we applied single-locus MK (McDonald and Kreitman 1991) tests (table 1). The highest proportion of significant MK tests was found for male-biased genes (7/17), all of which deviated in the direction of positive selection (i.e., a relative excess of nonsynonymous divergence; table 2). Female-biased and unbiased genes displayed lower proportions of significant deviations from neutrality (3/13 and 3/15, respectively) and were less consistent in the direction of their departure (2 and 1 departed in the direction of positive selection, respectively; table 2). The 3 genes with significant MK tests that were inconsistent with positive selection (i.e., those with a relative excess of nonsynonymous polymorphism) appear to be cases of weak purifying selection (see below). Application of the MK test to the summed values of polymorphism and divergence within each class revealed a significant departure from neutrality in the direction of positive selection for both male- and female-biased genes, whereas the unbiased genes did not differ from the neutral expectation (table 1). Although the use of summed MK tables can potentially lead to a false signal of positive selection (Shapiro et al. 2007), this does not appear to be the case for our data, as the individual MK tables for male- and female-biased genes show a consistent trend toward a relative excess of nonsynonymous divergence (see supplementary table S1, Supplemental Materials online).
Table 1.
Summary of Polymorphism and Divergence Statistics
| Bias | Number of Genes | Significant MK Testsa | Positive Selectionb | Dsc | Psd | Dne | Pnf | P Valueg |
| Male | 17 | 7 | 7 | 464 | 235 | 645 | 96 | <0.0001 |
| Female | 13 | 3 | 2 | 256 | 129 | 185 | 44 | 0.0001 |
| Unbiased | 15 | 3 | 1 | 227 | 182 | 129 | 96 | 0.66 |
MK tests were performed for each gene and considered significant if P < 0.05.
Genes with significant MK tests showing a relative excess of nonsynonymous fixed differences.
The total number of synonymous fixed differences.
The total number of synonymous polymorphisms.
The total number of nonsynonymous fixed differences.
The total number of nonsynonymous polymorphisms.
The P value of the summed data as determined by a χ2 test.
Table 2.
Genes with Significant MK Tests
| Gene | Bias | Ds | Ps | Dn | Pn | P Valuea | Positive Selection? | TDsynb | TDnonc |
| CG1314 | Male | 33 | 12 | 80 | 4 | 0.001 | Yes | −1.30 | −0.82 |
| CG12681 | Male | 47 | 5 | 120 | 3 | 0.049 | Yes | 0.98 | 0.60 |
| CG1950 | Male | 33 | 20 | 64 | 15 | 0.018 | Yes | 0.12 | −0.80 |
| CG5662 | Male | 15 | 23 | 20 | 8 | 0.013 | Yes | −0.01 | 0.23 |
| CG5334 | Male | 21 | 5 | 63 | 3 | 0.034 | Yes | 0.25 | −1.61 |
| CG18341 | Male | 26 | 22 | 18 | 2 | 0.003 | Yes | −0.08 | −1.42 |
| CG10920 | Male | 43 | 24 | 56 | 11 | 0.010 | Yes | −0.06 | −0.27 |
| CG9125 | Female | 34 | 11 | 62 | 5 | 0.013 | Yes | −0.70 | 0.52 |
| CG3004 | Female | 23 | 18 | 7 | 0 | 0.036 | Yes | −0.43 | — |
| CG1749 | Female | 26 | 5 | 4 | 9 | 0.001 | No | −0.94 | −0.62 |
| CG1751 | Unbiased | 10 | 1 | 1 | 2 | 0.045 | No | −1.14 | −1.45 |
| CG14772 | Unbiased | 10 | 2 | 3 | 5 | 0.033 | No | −1.46 | −1.12 |
| CG9723 | Unbiased | 36 | 18 | 61 | 5 | 0.001 | Yes | −0.46 | −1.01 |
Symbols are the same as in table 1.
P value was determined by G-test when applicable, otherwise by Fisher's exact test.
Tajima's D for synonymous sites.
Tajima's D for nonsynonymous sites.
Estimation of the Proportion of Adaptive Amino Acid Replacements, α
The maximum likelihood method of Bierne and Eyre-Walker (2004) extends the MK test to multiple loci to estimate the fraction of amino acid replacements between species that were fixed by positive selection (α). In the case of neutral evolution, α is expected to be 0. A positive value of α indicates positive selection, whereas a negative value can indicate either balancing or weak purifying selection. For male-biased genes, the estimated α was 62% and was significantly greater than 0 (fig. 1A). The estimated α for female-biased genes was 33% but did not differ significantly from 0. In the case of unbiased genes, the estimated α was negative (fig. 1A). This appears to be the result of weak purifying selection, which allows slightly deleterious nonsynonymous mutations to persist at low frequency in a population but prevents them from reaching fixation. This interpretation is supported by the overall low frequency of nonsynonymous polymorphisms, as measured by Tajima's (1989) D statistic (table 3). The average Tajima's D for nonsynonymous sites was negative and consistently lower than the corresponding average for synonymous sites in all 3 groups of genes (table 3). This suggests that all the above α values may be underestimates. In an effort to reduce this effect, we repeated the analysis after removing low-frequency (singleton) polymorphisms at both synonymous and nonsynonymous sites. This led to higher estimates of α (76%, 31%, and 30% for male-, female-, and unbiased genes, respectively), but again only male-biased genes showed significant evidence of positive selection (fig. 1B).
FIG. 1.—
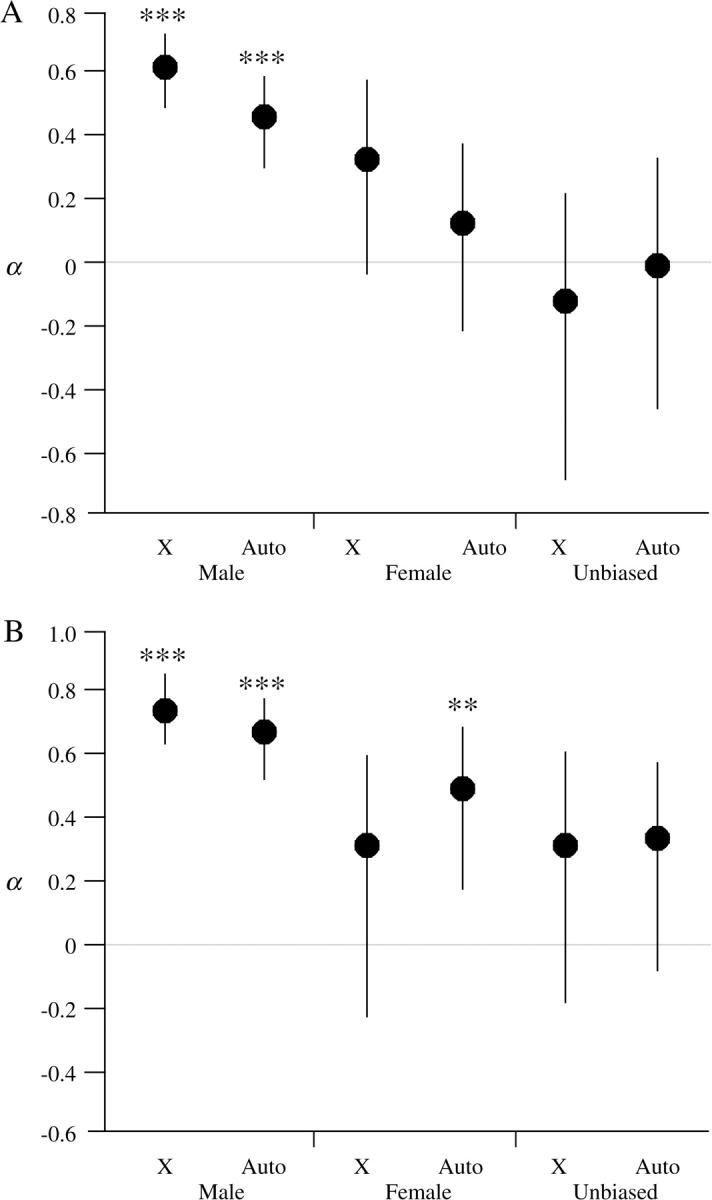
Maximum likelihood estimates of the fraction of positively selected amino acid replacements (α) for X-linked and autosomal genes with male-, female-, or unbiased expression using the method of Bierne and Eyre-Walker (2004). (A) Estimates of α using all polymorphic sites. (B) Estimates of α after removal of low-frequency (singleton) polymorphisms. Error bars indicate 95% CI. Asterisks indicate genes with a significant signal of positive selection. **P < 0.01, ***P < 0.001.
Table 3.
Average Values of Tajima's D
| Bias | Synonymous | Nonsynonymous |
| Male | −0.13 (0.368) | −0.61 (0.013) |
| Female | −0.29 (0.214) | −0.46 (0.091) |
| Unbiased | −0.54 (0.029) | −0.77 (0.002) |
P values (in parentheses) were determined as the proportion of 1000 simulations giving a D value lower than or equal to the observed.
For comparison, figure 1 also shows the α estimates for autosomal genes of the 3 expression categories (Pröschel et al. 2006). Overall, the X-linked and autosomal genes showed a similar pattern, with the strongest signal of positive selection in the male-biased genes. In addition, the male-biased genes showed a consistent difference between the X chromosome and autosomes, with a higher α for X-linked genes. However, in all cases there was considerable overlap in the 95% confidence intervals (CIs) of α for X-linked and autosomal genes (fig. 1).
Estimation of the Selection Parameter, γ
The Bayesian analysis method of Bustamante et al. (2002) uses MK table data to estimate the selection parameter γ = 2Nes (where Ne is the effective population size and s is the selection coefficient) for amino acid replacements in a group of genes, under the assumption that γ is normally distributed among genes. In the case of neutral evolution, γ is expected to be 0. A positive γ indicates positive selection, whereas a negative γ can indicate either balancing or weak purifying selection. Application of this method to our data produced γ estimates of 4.7 and 2.5 for male- and female-biased genes, respectively (fig. 2A). In both cases, the proportion of the distribution falling below 0 was less than 1% [P(γ ≤ 0) = 0.0001 and P(γ ≤ 0) = 0.0095, for male- and female-biased genes, respectively], indicating positive selection favoring amino acid replacements. In contrast, the mean γ for unbiased genes was −0.8 [P(γ ≤ 0) = 0.92], indicating weak purifying selection against nonsynonymous mutations. After repeating the analysis with singleton polymorphisms removed as above, the estimates for male-, female- and unbiased genes were 7.4, 2.1, and 0.5 [P(γ ≤ 0) < 0.0001, P(γ ≤ 0) = 0.0019, and P(γ ≤ 0) = 0.19, respectively; fig. 2B].
FIG. 2.—
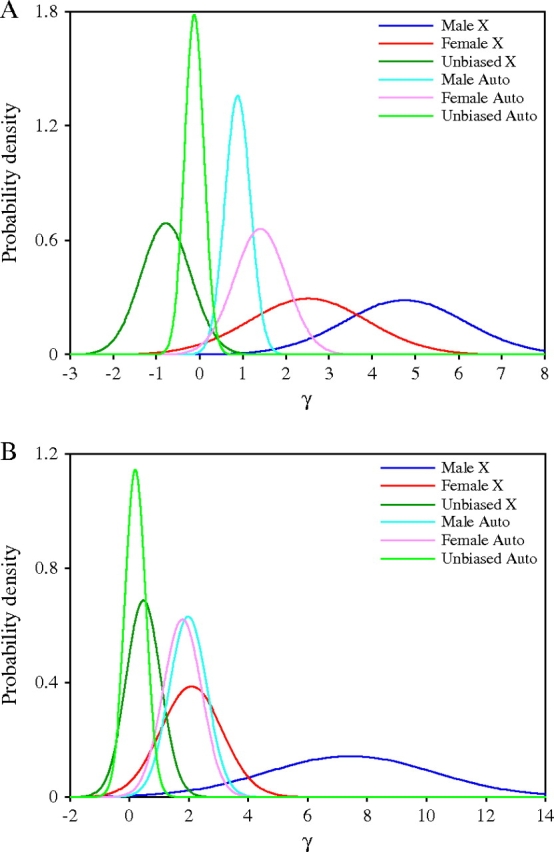
Posterior distribution of the selection parameter γ for X-linked and autosomal genes with male-, female-, or unbiased expression as determined by the method of Bustamante et al. (2002). (A) Distribution of γ using all polymorphic sites. (B) Distribution of γ after removal of low-frequency (singleton) polymorphisms.
Figure 2 also shows the γ distributions of the autosomal genes surveyed by Pröschel et al. (2006). A large difference between X-linked and autosomal male-biased genes is evident, with the average γ of the X-linked genes being 5-fold higher than that of the autosomal genes [P(X ≤ Auto) < 0.0001]. Interestingly, after correcting for weak purifying selection, both the X-linked and autosomal unbiased genes displayed similar distributions centered on 0, whereas X-linked and autosomal female-biased and autosomal male-biased genes displayed similar distributions centered on γ ∼ 2 (fig. 2B). The X-linked male-biased genes had by far the highest mean γ, which was 3-fold greater than that of the autosomal male-biased genes [P(X ≤ Auto) < 0.0001].
Joint Estimation of α and γ
Sawyer et al. (2003, 2007) extended the approach of Bustamante et al. (2002) to estimate the distribution of selection parameters associated with nonsynonymous mutations within and among genes. The underlying model assumes that the selective effects of nonsynonymous mutations are normally distributed within genes but that their mean may vary among genes. There are several advantages to this approach. First, it allows the estimation of the selection parameter (γ) for several classes of nonsynonymous mutations, including those newly arising in a population, those currently segregating in a population, and those fixed between species. Second, because the distribution of γ for all nonsynonymous fixed differences is inferred, it is possible to estimate the proportion of positively selected amino acid replacements (α) using different definitions of positive selection (e.g., γ > 0, γ > 2). Finally, because the selective effects of segregating polymorphisms are estimated (including those that are slightly deleterious), it is not necessary to remove low-frequency polymorphisms from the data.
We modified the approach of Sawyer et al. (2007) to include a single sequence of each gene from a third species (D. yakuba) that serves as an outgroup to D. melanogaster and D. simulans and allows for better estimation of the selective constraint on each gene over the 2 in-group lineages. Application of this method to the 45 X-linked genes revealed several differences between sex-biased and unbiased genes. Both male- and female-biased genes had a significantly higher proportion of positively selected new and segregating mutations, as well as fixed differences between species, than unbiased genes (table 4). In all of the above cases, male-biased genes had a higher proportion of positively selection mutations than female-biased genes, although none of these comparisons was significant. Consistent with previous findings for autosomal genes (Sawyer et al. 2007), a strict definition of positive selection (γ > 0) produced α estimates of 95% or higher for all groups of genes. This value drops fairly rapidly as the cutoff γ value for positive selection increases, making the differences among male-, female-, and unbiased genes more apparent (fig. 3) and suggesting that most amino acid replacements are weakly beneficial.
Table 4.
Summary of the Estimated Mutational Distribution
| Feature | Male | Female | Unbiased |
| Mean γ of estimated mutational distribution | −14.24** | −17.14 | −22.35 |
| Proportion of new mutations with γ > 0 | 0.105*** | 0.067* | 0.028 |
| Proportion of sample polymorphisms with γ > 0 | 0.466*** | 0.380** | 0.216 |
| Proportion of fixed differences with γ > 0 | 0.982*** | 0.978** | 0.968 |
| Proportion of fixed differences with γ > 2 | 0.906*** | 0.887** | 0.849 |
| Proportion of fixed differences with γ > 8 | 0.447*** | 0.397** | 0.310 |
| Proportion of fixed differences with γ > 12 | 0.214*** | 0.174** | 0.117 |
| Mean γ of fixed differences | 8.15*** | 7.48** | 6.42 |
| Proportion of “neutral” fixed differences (−2 < γ < 2) | 0.092*** | 0.111** | 0.148 |
Asterisks indicate values differing significantly from unbiased genes, as determined from 10,000 random permutations of the data. *P < 0.05, **P < 0.01, and ***P < 0.001.
FIG. 3.—

Distribution of fitness effects for amino acid replacements in X-linked and autosomal genes with male-, female-, or unbiased expression as determined by a modified version of the method of Sawyer et al. (2007). The y axis shows the fraction of amino acid replacements with selection parameter γ greater than the value given on the x axis.
Application of the above method to the 91 autosomal genes of Pröschel et al. (2006) gave a result similar to that of the X-linked genes with respect to sex-biased and unbiased genes, although in all cases the proportion of positively selected amino acid replacements was lower in the autosomal genes (fig. 3). To investigate this further, we calculated the rate of adaptive substitution for each group of genes, which we defined as the number of positively selected amino acid replacements per 1000 nonsynonymous sites. This measure takes into account not only the proportion of amino acid replacements that are adaptive but also the total number of replacements that have occurred between species. Figure 4 shows the X:autosome ratio of adaptive substitution for male-, female-, and unbiased genes. In all cases, the ratio is greater than 1, indicating an increased rate of adaptive evolution for X-linked genes (i.e., a fast-X effect). The largest effect is seen for male-biased genes, whereas the smallest is seen for female-biased genes.
FIG. 4.—

Ratio of the adaptive substitution rate of X-linked to autosomal genes with male-, female-, or unbiased expression. The rate of adaptive substitution was defined as the number of positively selected amino acid replacements per 1000 nonsynonymous sites. Three different γ cutoffs used to define positive selection are shown on the x axis.
Whole-Genome Comparisons
The above analyses indicate that X-linked genes, especially those with male-biased expression, experience greater rates of adaptive evolution than their autosomal counterparts. To see if this difference is reflected in overall sequence divergence between species, we examined the ratio of nonsynonymous to synonymous divergence (dN/dS) in male-, female-, and unbiased genes for whole-genome comparisons of D. melanogaster and either D. simulans or D. yakuba (fig. 5). In general, male-biased genes were significantly more divergent between species than either female- or unbiased genes (Mann–Whitney U test, P < 0.002 for all comparisons). Furthermore, X-linked male-biased genes had significantly higher dN/dS than autosomal male-biased genes, which is consistent with a greater rate of adaptive amino acid sequence replacement in the X-linked genes. There was also a small, but significant, increase in dN/dS for X-linked relative to autosomal unbiased genes. There was no difference in dN/dS between X-linked and autosomal female-biased genes. These results are in qualitative agreement with the X:autosome ratios of adaptive substitution rates inferred from the polymorphism and divergence data (fig. 4).
FIG. 5.—
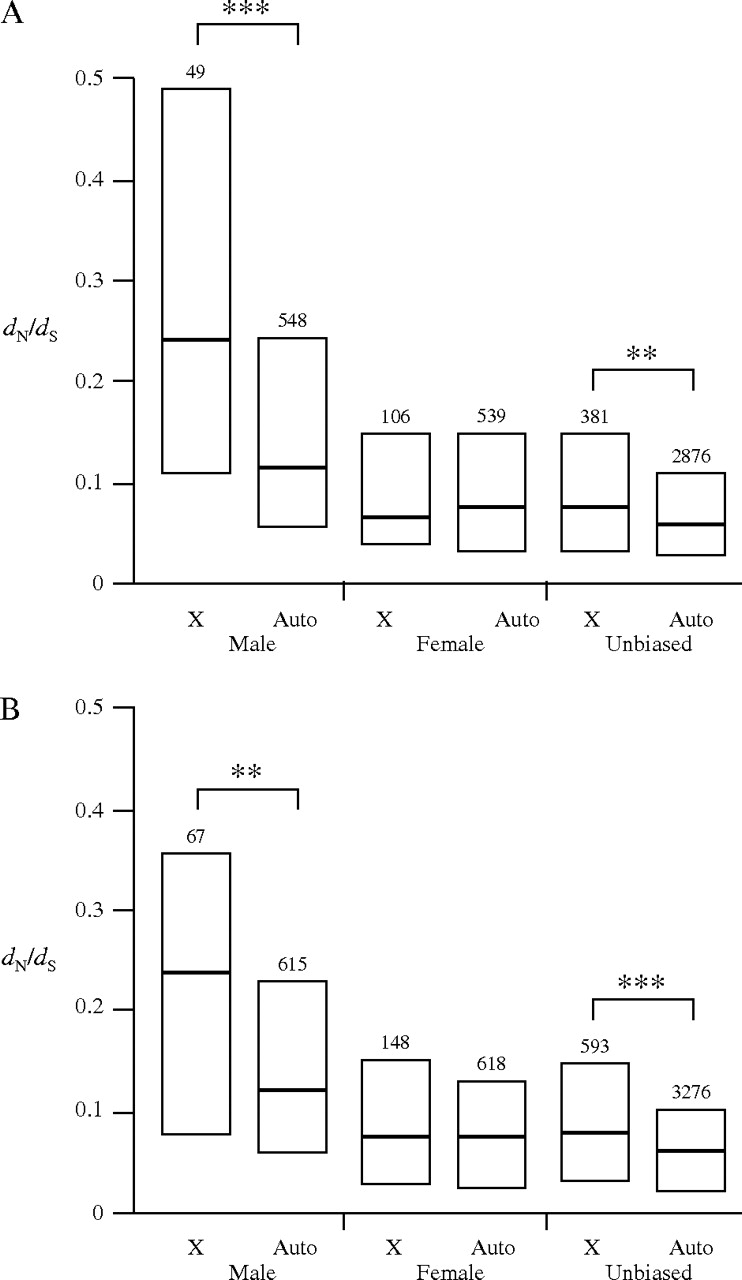
Evolutionary rates of X-linked and autosomal genes with male-, female-, or unbiased expression for whole-genome comparisons of (A) Drosophila melanogaster and Drosophila simulans or (B) D. melanogaster and Drosophila yakuba. Boxes represent the interquartile range, and heavy lines indicate the median. The number of genes in each class is given above the box. Brackets indicate significant differences between X-linked and autosomal genes within a given expression class. **P < 0.01, ***P < 0.001.
Discussion
Selection on X-linked Sex-Biased Genes
Our analysis of X-linked protein-coding genes by individual MK tests and the multilocus tests of Bierne and Eyre-Walker (2004) and Bustamante et al. (2002) revealed a strong and significant signal of positive selection in male-biased genes. Female-biased genes showed a weaker signal of positive selection that was significant by the test of Bustamante et al. (2002) and marginally significant by the test of Bierne and Eyre-Walker (2004). Unbiased genes showed little or no evidence for adaptive evolution by the above tests. These results match those reported previously for a similarly selected set of autosomal sex-biased genes (Pröschel et al. 2006). Taken together, this demonstrates that regardless of sex linkage, male-biased genes experience the greatest rate of adaptive protein evolution, followed by female-biased genes, and then unbiased genes. Although male-biased genes are significantly underrepresented on the X chromosome, those that remain X-linked do not appear to be at a disadvantage with respect to their opportunities for adaptive evolution. Instead, X-linked male-biased genes appear to experience more adaptive evolution than their autosomal counterparts (see below).
Analysis of the polymorphism and divergence data using a modified version of the method of Sawyer et al. (2007) provided a more detailed picture of the distribution of mutational effects. We find that in all 3 groups of genes, over 95% of the amino acid differences fixed between species can be attributed to positive selection if one uses the strict definition of γ > 0. However, the selective advantage of these amino acid replacements is typically quite small, rarely exceeding γ > 20 (fig. 3). Thus, the vast majority of these replacements could be considered “nearly neutral.” This result is consistent with a previous analysis of autosomal genes, which found a large excess of positively selected changes among nearly neutral amino acid replacements in Drosophila (Sawyer et al. 2007). However, we do observe some differences in the distributions of the fitness effects of amino acid replacements related to differences in sex-biased expression. For example, under a more conservative definition of positive selection (e.g., γ > 8 or γ > 12), male-biased genes clearly show the highest rate of adaptive evolution, followed by female-biased genes, and unbiased genes (table 4 and fig. 3). These results are consistent with those produced the methods of Bierne and Eyre-Walker (2004) and Bustamante et al. (2002) and suggest that an advantage of the Sawyer et al. (2007) method (and its extension presented here) is its ability to distinguish weakly selected amino acid replacements.
Also consistent with Sawyer et al. (2007), we find that the majority (around 70%) of nonsynonymous mutations segregating within a population are deleterious, although this differs significantly among the different sex-bias classes. Male-biased genes had the lowest frequency of deleterious segregating amino acid mutations (γ < 0), whereas unbiased genes had the highest (table 4). Conversely, male-biased genes had the highest frequency of positively selected segregating amino acid mutations (γ > 0), whereas unbiased genes had the lowest.
Faster-X Evolution
Comparison of polymorphism and divergence in 45 X-linked genes with that in 91 similarly chosen autosomal genes revealed evidence for a fast-X effect, which was particularly strong for male-biased genes. For example, 41% (7 out of 17) of the X-linked male-biased genes gave a significant MK test that was consistent with the action of recurrent positive selection, whereas only 21% (7 out of 33) of the autosomal male-biased genes did so. Similar results were obtained from the multilocus implementations of the MK test, with X-linked male-biased genes consistently showing the strongest signal of positive selection (figs. 1–3). Two main factors are likely to contribute to the increased size of the fast-X effect in male-biased genes. First, male-biased genes show the overall highest rates of adaptive evolution. Thus, there are more opportunities for X-linked or autosomal mutations to become fixed through the action of positive selection, which would make differences in the adaptive substitution rate between the X chromosome and the autosomes easier to detect. Second, male-biased genes primarily experience selection while in the heterogametic sex. As an example, consider a gene that is expressed exclusively in males. If new beneficial mutations tend to be recessive, then those occurring in an X-linked gene will have a higher fixation probability than those occurring in an autosomal gene because the X-linked mutations always experience selection in a hemizygous state. In contrast, a mutation occurring in an X-linked gene expressed exclusively in females will never be exposed to selection in a hemizygous state. Thus, new beneficial mutations that are recessive will always be hidden from selection until they drift to high enough frequency to be present in homozygotes. Consistent with this expectation, our results indicate that the female-biased genes show the weakest fast-X effect (fig. 4). The presence of a weak fast-X effect for female-biased genes (fig. 4) may be explained by the fact that these genes do not necessarily have female-exclusive expression. Thus, they may experience some selection while in the male genetic background. It is also possible that the fixation of dominant mutations that are beneficial to females, but harmful to males, contributes to the increased rate of adaptive evolution of X-linked, female-biased genes (Rice 1984; Charlesworth et al. 1987).
The fast-X effect should be strongest for genes with male-specific expression. Thus, one might expect a positive correlation between the male/female expression ratio and the rate of adaptive evolution of X-linked genes. Indeed, for the X-linked male-biased genes, we detect a significantly positive correlation between the male/female expression ratio and the selection parameter of amino acid replacements, γ (Pearson's R = 0.55, P = 0.02). There is also a positive correlation between the male/female expression ratio and dN/dS, although this is not significant (R = 0.12, P = 0.65). For the autosomal male-biased genes, correlations between the male/female expression ratio and either γ (R = 0.12, P = 0.53) or dN/dS (R = −0.03, P = 0.85) are not significant. The increased rate of adaptive evolution detected for the X-linked genes is not due to a greater male bias in their expression relative to the autosomal genes. On average, the autosomal genes show stronger male-biased expression than the X-linked genes (15-fold vs. 6-fold).
Microarray data indicate that all the genes in our survey are expressed in male reproductive tissues and that there is strong enrichment is testis (see Materials and Methods). Because the X chromosome is thought to become transcriptionally inactive during spermatogenesis (Lifschytz and Lindsley 1972; Hense et al. 2007), it may be that the X-linked genes differ from the autosomal genes in their function or in the timing of their expression (e.g., early vs. late spermatogenesis). At present, the functional annotation of the genes and the profiles of their expression during spermatogenesis are not known well enough to determine if such differences between the X and the autosomes exist. However, it is known that male-biased genes are underrepresented on the X and that they show a relatively high turnover rate between species (Zhang et al. 2007). Using the strict statistical criteria of Zhang et al. (2007), we find that 4/17 (24%) of the X-linked male-biased genes and 16/33 (48%) of the autosomal male-biased genes have conserved sex bias across the 3 species used in our analyses (D. melanogaster, D. simulans, and D. yakuba). The selection parameter of amino acid replacements, γ, does not differ significantly between conserved and nonconserved genes on the X (9.34 vs. 7.79, Mann–Whitney U test, P > 0.5) or the autosomes (5.56 vs. 5.62, P > 0.5). Similarly, dN/dS does not differ significantly between conserved and nonconserved genes on either the X (0.52 vs. 0.34, P > 0.5) or the autosomes (0.14 vs. 0.12, P > 0.5). However, the conserved genes tend to have higher mean γ and dN/dS than the nonconserved genes. This may be because the conserved genes have spent more of their evolutionary history as male-biased and, thus, have been subject to positive selection over a longer time scale. If so, this would make our detection of a fast-X effect conservative, as the X-linked genes have, on average, spent less of their evolutionary history as male-biased genes.
For all the multilocus tests of selection, we performed our analyses separately on X-linked and autosomal genes to avoid making any a priori assumptions about the relative effective population size Ne of X-linked and autosomal genes. For methods that estimate selection parameters in terms of γ = 2Nes, there could be a systematic bias if the X and autosomes differ in Ne. One might expect the X to have a smaller Ne because, in a population with equal numbers of males and females, there are 3 X chromosomes for every 4 copies of an autosome. However, several population genetic surveys have found no reduction of X-linked relative to autosomal polymorphism in African populations, including the population used here (Andolfatto 2001; Kauer et al. 2002; Hutter et al. 2007), which suggests that the X and autosomes have nearly equal Ne. Our own data also suggest an equal Ne for X chromosomes and autosomes: when combined with the data of Pröschel et al. (2006) the X:autosome ratio of synonymous polymorphism is 1.1, which does not differ significantly from 1.0 (χ2 = 2.76, P = 0.10).
Genome-Wide Divergence
In the above discussion, the fast-X effect refers to a greater rate of adaptive evolution for X-linked genes, which is inferred from ratios of polymorphism and divergence at synonymous and nonsynonymous sites. This does not necessarily mean that these genes have a higher rate of nonsynonymous substitution between species. As an example, consider the case in which gene A has 10 nonsynonymous fixed differences between species and an α of 0.5, whereas gene B has 40 nonsynonymous fixed differences and an α of 0.1. If the 2 genes have similar rates of synonymous substitution, then gene B would appear to evolve faster, whereas gene A would have the higher rate of adaptive evolution. However, much of the difference in rates of adaptive evolution between X-linked and autosomal genes in our data results from differences in the ratio of the number of nonsynonymous to synonymous fixed differences (Dn/Ds). For example, Dn/Ds for X-linked male-biased genes is 1.4, whereas that for autosomal male-biased genes is 0.5. This suggests that we should be able to detect a signal for faster-X evolution from whole-genome comparisons of nonsynonymous/synonymous substitution rates (dN/dS), especially when partitioning genes into sex-bias classes. This is exactly what we see in our whole-genome comparisons. There is a significant fast-X effect for male-biased and unbiased genes (fig. 5), but not for female-biased genes. These results match the expectation for the frequent occurrence of recessive beneficial mutations, which should have the strongest effect in male-biased genes, the weakest effect in female-biased genes, and an intermediate effect for unbiased genes (see above).
Our detection of the fast-X effect depends on the partitioning of genes into sex-bias classes. For example, although the effect is strong for male-biased genes, there are very few of these genes on the X chromosome (fig. 5). If one were to pool all X-linked and autosomal genes in the genome, the X-linked male-biased genes would cause only a small increase in overall X-linked divergence, whereas the autosomal male-biased genes would have a larger effect on autosomal divergence. Because both X-linked and autosomal male-biased genes have higher divergence than their female-biased and unbiased counterparts (fig. 5), this would work to obscure the fast-X effect. Similarly, the inclusion of female-biased genes would tend to obscure the effect, as these genes show no significant difference in dN/dS between the X and the autosomes (fig. 5). Finally, it should be noted that the unbiased genes were carefully chosen to show no male or female expression bias in any of 3 microarray data sets (Gnad and Parsch 2006). Thus, many genes that show conflicting sex-bias among experiments were not included in our analysis. If we repeat the analysis by pooling all genes in the genome, without consideration of sex bias, there is no evidence for a fast-X effect (Mann–Whitney U test, P = 0.10 for D. melanogaster/D. simulans, P = 0.49 for D. melanogaster/D. yakuba).
Comparison with Previous Studies
Our observation of a fast-X effect, both from analysis of polymorphism/divergence data and from whole-genome comparisons, runs counter to several recently published studies, most notably those of Connallon (2007) and Thornton et al. (2006), both of which found no evidence for faster-X evolution. Below we discuss several factors that may contribute to these differences.
Connallon (2007) used previously published polymorphism and divergence data from 337 D. melanogaster genes, including the autosomal data of Pröschel et al. (2006) used here, to show that α (measured by the method of Bierne and Eyre-Walker [2004]) is not significantly higher for X-linked than for autosomal genes. However, this data set contained only 2 X-linked male-biased genes (defined as having greater than a 2-fold male bias in a single genome-tiling microarray experiment [Stolc et al. 2004]) and thus was not suitable for testing faster-X evolution in male-biased genes, which are the group that show the strongest effect (fig. 4). To avoid problems of the unequal representation of X-linked and autosomal male-biased genes, Connallon (2007) removed all male-biased genes and analyzed a pooled set of female-biased and unbiased genes. This data set also showed no significant difference in α between X-linked and autosomal genes. A limitation of this approach is that the α estimates produced by the method of Bierne and Eyre-Walker (2004) typically have large 95% CIs, which makes it nearly impossible to detect significant differences between groups of genes when significance is defined by nonoverlapping 95% CIs. Our own data show that there is considerable overlap in the 95% CIs of α for X and autosomal genes (fig. 1) and also among male-biased, female-biased, and unbiased genes within the different chromosome groups. Thus, the inability to detect a fast-X effect by this approach may be a result of a lack of statistical power. Interestingly, the data of Connallon (2007) do show a consistent, but not significant, trend of higher α for X-linked genes (see his fig. 2).
Thornton et al. (2006) took advantage of an X-autosome translocation present in Drosophila pseudoobscura and Drosophila miranda, but absent in D. melanogaster and D. yakuba, to test for faster-X evolution. For this, they examined dN/dS of homologous genes that were X-linked in one species pair, but autosomal in the other. This analysis revealed no evidence for a fast-X effect. Additionally, these authors investigated dN/dS of X-linked and autosomal genes with male- and female-biased expression between D. melanogaster and D. yakuba. Here they found no significant difference in dN/dS between X-linked and autosomal genes within either expression class. For the male-biased genes, this contradicts our findings (fig. 5). There are 3 differences between our analysis and that of Thornton et al. (2006) that might explain this discrepancy. First, they used only a single microarray data set (Parisi et al. 2003) to classify sex-biased genes, wheras we used a consensus of this data set and 2 others (Ranz et al. 2003; Gibson et al. 2004). Thus, our data presumably contain fewer incorrectly classified genes. Second, Thornton et al. (2006) included only autosomal genes from chromosome arm 3L, whereas we include genes from all autosomal chromosome arms (except chromosome 4). The larger sample size of autosomal genes in our analysis should result in increased statistical power. Finally, Thornton et al. (2006) included only genes for which homologs could be aligned in D. melanogaster, D. yakuba, and D. pseudoobscura, whereas we did not require conservation in D. pseudoobscura. This is probably the major factor contributing to the difference between the 2 studies. The requirement of conservation in D. pseudoobscura eliminates the most rapidly evolving genes from the analysis, and it is known that male-biased genes, especially those that are X-linked, tend to be the least conserved between distantly related species (Parisi et al. 2003; Zhang et al. 2004; Musters et al. 2006; Haerty et al. 2007; Sturgill et al. 2007; Zhang et al. 2007). Indeed, this can explain why the dN/dS values of Thornton et al. (2006; see their fig. 3) are much lower than the values in figure 5. If we repeat our analysis following the method of Thornton et al. (2006) as closely as possible, we get median dN/dS values of 0.061 and 0.058 for male-biased genes on chromosomes X and 3L, respectively, and this difference is not significant (Mann–Whitney U test, P = 0.44). Thus, the results presented by Thornton et al. (2006) appear to be valid for the data they used. However, we suggest that their approach was too conservative to detect a fast-X effect for male-biased genes.
Because detection of the fast-X effect appears to be sensitive to several factors described above, it is not surprising that its existence has been controversial and that different studies have reached opposite conclusions. Our results show that X-linked genes, especially those with male-biased expression, do evolve faster that autosomal genes and that this difference in evolutionary rate is due to increased adaptive evolution of X-linked genes. Detection of this effect, however, requires the accurate partitioning of genes by their expression level in the 2 sexes and the use of sufficiently powerful statistical methods.
Supplementary Material
Supplementary tables S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank K. Azadov, Y. Cämmerer, H. Gebhart, M. Pröschel, and Z. Zhang for help with sequencing. The manuscript was improved thanks to comments from D. Presgraves, the editor, and 2 anonymous reviewers. This work was supported by Deutsche Forschungsgemeinschaft grant PA 903/4 (to J.P. and J.F.B.), National Science Foundation grant DMS-0107420 (to S.A.S.), and National Institutes of Health grants GM68465 and GM61351 (to D.L.H.).
References
- Andolfatto P. Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Mol Biol Evol. 2001;18:279–290. doi: 10.1093/oxfordjournals.molbev.a003804. [DOI] [PubMed] [Google Scholar]
- Baines JF, Harr B. Reduced X-linked diversity in derived populations of house mice. Genetics. 2007;175:1911–1921. doi: 10.1534/genetics.106.069419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, et al. (13 co-authors) Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007;5:e310. doi: 10.1371/journal.pbio.0050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt AJ, Presgraves DC, Swanson W. A test for faster X evolution in Drosophila. Mol Biol Evol. 2002;19:1816–1819. doi: 10.1093/oxfordjournals.molbev.a004006. [DOI] [PubMed] [Google Scholar]
- Bierne N, Eyre-Walker A. The genomic rate of adaptive amino acid substitution in Drosophila. Mol Biol Evol. 2004;21:1350–1360. doi: 10.1093/molbev/msh134. [DOI] [PubMed] [Google Scholar]
- Bustamante CD, Nielsen R, Sawyer SA, Olsen KM, Purugganan MD, Hartl DL. The cost of inbreeding in Arabidopsis. Nature. 2002;416:531–534. doi: 10.1038/416531a. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Coyne JA, Barton NH. The relative rates of evolution of sex chromosomes and autosomes. Am Nat. 1987;130:113–146. [Google Scholar]
- Chintapalli VR, Wang J, Dow JAT. Using FlyAtlas to identify better Drosophila models of human disease. Nat Genet. 2007;39:715–720. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- Comeron JM, Guthrie TB. Intragenic Hill-Robertson interference influences selection intensity on synonymous mutations in Drosophila. Mol Biol Evol. 2005;22:2519–2530. doi: 10.1093/molbev/msi246. [DOI] [PubMed] [Google Scholar]
- Comeron JM, Kreitman M. Population, evolutionary and genomic consequences of interference selection. Genetics. 2002;161:389–410. doi: 10.1093/genetics/161.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connallon T. Adaptive protein evolution of X-linked and autosomal genes in Drosophila: implications for faster-X hypotheses. Mol Biol Evol. 2007;24:2566–2572. doi: 10.1093/molbev/msm199. [DOI] [PubMed] [Google Scholar]
- Connallon T, Knowles LL. Intergenomic conflict revealed by patterns of sex-biased gene expression. Trends Genet. 2005;21:495–499. doi: 10.1016/j.tig.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Counterman BA, Ortiz-Barrientos D, Noor MA. Using comparative genomic data to test for fast-X evolution. Evolution. 2004;58:656–660. [PubMed] [Google Scholar]
- Dorus S, Busby SA, Gerike U, Shabanowitz J, Hunt DF, Karr TL. Genomic and functional evolution of the Drosophila melanogaster sperm proteome. Nat Genet. 2006;38:1440–1445. doi: 10.1038/ng1915. [DOI] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 2007;8:689–698. doi: 10.1038/nrg2167. [DOI] [PubMed] [Google Scholar]
- Gibson G, Riley-Berger R, Harshman L, Kopp A, Vacha S, Nuzhdin S, Wayne M. Extensive sex-specific nonadditivity of gene expression in Drosophila melanogaster. Genetics. 2004;167:1791–1799. doi: 10.1534/genetics.104.026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka S, Ometto L, Mousset S, Stephan W, De Lorenzo D. Demography and natural selection have shaped genetic variation in Drosophila melanogaster: a multi-locus approach. Genetics. 2003;165:1269–1278. doi: 10.1093/genetics/165.3.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnad F, Parsch J. Sebida: a database for the functional and evolutionary analysis of genes with sex-biased expression. Bioinformatics. 2006;22:2577–2579. doi: 10.1093/bioinformatics/btl422. [DOI] [PubMed] [Google Scholar]
- Haddrill PR, Halligan DL, Tomaras D, Charlesworth B. Reduced efficacy of selection in regions of the Drosophila genome that lack crossing over. Genome Biol. 2007;8:R18. doi: 10.1186/gb-2007-8-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haerty W, Jagadeeshan S, Kulathinal RJ, et al. (11 co-authors) Evolution in the fast lane: rapidly evolving sex-related genes in Drosophila. Genetics. 2007;177:1321–1335. doi: 10.1534/genetics.107.078865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hense W, Baines JF, Parsch J. X chromosome inactivation during Drosophila spermatogenesis. PLoS Biol. 2007;5:e273. doi: 10.1371/journal.pbio.0050273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Kliman RM. Interactions between natural selection, recombination and gene density in the genes of Drosophila. Genetics. 2002;160:595–608. doi: 10.1093/genetics/160.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter S, Li H, Beisswanger S, De Lorenzo D, Stephan W. Distinctly different sex ratios in African and European populations of Drosophila melanogaster inferred from chromosomewide single nucleotide polymorphism data. Genetics. 2007;177:469–480. doi: 10.1534/genetics.107.074922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer M, Zangerl B, Dieringer D, Schlotterer C. Chromosomal patterns of microsatellite variability contrast sharply in African and non-African populations of Drosophila melanogaster. Genetics. 2002;160:247–256. doi: 10.1093/genetics/160.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, Franz H, Weiss G, Lachmann M, Pääbo S. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science. 2005;309:1850–1854. doi: 10.1126/science.1108296. [DOI] [PubMed] [Google Scholar]
- Lifschytz E, Lindsley DL. The role of X-chromosome inactivation during spermatogenesis. Proc Natl Acad Sci USA. 1972;69:182–186. doi: 10.1073/pnas.69.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Wu CI. Weak selection revealed by the whole-genome comparison of the X chromosome and autosomes of human and chimpanzee. Proc Natl Acad Sci USA. 2005;102:4063–4067. doi: 10.1073/pnas.0500436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE, Axelsson E, Ellegren H. Fast-X on the Z: rapid evolution of sex-linked genes in birds. Genome Res. 2007;17:618–624. doi: 10.1101/gr.6031907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, Kim Y, Hartl DL, Parsch J. Identification of a locus under complex positive selection in Drosophila simulans by haplotype mapping and composite-likelihood estimation. Genetics. 2004;168:265–279. doi: 10.1534/genetics.103.025494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metta M, Gudavalli R, Gibert JM, Schlötterer C. No accelerated rate of protein evolution in male-biased Drosophila pseudoobscura genes. Genetics. 2006;174:411–420. doi: 10.1534/genetics.106.057414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musters H, Huntley MA, Singh RS. A genomic comparison of faster-sex, faster-X, and faster-male evolution between Drosophila melanogaster and Drosophila pseudoobscura. J Mol Evol. 2006;62:693–700. doi: 10.1007/s00239-005-0165-5. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Bustamante C, Clark AG, et al. (13 co-authors) A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005;3:e170. doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ometto L, Glinka S, De Lorenzo D, Stephan W. Inferring the effects of demography and selection on Drosophila melanogaster populations from a chromosome-wide scan of DNA variation. Mol Biol Evol. 2005;22:2119–2130. doi: 10.1093/molbev/msi207. [DOI] [PubMed] [Google Scholar]
- Orr HA, Betancourt AJ. Haldane's sieve and adaptation from the standing genetic variation. Genetics. 2001;157:875–884. doi: 10.1093/genetics/157.2.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Nuttall R, Naiman D, Bouffard G, Malley J, Andrews J, Eastman S, Oliver B. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science. 2003;299:697–700. doi: 10.1126/science.1079190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Nuttall R, Edwards P, et al. (12 co-authors) A survey of ovary-, testis-, and soma-biased gene expression in Drosophila melanogaster adults. Genome Biol. 2004;5:R40. doi: 10.1186/gb-2004-5-6-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC. Recombination enhances protein adaptation in Drosophila melanogaster. Curr Biol. 2005;15:1651–1656. doi: 10.1016/j.cub.2005.07.065. [DOI] [PubMed] [Google Scholar]
- Pröschel M, Zhang Z, Parsch J. Widespread adaptive evolution of Drosophila genes with sex-biased expression. Genetics. 2006;174:893–900. doi: 10.1534/genetics.106.058008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science. 2003;300:1742–1745. doi: 10.1126/science.1085881. [DOI] [PubMed] [Google Scholar]
- Rice WR. Sex chromosomes and the evolution of sexual dimorphism. Evolution. 1984;38:735–742. doi: 10.1111/j.1558-5646.1984.tb00346.x. [DOI] [PubMed] [Google Scholar]
- Richards S, Liu Y, Bettencourt BR, et al. (52 co-authors) Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 2005;15:1–18. doi: 10.1101/gr.3059305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DW, Carr M, Pomiankowski A. Male genes: X-pelled or X-cluded? Bioessays. 2003;25:739–741. doi: 10.1002/bies.10323. [DOI] [PubMed] [Google Scholar]
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Sawyer SA, Hartl DL. Population genetics of polymorphism and divergence. Genetics. 1992;132:1161–1176. doi: 10.1093/genetics/132.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SA, Kulathinal RJ, Bustamante CD, Hartl DL. Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection. J Mol Evol. 2003;57(1 Suppl):S154–S164. doi: 10.1007/s00239-003-0022-3. [DOI] [PubMed] [Google Scholar]
- Sawyer SA, Parsch J, Zhang Z, Hartl DL. Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila. Proc Natl Acad Sci USA. 2007;104:6504–6510. doi: 10.1073/pnas.0701572104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenke TA, Begun DJ. Natural selection drives Drosophila immune system evolution. Genetics. 2003;164:1471–1480. doi: 10.1093/genetics/164.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JA, Huang W, Zhang C, et al. (12 co-authors) Adaptive genic evolution in the Drosophila genomes. Proc Natl Acad Sci USA. 2007;104:2271–2276. doi: 10.1073/pnas.0610385104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh ND, Larracuente AM, Clark AG. Contrasting the efficacy of selection on the X and autosomes in Drosophila. Mol Biol Evol. 2007;25:454–467. doi: 10.1093/molbev/msm275. [DOI] [PubMed] [Google Scholar]
- Stolc V, Gauhar Z, Mason C, et al. (12 co-authors) A gene expression map for the euchromatic genome of Drosophila melanogaster. Science. 2004;306:655–660. doi: 10.1126/science.1101312. [DOI] [PubMed] [Google Scholar]
- Sturgill D, Zhang Y, Parisi M, Oliver B. Demasculinization of X chromosomes in the Drosophila genus. Nature. 2007;450:238–242. doi: 10.1038/nature06330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson WJ, Clark AG, Waldrip-Dail HM, Wolfner MF, Aquadro CF. Evolutionary EST analysis identifies rapidly evolving male reproductive proteins in Drosophila. Proc Natl Acad Sci USA. 2001;98:7375–7379. doi: 10.1073/pnas.131568198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton AR. Contingency tests of neutrality using intra/interspecific gene trees: the rejection of neutrality for the evolution of the mitochondrial cytochrome oxidase II gene in the Hominoid primates. Genetics. 1996;144:1263–1270. doi: 10.1093/genetics/144.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton K, Long M. Rapid divergence of gene duplicates on the Drosophila melanogaster X chromosome. Mol Biol Evol. 2002;19:918–925. doi: 10.1093/oxfordjournals.molbev.a004149. [DOI] [PubMed] [Google Scholar]
- Thornton K, Long M. Excess of amino acid substitutions relative to polymorphism between X-linked duplications in Drosophila melanogaster. Mol Biol Evol. 2005;22:273–284. doi: 10.1093/molbev/msi015. [DOI] [PubMed] [Google Scholar]
- Thornton K, Bachtrog D, Andolfatto P. X chromosomes and autosomes evolve at similar rates in Drosophila: no evidence for faster-X protein evolution. Genome Res. 2006;16:498–504. doi: 10.1101/gr.4447906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgerson DG, Singh RS. Sex-linked mammalian sperm proteins evolve faster than autosomal ones. Mol Biol Evol. 2003;20:1705–2179. doi: 10.1093/molbev/msg193. [DOI] [PubMed] [Google Scholar]
- Torgerson DG, Singh RS. Enhanced adaptive evolution of sperm-expressed genes on the mammalian X chromosome. Heredity. 2006;96:39–44. doi: 10.1038/sj.hdy.6800749. [DOI] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet. 2006;7:645–653. doi: 10.1038/nrg1914. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang J. Rapid evolution of mammalian X-linked testis-expressed homeobox genes. Genetics. 2004;167:879–888. doi: 10.1534/genetics.103.025072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature. 2007;450:233–238. doi: 10.1038/nature06323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Parsch J. Positive correlation between evolutionary rate and recombination rate in Drosophila genes with male-biased expression. Mol Biol Evol. 2005;22:1945–1947. doi: 10.1093/molbev/msi189. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hambuch TM, Parsch J. Molecular evolution of sex-biased genes in Drosophila. Mol Biol Evol. 2004;21:2130–2139. doi: 10.1093/molbev/msh223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.