

Abstract
Cancer-associated epithelial to mesenchymal transition (EMT) is crucial for invasion and metastasis. Molecular hallmarks of EMT include down-regulation of the epithelial adhesion protein E-cadherin, de-novo expression of N-cadherin and the mesenchymal intermediate filament proteins vimentin and fibronectin. Expression of HPV16 E7 in normal human epithelial cells caused increased levels of vimentin and fibronectin, whereas the epithelial adhesion protein E-cadherin was expressed at decreased levels. Similar patterns of vimentin, fibronectin and E-cadherin expression were also detected in cells expressing HPV16 E6 and E7 or the entire HPV16 early transcriptional unit. HPV16 E6 and E7 were each able to induce N-cadherin expression. Interestingly, these changes in expression levels of EMT associated proteins are not similarly reflected at the level of mRNA expression, suggesting that HPV16 oncoproteins may also modulate EMT through non-transcriptional mechanisms. Hence, HPV16 oncoproteins may contribute to malignant progression through EMT induction.
Introduction
Human papillomaviruses (HPVs) are small DNA viruses that infect basal epithelial cells and cause hyperproliferative lesions. Of the ∼140 described HPV genotypes approximately 40 are associated with infections of mucosal epithelia and are further classified into high- and low-risk groups based on the relative malignant potential of the lesions that they cause. Whereas low-risk HPVs, such as HPV6 and 11, cause benign genital warts, high-risk HPVs, such as HPV16 and HPV18, cause premalignant squamous intraepithelial neoplasias that can progress to cervical carcinomas (reviewed in zur Hausen, 2002). Integration of high-risk HPV genomes into a host cell chromosome is a frequent hallmark of malignant progression and leads to persistent, deregulated expression of the E6 and E7 oncoproteins, which is necessary and sufficient for induction and maintenance of the transformed phenotype (reviewed in Munger et al., 2004). High-risk HPV E6 and E7 oncoproteins have neither enzymatic nor specific DNA binding activities, and function by perturbing host cellular regulatory networks. High-risk HPV E7 proteins bind and induce the degradation of the hypophosphorylated, growth suppressive form of the retinoblastoma tumor suppressor protein (pRB) (Boyer et al., 1996; Dyson et al., 1992; Dyson et al., 1989; Jones et al., 1997) causing persistent activation of E2F transcription factors. This results in aberrant S-phase entry. In addition, the HPV E7 oncoprotein can dysregulate apoptosis, abrogate cell cycle arrest in response to DNA damage, differentiation or cytostatic cytokines and cause genomic instability through induction of centrosome duplication errors and other mechanisms (reviewed in McLaughlin-Drubin et al., 2009). High-risk HPV E6 proteins in complex with E6-associated protein (E6AP) target the p53 tumor suppressor protein for degradation (Scheffner et al., 1990). In addition, p53-independent activities of E6 such as telomerase activation (Klingelhutz et al., 1996), association with PDZ proteins (Kiyono et al., 1997; Lee et al., 1997) and other cellular target proteins (reviewed in Howie et al., 2009) also contribute to the oncogenic activities of high-risk HPV E6 proteins.
Epithelial cells, the targets of HPV infection, are connected by specialized membrane structures, such as adherens junctions, tight junctions and desmosomes, that are characterized by a localized distribution of adhesion molecules including cadherins, catenins and integrins. Thus, epithelial cells build a lateral layered belt by maintaining intimate contact with neighboring cells. In contrast, mesenchymal cells are spindle-shaped, anchorage-independent, motile cells that neither form tightly connected nor polarized, structures. Epithelial cells can convert into mesenchymal cells by a multi-step process known as epithelial to mesenchymal transition (EMT). Cells undergoing EMT lose their typical epithelial characteristics and acquire mesenchymal properties. This process is reversible and MET denotes the process whereby mesenchymal cells reacquire epithelial characteristics. EMT importantly contributes to several physiological and pathological processes, including embryogenesis, inflammation and cancer progression, respectively. Since EMT is integral to the degradation of the basal membrane, this process is also involved in the intravasation of cells into blood or lymphatic vessels in order to form micrometastases. Therefore, it has been postulated that EMT is necessary for tumor dedifferentiation, a step that is generally associated with high invasion potential and chemoresistance (reviewed in Thiery et al., 2006).
The onset of EMT as well as the spectrum of changes that subsequently occur, are triggered by extensive crosstalk between signaling pathways and have two common endpoints: (1) downregulation of E-cadherin (Hirohashi et al., 2003) and (2) expression of EMT associated genes. E-cadherin is a calcium-dependent integral membrane glycoprotein that connects via undercoating proteins such as catenins to actin filaments. E-cadherin can function as a tumor suppressor by mediating invasion-suppressing properties and maintaining the epithelial phenotype. While E-cadherin expression is detected in well-differentiated carcinomas, expression is reduced in many undifferentiated tumors (Hirohashi et al., 2003). A characteristic of EMT is a “cadherin switch” from expression of E-cadherin to N-cadherin. N-cadherin is a pro-migratory protein with expression restricted to neural tissues as well as fibroblasts, osteoblasts, endothelial, retinal and mesothelial cells. It is typically not expressed in epithelial cells but is detected in certain carcinomas (Derycke et al., 2004) where it is thought to promote angiogenesis and adhesion (reviewed in Jeanes et al., 2008). Another hallmark of EMT is the increased expression of intermediate filament proteins such as vimentin and fibronectin. Vimentin is involved in anchoring organelles in the cytoplasm and is often expressed in growth factor stimulated epithelial cells. In cancers, vimentin expression is associated with a dedifferentiated, malignant phenotype, increased motility, invasive ability and poor clinical prognosis (reviewed in Kokkinos et al., 2007). Fibronectin is a key component of extracellular matrix and acts as a binding platform for cell surface receptors; adhesion of cancer cells to fibronectin enhances their tumorigenicity and apoptosis resistance (Han et al., 2006).
Previous studies have suggested that high-risk HPV oncoproteins may contribute to EMT. For example, HPV16 E6/E7 immortalized human gingival keratinocytes display a fibroblast-like phenotype after ethanol treatment (Chamulitrat et al., 2003). HPV18 E6 expression was correlated with a fibroblastoid morphology in SV40-immortalized human keratinocytes (Watson et al., 2003). In addition, a microarray analysis indicated modulation of a significant number of genes involved in keratinocyte differentiation and EMT by HPV16 E6 (Duffy et al., 2003). Here we report that the HPV16 E6 and E7 oncoproteins can each independently contribute to induction of EMT-associated molecular changes. Since most of these some of the observed alterations are not observed at the mRNA level, non-transcriptional mechanisms likely contribute to HPV16 E6 and/or E7 induced EMT as well.
Materials and Methods
Plasmids
The following human β-actin expression plasmids were used: p1435 (HPV16 E7), p1436 (HPV16 E6), p1321 (HPV16 E6/E7), p1319 (HPV16 early region) and the parental vector p1318 (Munger et al., 1989).
Cell lines and culture
Primary human foreskin keratinocytes (HFK) were isolated from anonymous neonatal circumcisions. Two to five human foreskins per culture were washed with phosphate-buffered saline (PBS) and diced into small fragments. After incubation in 25mg dispase/ml PBS at 4°C overnight, the epidermis was separated from the dermis, minced and trypsinized into a single-cell suspension. HKFs were maintained in keratinocyte serum free medium (KSF-M; Invitrogen) supplemented with human recombinant epidermal growth factor 1-53, bovine pituitary extract, 100U/ml penicillin, 100 μg/ml streptomycin, 10 μg/ml gentamicin, and 0.5 μg/ml amphotericin B. HFKs with stable expression of HPV16 E6, E7, E6 and E7 or the early region (ER) were created by transfecting primary HFK populations with the appropriate expression plasmids and pCDNA3.1 (Invitrogen) at 5:1 ratio using the Amaxa Human Keratinocyte Nucleofector kit (Amaxa Biosystems) according to the manufacturer's instructions. Following selection with G418, the cells were cultured in the KSF-M. Normal oral keratinocytes (NOK) immortalized by human telomerase (hTERT) and NOK hTERT/E7 (Piboonniyom et al., 2003) were maintained in KSF-M.
Primary human foreskin fibroblasts (HFFs) were generated from anonymous newborn circumcisions. Dermis that was separated from the epidermis during keratinocyte preparation was minced and incubated in a 2mg/ml collagenase solution in serum free DMEM and trypsin (4:1) for four hours at 37°C. HFFs were maintained in DMEM supplemented with 10% calf serum, 50U/ml penicillin, and 50μg/ml streptomycin.
HPV16-positive CaSki human cervical carcinoma were obtained from ATCC and cultured in Dulbecco's modified Eagle medium (DMEM, Invitrogen), 10% fetal bovine serum, and 1% penicillin/streptomycin.
Immunoblotting
Cells were extracted in RIPA buffer (150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50mM Tris-HCl pH 8.0) supplemented with one complete EDTA-free protease inhibitor cocktail tablet (Roche) per 25 ml lysis buffer. Cells were scraped on ice and lysates were cleared by centrifugation at 16,000 × g for 15 min at 4°C. Protein concentrations were measured using the Bradford method (Bio-Rad). Samples containing 50 to 200μg of protein were boiled in SDS-containing sample buffer, separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride membranes (Millipore). Membranes were blocked for one hour in 5% nonfat dry milk and 1% BSA in TNET buffer (50 mM Tris HCl, 150 mM NaCl, 5 mM EDTA, 0.1% Tween-20) or TBST buffer (10 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20) and probed with the appropriate antibody. Primary antibodies were used at the following dilutions: a mixture of 8C9 (1:150; Zymed/Invitrogen) and ED17 (1:200; Santa Cruz) for HPV16 E7, fibronectin (sc-59826, 1:50; Santa Cruz), vimentin (V9, 1:1,000; Chemicon), N-cadherin (610920, 1:2,500; BD), E-cadherin (610181, 1:250; BD), p53 (OP43, 1:1,000; Oncogene), pRB (sc-7905, 1:500; Santa Cruz), β-actin (CP01, 1:1,000; Calbiochem) and α-tubulin (T6199, 1:1,000; Sigma). Appropriate secondary anti-mouse horseradish peroxidase-conjugated antibodies were used (1:10,000; Amersham). Proteins were visualized by enhanced chemiluminescence (Western Lightning Chemiluminescence Reagent Plus, Perkin-Elmer Life Science, Inc.) and exposed on BioMax XAR film (Kodak) or electronically acquired with a 4000R image station (Kodak). Band quantification analysis was performed using the Kodak imaging software, version 4.0.
Immunofluorescence
Cells were plated on glass coverslips, fixed with 3% paraformaldehyde in phosphate-buffered saline (PBS), washed with PBS, and permeabilized with 1% Triton X-100 in PBS for 10 min at room temperature. Following permeabilization the cells were washed with wash buffer (PBS) containing 0.02% saponin, 0.05% sodium azide, and 1% bovine serum albumin (BSA), blocked with 10% normal goat serum (Jackson Immunoresearch) at room temperature for an hour and incubated for 45 min with N-cadherin antibody (610920, BD) at 37°C. Blocking solution and antibodies were diluted in wash buffer. Secondary antibody was an Alexa Fluor 488-conjugated goat anti-mouse antibody (1:1,000; Invitrogen). Nuclei were counterstained with Hoechst 33258 (Sigma) and TO-PRO-3 (Invitrogen) dyes at a 1:2,000 dilution. Phalloidin staining was performed using a 1:100 dilution of rhodamine-conjugated phalloidin (Molecular Probes/Invitrogen) followed by counterstaining with Hoechst and TO-PRO-3.
siRNA experiments
HFFs were grown on coverslips for 24 hrs until they were approximately 40%confluent, followed by transfection with N-cadherin specific siRNA oligonucleotides (“SMARTpool”, Millipore) or a scrambled control siRNA (Millipore) using Lipofectamine2000 (Invitrogen). After incubation for 48hrs the cells were fixed and stained as described above.
Microarray analysis
Messenger RNA was isolated with the Agilent RNA Isolation kit accordingly to the manufacturer's protocol from four independent, passage and donor matched HFK populations with stable expression of HPV16 E6 or E7 and control vector transfected primary HFKs. RNA quality was evaluated using the Agilent BioAnalyzer 2100 and gene expression profiles were assayed using the Affymetrix U133Plus2.0 GeneChip™ according to the manufacturer's instructions. Data were normalized using RMA and array quality assessed using NUSE and RLE scores (Irizarry et al., 2003); a variety of statistical and data mining analyses were performed to identify genes and patterns of gene expression correlating with the various phenotypes. Log2 ratios of gene expression in HPV16 E6 versus the vector control cells, and HPV16 E7 expressing versus the empty vector control cells were calculated and mean-centered and subjected to average-linkage hierarchical clustering using a Euclidean distance metric (Eisen et al., 1998; Michaels et al., 1998; Wen et al., 1998) using the heatmap2 function in the gplots package (http://cran.r-project.org/web/packages/gplots/index.html) implemented in the open source statistical computing language R (http://www.r-project.org/). All gene expression data were collected in accordance with the MIAME standards (Brazma et al., 2001) and have been deposited with the ArrayExpress database under accession number (YYY).
Real-time PCR
Quantitative real time PCR was performed using the QuantiTect SYBRGreen PCR kit (Qiagen) following the manufacturer's instructions. Samples were analyzed on a 7300 Fast RT-PCR system (Applied BioSystems) in triplicate together with the matching negative controls. Primers were: SNAI1 forward ATG AGG AAT CTG GCT GCT GT and reverse CAG GAG AAA ATG CCT TTG GA; TWIST1 forward TGC ATG CAT TCT CAA GAG GT and reverse CTA TGG TTT TGC AGG CCA GT; FN1 forward GGA GTT GAT TAT ACC ATC ACT G and reverse TTT CTG TTT GAT CTG GAC CT; CDH2 forward TGG GAA TCC GAC GAA TGG and reverse TGC AGA TCG GAC CGG ATA CT; CDH1 forward TGA AGG TGA CAG AGC CTC TGG AT and reverse TGG GTG AAT TCG GGC TTG TT; GAP-DH forward GAT TCC ACC CAT GGC AAA TCC and reverse TGG GAT TTC CAT TGA TGA CAA G.
Results
HPV16 E6 and E7 modulate expression of genes involved in EMT and differentiation-associated processes in human foreskin keratinocytes
Whereas HPV16 E6 expression in epithelial cells has previously been shown to upregulate several genes that are normally expressed in mesenchymal lineages (Duffy et al., 2003), the role of HPV16 E7 in modulating the expression of EMT related genes has not been extensively studied. To address this issue in more detail, we analyzed mRNA microarray expression data obtained from HFK populations with stable expression of HPV16 E6 or E7 compared to donor and passage matched vector control cells. We focused on a selection of 26 genes representing epithelial and mesenchymal proteins and regulators involved in EMT associated processes or differentiation. Genes exhibiting similar patterns of expression changes in HPV16 E6 versus HPV16 E7 expressing HFKs, each compared to control HFKs were identified and grouped using a hierarchical clustering algorithm. This analysis suggested only subtle changes in the expression levels of these genes in E6 or E7 expressing cells (Figure 1A). This was surprising and we validated expression of small subgroup of these genes by quantitative reverse transcription PCR (Figure 1B). Collectively these analyses revealed that whereas HPV16 E6 and E7 expression each induce subtle alterations in gene expression levels of EMT associated genes, E6 or E7 expression does not result in dramatic alteration of mRNA levels of classical mesenchymal proteins, such as vimentin, fibronectin, fibroblast-surface-protein-1 (FSP-1, S100A4) and smooth-muscle actin. In some cases where mRNA levels changed, HPV16 E6 and E7 expression appeared to have opposing effects; expression of TGF-β receptor II mRNA is upregulated by HPV16 E7 and downregulated by HPV16 E6, whereas TGF-β receptor III mRNA is downregulated by HPV16 E7 and upregulated by HPV16 E6. Based on these findings we wanted to determine how expression of some of these EMT associated genes is modulated at the level of protein expression and when HPV16 E6 and E7 are co-expressed.
Figure 1.
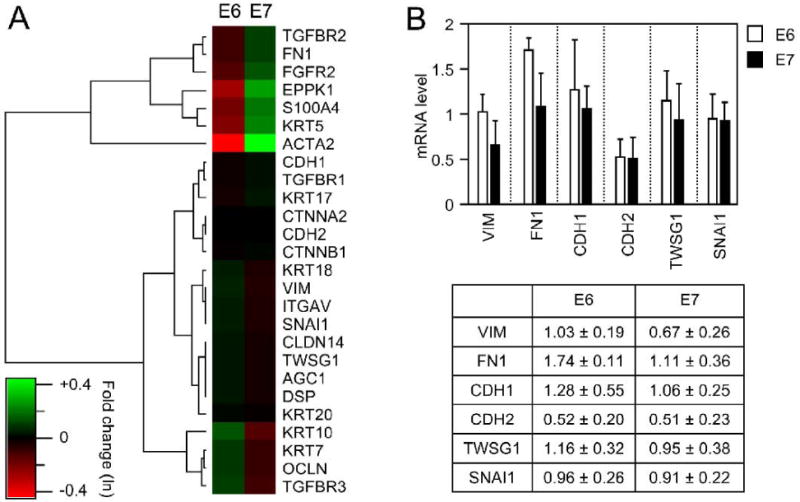
Differential expression of EMT associated genes in HPV16 E6 and HPV16 E7 expressing cells. A. GeneChip array analysis was performed with mRNA isolated from two different passage and donor matched populations. Heatmap representation of the mean-standardized log2 expression ratios of 26 EMT and keratinocyte differentiation specific genes expressed in HPV16 E6 expressing versus control (E6) and HPV16 E7 versus the control (E7) populations. Agglomerative hierarchical clustering based on Euclidean distance was used to identify genes with similar expression patterns in HPV16 E6 and E7 expressing cells. Genes are designated by official gene symbols: CTNNA2, α-catenin; CTNNBL1, β-catenin; CDH2, N-cadherin; FN1, fibronectin; VIM, vimentin; ACTA2, smooth muscle actin; S100A4, S100 calcium binding protein/ fibroblast-surface-protein-1; AGC1, aggrecan 1; FGFR2, fibroblast growth factor receptor 2; SNAI1, snail homolog 1; TWSG1, twisted gastrulation homolog 1; ITGAV, vitronectin; DSP, desmoplakin; OCLN, occluding; CLDN14, claudin 14; CDH1, E-cadherin; EPPK1, epiplakin; KRT, keratin; TGFBR, transforming growth factor beta receptor. B. Quantitative reverse transcription PCR analysis for a subset of EMT marker proteins. The columns in the bar graph represent the fold change of gene expression for vimentin (VIM), E-cad (CDH1), N-cad (CDH2), fibronectin (FN), twist (TWSG1) and snail (SNAI1) relative to control vector transfected cells.
HPV16 E7 is a modulator of EMT associated proteins in normal human epithelial cells
EMT is often manifested by morphological alterations whereby epithelial cells acquire a fibroblastoid phenotype. To be able to directly compare these alterations to our mRNA analysis we analyzed E-cadherin levels in a set of passage and donor matched populations of HFK with stable expression of HPV16 E6 and/or E7 by Western blot analyses. Control vector transfected HFKs were used as controls. Similar to what we observed with the NOKs, E-cadherin was expressed in HFKs and expression of HPV16 E6 or HPV16 E7 each resulted in reduced E-cadherin expression, and E-cadherin expression was even more dramatically reduced in HPV16 E6/E7 expressing HFKs (Figure 2A). The magnitude of these effects was quite surprising given the much more subtle changes in mRNA levels in HPV16 E7 expressing HFKs.
Figure 2.
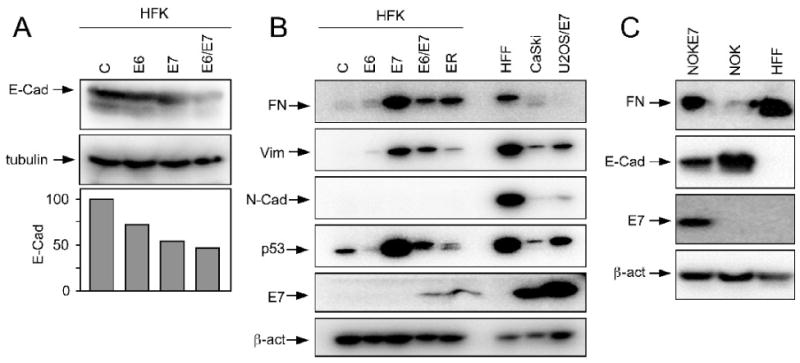
Expression of EMT associated proteins in HPV oncogene expressing human keratinocytes. A. Western blot analysis of E-cadherin (E-Cad) expression in HPV16 E6 and/or E7 expressing primary human keratinocytes (HFK) as compared to matched control HFKs. Primary human foreskin fibroblasts (HFF) are shown as a control. A tubulin blot is shown as a loading control. Quantification of E-cadherin levels normalized to tubulin expression is shown underneath B. Western blot analysis of fibronectin (FN), vimentin (Vim), and N-cadherin (N-Cad) expression in HPV16 E6 and/or E7 and early region (ER) expressing primary human keratinocytes (HFK) as compared to matched control HFKs. Primary human foreskin fibroblasts (HFF), HPV16 positive CaSki cervical carcinoma cells and HPV16 E7 expressing U2OS human osteosarcoma cells (U2OS/E7) were used as controls. An E7 blot documents HPV16 E7 expression in the appropriate cell populations; expression of p53 (destabilized by E6 and stabilized by E7) is shown as a surrogate marker for HPV16 oncogene expression. A β-actin blot is shown as a loading control. C. Western blot analysis of fibronectin (FN) and E-cadherin (E-Cad) expression in hTert immortalized normal human oral epithelial cells (NOK) and a matched population with HPV16 E7 expressing (NOK-E7). Primary human foreskin fibroblasts (HFF) are shown as a control. An E7 blot documents HPV16 E7 expression in NOK-E7, and β-actin expression is shown as a loading control.
Based on these results we also compared expression of the mesenchymal marker proteins fibronectin and vimentin as well as N-cadherin in HPV16 oncoprotein expressing HFKs. As expected, expression of these proteins was very low in control vector transfected HFKs, but they were robustly expressed in HFFs. HPV16 E6 expressing cells only showed a subtle increase in fibronectin and vimentin expression, but expression of these proteins was markedly increased in the HPV16 E7 expressing cells. While the levels were somewhat lower in HPV16 E6/E7 expressing HFKs or HFKs transfected with the entire early region of HPV16, they remained higher than in control HFKs or HFKs with expression of HPV16 E6 only. N-cadherin was not consistently expressed at readily detectable levels in any of the HFK populations (Figure 2B). It is worth noting that these alterations in EMT protein expression are not accompanied by similar changes in the abundance of the corresponding mRNAs (Figure 1). Moreover, HPV16 E7 expression in these cells is considerably lower than in the HPV16 positive CaSki cervical carcinoma cell line and hence the observed effects are not a consequence of high-level HPV oncoprotein expression. Of note, CaSki cells express detectable levels of fibronectin, vimentin as well as N-cadherin, suggesting that they show some evidence of EMT.
To validate the effect of HPV16 E7 expression causes alterations in levels of EMT-associated proteins, we analyzed expression of fibronectin and E-cadherin in a matched set of hTERT immortalized normal oral keratinocytes (NOK) and NOKs with stable expression of HPV16 E7 (NOKE7) (Piboonniyom et al., 2003). Primary human foreskin fibroblasts (HFFs) were used as controls. As expected, NOKs contained high levels of E-cadherin and barely detectable levels of fibronectin, whereas HFFs expressed no detectable E-cadherin but had high levels of fibronectin. NOK-E7 cells contained markedly decreased levels of E-cadherin and increased levels of fibronectin (Figure 2C).
HPV16 oncogenes E6 and E7 induce the de-novo expression of N-cadherin in primary epithelial cells
Since N-cadherin was expressed in the HFK populations at levels below the limit of clear detection by Western blotting (data not shown) we performed immunofluorescence experiments with these cell populations. As expected, N-cadherin was detected at extremely low levels in HFKs but was expressed at high levels in HFFs. HPV16 E6 as well as HPV16 E7 expressing keratinocytes also showed evidence for increased levels of N-cadherin, which was also observed in HFKs expressing HPV16 E6 and E7 or the entire early region (Figure 3; upper panels). To ascertain that the signal that we observe in these immunofluorescence experiments reflects N-cadherin expression, we transfected HFF, which express high levels of N-cadherin with an N-cadherin specific siRNA oligonucleotide pool or a scrambled control siRNA as a control. N-cadherin depletion caused a dramatic decrease in the immunofluorescence signal, consistent with the notion that we detect N-cadherin expression with this antibody. We also stained these cells with phalloidin and detected an increased appearance of filamentous actin that paralleled the increase in N-cadherin expression in HPV16 oncoprotein expressing cells (Figure 3; lower panels)
Figure 3.
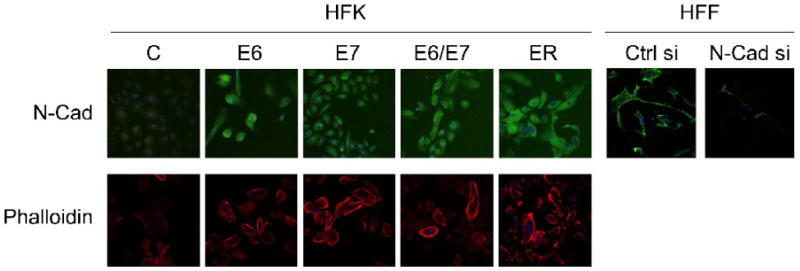
N-cadherin and filamentous actin expression of HPV16 E6 and/or E7 and early region (ER) in primary human keratinocytes (HFKs). N-Cad expression (upper panels) in a panel of passage and donor matched human foreskin keratinocyte (HFK) populations expressing HPV16 E6 and/or E7 oncoproteins or the HPV16 entire early transcription unit (ER), or transfected with empty vector (C) was assessed by immunofluorescence microscopy. The specificity of the antibody was assessed by transfection of primary human foreskin fibroblasts (HFFs) with N-cadherin siRNA (N-Cad si) or scrambled control siRNA (Ctrl si) followed by immunofluorescence analysis at 48 hours after siRNA transfection. Filamentous actin (lower panels) was visualized by fluorescence microscopy of cells stained with rhodamine-conjugated phalloidin.
Discussion
EMT is an essential process of transdifferentiation that establishes cell lineage identity during embryogenesis. EMT related processes are also triggered during carcinogenic progression. Cancer-associated EMT, however, does not generally involve a complete lineage switch but endows emerging tumor cells with migratory and invasive properties (reviewed in Thiery et al., 2006) and may also contribute to the cancer-stem cell phenotype (Mani et al., 2008). Hence tumor-associated EMT is key to acquisition of an invasive and metastatic phenotype and therefore to malignant progression. Importantly, however, tumor-associated EMT is reversible through a process that is referred to mesenchymal to epithelial transition (MET) (reviewed in Thiery et al., 2006). This may account for some of the inconsistencies in studies with clinical materials that have focused on determining tumor grade specific expression of certain EMT markers.
EMT has been detected in cervical carcinomas (Hagemann et al., 2007) and was correlated to EGF receptor overexpression and increased expression and nuclear localization of the transcription factor snail (Lee et al., 2008). HPV oncoproteins may directly contribute to EMT induction (Duffy et al., 2003) and expression of the HPV18 E6 oncoprotein in SV40 immortalized keratinocytes caused morphological conversion to a fibroblastoid morphology (Watson et al., 2003). In this study we did not detect dramatic morphological alterations in HPV16 E6 and/or E7 expressing primary human foreskin keratinocytes and there were only subtle differences (less than 2 fold) in mRNA levels of EMT and differentiation associated genes in the different cell populations. The mRNA levels of many EMT genes, including the two transcription factors and EMT master regulators, twist and snail, were virtually unaltered. This is in contrast to the a previous study (Duffy et al., 2003), which may reflect the different culture conditions that were used in the two studies. We were thus surprised to detect much more dramatic changes at the level of protein expression. The HPV16 E7 oncoprotein importantly contributed to induction of these changes; HPV16 E7 expressing cells displayed markedly decreased levels of E-cadherin and increased levels of fibronectin as well as vimentin (Figure 2).
Vimentin expression has previously been associated with acquisition of invasive properties in HPV33 transformed keratinocytes (Gilles et al., 1994a; Gilles et al., 1994b). In studies with clinical specimens, vimentin expression was detected in invasive cervical carcinoma but not in high-grade cervical intraepithelial neoplasias (CIN III) (Gilles et al., 1996). Another study revealed vimentin expression in ∼60% of a total of 120 cervical carcinoma specimens that were analyzed (Schaafsma et al., 1993). Vimentin staining was extensive and throughout large portions of the tumor in some samples, whereas in others staining was confined to small clusters of basal and parabasal cells at or near the tumor-stromal interface. This suggests that vimentin expression may be particularly high at the “invasive front” of the tumor. Fibronectin staining has also been documented in some cervical carcinoma, but staining was diffuse and not confined to the invasive front. Moreover, there was no correlation of fibronectin staining with tumor grade (Goldberg et al., 1998). HPV16 E7 has been reported to repress the fibronectin promoter (Rey et al., 2000), but we observed a slight increase in fibronectin mRNA expression (Figure 1) and dramatically increased fibronectin protein levels in HPV16 E7 expressing cells (Figure 2).
Cancer-associated EMT generally involves a cadherin switch; whereas there is decreased E-cadherin expression, N-cadherin is expressed at higher levels. Loss of E-cadherin expression has been reported in many tumors. In some tumor types, including cervical carcinomas, E-cadherin may be transcriptionally silenced by DNA hypermethylation (Chen et al., 2003; Dong et al., 2001; Kang et al., 2005; Narayan et al., 2003; Shivapurkar et al., 2007; Terra et al., 2007; Yang et al., 2006). While the mechanism of the decreased E-cadherin expression in HPV oncoprotein expressing HFKs is not clear, our results are consistent with a recent report that depletion of HPV16 E7 by siRNA in HPV16 transformed human keratinocytes restored normal E-cadherin expression through a mechanism that is likely independent of the transcription factors slug and snail (Caberg et al., 2008). Furthermore, expression of the HPV16 E7 related SV40 large tumor antigen in MDCK cells induced EMT and loss of E-cadherin and keratin expression (Martel et al., 1997). Moreover, siRNA mediated depletion of pRB, which is targeted for degradation by HPV16 E7, resulted in deregulated E-cadherin expression, reduced cell-to-cell adhesion and EMT-related morphological alterations in MCF7 breast cancer cells. Most strikingly, there was concurrent down-regulation of pRB and E-cadherin expression in mesenchymal-like invasive cancers breast cancer specimens (Arima et al., 2008). It will be interesting to determine whether the ability of HPV16 E7 to reduce E-cadherin expression is linked to the ability to destabilize pRB.
N-cadherin expression is linked to increased cell motility and invasiveness in tumors (Derycke et al., 2004) and ectopic N-cadherin expression in epithelial cells causes morphological alterations and increased motility (Hazan et al., 1997; Islam et al., 1996). In our experiments N-cadherin mRNA levels were unaltered in HPV16 oncogene expressing cells and the protein levels remained below the level of consistent, reliable detection by Western blotting in multiple HFK populations that we tested. However, we consistently observed increased N-cadherin levels in HPV16 E6 and/or E7 expressing HFKs as compared to control cells by immunofluorescence (Figure 3).
In summary, our data suggests that HPV16 E7 importantly contributes to induction of an EMT related process in epithelial cells. Consistent with this notion, previous studies with transgenic mice showed that HPV16 E7 expression in combination with low dose estrogen administration is sufficient for induction of invasive cervical carcinomas (Riley et al., 2003), which conceivably involves EMT. While co-expression of the HPV16 E6 oncogene resulted in the formation of larger, more extensive carcinomas, HPV16 E6 expressing mice only developed precancerous lesions (Riley et al., 2003). Since the observed changes in E-cadherin, fibronectin and vimentin expression in HPV16 E7 proteins were much more dramatic than what would have been predicted from our mRNA expression analysis, we hypothesize that HPV16 E7 may affect EMT, at least in part through non-transcriptional mechanisms. Indeed, HPV16 E7 causes stabilization of the p53 tumor suppressor and the cdk inhibitor p21CIP1 (Jones et al., 1997; Jones et al., 1999) and targets the tumor suppressor pRB and the related p107 and p130 proteins for degradation (Boyer et al., 1996; Gonzalez et al., 2001; Jones et al., 1997; Zhang et al., 2006). Hence, it is conceivable that E7 may either directly alter the stability of these EMT related proteins or modulate protein levels indirectly through (de)stabilization of factors that regulate their expression.
Acknowledgments
We thank Amy Baldwin for advice with generating HPV16 oncogene expressing keratinocyte cultures, Kristina M. Holton and Renee Rubio for assistance in generating microarray data, Adrian Marino-Enriquez for technical advice and Margaret E. McLaughlin-Drubin for helpful suggestions on this manuscript. We also thank the two anonymous reviewers for their constructive criticisms on an earlier version of this manuscript. KM and KH gratefully acknowledge the invaluable support of Kay Feenby-Skcubrats throughout this study. Supported by PHS grants HG004233 (JQ, KM), CA081135 (KM) and CA06698 (KM). KH was supported by a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft (DFG HE5494/1-1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arima Y, Inoue Y, Shibata T, Hayashi H, Nagano O, Saya H, Taya Y. Rb depletion results in deregulation of E-cadherin and induction of cellular phenotypic changes that are characteristic of the epithelial-to-mesenchymal transition. Cancer Res. 2008;68(13):5104–12. doi: 10.1158/0008-5472.CAN-07-5680. [DOI] [PubMed] [Google Scholar]
- Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56(20):4620–4. [PubMed] [Google Scholar]
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FC, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze-Kremer S, Stewart J, Taylor R, Vilo J, Vingron M. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat Genet. 2001;29(4):365–71. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- Caberg JH, Hubert PM, Begon DY, Herfs MF, Roncarati PJ, Boniver JJ, Delvenne PO. Silencing of E7 oncogene restores functional E-cadherin expression in human papillomavirus 16-transformed keratinocytes. Carcinogenesis. 2008;29(7):1441–7. doi: 10.1093/carcin/bgn145. [DOI] [PubMed] [Google Scholar]
- Chamulitrat W, Schmidt R, Chunglok W, Kohl A, Tomakidi P. Epithelium and fibroblast-like phenotypes derived from HPV16 E6/E7-immortalized human gingival keratinocytes following chronic ethanol treatment. Eur J Cell Biol. 2003;82(6):313–22. doi: 10.1078/0171-9335-00317. [DOI] [PubMed] [Google Scholar]
- Chen CL, Liu SS, Ip SM, Wong LC, Ng TY, Ngan HY. E-cadherin expression is silenced by DNA methylation in cervical cancer cell lines and tumours. Eur J Cancer. 2003;39(4):517–23. doi: 10.1016/s0959-8049(02)00175-2. [DOI] [PubMed] [Google Scholar]
- Derycke LD, Bracke ME. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int J Dev Biol. 2004;48(56):463–76. doi: 10.1387/ijdb.041793ld. [DOI] [PubMed] [Google Scholar]
- Dong SM, Kim HS, Rha SH, Sidransky D. Promoter hypermethylation of multiple genes in carcinoma of the uterine cervix. Clin Cancer Res. 2001;7(7):1982–6. [PubMed] [Google Scholar]
- Duffy CL, Phillips SL, Klingelhutz AJ. Microarray analysis identifies differentiation-associated genes regulated by human papillomavirus type 16 E6. Virology. 2003;314(1):196–205. doi: 10.1016/s0042-6822(03)00390-8. [DOI] [PubMed] [Google Scholar]
- Dyson N, Guida P, Munger K, Harlow E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J Virol. 1992;66(12):6893–902. doi: 10.1128/jvi.66.12.6893-6902.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243(4893):934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles C, Piette J, Peter W, Fusenig NE, Foidart JM. Differentiation ability and oncogenic potential of HPV-33- and HPV-33 + ras-transfected keratinocytes. Int J Cancer. 1994a;58(6):847–54. doi: 10.1002/ijc.2910580617. [DOI] [PubMed] [Google Scholar]
- Gilles C, Polette M, Piette J, Birembaut P, Foidart JM. Epithelial-to-mesenchymal transition in HPV-33-transfected cervical keratinocytes is associated with increased invasiveness and expression of gelatinase A. Int J Cancer. 1994b;59(5):661–6. doi: 10.1002/ijc.2910590514. [DOI] [PubMed] [Google Scholar]
- Gilles C, Polette M, Piette J, Delvigne AC, Thompson EW, Foidart JM, Birembaut P. Vimentin expression in cervical carcinomas: association with invasive and migratory potential. J Pathol. 1996;180(2):175–80. doi: 10.1002/(SICI)1096-9896(199610)180:2<175::AID-PATH630>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Goldberg I, Davidson B, Lerner-Geva L, Gotlieb WH, Ben-Baruch G, Novikov I, Kopolovic J. Expression of extracellular matrix proteins in cervical squamous cell carcinoma--a clinicopathological study. J Clin Pathol. 1998;51(10):781–5. doi: 10.1136/jcp.51.10.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75(16):7583–91. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann T, Bozanovic T, Hooper S, Ljubic A, Slettenaar VI, Wilson JL, Singh N, Gayther SA, Shepherd JH, Van Trappen PO. Molecular profiling of cervical cancer progression. Br J Cancer. 2007;96(2):321–8. doi: 10.1038/sj.bjc.6603543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SW, Roman J. Fibronectin induces cell proliferation and inhibits apoptosis in human bronchial epithelial cells: pro-oncogenic effects mediated by PI3-kinase and NF-kappa B. Oncogene. 2006;25(31):4341–9. doi: 10.1038/sj.onc.1209460. [DOI] [PubMed] [Google Scholar]
- Hazan RB, Kang L, Whooley BP, Borgen PI. N-cadherin promotes adhesion between invasive breast cancer cells and the stroma. Cell Adhes Commun. 1997;4(6):399–411. doi: 10.3109/15419069709004457. [DOI] [PubMed] [Google Scholar]
- Hirohashi S, Kanai Y. Cell adhesion system and human cancer morphogenesis. Cancer Sci. 2003;94(7):575–81. doi: 10.1111/j.1349-7006.2003.tb01485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie HL, Katzenellenbogen RA, Galloway DA. Papillomavirus E6 proteins. Virology. 2009;384(2):324–34. doi: 10.1016/j.virol.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam S, Carey TE, Wolf GT, Wheelock MJ, Johnson KR. Expression of N-cadherin by human squamous carcinoma cells induces a scattered fibroblastic phenotype with disrupted cell-cell adhesion. J Cell Biol. 1996;135(6 Pt 1):1643–54. doi: 10.1083/jcb.135.6.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27(55):6920–9. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Munger K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J Virol. 1997;71(4):2905–12. doi: 10.1128/jvi.71.4.2905-2912.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Thompson DA, Suh-Burgmann E, Grace M, Munger K. Expression of the HPV E7 oncoprotein mimics but does not evoke a p53-dependent cellular DNA damage response pathway. Virology. 1999;258(2):406–14. doi: 10.1006/viro.1999.9733. [DOI] [PubMed] [Google Scholar]
- Kang S, Kim JW, Kang GH, Park NH, Song YS, Kang SB, Lee HP. Polymorphism in folate- and methionine-metabolizing enzyme and aberrant CpG island hypermethylation in uterine cervical cancer. Gynecol Oncol. 2005;96(1):173–80. doi: 10.1016/j.ygyno.2004.09.031. [DOI] [PubMed] [Google Scholar]
- Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A. 1997;94(21):11612–6. doi: 10.1073/pnas.94.21.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380(6569):79–82. doi: 10.1038/380079a0. [DOI] [PubMed] [Google Scholar]
- Kokkinos MI, Wafai R, Wong MK, Newgreen DF, Thompson EW, Waltham M. Vimentin and epithelial-mesenchymal transition in human breast cancer--observations in vitro and in vivo. Cells Tissues Organs. 2007;185(13):191–203. doi: 10.1159/000101320. [DOI] [PubMed] [Google Scholar]
- Lee MY, Chou CY, Tang MJ, Shen MR. Epithelial-mesenchymal transition in cervical cancer: correlation with tumor progression, epidermal growth factor receptor overexpression, and snail up-regulation. Clin Cancer Res. 2008;14(15):4743–50. doi: 10.1158/1078-0432.CCR-08-0234. [DOI] [PubMed] [Google Scholar]
- Lee SS, Weiss RS, Javier RT. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A. 1997;94(13):6670–5. doi: 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel C, Harper F, Cereghini S, Noe V, Mareel M, Cremisi C. Inactivation of retinoblastoma family proteins by SV40 T antigen results in creation of a hepatocyte growth factor/scatter factor autocrine loop associated with an epithelial-fibroblastoid conversion and invasiveness. Cell Growth Differ. 1997;8(2):165–78. [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Munger K. The human papillomavirus E7 oncoprotein. Virology. 2009;384(2):335–44. doi: 10.1016/j.virol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels GS, Carr DB, Askenazi M, Fuhrman S, Wen X, Somogyi R. Cluster analysis and data visualization of large-scale gene expression data. Pac Symp Biocomput. 1998:42–53. [PubMed] [Google Scholar]
- Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78(21):11451–60. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989;63(10):4417–21. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan G, Arias-Pulido H, Koul S, Vargas H, Zhang FF, Villella J, Schneider A, Terry MB, Mansukhani M, Murty VV. Frequent promoter methylation of CDH1, DAPK, RARB, and HIC1 genes in carcinoma of cervix uteri: its relationship to clinical outcome. Mol Cancer. 2003;2:24. doi: 10.1186/1476-4598-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piboonniyom SO, Duensing S, Swilling NW, Hasskarl J, Hinds PW, Munger K. Abrogation of the retinoblastoma tumor suppressor checkpoint during keratinocyte immortalization is not sufficient for induction of centrosome-mediated genomic instability. Cancer Res. 2003;63(2):476–83. [PubMed] [Google Scholar]
- Rey O, Lee S, Park NH. Human papillomavirus type 16 E7 oncoprotein represses transcription of human fibronectin. J Virol. 2000;74(10):4912–8. doi: 10.1128/jvi.74.10.4912-4918.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley RR, Duensing S, Brake T, Munger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63(16):4862–71. [PubMed] [Google Scholar]
- Schaafsma HE, Van Der Velden LA, Manni JJ, Peters H, Link M, Rutter DJ, Ramaekers FC. Increased expression of cytokeratins 8, 18 and vimentin in the invasion front of mucosal squamous cell carcinoma. J Pathol. 1993;170(1):77–86. doi: 10.1002/path.1711700113. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63(6):1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Shivapurkar N, Sherman ME, Stastny V, Echebiri C, Rader JS, Nayar R, Bonfiglio TA, Gazdar AF, Wang SS. Evaluation of candidate methylation markers to detect cervical neoplasia. Gynecol Oncol. 2007;107(3):549–53. doi: 10.1016/j.ygyno.2007.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terra AP, Murta EF, Maluf PJ, Caballero OL, Brait M, Adad SJ. Aberrant promoter methylation can be useful as a marker of recurrent disease in patients with cervical intraepithelial neoplasia grade III. Tumori. 2007;93(6):572–9. doi: 10.1177/030089160709300610. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Watson RA, Thomas M, Banks L, Roberts S. Activity of the human papillomavirus E6 PDZ-binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J Cell Sci. 2003;116(Pt 24):4925–34. doi: 10.1242/jcs.00809. [DOI] [PubMed] [Google Scholar]
- Wen X, Fuhrman S, Michaels GS, Carr DB, Smith S, Barker JL, Somogyi R. Large-scale temporal gene expression mapping of central nervous system development. Proc Natl Acad Sci U S A. 1998;95(1):334–9. doi: 10.1073/pnas.95.1.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HJ, Liu VW, Wang Y, Tsang PC, Ngan HY. Differential DNA methylation profiles in gynecological cancers and correlation with clinico-pathological data. BMC Cancer. 2006;6:212. doi: 10.1186/1471-2407-6-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chen W, Roman A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc Natl Acad Sci U S A. 2006;103(2):437–42. doi: 10.1073/pnas.0510012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]