

Abstract
Although interferon β (IFNβ) decreases relapse rate and disease activity in multiple sclerosis (MS), the mechanisms involved have not been elucidated. The present study is the first report on the apoptotic effect of IFNβ in mature, but not immature, myeloid dendritic cells (DCs). Both exogenous IFNβ added to DCs matured through exposure to proinflammatory cytokines and endogenous IFNβ secreted after lipopolysaccharide stimulation induced DC cell death. Apoptosis of mature DCs required both NF-κB and STAT-1 activation, and was mediated through the induction of caspase-11 expression and activation of caspase-3. In vivo, we observed increased caspase-11 expression and a significant decrease in the number of splenic DCs after lipopolysaccharide administration in wt but not in STAT-1–deficient mice. Since mature DCs are major contributors to the inflammatory response and essential partners in the induction of adaptive immunity, IFNβ-dependent elimination of activated DCs could play an essential role in re-establishing homeostasis, and might represent a new molecular mechanism for the therapeutic effect of IFNβ in MS.
Introduction
Recombinant interferon β (IFNβ) is one of the few therapeutic interventions found to be effective in remitting-relapsing multiple sclerosis (MS), although the response to therapy is only partial and does not extend to all patients (reviewed in Weinstock-Guttman et al1). The molecular mechanisms responsible for the beneficial effect of IFNβ in MS are not fully elucidated. Antiviral, antiproliferative as well as immunomodulatory effects have been described for IFNβ. The most relevant in terms of MS therapy are the immunoregulatory effects, which include inhibition of antigen presentation and T-cell proliferation, altered cytokine and matrix metalloproteinase expression, and induction of suppressor function (reviewed in Markowitz2)
Dendritic cells (DCs) are major producers of IFN type I. Whereas plasmacytoid DCs (pDCs) function as major producers of both IFNα and IFNβ after activation through TLR7, TLR8 and TLR9, myeloid DCs (mDCs) produce mostly IFNβ in response to signaling through TLR3 and TLR4 (reviewed in Coccia3). Although both IFNα and IFNβ have antiviral activity and bind to the same receptor, their biologic activities are different, and in some cases opposite. For example, IFNα appears to be mostly proinflammatory, especially in systemic lupus erythematosus where it activates DCs and contributes to the expansion of autoreactive T cells (reviewed in Coccia3 and Blanco et al4). In contrast, IFNβ prevents the maturation and activation of pDCs in MS patients, reduces proinflammatory cytokine release, including IFNα from human pDCs, and inhibits IL-12 production while promoting IL-10 and IL-27 expression in mDCs.3–5
DC maturation is necessary for activation of naive T cells and is regulated by interactions with pathogens and by the cytokine microenvironment. Timely removal of mature DCs is equally important to prevent hyperactivation of the immune system. Little is known about the factors that control mature DC survival and/or apoptosis. Previous reports showed that defective DC apoptosis, resulting in DC accumulation, led to systemic autoimmune manifestations, and that DCs matured through exposure to TLR ligands or proinflammatory cytokines undergo apoptosis.6–8 With the exception of MHCII signaling,6 the factors involved in the initiation of mature DC apoptosis have not been identified.
In contrast to IFNα, IFNβ has been reported to induce apoptosis in certain cell types, such as pro-B cells, human adrenocortical carcinomas, and hepatoma cell lines, and in TLR-4–activated microglia.9–12 The signaling pathway initiated by IFNβ through IFN type I receptors involves STAT1 homodimers and STAT1/STAT2 heterodimers. STAT-1 has been reported to have a proapoptotic effect through transcription-independent interactions with p53, TRADD, and NF-κBp65, and through transcription-dependent activation of caspases, death receptors and ligands, and iNOS (reviewed in Kim and Lee13). Most studies linked STAT-1 to expression of the inflammatory caspases-1 and -11 leading to activation of the effector caspases.14–17 Mouse caspase-11, an ortholog of human caspase-4, is induced in hematopoietic cells by lipopolysaccharide (LPS) and IFNs, and activates caspases-1 and -3 (reviewed in Martinon and Tschopp18 and Nadiri et al19).
In the present study, we examined the role of IFNβ in the apoptosis of bone marrow–derived mDCs matured in the presence of a proinflammatory cytokine cocktail containing TNF-α, IL-1β, IL-6, and PGE2 or in the presence of LPS. Our results indicate that exogenous IFNβ added to DCs matured with the cytokine cocktail and endogenous IFNβ generated during LPS-mediated DC maturation induce cell death. IFNβ-induced DC apoptosis is mediated by STAT-1 and NF-κB induction of caspase-11 expression, which subsequently activates caspase-3. This study identifies IFNβ as an endogenous factor controlling mature DC survival, and provides a new cellular mechanism for the therapeutic effect of IFNβ in MS. This is particularly relevant since recent studies identified mDCs as essential contributors to the reactivation of myelin-specific T cells in the CNS perivascular space in experimental autoimmune encephalomyelitis (EAE) models of MS.20,21
Methods
Mice
Six- to 8-week-old male B10.A and BALB/c mice from The Jackson Laboratory, STAT-1−/− mice and corresponding wild-type controls from Taconic, and caspase-11−/− mice (Dr Junying Yuan, Harvard Medical School, Boston, MA) and corresponding wild-type controls were maintained in the Temple University School of Medicine animal facility under pathogen-free conditions. Mice were handled and housed in accordance with the guidelines of the Temple University Animal Care and Use Committee.
Reagents
LPS (Escherichia coli 055:B5) and prostaglandin E2 (PGE2) were purchased from Sigma. GM-CSF, TNF-α, IL-1β, and IL-6 were purchased from PeproTech Inc. IFNβ and neutralizing IFNβ antibody were purchased from PBL Biomedical Laboratories. NF-κB activation inhibitor, JSH-23, was purchased from Calbiochem. IFNγ, annexin V, propidium iodine, PE anti–active caspase-3, PE anti–phospho-STAT-1, PE anti–STAT-1, and PE anti-CD11c were purchased from BD PharMingen.
Generation and purification of DCs from bone marrow
DCs were generated from bone marrow. Briefly, 2 × 106 bone marrow cells flushed out from femur and tibiae were cultured in 100-mm Petri dishes containing 10 mL RPMI 1640 medium supplemented with 10% heat-inactivated FBS (Atlanta Biologicals), 2 mM l-glutamine, and 20 ng/mL recombinant GM-CSF. After 3 days, another 10 mL complete medium containing GM-CSF was added to each dish. On day 8, the nonadherent cells were harvested and purified by immunomagnetic sorting with anti-CD11c–coated magnetic beads using the autoMACS system according to the manufacturer's instructions (Miltenyi Biotec). The purity of the sorted cells was determined by fluorescence-activated cell sorting (FACS) analysis (> 96% for CD11c+ cells).
FACS analysis
Cells were subjected to FACS analysis in a 4-color FACSCalibur (BD Biosciences). Data were collected for 10 000 cells and analyzed using CellQuest software from BD PharMingen. For active caspase-3 expression, the cells were fixed and permeabilized followed by active caspase-3 antibody from BD PharMingen. For FACS analysis of phosphorylated NF-κBp65 and STAT-1, DCs were treated with indicated for different time points. After incubation, the cells were fixed and permeabilized with Cytofix/Cytoperm according to the manufacturer's instructions (BD Biosciences). The cells were incubated with rabbit polyclonal antiphosphorylated NF-κBp65 or total NF-κBp65 Abs (Cell Signaling Technology) at room temperature for 60 minutes, followed by the appropriate Alexa-conjugated goat anti–rabbit IgG for 30 minutes (Invitrogen). For phosphorylated and total STAT-1, fixed and permeabilized DCs were incubated with PE anti–phopho-STAT-1 or PE anti–STAT-1 PE Abs (BD Biosciences) at room temperature for 1 hour. After extensive washing, the cells were analyzed by FACS.
Real-time RT-PCR
Caspase-11 and IFNβ expression were detected by SYBR green–based real-time reverse-transcription–polymerase chain reaction (RT-PCR). RNA was prepared from purified CD11c+ DCs treated as indicated in the figures for 24 hours and cDNA was prepared. The 20 μL (total volume) of the PCR mixture consists of 4 μL diluted cDNA, 10 μL SYBR green–containing PCR master mixture (2×), and 150 nM of each primer. The primers for real-time RT-PCR were designed using the Primer Express software from Applied Biosystems, and are as follows: caspase-11: sense 5′-GCCACTTGCCAGGTCTACGAG-3′ and antisense 5′-AGGCCTGCACAATGATGACTTT-3′; IFNβ: sense 5′-CCCTATGGAGATGACGGAGA-3′ and antisense 5′-ACCCAGTGCTGGAGAAATTG-3′. Real-time RT-PCR was performed using the Stratagene Mx3005P, and the cycling conditions used were 95°C for 30 seconds, 55°C for 1 minute, 72°C for 30 seconds, for 40 cycles, followed by a melting point determination or dissociation curves. The expression level of each gene is indicated by the cycle numbers needed for the cDNA to be amplified to reach a threshold. The amount of DNA is calculated from the cycle numbers using standard curves and the results are normalized to the housekeeping gene β-actin from the same sample.
Apoptosis assay
DCs were collected, washed, and analyzed using the FITC annexin V/PI apoptosis assay (BD Biosciences) or terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (Roche Applied Sciences). Staining was performed as recommended by the manufacturers and the cells were analyzed immediately by flow cytometry.
IFNβ enzyme-linked immunosorbent assay
Purified CD11c+ DCs (106 cells/mL) were seeded in 12-well plates and treated as described in “Results.” The amounts of IFNβ released in the medium 24 hours later were measured using the mouse IFNβ enzyme-linked immunosorbent assay (ELISA) Kit (PBL Biomedical Laboratories) according to the manufacturer's instructions. The absorbance was determined using a Polarstar Optima Plate Reader at the wavelength of 450 nm.
Chromatin immunoprecipitation assay
After various treatments, DCs were fixed with 1% formaldehyde (final concentration) for 15 minutes, followed by 125 mM glycine (final concentration) for 5 minutes. Cells were washed twice with ice-cold phosphate-buffered saline containing protease inhibitors, collected and resuspended in SDS lysis buffer containing protease inhibitors, incubated for 10 minutes on ice, and sonicated to shear DNA. After sonication, the lysates were centrifuged at 12 000g for 10 minutes at 4°C and the supernatants were diluted in chromatin immunoprecipitation (ChIP) dilution buffer (0.01% SDS, 1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl, plus protease inhibitors). The chromatin fraction was precleared with protein A/G agarose followed by immunoprecipitation with anti–NF-κBp65, anti–STAT-1, or control IgG (Santa Cruz Biotechnology). After overnight incubation at 4°C, protein A/G agarose was added for 2 hours, and the immune complexes were washed sequentially with low-salt wash buffer once (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, and 150 mM NaCl), high-salt wash buffer once (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, and 500 mM NaCl), LiCl wash buffer once (0.25 M LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid, 1 mM EDTA, and 10 mM Tris-HCl, pH 8.1), and Tris/EDTA buffer twice (10 mM Tris-HCl and 1 mM EDTA, pH 8.0). Abs were eluted from the immune complexes with elution buffer (1% SDS, 0.1 M NaHCO3, and 200 mM NaCl) and cross-linking was reversed by heating at 65°C for 5 hours. After RNAse A and Proteinase K digestion, input DNA and precipitated DNA were purified and PCR amplified with primers encompassing the caspase-11 promoter region containing the NF-κBp65 and STAT-1 sites (sense: ATTCAGGAAAAGGGAGGCCA and antisense: AGTTACTGGGCATT TCCTTAGCA). The PCR products were analyzed by 2% agarose gel electrophoresis.
In vivo treatment
BALB/c mice were injected intraperitoneally with LPS (50 μg or 100 μg in 400 μL PBS). Control animals were injected with the same volume of PBS. In one set of experiments, mice were injected with 100 μg LPS, and splenic CD11c+ DCs were obtained 24 hours later and subjected to real-time RT-PCR for IFNβ and caspase-11 mRNA expression. In a second set of experiments, mice were injected with 50 μg LPS, and splenocytes obtained 48 hours later were stained with PE anti-CD11c Ab to determine the number of CD11c+ cells by FACS.
Statistical analysis
Results are given as means (± SE). Comparisons between 2 groups were done using the Student t test, whereas comparisons between multiple groups were done by ANOVA. Statistical significance was determined as P values less than .05.
Results
IFNβ induces apoptosis in cytokine-matured DCs in a dose- and time-dependent manner
To investigate whether IFNβ induces DC apoptosis, immature and cytokine-matured DCs (TNF-α + IL-1β + IL-6 + PGE2) were treated with IFNβ for various periods of time and apoptosis was determined by flow cytometry after annexin V/PI staining and using the TUNEL assay (Figure 1A-B). Compared with control DCs (medium), immature DCs treated with IFNβ and mature DCs not treated with IFNβ did not undergo apoptosis. In contrast, we observed significant levels of apoptosis in mature DCs treated with IFNβ at 48 and 72 hours. By 72 hours, approximately 66% of mature DCs treated with IFNβ were apoptotic, compared with 1% to 5% in the absence of IFNβ. IFNβ induced apoptosis in mature DCs in a dose-dependent manner (Figure 1C). These results indicate that IFNβ induces apoptosis of cytokine-matured DCs, but not of immature DCs.
Figure 1.

IFNβ induces apoptosis in mature but not immature DCs. CD11c+ DCs were treated with IFNβ (1000 IU/mL) or the cytokine cocktail containing TNF-α (20 ng/mL), IL-1β (10 ng/mL), IL-6 (10 ng/mL), and PGE2 (10−6 M) in the presence or absence of IFNβ (1000 IU/mL). DCs were collected at different time points (24, 48, and 72 hours) and analyzed for apoptosis through annexin V and PI staining (A) or subjected to TUNEL assay (at 48 and 72 hours; B). DCs were matured with the cytokine cocktail in the presence of different concentrations of IFNβ for 48 hours followed by apoptosis assays (C). Percentage viability represents the percentage of annexin V– and PI-negative cells (lower left quadrant). Data are representative of 3 independent experiments.
In parallel experiments, we assessed the effects of IFNβ on DC costimulatory molecule expression, IL-12 production, and T-cell activation. Cytokine-matured DCs expressed higher levels of CD40, CD80, and CD86 compared with unstimulated DCs. IFNβ increased the expression of costimulatory molecules, particularly CD40 and CD86 (Supplemental Figure 1, available on the Blood website; see the Supplemental Materials link at the top of the online article). In contrast to the effect on costimulatory molecules, IFNβ treatment of DCs resulted in a complete shutoff of IL-12p70 production (Supplemental Figure 2) and a significant reduction in the DC stimulatory effect on T-cell proliferation as determined by CFSE staining and 3H-thymidine incorporation (Supplemental Figure 3A-B). In agreement with the IFNβ inhibitory effect on IL-12 production and on DC-induced proliferation of T cells, we also observed a significant reduction in IL-2 and IFNγ production when T cells were cocultured with IFNβ-treated DCs (Supplemental Figure 3C).
STAT-1 signaling is required for IFNβ-induced apoptosis in cytokine-matured DCs
The binding of IFNβ to the type I IFN receptor results in the phosphorylation and activation of TYK2 and JAK1, which in turn regulate the phosphorylation and activation of STAT-1, STAT-2, STAT-3, and STAT-5. STAT-1 has been previously reported to induce apoptosis in different cell types.13,22
To investigate whether IFNβ-induced STAT-1 signaling mediates apoptosis in cytokine-matured DCs, we treated immature and cytokine-matured wild-type (wt) and STAT-1−/− DCs with IFNβ. As expected, IFNβ induced STAT-1 phosphorylation in wt but not in STAT-1−/− DCs (Figure 2A). Apoptosis was detected in wt but not STAT-1−/− cytokine-matured DCs treated with IFNβ (Figure 2B), indicating that IFNβ-induced STAT-1 signaling is required for mature DC apoptosis.
Figure 2.

STAT-1 signaling is required for IFNβ-induced apoptosis. (A) wt and STAT-1−/− DCs were treated with the cytokine cocktail in the presence or absence of IFNβ (1000 IU/mL) for 30 minutes. DCs were fixed and permeabilized followed by intracellular staining for phospho–STAT-1 and total STAT-1; (B-C) wt and STAT-1−/− DCs were treated with the cytokine cocktail in the presence or absence of IFNβ (B) or IFNγ (C). Forty-eight hours later, DCs were subjected to apoptosis assays. Data are representative of 2 independent experiments with triplicate samples.
Since IFNγ also induces STAT-1 signaling, mature DCs were exposed to IFNγ. Similar to IFNβ, IFNγ also induced apoptosis in mature wt but not STAT-1−/− DCs (Figure 2C).
IFNβ induces caspase-11 expression and caspase-3 activation in cytokine-matured DCs
Murine caspase-11, an upstream caspase induced by proinflammatory cytokines, processes and activates caspase-3, leading to apoptosis.14,23,24 To investigate whether caspase-11 is involved in IFNβ-induced mature DC apoptosis, we examined the expression of caspase-11 mRNA in DCs. Expression of caspase-11 was very low in immature and cytokine-matured DCs (Figure 3A inset). In contrast, IFNβ induced caspase-11 expression in immature and particularly in cytokine-matured DCs (Figure 3A top panel). Since previous studies showed that caspase-11 directly mediates activation of caspase-3, we determined caspase-3 activation in immature and cytokine-matured DCs exposed to IFNβ. No active caspase-3 was detected in immature DCs treated or not treated with IFNβ, or in cytokine-matured DCs in the absence of IFNβ (Figure 3A bottom panels). In contrast, a significant number of cytokine-matured DCs (> 30%) expressed intracellular active caspase-3 after exposure to IFNβ (Figure 3A bottom panels).
Figure 3.

IFNβ induces caspase-11 expression and active caspase-3 in mature DCs. (A) B10.A DCs were treated with IFNβ or matured with the cytokine cocktail in the presence or absence of IFNβ (1000 IU/mL) and analyzed for caspase-11 expression by real-time RT-PCR (24 hours; top panel) and for levels of active caspase-3 by flow cytometry (48 hours; bottom panel). (B) wt and STAT-1−/− DCs were treated as in panel A and analyzed for caspase-11 expression and active caspase-3 levels. (C) wt and STAT-1−/− mice were matured with the cytokine cocktail in the presence or absence of IFNγ (100 ng/mL) and analyzed for levels of active caspase-3 by flow cytometry. Data are representative of 3 independent experiments.
To verify that IFNβ-induced STAT-1 activation is required for caspase-11 expression and caspase-3 activation, cytokine-matured wt and STAT-1−/− DCs were exposed to IFNβ. Caspase-11 expression was determined by real-time RT-PCR, and caspase-3 activation was evaluated by FACS. IFNβ induced caspase-11 expression and caspase-3 activation in wt but not STAT-1−/− cytokine-matured DCs (Figure 3B). Similar to IFNβ, IFNγ induced active caspase-3 expression in cytokine-matured wt but not STAT-1−/− DCs (Figure 3C). These results demonstrate that STAT-1 signaling is required to induce caspase-11 expression and caspase-3 activation.
Caspase-11 is required for caspase-3 activation and subsequent apoptosis in cytokine-matured DCs treated with IFNβ
To investigate the role of caspase-11 in caspase-3 activation and apoptosis of IFNβ-treated, cytokine-matured DCs, we used DCs generated from wt and caspase-11–deficient mice. DCs were cultured in medium or matured with the cytokine cocktail and treated with or without IFNβ. Caspase-11 expression was determined by real-time RT-PCR and activation of caspase-3 was evaluated by FACS. Increased caspase-11 expression, caspase-3 activation, and DC apoptosis were observed in IFNβ-treated, cytokine-matured wt DCs, but not in caspase-11−/− DCs (Figure 4). These results suggest that IFNβ-induced caspase-11 is responsible for caspase-3 activation and apoptosis in cytokine-matured DCs.
Figure 4.

IFNβ-induced DC apoptosis requires caspase-11 expression. CD11c+ DCs generated from wt or caspase-11−/− mice were treated with IFNβ (1000 IU/mL) or the cytokine cocktail in the presence or absence of IFNβ (1000 IU/mL). Caspase-11 mRNA expression was determined by real-time RT-PCR (24 hours); active caspase-3 levels and apoptosis were determined by FACS (48 hours). Data are representative of 2 independent experiments with triplicate samples.
LPS-induced DC apoptosis is mediated through expression and release of IFNβ
DCs matured with LPS secrete IFNβ, express active caspase-3, and undergo apoptosis (Figure 5A). To examine whether LPS-induced apoptosis is mediated through IFNβ, we examined viability and percentage of activated caspase-3 in the presence of neutralizing anti-IFNβ Abs. Neutralizing IFNβ Abs significantly reduce the percentage of active caspase-3 and partially prevent LPS-induced apoptosis (Figure 5A right and middle panels), indicating that IFNβ plays an important role in mature DC apoptosis.
Figure 5.

LPS-induced DC apoptosis requires IFNβ. (A) IFNβ production by DCs was measured by ELISA 24 hours after treatment with LPS (100 ng/mL; left panel). DCs were treated with LPS (100 ng/mL) and cultured in the presence or absence of neutralizing IFNβ Ab (10 μg/mL) and analyzed 48 hours later for levels of active caspase-3 and apoptosis by FACS (right and middle panels). (B) wt and STAT-1−/− DCs were treated with LPS (1 μg/mL) and analyzed for caspase-11 mRNA expression by real-time RT-PCR (24 hours), and for levels of active caspase-3 and viability (48 hours) by FACS. (C) wt and caspase-11−/− DCs were treated and analyzed as described in panel B. Data are representative of 2 independent experiments with triplicate samples.
To further investigate whether LPS-induced apoptosis depends on STAT-1 activation, we measured viability, caspase-11 expression, and percentage active caspase-3 in LPS-treated wt and STAT-1–deficient DCs. As expected from the involvement of LPS-induced IFNβ in DC apoptosis, LPS-treated STAT-1–deficient DCs do not undergo apoptosis, and do not express caspase-11 or active caspase-3 (Figure 5B). The direct involvement of caspase-11 in LPS-induced apoptosis was tested using caspase-11–deficient DCs. In contrast to wt DCs, LPS does not induce apoptosis or caspase-3 activation in caspase-11–deficient DCs (Figure 5C).
NF-κB activation plays a role in mature DC apoptosis
Our results indicate that STAT-1 signaling plays an important role in IFNβ-induced caspase activation and apoptosis in LPS- and cytokine-matured DCs (Figures 2–3, 5). However, treatment of immature DCs with IFNβ does not result in caspase-3 activation or apoptosis (Figures 1, 3), suggesting that, in addition to STAT-1, signals mediated through other pathways are required for caspase activation and apoptosis. Since both cytokines and LPS activate NF-κB, we measured first p65 phosphorylation in our experimental system. Treatment of DCs with the cytokine cocktail increased phospho-p65 levels at 15 minutes (Figure 6A). IFNβ treatment did not affect phospho-p65 levels either in immature DCs or in DCs treated with the cytokine cocktail (Figure 6A). In contrast, the cytokine cocktail did not induce STAT-1 phosphorylation, whereas IFNβ treatment of either immature DCs or DCs treated with the cytokine cocktail resulted in significant levels of STAT-1 phosphorylation (Figure 6B). LPS induced phosphorylation of both p65 and STAT-1 (delayed effect on STAT-1 compared with p65, in agreement with the required induction of IFNβ by LPS; Figure 6C).
Figure 6.

Involvement of NF-κB and STAT-1 in caspase-11 expression. (A-B) CD11c+ DCs were treated with IFNβ (1000 IU/mL), or cytokine cocktail in the presence or absence of IFNβ (1000 IU/mL). At 15, 30, and 60 minutes, DCs were fixed, permeabilized, and analyzed by intracellular staining with (A) phospho-p65, total p65, (B) phospho-STAT-1, and total-STAT-1 antibody. (C) CD11c+ DCs were treated with LPS (1 μg/mL) for different time periods and analyzed for phospho-p65 and phospho–STAT-1 intracellular levels. (D-E) CD11c+ DCs were treated with IFNβ (1000 IU/mL), LPS (1 μg/mL), cytokine cocktail in the presence or absence of IFNβ (1000 IU/m) for 1.5 hours (D), or with LPS (1 μg/mL) for 4, 6, and 8 hours (E). Cells were fixed, sonicated, and subjected to ChIP analysis using antibodies to STAT-1, NF-κBp65, or control IgG. Precipitated DNA was isolated and evaluated by PCR using specific primers for the proximal regulatory region of the caspase-11 promoter. PCR products were fractionated through 2% agarose gel electrophoresis. Data are representative of 3 independent experiments (A-C) and 2 representative experiments (D-E).
The possible role of NF-κB in IFNβ-induced apoptosis of mature DCs was further investigated using the NF-κB inhibitor JSH-23. DCs were pretreated with JSH-23 followed by treatment with the cytokine cocktail (TNF-α + IL-1β + IL-6 + PGE2) and IFNβ or LPS. JSH-23 reduced apoptosis in a dose-dependent manner (Table 1).
Table 1.
NF-κB inhibition prevents apoptosis in cytokine-matured DCs treated with IFNβ and in LPS-matured DCs
| JSH-23, μM | % Viability |
|
|---|---|---|
| TNF-α + IL-1β + IL-6 + PGE2 + IFNβ | LPS | |
| 0 | 44 ± 1 | 61 ± 3 |
| 0.1 | 54 ± 2 | 66 ± 1 |
| 1 | 71 ± 7 | 90 ± 1 |
| 10 | 95 ± 1 | 94 ± 1 |
CD11c+ DCs were pretreated with different concentrations of JSH-23 for 1 hour followed by treatment with the cytokine cocktail (TNF-α + IL-1β + IL-6 + PGE2) in the presence of IFNβ (500 IU/mL), or with LPS (100 ng/mL). A second dose of JSH-23 was added 24 hours later, and apoptosis was assessed 48 hours after the initiation of cultures by flow cytometry after annexin V/PI staining. Medium controls (no treatment) and cells treated with the cytokine cocktail in the absence of IFNβ averaged 90% to 95% viability.
These results suggest that both STAT-1 (activated by IFNβ) and NF-κB (activated during DC maturation induced by cytokines or LPS) are required for IFNβ-induced apoptosis of mature DCs.
NF-κB and STAT-1 binding to the caspase-11 promoter
The caspase-11 promoter region includes NF-κB and STAT-1 binding sites.25 To investigate whether treatment with the cytokine cocktail plus IFNβ or with LPS induces in vivo interactions of NF-κB p65 and STAT-1 with the caspase-11 promoter, we performed ChIP assays. Lysates from untreated DCs (medium), DCs treated with IFNβ, DCs treated with the cytokine cocktail in the presence or absence of IFNβ, or with LPS, were immunoprecipitated with anti-NF-κBp65 and anti-STAT-1 Abs, followed by PCR amplification using primers encompassing the NF-κB and STAT-1 sites in the murine caspase-11 promoter. IFNβ treatment of both immature and cytokine-matured DCs induced STAT-1 binding at an early time point (90 minutes), and NF-κB binding was observed for cytokine- and LPS-matured DCs (Figure 6D). In agreement with the later induction of IFNβ by LPS treatment, we observed STAT-1 binding in LPS-matured DCs at 6 and 8 hours (Figure 6E).
In vivo administration of LPS induces caspase-11 expression and causes a decrease in splenic DCs
To determine whether LPS induces caspase-11 in vivo, mice were injected intraperitoneally with LPS. CD11c+ DCs were purified from spleen 24 hours later and analyzed for IFNβ and caspase-11 mRNA expression. A significant increase in IFNβ (Figure 7A) and caspase-11 (Figure 7B) expression was observed in CD11c+ splenic DCs from mice injected with LPS compared with controls.
Figure 7.

In vivo effects of LPS administration on caspase-11 expression and numbers of CD11c+ splenic DCs. BALB/c mice (males; n = 6) were injected with LPS (100 μg). Twenty-four hours later, splenic CD11c+ DCs were purified and subjected to real-time RT-PCR for (A) IFNβ or (B) caspase-11. (C) wt and STAT-1−/− mice were injected intraperitoneally with LPS (50 μg). Forty-eight hours later, splenocytes were analyzed for CD11c expression. (D) Model for IFNβ-induced apoptosis in mature DCs. Maturation by the cytokine cocktail activates NF-κB; IFNβ activates STAT-1. The 2 transcription factors bind to caspase-11 promoter. Caspase-11 subsequently activates caspase-3 and results in apoptosis.
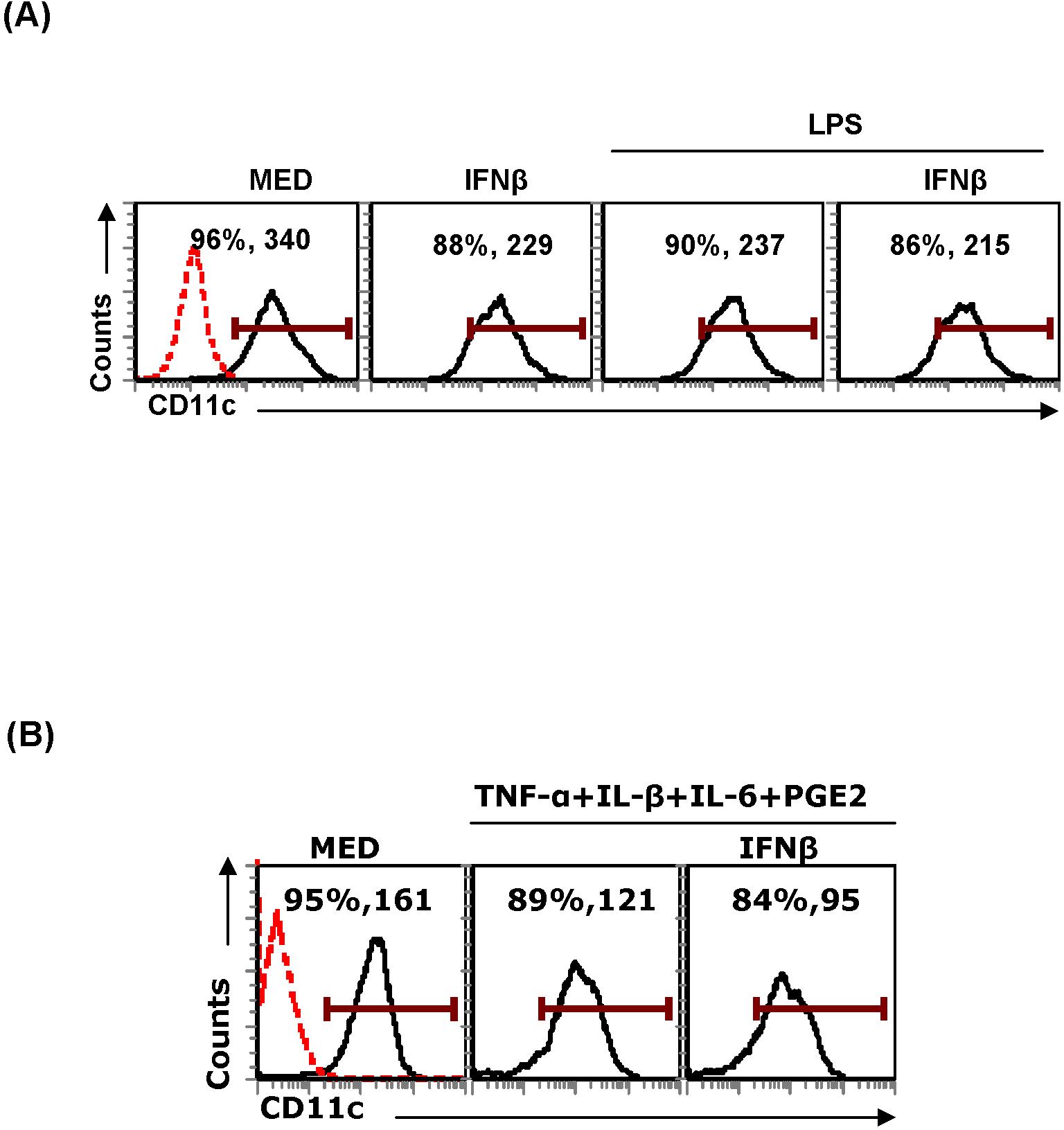
To evaluate DC apoptosis in vivo, we injected wt and STAT-1−/− mice with LPS. Spleen cells were harvested 48 hours later and the numbers of CD11c+ DCs were determined by FACS. Splenic DCs consist of 2 CD11c+ populations: CD11chi and CD11cint. After LPS treatment, the CD11chi population disappeared in both wt and STAT-1−/− mice (Figure 7C). However, we also observed a reduction in CD11cint splenic DCs in LPS-treated wt but not in STAT-1−/− mice (Figure 7C). Since the disappearance of the CD11chi-expressing splenic DCs could be due to the LPS-induced down-regulation of CD11c expression, we measured CD11c expression in an in vitro system consisting of conventional bone marrow–derived DCs. Indeed, we observed a significant decrease in the level of CD11c expression in DCs treated with LPS, cytokine cocktail, and IFNβ (Supplemental Figure 4A-B).
These results indicate that in vivo treatment with LPS results in a STAT-1–independent reduction in the level of CD11c expression, followed by a STAT-1–dependent reduction in the numbers of splenic DCs. Taken together with the LPS-induced increase in IFNβ and caspase-11 expression, the results suggest that the decrease in the numbers of splenic DCs is due to apoptosis mediated through the induction of endogenous IFNβ and caspase-11.
Discussion
Although IFN type I and II have been reported to induce apoptosis in specific cell lines (reviewed in Maher et al26), there is no information on IFN-induced apoptosis in DCs. The present study is the first report on the apoptotic effect of IFNβ in mature myeloid DCs. In the present study, we examined the role of exogenous IFNβ on DCs matured with the proinflammatory cytokine cocktail consisting of TNF-α, IL-1β, IL-6, and PGE2 as previously described.27,28 IFNβ induced cell death at 48 and 72 hours in a dose-dependent manner. In addition, LPS-treated DCs secreted IFNβ as early as 24 hours, and DC apoptosis observed 48 hours after LPS treatment was partially prevented in the presence of neutralizing IFNβ Abs. This suggests a role for endogenous IFNβ released after LPS stimulation in mature DC apoptosis. Apoptosis of mature DCs exposed to IFNβ requires both NF-κB and STAT-1 activation, and is mediated through the induction of caspase-11 expression, and activation of caspase-3. In vivo, we observed IFNβ and caspase-11 expression after LPS administration in splenic DCs and a significant decrease in the number of CD11cint splenic DCs in wt but not in STAT-1–deficient mice.
An interesting observation was the disappearance of CD11chi splenic DCs in both wt and STAT-1–deficient mice after LPS administration. In spleen, the 2 CD11chi and CD11cint populations represent conventional and plasmacytoid DCs, respectively. Our in vitro experiments with conventional bone marrow–derived DCs treated with LPS or cytokine cocktail showed a reduction in CD11c expression. Therefore, the splenic CD11cint cells observed after in vivo LPS administration presumably represent a mix of conventional and plasmacytoid DCs. Therefore, the reduction in CD11cint DCs could be due to IFNβ/caspase-11–mediated apoptosis of conventional DCs, plasmacytoid DCs, or both.
At earlier time points (24 or 48 hours) IFNβ reduced or eliminated IL-12 production and reduced the stimulatory capacity of DCs for T-cell proliferation and IFNγ/IL-2 production, although it up-regulated DC expression of CD40 and CD86. This apparent contradictory effect might be because IFNβ also up-regulates DC expression of the inhibitory molecules PD-L1 and PD-L2 (results not shown), which might cancel the effects on CD40 and CD86.
IFNβ decreases relapse rate and disease activity in MS.29–31 In EAE, administration of IFNβ had a beneficial effect, and IFNβ-deficient mice exhibited exacerbated disease.32 The molecular mechanisms involved in the beneficial effect of IFNβ in MS/EAE have not been yet elucidated. Several reports indicate effects on the expression of adhesion molecules, metalloproteinases, and cytokines; alterations of the Th1/Th2 balance in favor of Th2 differentiation; a decrease in blood-brain barrier permeability; and induction of suppressor T cells.29–31,33,34 Since mDCs play an important role in the reactivation of myelin-specific CD4+ T cells in EAE,20,21 a potential effect of IFNβ on DC activation and function is of significant interest.
In contrast to IFNα, which contributes to DC maturation by inducing higher levels of MHCII and costimulatory molecules,35–37 IFNβ was reported to have anti-inflammatory effects, reducing CD40-dependent IL-12 production, and inducing the expression of the anti-inflammatory cytokines IL-10 and IL-27.38–40 The different biologic effects of IFNα and β are difficult to explain since they bind to the same receptor. One possible explanation is their different affinities for the 2 receptor subunits, leading to qualitative differences in downstream signaling pathways and to differential gene activation.41–43
IFNβ signals through STAT-1 homodimers or heterodimers. STAT-1 has been directly implicated in TNF-α–, hypoxia-, and ischemia reperfusion–induced apoptosis through the up-regulation of caspases and Fas/FasL (reviewed in Kim and Lee,13 Stephanou and Latchman,22 and Maher et al26). Since neither IFNβ nor LPS induced apoptosis in STAT-1–deficient DCs, our results indicated that IFNβ-induced STAT-1 signaling was essential for DC apoptosis. In agreement with the fact that IFNγ also signals through STAT-1 homodimers, we observed that exogenous IFNγ also induced apoptosis in wt but not in STAT-1–deficient DCs matured with the proinflammatory cytokine cocktail. However, in contrast to IFNβ, IFNγ therapy proved to be detrimental in MS.44–46 The effects of IFNγ and IFNβ on T-cell differentiation, with IFNγ promoting Th1 and IFNβ promoting Th2 differentiation, certainly play an essential role in their opposite therapeutic actions.
STAT-1–dependent activation of inflammatory caspases, particularly caspase-11, has been reported in fibroblasts, cardiomyocytes, and microglia (reviewed in Kim and Lee13). In microglia, TLR4 but not TLR2 ligands were shown to induce caspase-11 expression and apoptosis in an IFNβ-dependent manner.10 Murine caspase-11, a functional orthologue of human caspase-4, belongs to the subfamily of inflammatory caspases (reviewed in Martinon and Tschopp,18 Nadiri et al,19 and Yeretssian47). The main substrates of inflammatory caspases, activated through intracellular NOD-like receptors organized in inflammasomes, are cytokines such as pro–IL-1β, pro–IL-18, and IL-33, and executioner caspases such as caspase-3. Murine caspase-11 is highly induced in hematopoietic cells by LPS and IFNβ or IFNγ,10,25,48 leading to apoptosis through activation of caspase-3.10,14,23,24,49,50 Functional studies of the caspase-11 promoter identified 2 essential sites, an NF-κB and a STAT-1 binding site.25 The present study is in agreement with the previously reported observations, since both LPS treatment and maturation with the cytokine cocktail in the presence of IFNβ induced STAT-1 and NF-κB binding to caspase-11 promoter in ChIP assays. The fact that STAT-1 binding is observed at later time points in DCs treated with LPS is in agreement with the requirement for IFNβ induction by LPS.
Based on our results we propose that DC apoptosis requires 2 separate signals: activation of NF-κB provided by ligands inducing maturation such as inflammatory cytokines or TLR ligands activating the MyD88 signaling pathway, and activation of STAT-1 resulting from signaling through interferon receptors. The TLR4 ligand LPS activates NF-κB through the MyD88 pathway and induces IFNβ expression through IRF3, resulting in subsequent STAT-1 activation. The 2 transcription factors (NF-κB and STAT-1) induce expression of caspase-11 and subsequent activation of caspase-3, leading to apoptosis of mature DCs (Figure 7D). Because mature DCs are major contributors to innate inflammatory responses and to the initiation of adaptive immunity through the activation and differentiation of CD4+ T cells, IFNβ-dependent elimination of activated DCs could play an essential role in re-establishing homeostasis after the elimination of pathogens, and represent a new molecular mechanism for the therapeutic effect of IFNβ in autoimmune diseases such as MS.
Supplementary Material
Acknowledgments
We thank Dr Junying Yuan (Harvard Medical School) for providing us with the caspase-11–deficient mice.
This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases grant RO1AI052306 (D.G.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.-H.Y. designed and performed experiments and analyzed data; and D.G. designed experiments, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Doina Ganea, Temple University School of Medicine, Department of Microbiology and Immunology, 3400 N Broad St, Philadelphia, PA 19140; e-mail: doina.ganea@temple.edu.
References
- 1.Weinstock-Guttman B, Ramanathan M, Zivadinov R. Interferon-beta treatment for relapsing multiple sclerosis. Expert Opin Biol Ther. 2008;8:1435–1447. doi: 10.1517/14712598.8.9.1435. [DOI] [PubMed] [Google Scholar]
- 2.Markowitz CE. Interferon-beta: mechanism of action and dosing issues. Neurology. 2007;68:S8–11. doi: 10.1212/01.wnl.0000277703.74115.d2. [DOI] [PubMed] [Google Scholar]
- 3.Coccia EM. IFN regulation and functions in myeloid dendritic cells. Cytokine Growth Factor Rev. 2008;19:21–32. doi: 10.1016/j.cytogfr.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008;19:41–52. doi: 10.1016/j.cytogfr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lande R, Gafa V, Serafini B, et al. Plasmacytoid dendritic cells in multiple sclerosis: intracerebral recruitment and impaired maturation in response to interferon-beta. J Neuropathol Exp Neurol. 2008;67:388–401. doi: 10.1097/NEN.0b013e31816fc975. [DOI] [PubMed] [Google Scholar]
- 6.Bertho N, Blancheteau VM, Setterblad N, et al. MHC class II-mediated apoptosis of mature dendritic cells proceeds by activation of the protein kinase C-delta isoenzyme. Int Immunol. 2002;14:935–942. doi: 10.1093/intimm/dxf058. [DOI] [PubMed] [Google Scholar]
- 7.Chen M, Wang YH, Wang Y, et al. Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 2006;311:1160–1164. doi: 10.1126/science.1122545. [DOI] [PubMed] [Google Scholar]
- 8.Dubois SP, Waldmann TA, Muller JR. Survival adjustment of mature dendritic cells by IL-15. Proc Natl Acad Sci U S A. 2005;102:8662–8667. doi: 10.1073/pnas.0503360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gamero AM, Potla R, Wegrzyn J, et al. Activation of Tyk2 and Stat3 is required for the apoptotic actions of interferon-beta in primary pro-B cells. J Biol Chem. 2006;281:16238–16244. doi: 10.1074/jbc.M509516200. [DOI] [PubMed] [Google Scholar]
- 10.Jung DY, Lee H, Jung BY, et al. TLR4, but not TLR2, signals autoregulatory apoptosis of cultured microglia: a critical role of IFN-beta as a decision maker. J Immunol. 2005;174:6467–6476. doi: 10.4049/jimmunol.174.10.6467. [DOI] [PubMed] [Google Scholar]
- 11.Khvalevsky E, Rivkin L, Rachmilewitz J, Galun E, Giladi H. TLR3 signaling in a hepatoma cell line is skewed towards apoptosis. J Cell Biochem. 2007;100:1301–1312. doi: 10.1002/jcb.21119. [DOI] [PubMed] [Google Scholar]
- 12.van Koetsveld PM, Vitale G, de Herder WW, et al. Potent inhibitory effects of type I interferons on human adrenocortical carcinoma cell growth. J Clin Endocrinol Metab. 2006;91:4537–4543. doi: 10.1210/jc.2006-0620. [DOI] [PubMed] [Google Scholar]
- 13.Kim HS, Lee MS. STAT1 as a key modulator of cell death. Cell Signal. 2007;19:454–465. doi: 10.1016/j.cellsig.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Kang SJ, Wang S, Hara H, et al. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol. 2000;149:613–622. doi: 10.1083/jcb.149.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee J, Hur J, Lee P, et al. Dual role of inflammatory stimuli in activation-induced cell death of mouse microglial cells. Initiation of two separate apoptotic pathways via induction of interferon regulatory factor-1 and caspase-11. J Biol Chem. 2001;276:32956–32965. doi: 10.1074/jbc.M104700200. [DOI] [PubMed] [Google Scholar]
- 16.Suk K, Chang I, Kim YH, et al. Interferon gamma (IFNgamma) and tumor necrosis factor alpha synergism in ME-180 cervical cancer cell apoptosis and necrosis. IFNgamma inhibits cytoprotective NF-kappa B through STAT1/IRF-1 pathways. J Biol Chem. 2001;276:13153–13159. doi: 10.1074/jbc.M007646200. [DOI] [PubMed] [Google Scholar]
- 17.Suk K, Kim S, Kim YH, et al. IFN-gamma/TNF-alpha synergism as the final effector in autoimmune diabetes: a key role for STAT1/IFN regulatory factor-1 pathway in pancreatic beta cell death. J Immunol. 2001;166:4481–4489. doi: 10.4049/jimmunol.166.7.4481. [DOI] [PubMed] [Google Scholar]
- 18.Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10–22. doi: 10.1038/sj.cdd.4402038. [DOI] [PubMed] [Google Scholar]
- 19.Nadiri A, Wolinski MK, Saleh M. The inflammatory caspases: key players in the host response to pathogenic invasion and sepsis. J Immunol. 2006;177:4239–4245. doi: 10.4049/jimmunol.177.7.4239. [DOI] [PubMed] [Google Scholar]
- 20.Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- 21.Miller SD, McMahon EJ, Schreiner B, Bailey SL. Antigen presentation in the CNS by myeloid dendritic cells drives progression of relapsing experimental autoimmune encephalomyelitis. Ann N Y Acad Sci. 2007;1103:179–191. doi: 10.1196/annals.1394.023. [DOI] [PubMed] [Google Scholar]
- 22.Stephanou A, Latchman DS. Opposing actions of STAT-1 and STAT-3. Growth Factors. 2005;23:177–182. doi: 10.1080/08977190500178745. [DOI] [PubMed] [Google Scholar]
- 23.Kang SJ, Wang S, Kuida K, Yuan J. Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ. 2002;9:1115–1125. doi: 10.1038/sj.cdd.4401087. [DOI] [PubMed] [Google Scholar]
- 24.Suk K, Kim SY, Kim H. Essential role of caspase-11 in activation-induced cell death of rat astrocytes. J Neurochem. 2002;80:230–238. doi: 10.1046/j.0022-3042.2001.00705.x. [DOI] [PubMed] [Google Scholar]
- 25.Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem. 2002;277:41624–41630. doi: 10.1074/jbc.M207852200. [DOI] [PubMed] [Google Scholar]
- 26.Maher SG, Romero-Weaver AL, Scarzello AJ, Gamero AM. Interferon: cellular executioner or white knight? Curr Med Chem. 2007;14:1279–1289. doi: 10.2174/092986707780597907. [DOI] [PubMed] [Google Scholar]
- 27.Luft T, Jefford M, Luetjens P, et al. Functionally distinct dendritic cell (DC) populations induced by physiologic stimuli: prostaglandin E(2) regulates the migratory capacity of specific DC subsets. Blood. 2002;100:1362–1372. doi: 10.1182/blood-2001-12-0360. [DOI] [PubMed] [Google Scholar]
- 28.Scandella E, Men Y, Gillessen S, Forster R, Groettrup M. Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood. 2002;100:1354–1361. doi: 10.1182/blood-2001-11-0017. [DOI] [PubMed] [Google Scholar]
- 29.Adorini L. Immunotherapeutic approaches in multiple sclerosis. J Neurol Sci. 2004;223:13–24. doi: 10.1016/j.jns.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 30.Neuhaus O, Archelos JJ, Hartung HP. Immunomodulation in multiple sclerosis: from immunosuppression to neuroprotection. Trends Pharmacol Sci. 2003;24:131–138. doi: 10.1016/S0165-6147(03)00028-2. [DOI] [PubMed] [Google Scholar]
- 31.Polman CH, Uitdehaag BM. New and emerging treatment options for multiple sclerosis. Lancet Neurol. 2003;2:563–566. doi: 10.1016/s1474-4422(03)00505-2. [DOI] [PubMed] [Google Scholar]
- 32.Teige I, Treschow A, Teige A, et al. IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol. 2003;170:4776–4784. doi: 10.4049/jimmunol.170.9.4776. [DOI] [PubMed] [Google Scholar]
- 33.Floris S, Ruuls SR, Wierinckx A, et al. Interferon-beta directly influences monocyte infiltration into the central nervous system. J Neuroimmunol. 2002;127:69–79. doi: 10.1016/s0165-5728(02)00098-x. [DOI] [PubMed] [Google Scholar]
- 34.Harzheim M, Stepien-Mering M, Schroder R, Schmidt S. The expression of microfilament-associated cell-cell contacts in brain endothelial cells is modified by IFN-beta1a (Rebif). J Interferon Cytokine Res. 2004;24:711–716. doi: 10.1089/jir.2004.24.711. [DOI] [PubMed] [Google Scholar]
- 35.Dauer M, Pohl K, Obermaier B, et al. Interferon-alpha disables dendritic cell precursors: dendritic cells derived from interferon-alpha-treated monocytes are defective in maturation and T-cell stimulation. Immunology. 2003;110:38–47. doi: 10.1046/j.1365-2567.2003.01702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santini SM, Di Pucchio T, Lapenta C, Parlato S, Logozzi M, Belardelli F. The natural alliance between type I interferon and dendritic cells and its role in linking innate and adaptive immunity. J Interferon Cytokine Res. 2002;22:1071–1080. doi: 10.1089/10799900260442494. [DOI] [PubMed] [Google Scholar]
- 37.Tough DF. Type I interferon as a link between innate and adaptive immunity through dendritic cell stimulation. Leuk Lymphoma. 2004;45:257–264. doi: 10.1080/1042819031000149368. [DOI] [PubMed] [Google Scholar]
- 38.McRae BL, Beilfuss BA, van Seventer GA. IFN-beta differentially regulates CD40-induced cytokine secretion by human dendritic cells. J Immunol. 2000;164:23–28. doi: 10.4049/jimmunol.164.1.23. [DOI] [PubMed] [Google Scholar]
- 39.Remoli ME, Gafa V, Giacomini E, Severa M, Lande R, Coccia EM. IFN-beta modulates the response to TLR stimulation in human DC: involvement of IFN regulatory factor-1 (IRF-1) in IL-27 gene expression. Eur J Immunol. 2007;37:3499–3508. doi: 10.1002/eji.200737566. [DOI] [PubMed] [Google Scholar]
- 40.Yasuda CL, Al-Sabbagh A, Oliveira EC, Diaz-Bardales BM, Garcia AA, Santos LM. Interferon beta modulates experimental autoimmune encephalomyelitis by altering the pattern of cytokine secretion. Immunol Invest. 1999;28:115–126. doi: 10.3109/08820139909061141. [DOI] [PubMed] [Google Scholar]
- 41.Coelho LF, Magno de Freitas Almeida G, Mennechet FJ, Blangy A, Uze G. Interferon-alpha and -beta differentially regulate osteoclastogenesis: role of differential induction of chemokine CXCL11 expression. Proc Natl Acad Sci U S A. 2005;102:11917–11922. doi: 10.1073/pnas.0502188102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gavutis M, Lata S, Lamken P, Muller P, Piehler J. Lateral ligand-receptor interactions on membranes probed by simultaneous fluorescence-interference detection. Biophys J. 2005;88:4289–4302. doi: 10.1529/biophysj.104.055855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaitin DA, Roisman LC, Jaks E, et al. Inquiring into the differential action of interferons (IFNs): an IFN-alpha2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-beta. Mol Cell Biol. 2006;26:1888–1897. doi: 10.1128/MCB.26.5.1888-1897.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson KP. Treatment of multiple sclerosis with various interferons: the cons. Neurology. 1988;38:62–65. [PubMed] [Google Scholar]
- 45.Panitch HS, Bever CT., Jr Clinical trials of interferons in multiple sclerosis: what have we learned? J Neuroimmunol. 1993;46:155–164. doi: 10.1016/0165-5728(93)90245-t. [DOI] [PubMed] [Google Scholar]
- 46.Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology. 1987;37:1097–1102. doi: 10.1212/wnl.37.7.1097. [DOI] [PubMed] [Google Scholar]
- 47.Yeretssian G, Labbe K, Saleh M. Molecular regulation of inflammation and cell death. Cytokine. 2008;43:380–390. doi: 10.1016/j.cyto.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 48.Arai H, Furuya T, Yasuda T, Miura M, Mizuno Y, Mochizuki H. Neurotoxic effects of lipopolysaccharide on nigral dopaminergic neurons are mediated by microglial activation, interleukin-1beta, and expression of caspase-11 in mice. J Biol Chem. 2004;279:51647–51653. doi: 10.1074/jbc.M407328200. [DOI] [PubMed] [Google Scholar]
- 49.Hisahara S, Yuan J, Momoi T, Okano H, Miura M. Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J Exp Med. 2001;193:111–122. doi: 10.1084/jem.193.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sasagawa T, Hlaing M, Akaike T. Synergistic induction of apoptosis in murine hepatoma Hepa1-6 cells by IFN-gamma and TNF-alpha. Biochem Biophys Res Commun. 2000;272:674–680. doi: 10.1006/bbrc.2000.2835. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}