

Abstract
Synthetic triterpenoids, such as 2-cyano-3,12-dioxoolean-1, 9-dien-28-oic acid (CDDO) and its derivatives, are an extremely potent class of new anti-cancer therapeutic agents, characterized by high anti-tumor potency and low toxicity to normal tissues. This report is the first to investigate the effects of C-28 derivatives of CDDO on 22 pediatric solid tumor cell lines, including neuroblastoma, rhabdomyosarcoma, osteosarcoma, and Ewing's sarcoma. We determined IC50s in the range of 5–170 nM for inhibition of colony formation and DNA synthesis, and 110–630 nM for metabolic cell death and decrease in cell number, using the C-28 CDDO analogs, CDDO methyl ester (CDDO-Me), CDDO imidazolide (CDDO-Im), CDDO ethyl amide (CDDO-EA), CDDO trifluoroethyl amide (CDDO-TFEA), and CDDO diethylamide (CDDO-DE). After treatment of human neuroblastoma cells with CDDO-Me, cell cycle studies show depletion of the S-phase, while apoptosis studies show conformational activation and mitochondrial translocation of Bax protein, as well as activation of caspases -3 and -8. These data demonstrate the potential utility of CDDO analogs as promising novel therapeutic agents for high-risk pediatric solid tumors.
Keywords: triterpenoid, CDDO, Bax, apoptosis, neuroblastoma
Introduction
Neuroblastoma is the most common extra-cranial solid tumor of childhood and accounts for 7–10% of all pediatric cancers 1. Advances in early detection and surgery have largely been responsible for improved long-term survival rates, which plateau at 60% at 5 years.2 However, disease is metastatic at diagnosis in more than 70% of cases, and 85% of the tumors are high grade. The outlook for these high-risk patients remains poor, and prompts the development of novel agents to treat this disease. Currently, there is a great emphasis on the development of molecularly targeted therapies for cancer, including neuroblastoma.3,4 However, there is abundant evidence that multiple signaling pathways, including NFκB,5 Jak/STAT,6 cyclooxygenase,7,8 p539,10 p38-MAPK and others are active and linked directly to the viability,11 progression, and chemotherapy-resistance of neuroblastoma. These pathways work in interconnected networks, suggesting that multifunctional agents capable of targeting multiple pathways, rather than specific molecules, would be a more effective strategy to inhibit growth and induce cell death in neuroblastoma.
Synthetic triterpenoids have emerged recently as a potent class of small molecules that are highly effective in both the prevention and treatment of cancer.12 An oleanane triterpenoid, 2-cyano-3-,12-dioxoolean-1,9-dien-28-oic acid (CDDO) and its derivatives were originally developed as inhibitors of nitric oxide production in macrophages13-15 and as anti-proliferative agents for tumor cells.16 C-28 methyl ester (CDDO-Me) and C-28 imidazolide (CDDO-Im) derivatives of CDDO have demonstrated significant anti-tumor activities for various tumor types with IC50s in the nanomolar range.12 At the same time, very low, if any, side effects were detected at clinically-relevant concentrations of CDDO-Me and CDDO-Im.17 Other C-28 amide derivatives have also been synthesized;18 new compounds studied here include CDDO-Ethyl amide (CDDO-EA), CDDO-Diethyl amide (CDDO-DE) and CDDO-Trifluoroethyl amide (CDDO-TFEA).
It is now clear that the synthetic triterpenoids have the ability to modulate the function and expression of many relevant targets. This multifunctional behavior is in part based on the capacity of the triterpenoids to react with thiol groups by Michael addition,19 and underlies the ability of these agents to directly affect both NFκB20,21 and Jak/STAT signaling pathways,22 and to modulate expression of genes linked to oxidative stress and cell survival through the Nrf2-Keap1 signaling pathway.23 Interaction of CDDO triterpenoids with peroxisome proliferator-activated receptor gamma,24 alteration in Her-2/neu protein phosphorylation,25 inhibition of p-Akt, Notch-126, and direct interaction with mitochondrial membranes27 have also been described.
The efficacy of the triterpenoids has been demonstrated in preclinical studies that include models of prevention or treatment in various types of cancer,23,28-35 either alone or in combination with conventional chemotherapy in cancers that exhibit resistance to standard agents.
To date, no published reports have examined the activity of the synthetic triterpenoids in the spectrum of solid tumors that are most common to the pediatric age group. A recent report26 describes the effect of CDDO-Im and CDDO-Me in the SK-N-MC Askin's tumor cell line, described previously as neuroblastoma.36 Our current article is the first to describe the potency and mechanisms of activity of several novel synthetic triterpenoids in human neuroblastoma and other pediatric solid tumors, including rhabdomyosarcoma, Ewing's sarcoma and osteosarcoma. Exposure to the synthetic triterpenoids induces a rapid and dose-dependent induction of apoptosis that follows an invariant and early induction of phase II response genes linked to oxidative stress pathways in neuroblastoma. Cell death in neuroblastoma is associated with both the activation and mitochondrial accumulation of the pro-apoptotic protein Bax, and activation of the caspase family of cysteine proteases. The results presented here support the further development of synthetic triterpenoids for clinical evaluation as a new and needed approach to the treatment of these common forms of childhood malignancy.
Results
CDDO and derivatives inhibit proliferation and viability of neuroblastoma and Ewing's sarcoma cells at nanomolar concentrations. Using the RT-CES system, we tested the effect of CDDO-Me in a total of thirteen neuroblastoma cell lines, as well as in two rhabdomyosarcoma, two osteosarcoma and three Ewing's sarcoma cell lines. CDDO-Me was chosen as the compound being currently investigated in Phase I Clinical Trial. We found all tested cell lines to be sensitive to CDDO-Me in a dose and time dependent manner. The IC50 values determined from dose response curves generated at 24 h by this method ranged from 160 nM to 630 nM for all pediatric solid tumors that were evaluated (Table 1).
Table 1. IC50 (nM) of CDDO and its derivatives in pediatric solid tumor cell lines.
| Cell line | Type | RT-CES | CDDO-Me Colony forming |
3H-thymidine | CDDO Colony forming |
CDDO-Im Colony forming |
CDDO-TFEA Colony forming |
CDDO-EA Colony forming |
CDDO-DE Colony forming |
|---|---|---|---|---|---|---|---|---|---|
| HUVEC | Umbilical vein endothelial cells | 110 | |||||||
| RD | Rhabdomyosarcoma | 280 | |||||||
| RH41 | 219 | ||||||||
| SA-OS | Osteosarcoma | 300 | |||||||
| U2-OS | 415 | ||||||||
| RD-ES | Ewing's sarcoma | 309 | |||||||
| TC32 | 402 | ||||||||
| TC71 | 373 | ||||||||
| TC106 | 117 | 43 | >200 | 37 | 95 | 195 | 125 | ||
| CHP134 | Neuroblastoma | 201 | |||||||
| IMR32 | 232 | ||||||||
| IMR5 | 610 | ||||||||
| LAN1 | 160 | 35 | 75 | >200 | 35 | 85 | |||
| LAN5 | 250 | ||||||||
| NB-EB | 305 | ||||||||
| NB-1691 | 283 | 8 | 137 | >200 | 5 | 125 | |||
| SKNAS | 418 | 115 | 76 | >200 | 30 | 170 | 95 | 35 | |
| SKNSH | 630 | ||||||||
| SKNBE2 | 273 | ||||||||
| SKNDZ | 525 | ||||||||
| SKNFI | 465 | ||||||||
| SY5Y | 181 | ||||||||
| 15N | 25 | 30 | 25 | 5 | 35 |
IC50 are determined by various methods: RT-CES, 3H-thymidine incorporations and colony forming assays. The following compounds were used: CDDO-Me, CDDO, CDDO-Im, CDDO-TFEA, CDDO-EA and CDDO-DE.
The principle of RT-CES is based on an impedance measurement by electrodes placed in each well with cultured cells. Impedance is proportional to the area occupied by attached cells, which increases as cell proliferate and decreases when they die and detach during either late apoptosis or necrosis. The IC50 determined by the RT-CES assay indicates the concentration of the compound required to reduce the area occupied by attached cells by 50% from the time point when the drug is added (Table 1). An alternate assessment of the dose-dependent effects of CDDO-Me on SK-N-AS cell using a spectrophotometric assay with the WST-1 proliferation reagent (Roche) produced similar IC50 values (data not shown), suggesting that a decrease in the area occupied by cell population due to death correlates with the drop in metabolic viability of the cells.
Out of all cell lines tested in RT-CES assay, we chose three representative human neuroblastoma cell lines with various IC50 ranging from 160 to 418 nM (LAN-1, NB1691 and SK-N-AS). We added previously untested 15N neuroblastoma and TC106 Ewing's sarcoma cell lines to be tested for colony formation in the presence of four compounds: CDDO, CDDO-Im, CDDO-Me and CDDO-TFEA (Table 1, Fig. 1 and Fig. 2A-C). In addition, SK-N-AS and TC106 cells were tested for colony forming in the presence of CDDO-EA and CDDO-DE (Table 1 and Fig. 1). Neuroblastoma cell lines displayed significant sensitivity to CDDO-Im, CDDO-TFEA and CDDO-Me at concentrations less than 150 nM (see Table 1 for IC50s) Survival curves of and Fig. 2A–C for survival curves). SK-N-AS and TC106 cells were also sensitive to CDDO-EA and CDDO-DE at doses less than 200 nM. The parent compound, CDDO, had no inhibitory activity on colony formation by NB1691, LAN-1, SK-N-AS and TC106 cells when tested at concentrations less than 200 nM. However, it did inhibit colony formation by 15N cells within this concentration range.
Figure 1.
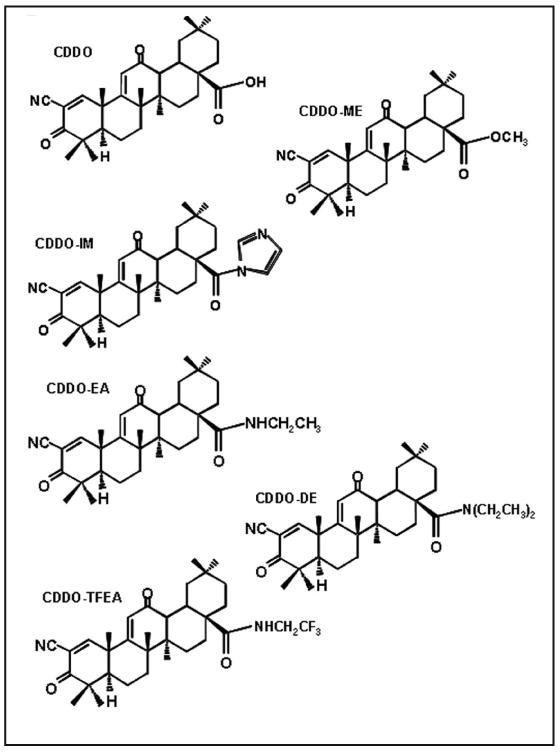
Chemical structures of studied triterpenoids: CDDO, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid; CDDO-Me, CDDO-28-methyl ester; CDDO-Im, CDDO-28-imidazolamide; CDDO-TFEA, CDDO-28-trifluoroethylamide; CDDO-EA, CDDO-28-ethylamide; and CDDO-DE, CDDO-28-diethylamide.
Figure 2.
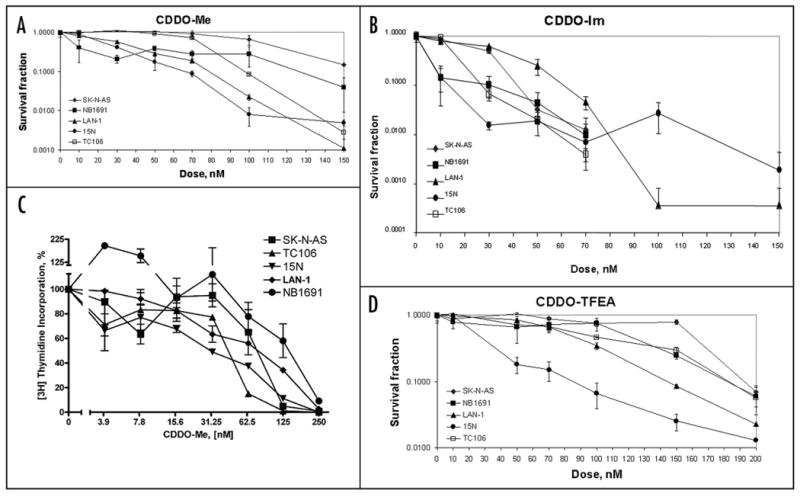
CDDO derivatives suppress the colony forming abilities of neuroblastoma cell lines. (A–C) colony forming assays were carried out as described in Materials and Methods. The following cell lines were tested: NB1691, 15N, LAN-1, SK-N-AS and TC106. The following triterpenoids were used: CDDO-Me (A), CDDO-Im (B) and CDDO-TFEA (C). Experiments were performed in triplicates. Error bars represent standard errors of survival fraction for each dose of the drug. (D) Inhibition of DNA synthesis by CDDO-Me in pediatric cancer cell lines as determined by thymidine incorporation assay. Thymidine incorporation assay was performed as described in Materials and Methods. The following cell lines were tested: NB1691, TC106, 15N, LAN-1 and SK-N-AS. Experiments were performed in triplicate. Error bars represent standard errors of means survival fraction for each dose of the drug.
The same five cell lines studied in the colony forming assay: neuroblastoma (NB1691, 15N, LAN-1 and SK-N-AS) and Ewing's sarcoma (TC106) were tested for 3H-thymidine incorporation into DNA after treatment with 0–250 nM CDDO-Me (Fig. 2D). In all cell lines, CDDO-Me decreased 3H-thymidine incorporation by 50% at concentrations of 30–150 nM, and by greater than 95% at 250 nM. As in the colony formation assay, the 15N cell line showed the highest sensitivity to CDDO-Me with an IC50 of ∼30 nM.
Neuroblastoma cells exposed to CDDO-Me, -Im and -TFEA show depletion of the S-phase population
To determine the effect of CDDO derivatives on cell cycle distribution, standard cell cycle analysis was performed on the neuroblastoma SK-N-AS cell line treated with CDDO-Me, -Im and -TFEA. Cells were exposed to 200 nM of triterpenoid for 24 h, at which time a BrdU incorporation assay was performed as described in Materials and Methods (Fig. 3A). A decrease of approximately 60 fold in S phase was found for all three compounds. Also, increases of approximately 1.5 fold in sub-G1/G0, which was indicative of DNA degradation and apoptosis, was observed (Fig. 3A). Cells treated with CDDO-Im, the most potent of all three compounds, showed 1.5 fold decreases in the G1/G0 phase.
Figure 3.

CDDO C-28 derivatives deplete the S-phase in SK-N-AS neuroblastoma cells. (A) Cell cycle profile (percentage of cells in each cell cycle phase) of SK-N-AS cells treated and control by FACS analysis. SK-N-AS cells were exposed to 200 nM of CDDO-Me, CDDO-IM, CDDO-TFEA or DMSO control for 24 hours. Cells were then pulsed with BrdU for 3 h. BrdU and total DNA content were evaluated by FACS analysis. Experiments were performed in triplicate. Error bars represent standard errors of means for each set of data. *p < 0.05, **p < 0.01, when compared to control. (B) CDDO-Me induces apoptotic morphological changes in neuroblastoma cells. Neuroblastoma cell lines 15N and SK-N-AS) were treated with 250 nM CDDO-Me for 10 hours. After treatment, cells were analyzed by cytospin. Black arrows, membrane blebbing; black arrowheads, nuclear fragmentation.
Neuroblastoma cells exhibit apoptotic morphology after CDDO-Me treatment. To study morphological and molecular apoptotic changes after treatment with triterpenoids, we chose CDDO-Me, since it displayed high biological activity in RT-CES, colony forming and 3H-thymidine incorporation assays and is currently studied in Phase I Clinical Trial. Neuroblastoma cell lines (15N and SK-N-AS) were treated with 250 nM CDDO-Me for 6 h. After treatment, cytospin slides were prepared and stained. Microscopy analysis revealed extensive nuclear fragmentation and membrane blebbing in treated 15N cells as early as 10 h after treatment (Fig. 3B). Similar membrane blebbing was detected in SK-N-AS cells, however, no significant nuclear fragmentation, at the 10 h time point was visible, although, some nuclei were pycnotic and had prominent lobes in the treated cells (Fig. 3B, arrows). This difference in responses to CDDO-Me in two cell lines correlates with higher sensitivity of 15N cells to CDDO-Me in comparison to SK-N-AS cells as determined by colony forming assay (Table 1, Fig. 2A).
CDDO-Me activates the Bax protein in neuroblastoma cells
Previous studies have described the pro-apoptotic activity of the synthetic triterpenoids, through mechanisms that include altered mitochondrial membrane potential and caspase activation.27,31,35,37,38 To examine the effects of CDDO-Me on mitochondrial membrane proteins and neuroblastoma viability, we first utilized the property of the 6A7 anti-Bax monoclonal antibody to bind the activated conformation of Bax protein.39,40 SK-N-AS and 15N cells were treated for 6 h with CDDO-Me (0–100 nM) (Fig. 4A). Staurosporine (1 μM) was used as a positive control. Cells were harvested and activated Bax was immunoprecipitated using the 6A7 monoclonal antibody. Western blot analysis of the immunoprecipitates demonstrated an approximate 10 to 15-fold increase in activated Bax with exposure to 10 and 50 nM of CDDO-Me in SK-N-AS, respectively, and approximately 15-fold increase after treatment with 1 μM staurosporine in comparison to untreated samples. These data indicate that Bax changes conformation and becomes active at as early as 6 h when cells are treated with as low as 10 nM of CDDO-Me (Fig. 4A). Interestingly, Bax activation in 15N was only detectable at 10 nM, 6h after treatment with CDDO-Me and not detectable at higher concentrations of CDDO-Me or 1 μM of staurosporine (Fig. 4A). This observation might reflect a complete translocation of the active form of Bax to the membrane (mitochondrial) fraction (Fig. 4B) in 15N cells, which was excluded from the immunoprecipitation reaction.
Figure 4.
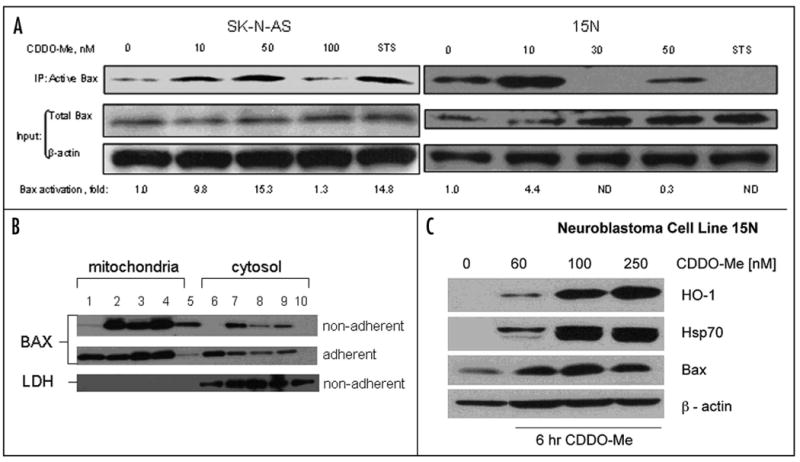
Exposure to CDDO-Me results in Bax conformation change and activation in SK-N-AS and 15N (A) neuroblastoma cells. Cells were treated and harvested as described in Material and Methods. Equal amount of total protein (1 mg) was used for IP with 6A7 Bax antibody. Whole cell extract (20 μg per sample) was analyzed in Input. Western blot analysis was performed using N20 Bax antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Equal loading in input and specificity of IP was verified using β-actin antibody. (B) CDDO-Me induces translocation of Bax protein into mitochondria in 15N neuroblastoma cells. 15N cells were treated with the following concentrations of CDDO-Me: 0 nM (lanes 1 and 6), 10 nM (lanes 2 and 7), 100 nM (lanes 3 and 8), 250 nM (lanes 4 and 9) and 1 μM of staurosporine (lanes 5 and 10) for 6 h. Floating (top) and adherent (bottom) cells were collected separately and mitochondrial and cytolic fractions were prepared and Western blotting was performed using anti-Bax antibody as described in Materials and Methods. (C) CDDO-Me induces steady-state levels of stress response proteins. Whole cell extracts of 15N cells prepared 6 h after addition of 0, 60, 100 or 250 nM of CDDO-Me. The membrane was probed with antibodies to the following proteins: HO-1 (top), Hsp70 (second from above), Bax (third from above), β-actin (bottom).
To monitor Bax translocation to mitochondria, we treated 15N cells with 10, 100 and 250 nM CDDO-Me or 1 μM staurosporine (Fig. 4B). Adherent (alive or early apoptotic) and floating (late apoptotic, dead or mitotic) cells were collected separately 6 h after treatment. Mitochondrial and cytosolic fractions were prepared and resolved on SDS-PAGE. Bax protein levels in all fractions were determined using Western blotting. A ∼2.7 fold decrease in Bax levels was detected in the cytosolic fraction of adherent 15N cells at doses of CDDO-Me as low as 10 nM, and a significant increase (∼23 fold) in Bax protein was noted in the mitochondrial fraction of the floating cells at 10 nM or higher of CDDO-Me. We observed similar Bax translocation in SK-N-AS cells (data not shown). Interestingly, there was an increase in Bax protein level in the cytosolic fraction of the floating cells (Fig. 4B, top panel, lines 6, 7, 8 and 9) after treatment with CDDO-Me, but not with staurosporine. This may be due to de novo synthesis of the Bax protein, and though characteristic of 15N cells, was not observed in SK-N-AS cells. It is noteworthy that 15N cells exhibit a relatively modest response to staurosporine in comparison to CDDO-Me (Fig. 4B, lanes 5 and 10). 15N cells may not activate Bax significantly in response to staurosporine (as observed in the Bax activation experiment, Fig. 4A), even though the total steady-state level of Bax is increased by 6 h after treatment with 1 μM of staurosporine (Fig. 4A, right panel).
To identify biomarkers of CDDO-Me activity in the treated cancer cells, we performed Western blot analyses of whole cell extracts of 15N cells (Fig. 4C), with antibodies to commonly induced stress-response proteins: HO-1, Hsp70 and Bax.41 CDDO-Me was added at 0, 60, 100 and 250 nM concentrations, cells were harvested 6 h later and whole cell extracts were prepared. We observed as much as a 100 fold increase in the steady-state levels of HO-1 and Hsp70 at 100 and 250 nM of CDDO-Me and ∼ 3–6 fold induction of Bax protein level at 60, 100 and 250 nM of CDDO-Me.
CDDO-Me induces dose-dependent activation of caspases and apoptosis in neuroblastoma cells
To determine whether G2/M arrest and Bax activation were coupled to caspase activation, caspase 3 and 8 activities were measured in SK-N-AS cells as described in Materials in Methods, and both time course and dose response curves were determined (Fig. 5A and B). Both caspase 3 and 8 were activated as early as 30 min after treatment with 250 nM CDDO-Me. Concentrations of 10–100 nM CDDO-Me were sufficient to induce both caspase 3 and 8 at the 18 h time point. However, concentrations in the range from 250 nM–1 μM CDDO-Me produced a greater effect (∼6–20 fold induction in caspase activity).
Figure 5.
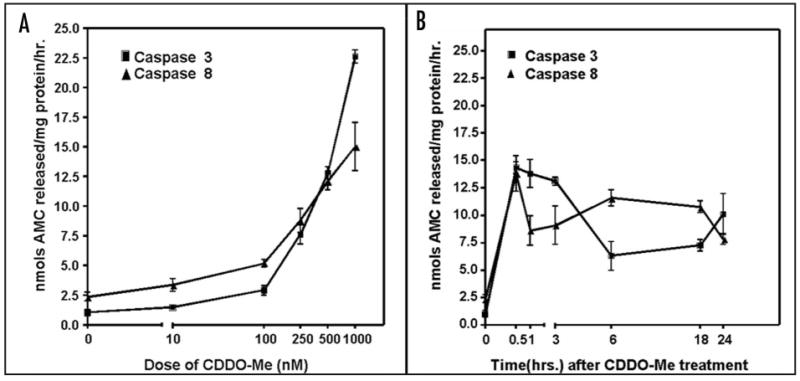
Activation of caspases 3 and 8 in SK-N-AS neuroblastoma cells following treatment with CDDO-Me. (A) dose-dependence after 6 h exposure to 0–250 nM of CDDO-Me; (B) time-courses 0–24 h of caspases 3 and 8 activation by 250 nM CDDO-Me. Experiments were performed in triplicate. Error bars represent standard errors of means for each point. AMC: 7-amino-4-methoxy coumarin.
Discussion
The synthetic triterpenoids are a promising class of new agents currently under development for the treatment and prevention of cancer, with CDDO-Me now in Phase II clinical trials in adults. Extensive efforts directed towards elucidating the molecular targets mediating effects on tumor cell growth and viability have uncovered the capacity of these agents to modulate an array of oncogenic, tumor suppressor, and stress response pathways that are also clearly involved in the pathogenesis of pediatric solid tumors. However, little or no attention has been directed at defining the activity of the triterpenoids in any preclinical model of childhood cancer. Our work is the first to describe cytotoxic activities of CDDO-derived triterpenoids in solid pediatric tumors.
We tested a total of 21 pediatric solid tumor cell lines for their responses to CDDO-derived triterpenoids in vitro. Three different methods were used (colony formation, thymidine incorporation and RT-CES) to determine the IC50 values for several derivatives. The concentration of synthetic triterpenoid required for the inhibition of colony formation, DNA replication and cell proliferation was consistently in the range of 5 to 130 nM, depending on the cell line and CDDO derivative.
A second set of IC50 values reflected the drug concentration required for 50% reduction in cell population determined by RT-CES system. These values ranged from 110 to 630 nM for CDDO-Me in all cell lines examined. In comparison, the IC50 defined by the WST-1 metabolic death assay for the commonly used drug doxorubicin is in the range of 1.5 to 10 μM (1–5 μg/ml) in neuroblastoma cell lines.42 Likewise, IC50 for cisplatin in primary neuroblastoma is in the range of 0.6–50 μM in the WST-1 assay.43,44 The IC50 values for CDDO-Me determined by both the colony forming and 3H-thymidine incorporation assays were significantly (4–35 fold) lower than the IC50 determined by RT-CES assay in the same cell lines. This indicates that the inhibition of proliferation and DNA replication determined using colony forming and 3H-thymidine incorporation assays requires significantly lower concentrations than cell detachment from substrate that are determined by the RT-CES and WST-1 assays.
It is important to note that the inhibitory activity of CDDO-Me, CDDO-TFEA, CDDO-DE, CDDO-EA and CDDO-Im is uniformly observed at nanomolar concentrations that are readily achieved in vivo without toxicity. CDDO-Im was the most potent of the whole panel of drugs, followed by CDDO-Me, CDDO-DE, CDDO-EA and CDDO-TFEA. The parental compound, CDDO, did not exhibit significant cytotoxicity at the doses used. This makes synthetic triterpenoids extremely attractive candidates to develop as potential adjuncts to standard chemotherapy agents currently in routine use for pediatric solid tumors.
Previous studies indicated a decrease in the cytosolic level of Bax protein following exposure with CDDO-Me.32 Here we investigated changes in Bax conformation and sub-cellular distribution. Conformational activation of the Bax protein was detected using the specific 6A7 antibody, which recognizes an epitope hidden in the inactive Bax form,45 but exposed when Bax is activated by an apoptotic stimulus. As little as 10 nM CDDO-Me was sufficient to rapidly activate Bax in neuroblastoma cells, resulting in translocation of Bax from cytosol to mitochondria. The activation of caspases 3 and 8 by sub-micromolar doses of CDDO-Me occurred rapidly and detected as early as 30 min post-treatment in neuroblastoma cells. Activation of caspases 8 and 3 by CDDO has been previously described to be cytochrome c-independent,34,46 which raises a question whether Bax activation by CDDO-Me is absolutely required for the induction of apoptosis. Such early activation of caspase-8 has been described previously47 and was shown to precede apoptotic alterations in mitochondrial membrane potential. Further studies required to determine whether CDDO-Me activates Bax and caspases independently or these two events depend on each other. Also, the event that results in Bax activation and precise mechanism of Bax activation by CDDO-Me remains to be identified. These events lead to classical morphologic changes of apoptotic cell death. We have not detected, however, any DNA laddering (data not shown), which suggested that a calcium-dependent DNAse is not activated by CDDO-Me and is not required for triterpenoid-induced apoptosis.
We observed an increase in sub-G1/G0 and severe depletion in the S-phase populations following treatment with CDDO-Me, -Im and -TFEA. A similar profile for cell cycle distribution has been observed in neuroblastoma cells when exposed to other agents that induced apoptosis, including N-(4 hydroxyphenyl)retinamide.48 Severe depletion in S-phase indicates that DNA synthesis is extremely sensitive to all studied triterpenoids. We also cannot rule out G2/M block in addition to the S-phase depletion, since cells trapped in the G2/M at the time of treatment by triterpenoids did not progress into G1/G0 phase.
The suppression of tumor growth in vivo by the triterpenoids could be a consequence of either direct anti-tumor effects and of indirect effects on the tumor microenvironment.48 CDDO-Me has been shown to inhibit angiogenesis in vivo,49 which may reflect inhibition of endothelial cell proliferation, a physiological process that happens in the early stages of angiogenesis, and is critical for tumor growth. HUVEC cells, which have been used as a surrogate marker for angiogenesis, are highly sensitive to CDDO-Me, with an IC50 of 110 nM after 24 h treatment. By now, it is well established that both non-human primates and human patients are much less susceptible than rodents to any toxic effects of CDDO-Me; in 28-day studies, monkeys will tolerate oral doses of CDDO-Me that are 10 times greater than those which cause significant toxicity in rodents.50 Regardless, the significant inhibition of tumor growth reported here supports the concept that derivatives of CDDO are a relevant class of new agents that should be moved into clinical development and evaluation for their activity against pediatric solid tumors.
Materials and Methods
Synthetic Triterpenoids
The triterpenoids were made as described.15,18,28 CDDO-TFEA, CDDO-EA, and CDDO-DE were synthesized at Ash-Stevens (Detroit, MI). Stock solutions were made (.01 M) in DMSO and frozen until used.
Cell culture
The following cell lines (American Type Culture Collection) were used: human umbilical vein endothelial cell line, HUVEC; rhabdomyosarcoma, RD and RH41; osteosarcoma, SA-OS and U2-OS; Ewing's sarcoma, RD-ES, TC32, TC106 and TC71; neuroblastoma, CHP-134, IMR5, NB1691, IMR32, LAN1, NB-EB, SK-N-AS, SY5Y, SK-N-SH, SK-N-BE2, SK-N-FI, 15N and SK-N-DZ, were grown 37°C, 5% CO2 in 1× RPMI 1640 with L-glutamine (Invitrogen, Carlsbad, CA) with 100 units/ml penicillin, 100μg/ml streptomycin, and 10% FBS (Gemini Bio-Products, West Sacramento, CA).
3H-thymidine incorporation assay
Cells were plated in triplicate at 1 × 105 cells per well in 96-well plates and cultured in 200 μl of medium containing CDDO-Me (0–250 nM) overnight. After 22 h, cells were pulsed for 2 h with 3H-thymidine (Perkin Elmer, Wellesley, MA) at 1 μCi/25 μl of medium per well. Samples were harvested onto a filter with the MicroBeta FilterMate (Perkin Elmer), prepared with Betaplate Scint (Perkin Elmer) and read with the Wallac MicroBeta Tri-Lux (Perkin Elmer).
Colony Forming Assay
Cells were plated in triplicate in 60 mm dishes at 4 × 103 cells/dish with serial dilutions of triterpenoid (0–200 nM) and incubated for least seven days until colonies of 50–100 cells/colony were formed. Medium was removed, colonies were stained with 1% Crystal Violet in 10% ethanol, rinsed twice with water, and counted using Quantity One Software (BioRad, Hercules, CA).
RT-CES assay
Continuous cell proliferation was measured by RT-CES (ACEA Biosciences Inc., San Diego, CA). Briefly, cells were seeded at 104 cells/well on 16-well e-plates (ACEA) in 200μl of medium. 20 h later, 20 μl of medium containing either CDDO-Me from 0.13 μM to 1 μM or DMSO was added. Proliferation was monitored every hour. Dose response curves were plotted using the ACEA RT-CES SP software (ACEA), and IC50s were calculated after 24 h exposure to drug.
Analysis of apoptotic morphology
15N and SK-N-AS cells were treated with CDDO-Me (250 nM) for 10 h, harvested and washed twice with cold PBS. Cells were then used to make cytospins and stained with Hema 3 Stain Set (Biochemical Sciences Inc., Swedesboro, NJ).
Western blot analysis
Cells were treated with 250 nM CDDO-Me for 6h and then harvested using 1× RIPA buffer containing Protease Inhibitor Cocktail (Roche, Indianapolis, IN) on ice for 20 min. Extracts (50 μg) were resolved by Novex Tris-glycine gels (4–20%; Invitrogen Corporation, Carlsbad, CA) and transferred to nitrocellulose membranes (Invitrogen), followed by Ponceau S staining and blocking by 5% non-fat milk in TBST (0.05% Tween 20, FisherBiotech, Fair Lawn, NJ). Membranes were probed with HO-1, Bax (Santa Cruz Biotechnology, Santa Cruz, CA), or Hsp70 (Cell Signaling Technology, Danvers, MA) antibodies overnight at 4°C. HRP-conjugated secondary antibodies (SouthernBiotech, Birmingham, AL) were then used followed by ECL detection (Pierce). Membranes were stripped (Pierce) and re-probed using β-actin antibody (Sigma, St. Louis, MI).
Immunoprecipitation of activated bax
Protein G Sepharose beads (20 μl, Pharmacia) were pre-incubated with 2 μg of anti-Bax monoclonal antibody (6A7) overnight on a rotating disk at 4°C. SK-N-AS cells (1 × 106 cells/100 mm dish) were treated with either CDDO-Im, CDDO-Me, staurosporine (STS) or an equal amount of DMSO and harvested in CHAPS buffer after either 6 or 24 h. Supernatants were collected (30 min, 14,000 rpm, benchtop centrifuge, 4°C). Beads were washed three times with 200 μl CHAPS buffer, and cell extracts (1 mg of total protein) were added to the 6A7 antibody/beads complexes. After 2 h incubation on a rotating disk at 4°C, beads were collected by centrifugation (1000 rpm, benchtop centrifuge, 3 sec, 4°C), washed X3 with 100 μl of CHAPS buffer, re-suspended in 40 μl of 2X Laemmli buffer, boiled for 5 min and resolved on SDS-PAGE alongside with the input samples (20 μg of total protein in CHAPS lysis buffer). Western blot analysis was performed using anti-Bax (N20) antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
Preparation of mitochondrial and cytosolic fractions
Cells were incubated for 6 h with 0, 10, 100, or 250 nM CDDO-Me or 1 μM of staurosporine in complete RPMI medium. Cells were then harvested and homogenized in 10 mM Tris HCl (pH 7.4) containing 0.32 M sucrose and 1 mM potassium EDTA using a Dounce homogenizer with four strokes of loosely fitting pestle. The homogenate was centrifuged at 1,000 g for 5 min to remove cell debris and nuclei. The mitochondria were collected by centrifuging the supernatant at 13,000 g for 5 minutes, with the resulting supernatant used as the source of cytosol. Both pellet and supernatant were subjected to Western blotting, as described above.
Caspase activity
15N cells were treated for 6 h with 0–250 nM of CDDO-Me, as well as a fixed dose of 250 nM over a time range 0–24 hours. Cells were collected by centrifugation at 600 g for 5 min at 4°C and then washed once with PBS. Cell pellets were re-suspended in 1× lysis buffer (250 mM HEPES, pH 7.4, 25 mM CHAPS, 25 mM DTT) at a concentration of 500 μl per 107 cells. The homogenates were centrifuged 14,000 g for 15 min at 4° and 5 μl cell extract was added to 5μl 1× assay buffer (20 mM HEPES, pH 7.4, 0.1% CHAPS, 5 mM DTT, 2 mM EDTA and 50% sucrose). Specific caspase inhibitors were added to the extracts and manufacturers' procedures were carried out from the Caspase 3 (or 8) Assay Kit (Sigma, St. Louis, MI). AMC release was determined by measuring fluorescence with excitation at 360 nm and emission at 460 nm using Victor3 1420 Multilabel Counter (PerkinElmer, Wellesley, MA).
Cell cycle analysis
SK-N-AS cells were treated with 200 nM CDDO-Me, CDDO-Im, or CDDO-TFEA for 24 h. Cells were treated with BrdU for three hours to allow for BrdU incorporation prior to harvesting. Cells were prepared for FACS with the BrdU Flow Kit (BD Pharmingen) following the manufactures procedures. Cells were stained with FITC anti-BrdU and 7-AAD. Cells were analyzed by FACS (FACSCalibur, BD Pharmingen).
Statistical Analysis
All experiments were performed a minimum of three times. Statistical evaluation was done with GRAPHPAD PRISM software, version 4.0 (GraphPad, San Diego). Significant differences between experiments were assessed by Student t-test. Standard errors of means were determined.
Acknowledgments
We thank Megan Padgett for expert assistance in the organization and preparation of the manuscript and Dr. Shigemi Matsuyama from CWRU for his advice on Bax immunoprecipitation. Support of our research by the NIH (R01 CA78814), National Foundation for Cancer Research, members of the Dartmouth Class of 1934, and Reata Pharmaceuticals is gratefully acknowledged.
References
- 1.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–16. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 2.Gutierrez JC, Fischer AC, Sola JE, Perez EA, Koniaris LG. Markedly improving survival of neuroblastoma: a 30-year analysis of 1,646 patients. Pediatr Surg Int. 2007;23:637–46. doi: 10.1007/s00383-007-1933-7. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe N, Sawai H, Ogihara-Umeda I, Tanada S, Kim EE, Yonekura Y, Sasaki Y. Molecular therapy of human neuroblastoma cells using Auger electrons of 111In-labeled N-myc antisense oligonucleotides. J Nucl Med. 2006;47:1670–7. [PubMed] [Google Scholar]
- 4.McKee AE, Thiele CJ. Targeting caspase 8 to reduce the formation of metastases in neuroblastoma. Expert Opin Ther Targets. 2006;10:703–8. doi: 10.1517/14728222.10.5.703. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong MB, Bian X, Liu Y, Subramanian C, Ratanaproeksa AB, Shao F, Yu VC, Kwok RP, Opipari AW, Castle VP. Signaling from p53 to NFkappa B determines the chemotherapy responsiveness of neuroblastoma. Neoplasia. 2006;8:964–74. [PMC free article] [PubMed] [Google Scholar]
- 6.Kuroda H, Sugimoto T, Horii Y, Sawada T. Signaling pathway of ciliary neurotrophic factor in neuroblastoma cell lines. Med Pediatr Oncol. 2001;36:118–21. doi: 10.1002/1096-911X(20010101)36:1<118::AID-MPO1028>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 7.Lau L, Hansford LM, Cheng LS, Hang M, Baruchel S, Kaplan DR, Irwin MS. Cyclooxygenase inhibitors modulate the p53/HDM2 pathway and enhance chemotherapy-induced apoptosis in neuroblastoma. Oncogene. 2007;26:1920–31. doi: 10.1038/sj.onc.1209981. [DOI] [PubMed] [Google Scholar]
- 8.Pyrko P, Soriano N, Kardosh A, Liu YT, Uddin J, Petasis NA, Hofman FM, Chen CS, Chen TC, Schonthal AH. Downregulation of survivin expression and concomitant induction of apoptosis by celecoxib and its non-cyclooxygenase-2-inhibitory analog, dimethyl-celecoxib (DMC), in tumor cells in vitro and in vivo. Mol Cancer. 2006;5:19. doi: 10.1186/1476-4598-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Becker K, Marchenko ND, Maurice M, Moll UM. Hyperubiquitylation of wild-type p53 contributes to cytoplasmic sequestration in neuroblastoma. Cell Death Differ. 2007 doi: 10.1038/sj.cdd.4402126. [DOI] [PubMed] [Google Scholar]
- 10.Slack AD, Chen Z, Ludwig AD, Hicks J, Shohet JM. MYCN-directed centrosome amplification requires MDM2-mediated suppression of p53 activity in neuroblastoma cells. Cancer Res. 2007;67:2448–55. doi: 10.1158/0008-5472.CAN-06-1661. [DOI] [PubMed] [Google Scholar]
- 11.Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 2007;67:735–45. doi: 10.1158/0008-5472.CAN-06-2201. [DOI] [PubMed] [Google Scholar]
- 12.Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7:357–69. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- 13.Honda T, Rounds BV, Gribble GW, Suh N, Wang Y, Sporn MB. Design and synthesis of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages. Bioorganic & medicinal chemistry letters. 1998;8:2711–4. doi: 10.1016/s0960-894x(98)00479-x. [DOI] [PubMed] [Google Scholar]
- 14.Honda T, Rounds BV, Bore L, Favaloro FG, Jr, Gribble GW, Suh N, Wang Y, Spor MB. Novel synthetic oleanane triterpenoids: a series of highly active inhibitors of nitric oxide production in mouse macrophages. Bioorganic & medicinal chemistry letters. 1999;9:3429–34. doi: 10.1016/s0960-894x(99)00623-x. [DOI] [PubMed] [Google Scholar]
- 15.Honda T, Rounds BV, Bore L, Finlay HJ, Favaloro FG, Jr, Suh N, Wang Y, Sporn MB, Gribble GW. Synthetic oleanane and ursane triterpenoids with modified rings A and C: a series of highly active inhibitors of nitric oxide production in mouse macrophages. Journal of medicinal chemistry. 2000;43:4233–46. doi: 10.1021/jm0002230. [DOI] [PubMed] [Google Scholar]
- 16.Suh N, Wang Y, Honda T, Gribble GW, Dmitrovsky E, Hickey WF, Maue RA, Place AE, Porter DM, Spinella MJ, Williams CR, Wu G, Dannenberg AJ, Flanders KC, Letterio JJ, Mangelsdorf DJ, Nathan CF, Nguyen L, Porter WW, Ren RF, Roberts AB, Roche NS, Subbaramaiah K, Sporn MB. A novel synthetic oleanane triterpenoid, 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, with potent differentiating, antiproliferative, and anti-inflammatory activity. Cancer Res. 1999;59:336–41. [PubMed] [Google Scholar]
- 17.Hyer ML, Croxton R, Krajewska M, Krajewski S, Kress CL, Lu M, Suh N, Sporn MB, Cryns VL, Zapata JM, Reed JC. Synthetic triterpenoids cooperate with tumor necrosis factor-related apoptosis-inducing ligand to induce apoptosis of breast cancer cells. Cancer Res. 2005;65:4799–808. doi: 10.1158/0008-5472.CAN-04-3319. [DOI] [PubMed] [Google Scholar]
- 18.Honda T, Honda Y, Favaloro FG, Jr, Gribble GW, Suh N, Place AE, Rendi MH, Sporn MB. A novel dicyanotriterpenoid, 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-onitrile, active at picomolar concentrations for inhibition of nitric oxide production. Bioorganic & medicinal chemistry letters. 2002;12:1027–30. doi: 10.1016/s0960-894x(02)00105-1. [DOI] [PubMed] [Google Scholar]
- 19.Couch RD, Browning RG, Honda T, Gribble GW, Wright DL, Sporn MB, Anderson AC. Studies on the reactivity of CDDO, a promising new chemopreventive and chemotherapeutic agent: implications for a molecular mechanism of action. Bioorg Med Chem Lett. 2005;15:2215–9. doi: 10.1016/j.bmcl.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 20.Yore MM, Liby KT, Honda T, Gribble GW, Sporn MB. The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor-kappaB activation through direct inhibition of IkappaB kinase beta. Mol Cancer Ther. 2006;5:3232–9. doi: 10.1158/1535-7163.MCT-06-0444. [DOI] [PubMed] [Google Scholar]
- 21.Ahmad R, Raina D, Meyer C, Kharbanda S, Kufe D. Triterpenoid CDDO-Me blocks the NFkappaB pathway by direct inhibition of IKKbeta on Cys-179. The Journal of biological chemistry. 2006;281:35764–9. doi: 10.1074/jbc.M607160200. [DOI] [PubMed] [Google Scholar]
- 22.Liby K, Voong N, Williams CR, Risingsong R, Royce DB, Honda T, Gribble GW, Sporn MB, Letterio JJ. The synthetic triterpenoid CDDO-Imidazolide suppresses STAT phosphorylation and induces apoptosis in myeloma and lung cancer cells. Clin Cancer Res. 2006;12:4288–93. doi: 10.1158/1078-0432.CCR-06-0215. [DOI] [PubMed] [Google Scholar]
- 23.Yates MS, Kwak MK, Egner PA, Groopman JD, Bodreddigari S, Sutter TR, Baumgartner KJ, Roebuck BD, Liby KT, Yore MM, Honda T, Gribble GW, Sporn MB, Kensler TW. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res. 2006;66:2488–94. doi: 10.1158/0008-5472.CAN-05-3823. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Porter WW, Suh N, Honda T, Gribble GW, Leesnitzer LM, Plunket KD, Mangelsdorf DJ, Blanchard SG, Willson TM, Sporn MB. Molecular endocrinology. Vol. 14. Baltimore, Md: 2000. A synthetic triterpenoid, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), is a ligand for the peroxisome proliferator-activated receptor gamma; pp. 1550–6. [DOI] [PubMed] [Google Scholar]
- 25.Konopleva M, Zhang W, Shi YX, McQueen T, Tsao T, Abdelrahim M, Munsell MF, Johansen M, Yu D, Madden T, Safe SH, Hung MC, Andreeff M. Synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces growth arrest in HER2-overexpressing breast cancer cells. Mol Cancer Ther. 2006;5:317–28. doi: 10.1158/1535-7163.MCT-05-0350. [DOI] [PubMed] [Google Scholar]
- 26.Gao X, Deeb D, Jiang H, Liu Y, Dulchavsky SA, Gautam SC. Synthetic triterpenoids inhibit growth and induce apoptosis in human glioblastoma and neuroblastoma cells through inhibition of prosurvival Akt, NFkappaB and Notch1 signaling. J Neurooncol. 2007;84:147–57. doi: 10.1007/s11060-007-9364-9. [DOI] [PubMed] [Google Scholar]
- 27.Samudio I, Konopleva M, Pelicano H, Huang P, Frolova O, Bornmann W, Ying Y, Evans R, Contractor R, Andreeff M. A novel mechanism of action of methyl-2-cyano-3,12 dioxoolean-1,9 diene-28-oate: direct permeabilization of the inner mitochondrial membrane to inhibit electron transport and induce apoptosis. Mol Pharmacol. 2006;69:1182–93. doi: 10.1124/mol.105.018051. [DOI] [PubMed] [Google Scholar]
- 28.Liby K, Royce DB, Williams CR, Risingsong R, Yore MM, Honda T, Gribble GW, Dmitrovsky E, Sporn TA, Sporn MB. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res. 2007;67:2414–9. doi: 10.1158/0008-5472.CAN-06-4534. [DOI] [PubMed] [Google Scholar]
- 29.Chintharlapalli S, Papineni S, Konopleva M, Andreef M, Samudio I, Safe S. 2-Cyano-3,12-dioxoolean-1,9-dien-28-oic acid and related compounds inhibit growth of colon cancer cells through peroxisome proliferator-activated receptor gamma-dependent and -independent pathways. Mol Pharmacol. 2005;68:119–28. doi: 10.1124/mol.105.011437. [DOI] [PubMed] [Google Scholar]
- 30.Temmink OH, Hoebe EK, Fukushima M, Peters GJ. Irinotecan-induced cytotoxicity to colon cancer cells in vitro is stimulated by pre-incubation with trifluorothymidine. Eur J Cancer. 2007;43:175–83. doi: 10.1016/j.ejca.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 31.Konopleva M, Tsao T, Estrov Z, Lee RM, Wang RY, Jackson CE, McQueen T, Monaco G, Munsell M, Belmont J, Kantarjian H, Sporn MB, Andreeff M. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid induces caspase-dependent and -independent apoptosis in acute myelogenous leukemia. Cancer Res. 2004;64:7927–35. doi: 10.1158/0008-5472.CAN-03-2402. [DOI] [PubMed] [Google Scholar]
- 32.Konopleva M, Tsao T, Ruvolo P, Stiouf I, Estrov Z, Leysath CE, Zhao S, Harris D, Chang S, Jackson CE, Munsell M, Suh N, Gribble G, Honda T, May WS, Sporn MB, Andreeff M. Novel triterpenoid CDDO-Me is a potent inducer of apoptosis and differentiation in acute myelogenous leukemia. Blood. 2002;99:326–35. doi: 10.1182/blood.v99.1.326. [DOI] [PubMed] [Google Scholar]
- 33.Kim KB, Lotan R, Yue P, Sporn MB, Suh N, Gribble GW, Honda T, Wu GS, Hong WK, Sun SY. Identification of a novel synthetic triterpenoid, methyl-2-cyano-3,12-dioxooleana-1,9-dien-28-oate, that potently induces caspase-mediated apoptosis in human lung cancer cells. Mol Cancer Ther. 2002;1:177–84. [PubMed] [Google Scholar]
- 34.Ito Y, Pandey P, Sporn MB, Datta R, Kharbanda S, Kufe D. The novel triterpenoid CDDO induces apoptosis and differentiation of human osteosarcoma cells by a caspase-8 dependent mechanism. Mol Pharmacol. 2001;59:1094–9. doi: 10.1124/mol.59.5.1094. [DOI] [PubMed] [Google Scholar]
- 35.Samudio I, Konopleva M, Hail N, Jr, Shi YX, McQueen T, Hsu T, Evans R, Honda T, Gribble GW, Sporn M, Gilbert HF, Safe S, Andreeff M. 2-Cyano-3,12-dioxooleana-1-,9-dien-28-imidazolide (CDDO-Im) directly targets mitochondrial glutathione to induce apoptosis in pancreatic cancer. The Journal of biological chemistry. 2005;280:36273–82. doi: 10.1074/jbc.M507518200. [DOI] [PubMed] [Google Scholar]
- 36.Dunn T, Praissman L, Hagag N, Viola MV. ERG gene is translocated in an Ewing's sarcoma cell line. Cancer genetics and cytogenetics. 1994;76:19–22. doi: 10.1016/0165-4608(94)90063-9. [DOI] [PubMed] [Google Scholar]
- 37.Brookes PS, Morse K, Ray D, Tompkins A, Young SM, Hilchey S, Salim S, Konopleva M, Andreeff M, Phipps R, Bernstein SH. The triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid and its derivatives elicit human lymphoid cell apoptosis through a novel pathway involving the unregulated mitochondrial permeability transition pore. Cancer Res. 2007;67:1793–802. doi: 10.1158/0008-5472.CAN-06-2678. [DOI] [PubMed] [Google Scholar]
- 38.Inoue S, Snowden RT, Dyer MJ, Cohen GM. CDDO induces apoptosis via the intrinsic pathway in lymphoid cells. Leukemia. 2004;18:948–52. doi: 10.1038/sj.leu.2403328. [DOI] [PubMed] [Google Scholar]
- 39.Hsu YT, Wolter KG, Youle RJ. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc Natl Acad Sci USA. 1997;94:3668–72. doi: 10.1073/pnas.94.8.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chitambar CR, Wereley JP, Matsuyama S. Gallium-induced cell death in lymphoma: role of transferrin receptor cycling, involvement of Bax and the mitochondria, and effects of proteasome inhibition. Mol Cancer Ther. 2006;5:2834–43. doi: 10.1158/1535-7163.MCT-06-0285. [DOI] [PubMed] [Google Scholar]
- 41.Crosby LM, Hyder KS, DeAngelo AB, Kepler TB, Gaskill B, Benavides GR, Yoon L, Morgan KT. Morphologic analysis correlates with gene expression changes in cultured F344 rat mesothelial cells. Toxicol Appl Pharmacol. 2000;169:205–21. doi: 10.1006/taap.2000.9049. [DOI] [PubMed] [Google Scholar]
- 42.Hamasaki K, Kogure K, Ohwada K. A biological method for the quantitative measurement of tetrodotoxin (TTX): tissue culture bioassay in combination with a water-soluble tetrazolium salt. Toxicon. 1996;34:490–5. doi: 10.1016/0041-0101(95)00151-4. [DOI] [PubMed] [Google Scholar]
- 43.Woessmann W, Chen X, Borkhardt A. Ras-mediated activation of ERK by cisplatin induces cell death independently of p53 in osteosarcoma and neuroblastoma cell lines. Cancer Chemother Pharmacol. 2002;50:397–404. doi: 10.1007/s00280-002-0502-y. [DOI] [PubMed] [Google Scholar]
- 44.Rosenhagen MC, Soti C, Schmidt U, Wochnik GM, Hartl FU, Holsboer F, Young JC, Rein T. The heat shock protein 90-targeting drug cisplatin selectively inhibits steroid receptor activation. Mol Endocrinol. 2003;17:1991–2001. doi: 10.1210/me.2003-0141. [DOI] [PubMed] [Google Scholar]
- 45.Peyerl FW, Dai S, Murphy GA, Crawford F, White J, Marrack P, Kappler JW. Elucidation of some Bax conformational changes through crystallization of an antibody-peptide complex. Cell Death Differ. 2007;14:447–52. doi: 10.1038/sj.cdd.4402025. [DOI] [PubMed] [Google Scholar]
- 46.Ito Y, Pandey P, Place A, Sporn MB, Gribble GW, Honda T, Kharbanda S, Kufe D. The novel triterpenoid 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid induces apoptosis of human myeloid leukemia cells by a caspase-8-dependent mechanism. Cell Growth Differ. 2000;11:261–7. [PubMed] [Google Scholar]
- 47.Weglarczyk K, Baran J, Zembala M, Pryjma J. Caspase-8 activation precedes alterations of mitochondrial membrane potential during monocyte apoptosis induced by phagocytosis and killing of Staphylococcus aureus. Infection and immunity. 2004;72:2590–7. doi: 10.1128/IAI.72.5.2590-2597.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–47. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 49.Vannini N, Lorusso G, Cammarota R, Barberis M, Noonan DM, Sporn MB, Albini A. The synthetic oleanane triterpenoid, CDDO-methyl ester, is a potent antiangiogenic agent. Mol Cancer Ther. 2007;6:3139–46. doi: 10.1158/1535-7163.MCT-07-0451. [DOI] [PubMed] [Google Scholar]
- 50.Kral R, M C, Abrahams R. Rodent and Primate Toxicity Studies of Oral RTA 402 (CDDO-Me), A Novel Agent with Anti-cancer and Anti-mucositis Activities. Abstracts of 2005 EORTC/AACR/NCI Meeting on Molecular Targets and Cancer Therapeutics; Philadelphia, PA. 2005. [Google Scholar]