

Summary
During the differentiation of muscle satellite cells, committed myoblasts respond to specific signalling cues by exiting the cell cycle, migrating, aligning, expressing muscle-specific genes and finally fusing to form multinucleated myotubes. The predominant foetal growth factor, IGF-2, initiates important signals in myogenesis. The aim of this study was to investigate whether ERK5 and its upstream MKK activator, MEK5, were important in the pro-myogenic actions of IGF-2. ERK5 protein levels, specific phosphorylation and kinase activity increased in differentiating C2 myoblasts. ERK5-GFP translocated from the cytoplasm to the nucleus after activation by upstream MEK5, whereas phospho-acceptor site mutated (dominant-negative) ERK5AEF-GFP remained cytoplasmic. Exogenous IGF-2 increased MHC levels, myogenic E box promoter-reporter activity, ERK5 phosphorylation and kinase activity, and rapidly induced nuclear localisation of ERK5. Transfection with antisense Igf2 decreased markers of myogenesis, and reduced ERK5 phosphorylation, kinase and transactivation activity. These negative effects of antisense Igf2 were rescued by constitutively active MEK5, whereas transfection of myoblasts with dominant-negative MEK5 blocked the pro-myogenic action of IGF-2. Our findings suggest that the MEK5-ERK5 pathway is a novel key mediator of IGF-2 action in myoblast differentiation.
Keywords: ERK5, IGF-2, MEK5, Myogenesis
Introduction
Classical MAPKs (mitogen-activated protein kinases) form a key part of signalling cascades that regulate many aspects of cell function. The classical pathway consists of an upstream MAPKKK, followed by MAPKK and finally MAPK; each component of the pathway phosphorylates and activates its respective downstream target in a linear manner, in a mechanism that allows amplification of the original signal (Johnson and Lapadat, 2002). ERK5 (extracellular signal-regulated kinase 5) is an intriguing and relatively new member of the MAPK family; it was first identified in 1995 and has a molecular weight that is at least twice that of ERK1, ERK2, p38 or JNK due to an approximately 400 amino acid C-terminal extension (Lee et al., 1995; Yan et al., 2001; Zhou et al., 1995). Unlike the classical MAPKs, ERK5 has two intrinsic catalytic functions: (1) conventional protein kinase activity in common with other MAPKs and (2) transactivation activity (reviewed by Nishimoto and Nishida, 2006; Wang and Tournier, 2006).
The N-terminal half of ERK5 has most in common with ERK1 and ERK2, with which it is 50% homologous. The phospho-target sequence for its upstream MKK, MEK5, is also in this region and, like the phospho-target for ERK1 and ERK2, is a TEY motif. Specificity of activation of the ERK5 cascade is achieved by a PB1 (Phox/Bem1p) domain in MEK5, which will only bind to a related domain in specific upstream MKKKs and in ERK5 (Nakamura et al., 2006; Seyfried et al., 2005); thus the ERK5 pathway is less promiscuous than most other MAPK cascades. The C-terminal half of ERK5 is responsible for its putative direct transactivation activity and is an active fragment alone, i.e. in the absence of the N-terminal domain (Kasler et al., 2000). Interaction between the C- and N-terminal domains has an important function in the regulation of ERK5 localisation and in the activation of its kinase activity (Kondoh et al., 2006). Thus, even though the N- and C-terminal multidomains of ERK5 can be segregated into unique actions, each can reciprocally influence the other.
ERK5 is activated both by stress (Abe et al., 1996) and by mitogens (English et al., 1999; Kamakura et al., 1999; Kato et al., 2000). Its precise function is not clear but it is abundantly expressed in skeletal muscle and heart (Dinev et al., 2001) and, with MEK5, is highly expressed in developing somites (the origin of precursor myoblasts) and in limb buds (Yan et al., 2003). Erk5-null mice have severe defects in angiogenesis and cardiac development; limb bud development is also arrested, although subsequent muscle development is difficult to assess as embryos die as soon as ∼E10.5 (Regan et al., 2002; Yan et al., 2003). Mek5-null mice are a close phenocopy of Erk5-null mice (Wang et al., 2005), providing indirect evidence to support the suggestion that ERK5 is the sole substrate for MEK5. A key target of the MEK5-ERK5 pathway is the MEF2 transcription factor family, and ERK5 directly phosphorylates MEF2, thereby increasing its transactivation activity (Kasler et al., 2000; Suzaki et al., 2002). MEF2 proteins perform an essential role in embryonic myogenesis by interacting with myogenic regulatory factors and facilitating muscle-specific gene expression (Black and Olson, 1998; Bour et al., 1995). Taken together, these findings suggest a potential role for ERK5 in myoblast differentiation and, in support of this, ERK5 has been reported to positively regulate myogenesis (Dinev et al., 2001).
Insulin-like growth factor 2 (IGF-2) is one of two secreted peptide hormones (the other being IGF-1) that act via both endocrine and local autocrine and paracrine mechanisms to activate the IGF type 1 receptor (IGF1R) and, in certain circumstances, the A isoform of the insulin receptor (Nakae et al., 2001). IGF-2 is abundantly expressed in the embryo, particularly in somites and muscle precursor cells, leading to the suggestion that IGF-2 has an important role in embryonic growth and muscle development (Stylianopoulou et al., 1988; Bondy et al., 1990). Indeed, mice that are null for Igf2 are only ∼60% of the weight of wild-type mice at birth, a defect that continues postnatally (DeChiara et al., 1990). Further, IGF-2 translation efficiency is regulated by the microRNA Lin-28, which positively regulates myogenesis (Polesskaya et al., 2007). The absolute importance of IGF signalling in muscle development is illustrated by the poor muscle hypertrophy and the dystrophic phenotype of Igf1r-null mice, which usually die at birth due to weakness of respiratory muscle (Liu et al., 1993). IGF-2 forms an autoregulatory feedback loop during myogenesis, its expression and activity being regulated by extracellular events such as cell-cell contact (Lovett et al., 2006) and secreted factors such as the IGF-binding protein, IGFBP-5 (Ren et al., 2008). IGF-activated PI3K-AKT and p38-MAPK pathways are both necessary for myoblast differentiation (Gonzalez et al., 2004) and converge on chromatin, thus targeting distinct components of transcriptional machinery and enabling upregulation of muscle-specific genes (Serra et al., 2007).
Even though different populations of precursor cells contribute to muscle formation pre- and postnatally (Buckingham, 2006), the key events that regulate myoblast differentiation are thought to utilise many common cellular mechanisms. Muscle fibre number is largely determined by birth and all postnatal muscle growth occurs via hypertrophy of existing fibres by activation and differentiation of adult muscle precursor cells, which are termed satellite cells (Le Grand and Rudnicki, 2007). Satellite cell myogenesis is a highly regulated process, necessitating strict temporal control of cell-cycle exit, coordinated expression of muscle regulatory and structural genes, and morphological transition from single cells to multinucleated myotubes (Berkes and Tapscott, 2005). Autocrine IGF-2 is a key regulator of satellite-cell differentiation and regulates the expression (Florini et al., 1991) and activity of myogenic regulatory factors such as myogenin and MyoD, via multiple mechanisms (e.g. Lovett et al., 2006; Wilson et al., 2003; Wilson and Rotwein, 2006) to coordinate myoblast differentiation. However, signalling pathways downstream of receptor activation by IGF-2 are poorly understood; the aim of this study was therefore to investigate the role of ERK5 in the regulation of satellite-cell myogenesis by IGF-2.
Results
Regulation of myogenesis by IGF-2
C2 myoblasts were induced to differentiate by transfer from high-serum growth medium (GM) to low-serum differentiation medium (DM) at an initial cell density of 2×105 cells per 100-mm dish, as described in the Materials and Methods and as previously reported by us (Gonzalez et al., 2004). Upregulation of mRNA levels for the key pro-myogenic growth factor Igf2 was readily observed within 24 hours, at the onset of myogenin mRNA upregulation (Fig. 1A). Protein levels of the late-stage myogenic marker MHC (myosin heavy chain, a muscle-specific structural protein) began to increase at 48 hours and were abundant by 72 hours (Fig. 1B), coincident with the onset of myoblast fusion into myotubes. By contrast, the levels of both total p38 MAPK and β-actin remained constant. These findings are in agreement with our previous studies (Gonzalez et al., 2004) and, as the aim of this study was to investigate IGF-2 signalling, we used total p38 levels as a loading control in subsequent studies. Transfection of myoblasts with an antisense Igf2 cDNA, which we (Cobb et al., 2004) and originally others (Stewart and Rotwein, 1996) have established as a means of decreasing IGF-2 protein levels and activity in myoblasts, inhibited differentiation, indicated by decreased MHC levels at 72 hours (Fig. 1C). In complementary studies, and as expected (Stewart et al., 1996), exogenous IGF-2 accelerated differentiation, as evidenced by increased p21 and MHC protein levels (Fig. 1C). These studies thus confirm the established importance of IGF-2 in the regulation of myoblast differentiation and provide models for further investigation of the mechanism of action of IGF-2.
Fig. 1.

Regulatory role of IGF-2 in myogenesis. C2 myoblasts were differentiated as described in Materials and Methods. (A) Igf2, Myogenin and Gapdh (as a loading control) mRNA levels were detected by northern blotting. (B) MHC, total p38 MAPK and β-actin protein levels were detected by western blotting. (C) MHC protein levels detected by western blotting in C2 myoblasts that were transfected with empty pcDNA vector or antisense Igf2 (Igf2as) and differentiated for 72 hours. (D) MHC, p21 and total p38 MAPK protein levels in differentiating C2 myoblasts treated with vehicle or exogenous IGF-2 (25 ng/ml) for 48 or 72 hours. Representative blots are shown from a minimum of three independent replicates.
ERK5 activity increases during myogenesis
The activation of ERK5 was investigated during C2 cell differentiation. Representative western blots to show changes in phospho-ERK5 and total ERK5 are shown in Fig. 2A (upper panels) and quantification of these is shown in Fig. 2B. Total ERK5 protein levels doubled 24 hours after the initiation of differentiation (P<0.01) and then gradually decreased during the remainder of the period investigated. By contrast, ERK5 phosphorylation increased fivefold 24 hours after the initiation of differentiation and this increase was maintained until at least 72 hours. The relative increase in ERK5 phosphorylation indicated that its activity should also increase, and this was supported by direct measurement of ERK5 kinase activity (Fig. 2A, lower panel, and mean values in Fig. 2C), which increased by almost fivefold at 24 hours (P<0.05) and peaked at 48-72 hours. Phosphorylation (indicated by the slower-migrating bands) and abundance of the ERK5 target MEF2C (myocyte enhancer factor 2C) broadly correlated with ERK5 kinase activity (Fig. 2A, third panel).
Fig. 2.
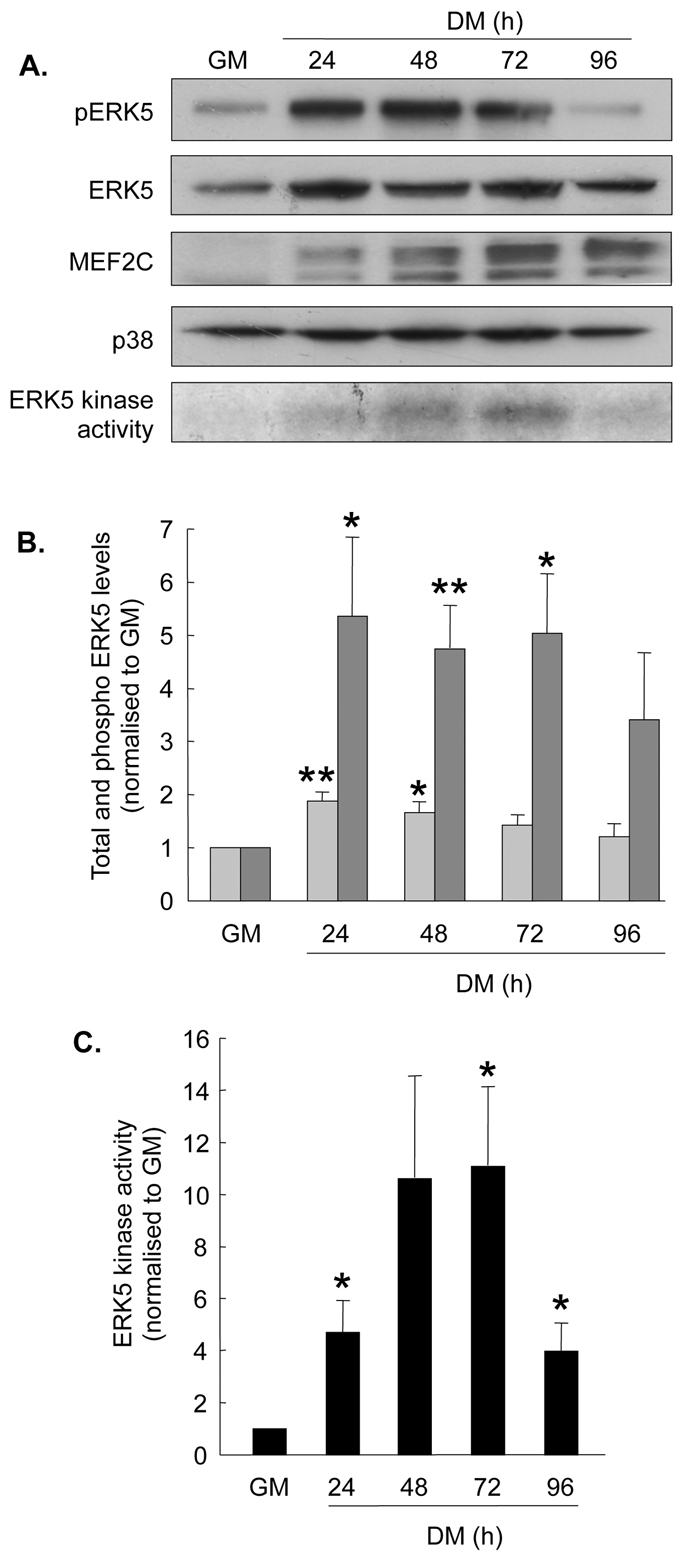
ERK5 activity increases during myogenesis. C2 myoblasts were differentiated as described in Materials and Methods. (A) Upper four panels show representative western blots (from n=5) to show phospho-ERK5 (pERK5), total ERK5, MEF2C (slower-migrating bands represent phosphorylated MEF2C) and total p38 MAPK protein levels. Lower panel shows a representative ERK5 kinase assay assessed by incorporation of [γ-32P]ATP into MBP. (B) Quantification of total (pale grey bars) and phospho-ERK5 (dark grey bars) western blots, normalised to growth medium (GM) values. (C) Quantification of ERK5 kinase activity during myogenesis, normalised to GM values. Data presented in B and C are the means ± s.e.m. of five individual experiments for all observations; *P<0.05, **P<0.01, compared with GM, assessed by Student's t-test.
We next investigated whether manipulation of ERK5 activity regulated myoblast differentiation. C2 myoblasts were transfected with either FLAG-tagged wild-type ERK5, HA-tagged constitutively active MEK5, (MEK5DD, the only direct upstream activator of ERK5 that has been identified to date), or FLAG-tagged dominant-negative ERK5 (ERK5AEF, which cannot be phosphorylated by MEK5). Successful transfection with these constructs was evidenced by the presence of HA and FLAG bands in western blots (Fig. 3A, upper panels) and, in the case of all forms of ERK5, by an increase in total ERK5 levels (Fig. 3A, fourth panel). Transfection with wild-type ERK5 alone only slightly increased ERK5 phosphorylation (Fig. 3A, third panel) and myogenic activity as assessed by an E box promoter-reporter assay (Fig. 3B). This construct consists of four E boxes upstream of a luciferase reporter, and is a well-established approach for measuring overall myogenic regulatory factor transcription activation (Wu et al., 2000). However, co-transfection of myoblasts with wild-type ERK5 and constitutively active MEK5 induced a clear increase in ERK5 phosphorylation and a threefold increase (P<0.01) in E box promoter-reporter activity. These increases were prevented when dominant-negative ERK5 was co-transfected with MEK5DD.
Fig. 3.

The MEK5-ERK5 pathway has a regulatory role in myogenesis. C2 myoblasts were transfected with pcDNA empty vector, FLAG-tagged wild-type ERK5 (ERK5/flag), HA-tagged constitutively active MEK5 (MEK5DD/HA), or FLAG-tagged dominant-negative ERK5 (ERK5AEF/flag) and differentiated for 48 hours. (A) Representative western blots (from n=4) to show FLAG and HA tags, phospho-ERK5 (pERK5), total ERK5 and total p38 MAPK protein levels. (B) Myogenic E box promoter-reporter activity in C2 myoblasts co-transfected with the constructs described in A. Data are normalised to pcDNA values and are means ± s.e.m.; **P<0.01 compared with pcDNA; #P<0.05 compared with MEK5DD/HA+ERK5FLAG. (C) Western blotting to detect fusion proteins of GFP and wild-type ERK5 (ERK-GFP) or dominant-negative ERK5 (ERK5AEF-GFP), and GFP alone, following transfection of C2 myoblasts with the appropriate constructs (as described in Materials and Methods), and total p38 MAPK. (D) C2 myoblasts were co-transfected with MEK5DD or empty vector and either ERK5-GFP or ERK5AEF-GFP, as indicated, and after 24 hours in GM were transferred to DM for 10 minutes. GFP fluorescence (green) was captured in successive confocal slices through cells (which have been combined into supplemental Movies 1-4) to show ERK5 localisation only (green, upper panels), or merged with DAPI (blue) staining to additionally display nuclei (lower panels); magnification 1000×; representative image from n=4.
To further examine ERK5 activation during myogenesis, we investigated the localisation of ERK5-GFP (green fluorescent protein) fusion proteins in transfected myoblasts. GFP (Fig. 3C, upper panel) and ERK5 (Fig. 3C, middle panel) bands were detected at the appropriate molecular weights for wild-type ERK5-GFP and dominant-negative ERK5AEF-GFP fusion proteins, together with a GFP-only control. Nuclear localisation of MAPKs is associated with their activation (Turjanski et al., 2007). Therefore myoblasts were transfected with ERK5-GFP or ERK5AEF-GFP in the presence or absence of MEK5DD and the localisation of ERK5-GFP examined after 10 minutes in DM. Serial stacked images were captured using confocal microscopy (Fig. 3D shows a single image through the nucleus and supplementary Movies 1-4 show complete stacked images). MEK5DD induced clear nuclear localisation of wild-type ERK5-GFP, which was not observed in cells transfected with the dominant-negative ERK5AEF-GFP.
Taken together, these findings suggest that ERK5 activity increases during C2 myoblast differentiation, and that forced activation of ERK5 increases myogenic potential, thus supporting a role for the MEK5-ERK5 pathway in myogenesis.
Exogenous IGF-2 enhances short-term and long-term ERK5 phosphorylation
Since Igf2 mRNA levels and ERK5 activation both increase during myoblast differentiation, we next investigated whether ERK5 was important for IGF-2 action in myogenesis. Treatment of differentiating myoblasts with IGF-2, concomitant with transfer to DM, modestly increased ERK5 phosphorylation within as little as 5 minutes and for a further 10 minutes by 20-40% (P<0.05; Fig. 4A); however, ERK5 phosphorylation returned to control levels 30-60 minutes after IGF-2 administration. Subsequently, ERK5 phosphorylation increased significantly and substantially by four- to eightfold (P<0.05 to P<0.01) longer term (24-72 hours) in response to exogenous IGF-2 (Fig. 4B). The role of ERK5 in myoblast differentiation might therefore be biphasic, with an initial rapid response, followed by a greater and more sustained activation and might reflect multiple roles for ERK5.
Fig. 4.

IGF-2 regulates ERK5 phosphorylation and localisation. (A) Left panel: representative western blot (from n=3) to detect phospho-ERK5 (pERK5), total ERK5 and total p38 MAPK in C2 myoblasts cultured in DM for 5-60 minutes and treated with vehicle or IGF-2 (25 ng/ml). Right panel: quantified ERK5 phosphorylation relative to total ERK5 levels; mean ± s.e.m.; n=3; *P<0.05, IGF-2 versus vehicle. (B) Left panel: representative western blot (from n=3) to detect pERK5, MHC, MEF2C, total ERK5 and total p38 MAPK in C2 myoblasts cultured for 24-72 hours in DM and treated with vehicle or IGF-2 (25 ng/ml). Right panel: quantified ERK5 phosphorylation relative to total ERK5 levels; n=3; *P<0.05, **P<0.01, IGF-2 versus vehicle. (C) C2 myoblasts were transfected with either wild-type ERK5-GFP or ERK5AEF-GFP and maintained in GM for 24 hours, when cells were treated with exogenous IGF-2 for 10 minutes. GFP fluorescence (green) was captured showing ERK5 localisation only (upper panels), or merged with DAPI (blue) staining to additionally display nuclei (lower panels); magnification 1000×; representative image from n=3.
To further investigate the role of ERK5 in IGF-2 action, the localisation of wild-type ERK5-GFP and dominant-negative ERK5AEF-GFP fusion proteins was determined after treatment of myoblasts with exogenous IGF-2. Myoblasts were transfected with these constructs and maintained in GM for 24 hours, after which exogenous IGF-2 was added. After only 10 minutes, nuclear localisation of wild-type ERK5-GFP was observed (Fig. 4C, left panels). However, nuclear ERK5AEF-GFP was not observed (Fig. 4C, right panels). Similar observations were made in myoblasts treated with IGF-1 (data not shown), implying IGF1R-mediated regulation.
These findings suggest that ERK5 forms part of a signalling pathway downstream of IGF-2.
ERK5 is required for the pro-myogenic actions of IGF-2
To observe the consequences of decreased IGF-2 signalling on ERK5 activity, myoblasts were transfected with the antisense Igf2 construct that inhibited myogenesis (IGF2as, presented in Fig. 1C). Total ERK5 levels were similar in myoblasts transfected with empty vector or IGF2as after 48 hours in DM, whereas ERK5 phosphorylation and kinase activity both decreased (Fig. 5A). As expected, treatment of myoblasts with exogenous IGF-2 accelerated myogenesis, assessed by MHC protein levels (Fig. 5B). By marked contrast, IGF-2-induced myogenesis was not as great when myoblasts had been co-transfected with a dominant-negative form of MEK5 (MEK5dn), evidenced by the more modest increase in MHC levels in the presence of MEK5dn, and also by the lack of increase in ERK5 phosphorylation and kinase activity induced by MEK5dn in the presence of IGF-2 (Fig. 5B).
Fig. 5.

MEK5 and ERK5 are essential for the pro-myogenic actions of IGF-2. (A) C2 myoblasts were transfected with pcDNA empty vector or antisense Igf2 (IGF2as). Phospho-ERK5 (pERK5), total ERK5 and total p38 levels were determined by western blotting, and ERK5 kinase activity was determined by incorporation of [γ-32P]ATP into MBP; representative blot from n=3. (B) C2 myoblasts were transfected with either empty pcDNA vector or MEK5dn and treated with exogenous IGF-2 (25 ng/ml) or vehicle. MHC, pERK5, total ERK5 and total p38 MAPK levels were determined by western blotting, and ERK5 kinase activity was determined by incorporation of [γ-32P]ATP into MBP after 72 hours in DM; representative blot of n=3. (C) Myogenic E box promoter-reporter activity in C2 myoblasts co-transfected with pcDNA, MEK5DD, MEK5dn or IGF2as together with GFP (expression shown by western blot in the bottom panel) and treated with vehicle or IGF-2 for 48 hours, as indicated in Materials and Methods; n=4; *P<0.05, **P<0.01, ***P<0.001, compared with pcDNA alone.
As a further indication of myogenic potential, the activity of the E box-luciferase promoter-reporter (4RE-luc) was measured. In support of a key myogenic role for IGF-2, the activity of this promoter-reporter increased significantly by 2.5-fold (P<0.001) upon addition of IGF-2 compared with vehicle and was significantly decreased by 50% (P<0.05) when cells were transfected with IGF2as (Fig. 5C). The myogenic role of MEK5 was also confirmed by the significant increase in 4RE-luc promoter-reporter activity of 3.5-fold (P<0.001) when transfected with MEK5DD, and decreased activity (P<0.05) after transfection with MEK5dn alone. The key observation was the prevention of IGF-2 action on promoter-reporter activity by transfection of myoblasts with MEK5dn before addition of exogenous IGF-2. These data therefore support the hypothesis that the MEK5 pathway is necessary for the myogenic actions of IGF-2 and that MEK5 is downstream of IGF-2.
IGF-2 has an established role in the regulation of cell migration (e.g. Guvakova, 2007; Kim et al., 2008) and, because migration and alignment are necessary before myoblast fusion during myogenesis, the migratory ability of myoblasts was investigated as a further physiological output of IGF-2 activity. This was achieved by assessing the number of cells migrating over a 6-hour period into a scratch wound (DeBiasio et al., 1987), as described in the Materials and Methods. Myoblasts were transfected with either empty pcDNA vector or MEK5dn, and subsequently treated with either IGF-2 or vehicle. As expected, exogenous IGF-2 significantly stimulated the number of cells migrating across the wound (1.57±0.16; mean ± s.e.m. relative to vehicle-treated and empty vector-transfected control cells; n=4; P<0.01). Treatment with MEK5dn significantly reduced the number of migrating cells compared with vector-alone transfected cells (0.72±0.07; n=4; P<0.05 compared with control cells). Importantly, MEK5dn also blocked the IGF-2-induced increase in the number of migrating cells (0.77±0.06; n=4; P<0.05 compared with controls), again suggesting that MEK5 lies on a key pathway downstream of IGF-2.
To further investigate the importance of the MEK5-ERK5 pathway in IGF-2 action, the ability of constitutively active MEK5DD to rescue the negative effects of antisense Igf2 was examined. IGF2as decreased 4RE-promoter-reporter activity; however, co-transfection of MEK5DD with IGF2as was able to reverse this decrease (Fig. 6A). As previously observed, IGF2as decreased protein levels of the late myogenic marker, MHC (Fig. 6B). Co-transfection with MEK5DD was able to partially rescue this decrease in MHC levels, implying that the MEK5 pathway is sufficient to rescue some of the actions of IGF-2 in myogenesis.
Fig. 6.
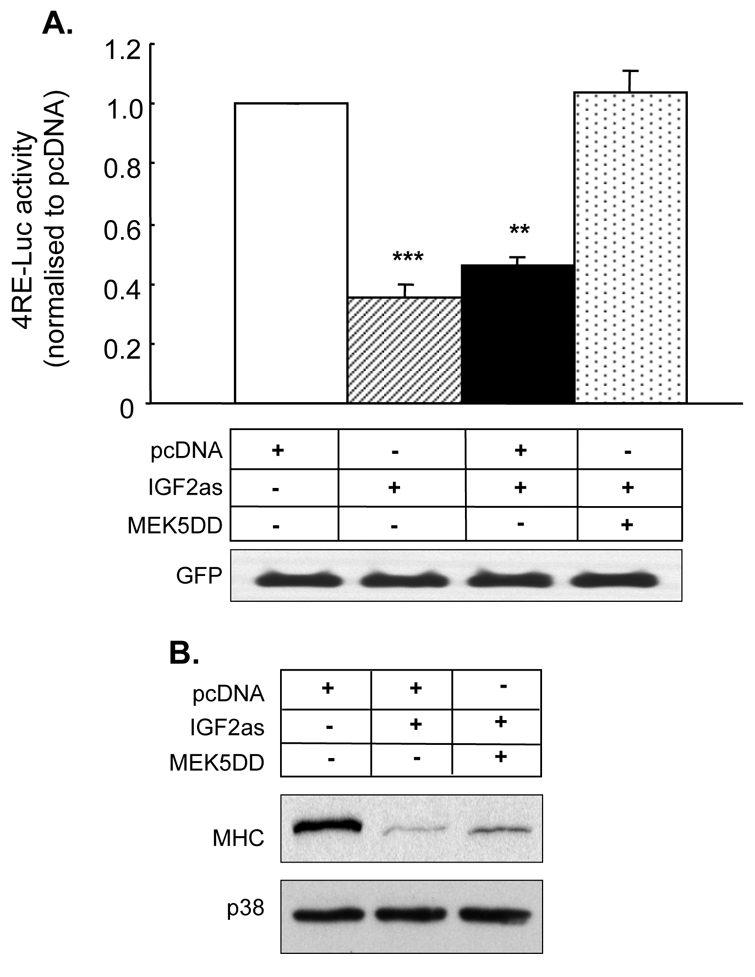
MEK5DD can rescue the anti-myogenic effects of antisense Igf2. (A) Myogenic E box promoter-reporter activity in C2 myoblasts co-transfected with either empty pcDNA, antisense Igf2 (IGF2as) or MEK5DD together with GFP; n=4; **P<0.01, ***P<0.001, compared with pcDNA. (B) Representative western blot (from n=4) to detect MHC and p38 MAPK levels in C2 myoblasts transfected as described for A.
Discussion
Myoblast differentiation requires the coordination of extracellular cues to regulate transcription and major morphological progression from single undifferentiated cells to the formation of specialised multinucleated myotubes; it thus provides an excellent paradigm in which to investigate key regulatory signalling pathways from the cell membrane to the nucleus, in which MAPKs have an important role. The significance of the current study is twofold. Firstly, we have demonstrated that the key foetal growth factor, IGF-2, activates the MEK5-ERK5 pathway: IGF-2 stimulates ERK5 nuclear localisation and increases its phosphorylation and kinase activity. Secondly, we have further demonstrated that MEK5 and ERK5 are essential mediators of the pro-myogenic actions of IGF-2. In the absence of MEK5 and/or ERK5, myogenic E box transcriptional activity is reduced, IGF-2-stimulated myoblast migration is inhibited and expression of the structural muscle-specific late differentiation marker MHC is decreased. These novel findings thus extend our knowledge of myogenic regulation, IGF-2 action and the function of MEK5 and ERK5.
IGF-2 has an important role in prenatal growth and development (DeChiara et al., 1990) and is additionally well established as a key pro-myogenic peptide. The possibility that IGFs might regulate ERK5 activity has been poorly investigated to date and only a few reports linking IGF signalling with ERK5 activation exist. IGFs are proposed to regulate vascular endothelial cell function, and Liu and colleagues observed transient (15-30 minute) stimulation of ERK5 kinase activity by IGF-1 in such cells (Liu et al., 2001). Linnerth and coworkers reported an essential role for ERK5 in IGF-2-stimulated phosphorylation of CREB in lung cancer cells (that had arisen in transgenic mice overexpressing IGF-2) and that this was important for the maintenance of the transformed phenotype (Linnerth et al., 2005). These findings and those presented here thus support the notion that the MEK5-ERK5 pathway is positively regulated by IGFs.
Even though the major upstream MAPKKKs that activate MEK5 and ERK5 include MEKK2, MEKK3 and Cot (Nishimoto and Nishida, 2006), the molecular mechanism(s) by which IGF-2 activates the MEK5-ERK5 pathway is not clear and few studies have addressed signalling pathways to MEK5-ERK5 via peptide growth factor receptors. Established mechanisms exist for the activation of ERK1 and ERK2 via the IGF1R and insulin receptor via Ras and Raf (LeRoith et al., 1995), but their putative involvement in ERK5 activation is controversial. English and coworkers suggested that Ras could regulate ERK5 activity (English et al., 1999) and Kamakura and coworkers proposed that Ras was important in EGF activation of ERK5 (Kamakura et al., 1999), in direct contrast to an earlier study investigating EGF activation of ERK5 (Kato et al., 1998). The non-receptor tyrosine kinase, Src, activates ERK5 (Schramp et al., 2008) and Src has been proposed as an intermediate in EGF stimulation of ERK5 activity (Sun et al., 2003). Recent studies investigating ERK5 as a downstream target of brain-derived neurotrophic factor (BDNF) activity (Wang et al., 2006b) have identified a Rap1-MEKK2 signalling cascade that activates ERK5; Ras and MEKK3 did not have a significant role. IGF-1 enhances the inflammatory response in vascular endothelial cells and one suggested mechanism for this is via the downregulation of Gab-1, which associates with and inhibits MEKK3 (Che et al., 2002). Overall, these findings do not suggest common pathways to MEK5 and ERK5 and thus an important future goal would be to identify the critical upstream signalling that links IGF-2 to MEK5 and ERK5 during myoblast differentiation.
Unusually for a MAPK, ERK5 protein levels doubled within the first 24 hours after myogenesis was initiated. This has also been observed in leukaemic cells, in which a role for Abl-kinase in the stabilisation of ERK5 protein (within hours) occurs by a mechanism that is independent of ERK5 phosphorylation; the authors further suggest that the abundance of ERK5 (in addition to phosphorylation) is important in oncogenesis (Buschbeck et al., 2005). Even though the responsiveness of myoblasts will be subject to some variability due to different concentrations of growth factors in different batches of serum, significant changes in ERK5 phosphorylation were observed. Similarly to ERK1 and ERK2, ERK5 appears to exhibit both short-term and longer-term increases in its activity. A rapid and modest increase in ERK5 phosphorylation was observed in myoblasts up to 15 minutes after IGF-2 stimulation, which then returned to baseline levels by 30 minutes; similar transient increases have also been observed in activated thymocytes (Sohn et al., 2008) and in vascular smooth muscle cells in response to insulin (Sharma and Goalstone, 2007). However, a consistent and greater increase in ERK5 phosphorylation was observed between 24 and 72 hours. Thus, in an analogous manner to ERK1 and ERK2, ERK5 could have different phospho-acceptor targets and regulate specific cellular events, depending on its temporal pattern of activation (Turjanski et al., 2007).
An intriguing aspect of ERK5 action is its dual function as a `conventional' MAPK, activating transcription factors such as the MEF2 family, c-fos, Fra-1, CREB and Sap1a (which regulates AP-1 function), coupled with its putative C-terminal-mediated transactivation activity (Kasler et al., 2000; Sohn et al., 2005). Studies using truncated forms of ERK5, in which the N- and C-terminal halves of the protein are discrete, demonstrate that its ability to phosphorylate downstream targets and to increase transactivation activity can be separate and independent (Buschbeck and Ullrich, 2005; Kasler et al., 2000). In vivo, however, these functions are likely to have some interdependence as it is thought that the N- and C-terminal regions of the molecule interact in inactive ERK5, masking a C-terminal nuclear localisation signal and allowing a nuclear export or cytoplasmic anchoring motif to be dominant. Additionally, when ERK5 is non-phosphorylated, C-terminal transactivation activity is suppressed (Kondoh et al., 2006). MEK5 activation of ERK5 kinase activity facilitates autophosphorylation in the C-terminus, facilitating nuclear localisation (Mody et al., 2003). Our findings support a positive relationship between ERK5 phosphorylation, localisation and transactivation activity: the form of ERK5 lacking a phospho-acceptor residue (ERK5AEF) was excluded from the nucleus and exhibited diminished ability to activate the E box promoter-reporter; and, conversely, constitutively active MEK5 induced increased ERK5 phosphorylation, nuclear localisation and E box promoter-reporter activity.
Myoblast differentiation cannot proceed without cell-cycle exit, which is also accompanied by increased resistance to apoptosis. The temporal activation of ERK5 would be compatible with a regulatory function in both of these events. However, current findings on such putative roles for ERK5 are conflicting. Initial observations on EGF-mediated proliferation in the MCF10A epithelial cell line indicated that ERK5 was an essential target for cell-cycle progression (Kato et al., 1998), but subsequent studies have not consistently supported the necessity for MEK5 and ERK5 in proliferation; the absence of MEK5 in null embryos does not affect cell-cycle progression (Wang et al., 2005). There is more evidence for an anti-apoptotic role for ERK5. Most convincing are the data from embryonic fibroblasts derived from either Erk5- or Mek5-null mice suggesting an essential cell survival role for MEK5 and ERK5 in both basal conditions and following osmotic stress (Wang et al., 2006a). A survival role for ERK5 is also supported by studies in immune function (Carvajal-Vergara et al., 2005; Garaude et al., 2006) and in neuronal cells (Sturla et al., 2005). However, there is some doubt whether the pro-survival effects of ERK5 are direct or indirect (Liu et al., 2006) and one study reports that it is a mediator of BDNF-promoted survival of developing neurons via activation of MEF2-mediated gene expression (Pi et al., 2004). In marked contrast to these findings is the report that ERK5 and MEF2 are important for medulloblastoma cell death (Sturla et al., 2005) and a recent suggestion that ERK5 is pro-apoptotic in thymocytes (Sohn et al., 2008). It is possible, therefore, that the function of ERK5 is cell-type specific and might indeed be different in normal versus transformed cells.
Materials and Methods
Reagents, plasmids and antibodies
Reagents were purchased as follows: myelin basic protein (MBP) from Sigma (Poole, UK); cell culture reagents from Invitrogen (Paisley, Scotland); and [γ-32P]ATP and [γ-32P]CTP from Amersham Pharmacia Biotech (Little Chalfont, UK).
Constructs and expression vectors were generous gifts or acquired as follows: myogenin cDNA probe (Eric N. Olson, University of Texas Southwestern Medical Center, Dallas, TX); Gapdh probe (Gavin Kelsey, Babraham Institute, Cambridge, UK); Igf2 cDNA probe (Miguel Constancia, Babraham Institute); Igf2 antisense cDNA (Peter Rotwein, Oregon Health Sciences University, Portland, OR); human ERK5 wild type, ERK5AEF [ERK5 TEY phospho-acceptor sites are mutated to AEF therefore acting as a dominant-negative], rat MEK5DD [constitutively active phosphomimetic MEK5 with phospo-acceptor serine sites mutated to acidic aspartate residues] and dominant-negative MEK5 [serine phospho-acceptor sites mutated to alanine residues] (Jiing-Dwan Lee, The Scripps Research Institute, La Jolla, CA); 4RE luciferase E box reporter from Pier Lorenzo Puri (University of California, San Diego, CA); GFP expression vector (pEGFP, Clontech Laboratories, Oxford, UK); pcDNA3 vector (Invitrogen). Primers were purchased from Sigma-Genosys (Cambridge, UK).
Sources of antibodies were as follows: mouse anti-MHC (MF20) from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA); rabbit anti-MEF2C (C-21) and goat anti-ERK5 (C-20) from Santa Cruz Biotechnology (Wembley, UK); rabbit anti-phospho-ERK5 (pT218/pY220) from Biosource International (Camarillo, CA); rabbit anti-GFP from Molecular Probes (Cambridge, UK); mouse anti-FLAG from Sigma (Dorset, UK); mouse anti-Haemaglutinin (HA) from Harlan Sera-Lab (Loughborough, UK); mouse anti-p21 from BD Pharmingen (San Diego, CA); rabbit anti-p38 from Cell Signalling Technology (supplied by NEB, Beverly, MA); anti-β-actin from Abcam (Cambridge, UK); horseradish peroxidase (HRP) conjugated goat anti-rabbit immunoglobulin G, goat anti-mouse IgG, donkey anti-goat IgG, Cy3 conjugated anti-goat and FITC conjugated anti-rabbit antibodies from Jackson Immunoresearch (Luton, UK).
Cloning
ERK5 and ERK5AEF were amplified from pcDNA3 vectors using primers designed to add the restriction sites XhoI (5′) and HindIII (3′), to create ERK5 in frame with GFP when ligated into pEGFP-N1, to distance ERK5 from GFP to minimise interference, and to mutate the stop codon to allow translation to continue through to GFP. Primer sequences were: sense, 5′-CCTCGAGACCATGGCCGAGCCTCTGAAGGAG-3′ and antisense, 5′-GGAAGCTTGGGGTCCTGGAGGTCAGGCAG-3′.
Amplification was performed using pfu polymerase to minimise errors. However, because pfu produces blunt-ended fragments, taq was added during the final extension cycle in order to add polyA and allow cloning into pGEMTeasy. The PCR products were sequenced around the TEY/AEF motif to check integrity. Sequencing primers were: sense, 5′-ATCATCCACTCCTCACAGCC-3′ and antisense, 5′-ATGCAGCCCACAGACCAGAG-3′.
ERK5 and ERK5AEF were digested from pGEMTeasy with XhoI/HindIII and ligated into similarly digested pEGFP-N1.
Cell culture
C2 myoblasts, derived from mouse muscle satellite cells, were seeded (2×105 cells into 100-mm plates) on to 2% gelatin-coated plates and initially cultured in growth medium [GM; Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum and 10% newborn calf serum]. At 80% confluence, differentiation was induced by replacing GM with differentiation medium (DM; DMEM with 2% horse serum). When indicated, exogenous IGF-2 (25 ng/ml) or vehicle was added to the medium immediately after cells were moved into DM; the medium was changed every 24 hours.
To examine cell migration, the classical `scratch-wound' assay (DeBiasio et al., 1987; Etienne-Manneville and Hall, 2001) was used. After 24 hours in DM, confluent myoblasts were scraped to form a break in the cell layer and were further incubated in DM for 6 hours. Myoblasts migrating across the `gap' to close the wound were analysed morphologically using Gill's haematoxylin and eosin (H and E) staining as described previously (Langley et al., 2002). The culture plates were washed three times with PBS and fixed with 4% paraformaldehyde for 10-15 minutes at room temperature. Cells were photographed using a Leica MZ6 microscope and the number of migrating cells per unit area was counted; data were expressed relative to pcDNA-only transfected cells. BrdU analyses demonstrated that changes in the number of migrating cells were not due to cell division over this time period.
Transfection
For transient transfections, 1×105 C2 myoblasts were seeded in 2% gelatin-coated dishes (60 mm diameter) 24 hours before transfection. Cells were transfected with 3 μg of either p38dn, ERK5, ERK5AEF, MEK5DD, MEK5dn or pcDNA3 using Effectene transfection reagent as recommended by the manufacturer (Qiagen, Crawley, UK). Transfected cells were cultured in GM for 24 hours and then transferred to DM for 48-96 hours. Cells were co-transfected with a GFP expression plasmid (pEGFP) to determine transfection efficiency between samples. Transfection efficiencies of 35-40% were obtained routinely, as assessed by fluorescence microscopy.
Whole-cell extract preparation
C2 cells were washed three times with PBS and harvested in 250 μl of lysis buffer (20 mM Tris-HCl, pH 7.5; 137 mM NaCl; 1 mM EGTA, pH 8; 1% Triton X-100; 10% glycerol; 1.5 mM MgCl2) containing protease and phosphatase inhibitors (10 mM NaF, 1 mM PMSF, 1 mM Na orthovanadate, 5 μg/ml aprotinin, 10 μg/ml leupeptin). Cellular debris was removed by centrifugation at 13000 g for 1 minute. Cell extracts were frozen in liquid nitrogen and stored at –80°C until use. Total protein in the extracts was determined by the Bradford assay (BioRad, Hemel Hempstead, UK).
Western blot analysis
Protein (50 μg) from cell lysates was resolved by SDS-PAGE and transferred to an Immobilon-P membrane (Millipore, Watford, UK) by electroblotting. The membranes were blocked with 0.2% I-block (Applied Biosystems, Warrington, UK) in 0.1% Tween20 in PBS for 1 hour at 37°C and probed with corresponding primary and secondary antibodies. Blots were washed in PBS plus 0.1% Tween20 and the antigen-antibody complexes visualised using enhanced chemiluminescence (ECL) reagents (Amersham Pharmacia Biotech) according to the manufacturer's instructions. When re-probing of the blots was performed, membranes were stripped with 5% acetic acid (pH 2.8) for 10 minutes at room temperature before being washed and blocked as described. Scanned autoradiographs were semi-quantified using AIDA 2D densitometry software.
Luciferase promoter reporter assays
C2 cells were co-transfected with 4RE-luc and pEGFP (for determination of transfection efficiency) exactly as described previously (Gonzalez et al., 2004; Wu et al., 2000) and harvested to measure luciferase activity using a luciferase assay kit (Promega, Southampton, UK). Where appropriate, the luciferase reporter was co-transfected with MEK5DD, MEK5dn, IGF2as or control empty vector (pcDNA3).
ERK5 immune complex kinase assay
Whole-cell extracts (50 μg) were incubated with 5 μl of total anti-ERK5 antibody for 12 hours at 4°C and subsequent kinase activity determined by incorporation of [γ-32P]ATP into myelin basic protein (MBP) exactly as described previously (Gonzalez et al., 2004). Scanned autoradiographs were semi-quantified using AIDA 2D densitometry software.
Fluorescence miscroscopy
C2 cells were seeded at 2×104 cells/ml onto single-well permanox slide chambers coated three times with 2% gelatin. After the appropriate incubation time, slides were washed carefully in PBS then fixed in 4% paraformaldehyde for 15 minutes. Slides were stored in PBS plus 0.05% Tween20 at 4°C. Fixed cells were permeabilised in PBS plus 0.2% (v/v) Triton X-100 (PBSTx) for 5 minutes and rinsed twice in PBS. Slides were mounted with Vectashield mounting medium with DAPI and stored at 4°C protected from light. Fluorescence was examined with an Olympus BX40 microscope.
Statistical analyses
Statistical differences between treatments were analysed using analysis of variance and/or Student's t-test, as indicated. Values are presented as means ± s.e.m. All experiments were performed independently at least three times.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/17/3104/DC1
This work was funded by the Biotechnology and Biological Sciences Research Council as a studentship (to E.J.C.) and Competitive Strategic Grant (to J.M.P.), and by Medical Research Council funding as a studentship (to F.A.L.). Deposited in PMC for release after 6 months.
References
- Abe, J., Kusuhara, M., Ulevitch, R. J., Berk, B. C. and Lee, J. D. (1996). Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J. Biol. Chem. 271, 16586-16590. [DOI] [PubMed] [Google Scholar]
- Berkes, C. A. and Tapscott, S. J. (2005). MyoD and the transcriptional control of myogenesis. Seminutes. Cell Dev. Biol. 16, 585-595. [DOI] [PubMed] [Google Scholar]
- Black, B. L. and Olson, E. N. (1998). Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 14, 167-196. [DOI] [PubMed] [Google Scholar]
- Bondy, C. A., Werner, H., Roberts, C. T., Jr and LeRoith, D. (1990). Cellular pattern of insulin-like growth factor-I (IGF-I) and type I IGF receptor gene expression in early organogenesis: comparison with IGF-II gene expression. Mol. Endocrinol. 4, 1386-1398. [DOI] [PubMed] [Google Scholar]
- Bour, B. A., O'Brien, M. A., Lockwood, W. L., Goldstein, E. S., Bodmer, R., Taghert, P. H., Abmayr, S. M. and Nguyen, H. T. (1995). Drosophila MEF2, a transcription factor that is essential for myogenesis. Genes Dev. 9, 730-741. [DOI] [PubMed] [Google Scholar]
- Buckingham, M. (2006). Myogenic progenitor cells and skeletal myogenesis in vertebrates. Curr. Opin. Genet. Dev. 16, 525-532. [DOI] [PubMed] [Google Scholar]
- Buschbeck, M. and Ullrich, A. (2005). The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J. Biol. Chem. 280, 2659-2667. [DOI] [PubMed] [Google Scholar]
- Buschbeck, M., Hofbauer, S., Di Croce, L., Keri, G. and Ullrich, A. (2005). Abl-kinase-sensitive levels of ERK5 and its intrinsic basal activity contribute to leukaemia cell survival. EMBO Rep. 6, 63-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Vergara, X., Tabera, S., Montero, J. C., Esparis-Ogando, A., Lopez-Perez, R., Mateo, G., Gutierrez, N., Parmo-Cabanas, M., Teixido, J., San Miguel, J. F. et al. (2005). Multifunctional role of Erk5 in multiple myeloma. Blood 105, 4492-4499. [DOI] [PubMed] [Google Scholar]
- Che, W., Lerner-Marmarosh, N., Huang, Q., Osawa, M., Ohta, S., Yoshizumi, M., Glassman, M., Lee, J. D., Yan, C., Berk, B. C. et al. (2002). Insulin-like growth factor-1 enhances inflammatory responses in endothelial cells: role of Gab1 and MEKK3 in TNF-alpha-induced c-Jun and NF-kappaB activation and adhesion molecule expression. Circ. Res. 90, 1222-1230. [DOI] [PubMed] [Google Scholar]
- Cobb, L. J., Salih, D. A., Gonzalez, I., Tripathi, G., Carter, E. J., Lovett, F., Holding, C. and Pell, J. M. (2004). Partitioning of IGFBP-5 actions in myogenesis: IGF-independent anti-apoptotic function. J. Cell Sci. 117, 1737-1746. [DOI] [PubMed] [Google Scholar]
- DeBiasio, R., Bright, G. R., Ernst, L. A., Waggoner, A. S. and Taylor, D. L. (1987). Five-parameter fluorescence imaging: wound healing of living Swiss 3T3 cells. J. Cell Biol. 105, 1613-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChiara, T. M., Efstratiadis, A. and Robertson, E. J. (1990). A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345, 78-80. [DOI] [PubMed] [Google Scholar]
- Dinev, D., Jordan, B. W., Neufeld, B., Lee, J. D., Lindemann, D., Rapp, U. R. and Ludwig, S. (2001). Extracellular signal regulated kinase 5 (ERK5) is required for the differentiation of muscle cells. EMBO Rep. 2, 829-834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English, J. M., Pearson, G., Hockenberry, T., Shivakumar, L., White, M. A. and Cobb, M. H. (1999). Contribution of the ERK5/MEK5 pathway to Ras/Raf signaling and growth control. J. Biol. Chem. 274, 31588-31592. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville, S. and Hall, A. (2001). Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell 106, 489-498. [DOI] [PubMed] [Google Scholar]
- Florini, J. R., Magri, K. A., Ewton, D. Z., James, P. L., Grindstaff, K. and Rotwein, P. S. (1991). “Spontaneous” differentiation of skeletal myoblasts is dependent upon autocrine secretion of insulin-like growth factor-II. J. Biol. Chem. 266, 15917-15923. [PubMed] [Google Scholar]
- Garaude, J., Cherni, S., Kaminski, S., Delepine, E., Chable-Bessia, C., Benkirane, M., Borges, J., Pandiella, A., Iniguez, M. A., Fresno, M. et al. (2006). ERK5 activates NF-kappaB in leukemic T cells and is essential for their growth in vivo. J. Immunol. 177, 7607-7617. [DOI] [PubMed] [Google Scholar]
- Gonzalez, I., Tripathi, G., Carter, E. J., Cobb, L. J., Salih, D. A., Lovett, F. A., Holding, C. and Pell, J. M. (2004). Akt2, a novel functional link between p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways in myogenesis. Mol. Cell. Biol. 24, 3607-3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvakova, M. A. (2007). Insulin-like growth factors control cell migration in health and disease. Int. J. Biochem. Cell Biol. 39, 890-909. [DOI] [PubMed] [Google Scholar]
- Johnson, G. L. and Lapadat, R. (2002). Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911-1912. [DOI] [PubMed] [Google Scholar]
- Kamakura, S., Moriguchi, T. and Nishida, E. (1999). Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases: identification and characterization of a signaling pathway to the nucleus. J. Biol. Chem. 274, 26563-26571. [DOI] [PubMed] [Google Scholar]
- Kasler, H. G., Victoria, J., Duramad, O. and Winoto, A. (2000). ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 20, 8382-8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, Y., Tapping, R. I., Huang, S., Watson, M. H., Ulevitch, R. J. and Lee, J. D. (1998). Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 395, 713-716. [DOI] [PubMed] [Google Scholar]
- Kato, Y., Chao, T. H., Hayashi, M., Tapping, R. I. and Lee, J. D. (2000). Role of BMK1 in regulation of growth factor-induced cellular responses. Immunol. Res. 21, 233-237. [DOI] [PubMed] [Google Scholar]
- Kim, J., Song, G., Gao, H., Farmer, J. L., Satterfield, M. C., Burghardt, R. C., Wu, G., Johnson, G. A., Spencer, T. E. and Bazer, F. W. (2008). Insulin-like growth factor II activates phosphatidylinositol 3-kinase-protooncogenic protein kinase 1 and mitogen-activated protein kinase cell Signaling pathways, and stimulates migration of ovine trophectoderm cells. Endocrinology 149, 3085-3094. [DOI] [PubMed] [Google Scholar]
- Kondoh, K., Terasawa, K., Morimoto, H. and Nishida, E. (2006). Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol. Cell. Biol. 26, 1679-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley, B., Thomas, M., Bishop, A., Sharma, M., Gilmour, S. and Kambadur, R. (2002). Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 277, 49831-49840. [DOI] [PubMed] [Google Scholar]
- Le Grand, F. and Rudnicki, M. (2007). Satellite and stem cells in muscle growth and repair. Development 134, 3953-3957. [DOI] [PubMed] [Google Scholar]
- Lee, J. D., Ulevitch, R. J. and Han, J. (1995). Primary structure of BMK1: a new mammalian map kinase. Biochem. Biophys. Res. Commun. 213, 715-724. [DOI] [PubMed] [Google Scholar]
- LeRoith, D., Werner, H., Beitner-Johnson, D. and Roberts, C. T., Jr (1995). Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 16, 143-163. [DOI] [PubMed] [Google Scholar]
- Linnerth, N. M., Baldwin, M., Campbell, C., Brown, M., McGowan, H. and Moorehead, R. A. (2005). IGF-II induces CREB phosphorylation and cell survival in human lung cancer cells. Oncogene 24, 7310-7319. [DOI] [PubMed] [Google Scholar]
- Liu, J. P., Baker, J., Perkins, A. S., Robertson, E. J. and Efstratiadis, A. (1993). Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75, 59-72. [PubMed] [Google Scholar]
- Liu, L., Cundiff, P., Abel, G., Wang, Y., Faigle, R., Sakagami, H., Xu, M. and Xia, Z. (2006). Extracellular signal-regulated kinase (ERK) 5 is necessary and sufficient to specify cortical neuronal fate. Proc. Natl. Acad. Sci. USA 103, 9697-9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W., Liu, Y. and Lowe, W. L., Jr (2001). The role of phosphatidylinositol 3-kinase and the mitogen-activated protein kinases in insulin-like growth factor-I-mediated effects in vascular endothelial cells. Endocrinology 142, 1710-1719. [DOI] [PubMed] [Google Scholar]
- Lovett, F. A., Gonzalez, I., Salih, D. A., Cobb, L. J., Tripathi, G., Cosgrove, R. A., Murrell, A., Kilshaw, P. J. and Pell, J. M. (2006). Convergence of Igf2 expression and adhesion signalling via RhoA and p38 MAPK enhances myogenic differentiation. J. Cell Sci. 119, 4828-4840. [DOI] [PubMed] [Google Scholar]
- Mody, N., Campbell, D. G., Morrice, N., Peggie, M. and Cohen, P. (2003). An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem. J. 372, 567-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae, J., Kido, Y. and Accili, D. (2001). Distinct and overlapping functions of insulin and IGF-I receptors. Endocr. Rev. 22, 818-835. [DOI] [PubMed] [Google Scholar]
- Nakamura, K., Uhlik, M. T., Johnson, N. L., Hahn, K. M. and Johnson, G. L. (2006). PB1 domain-dependent signaling complex is required for extracellular signal-regulated kinase 5 activation. Mol. Cell. Biol. 26, 2065-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto, S. and Nishida, E. (2006). MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 7, 782-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi, X., Yan, C. and Berk, B. C. (2004). Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ. Res. 94, 362-369. [DOI] [PubMed] [Google Scholar]
- Polesskaya, A., Cuvellier, S., Naguibneva, I., Duquet, A., Moss, E. G. and Harel-Bellan, A. (2007). Lin-28 binds IGF-2 mRNA and participates in skeletal myogenesis by increasing translation efficiency. Genes Dev. 21, 1125-1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan, C. P., Li, W., Boucher, D. M., Spatz, S., Su, M. S. and Kuida, K. (2002). Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. USA 99, 9248-9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, H., Yin, P. and Duan, C. (2008). IGFBP-5 regulates muscle cell differentiation by binding to IGF-II and switching on the IGF-II auto-regulation loop. J. Cell Biol. 182, 979-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramp, M., Ying, O., Kim, T. Y. and Martin, G. S. (2008). ERK5 promotes Src-induced podosome formation by limiting Rho activation. J. Cell Biol. 181, 1195-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra, C., Palacios, D., Mozzetta, C., Forcales, S. V., Morantte, I., Ripani, M., Jones, D. R., Du, K., Jhala, U. S., Simone, C. et al. (2007). Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol. Cell 28, 200-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried, J., Wang, X., Kharebava, G. and Tournier, C. (2005). A novel mitogen-activated protein kinase docking site in the N terminus of MEK5alpha organizes the components of the extracellular signal-regulated kinase 5 signaling pathway. Mol. Cell. Biol. 25, 9820-9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, G. and Goalstone, M. L. (2007). Regulation of ERK5 by insulin and angiotensin-II in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 354, 1078-1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn, S. J., Li, D., Lee, L. K. and Winoto, A. (2005). Transcriptional regulation of tissue-specific genes by the ERK5 mitogen-activated protein kinase. Mol. Cell. Biol. 25, 8553-8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn, S. J., Lewis, G. M. and Winoto, A. (2008). Non-redundant function of the MEK5-ERK5 pathway in thymocyte apoptosis. EMBO J. 27, 1896-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, C. E. and Rotwein, P. (1996). Insulin-like growth factor-II is an autocrine survival factor for differentiating myoblasts. J. Biol. Chem. 271, 11330-11338. [DOI] [PubMed] [Google Scholar]
- Stewart, C. E., James, P. L., Fant, M. E. and Rotwein, P. (1996). Overexpression of insulin-like growth factor-II induces accelerated myoblast differentiation. J. Cell Physiol. 169, 23-32. [DOI] [PubMed] [Google Scholar]
- Sturla, L. M., Cowan, C. W., Guenther, L., Castellino, R. C., Kim, J. Y. and Pomeroy, S. L. (2005). A novel role for extracellular signal-regulated kinase 5 and myocyte enhancer factor 2 in medulloblastoma cell death. Cancer Res. 65, 5683-5689. [DOI] [PubMed] [Google Scholar]
- Stylianopoulou, F., Efstratiadis, A., Herbert, J. and Pintar, J. (1988). Pattern of the insulin-like growth factor II gene expression during rat embryogenesis. Development 103, 497-506. [DOI] [PubMed] [Google Scholar]
- Sun, W., Wei, X., Kesavan, K., Garrington, T. P., Fan, R., Mei, J., Anderson, S. M., Gelfand, E. W. and Johnson, G. L. (2003). MEK kinase 2 and the adaptor protein Lad regulate extracellular signal-regulated kinase 5 activation by epidermal growth factor via Src. Mol. Cell. Biol. 23, 2298-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzaki, Y., Yoshizumi, M., Kagami, S., Koyama, A. H., Taketani, Y., Houchi, H., Tsuchiya, K., Takeda, E. and Tamaki, T. (2002). Hydrogen peroxide stimulates c-Src-mediated big mitogen-activated protein kinase 1 (BMK1) and the MEF2C signaling pathway in PC12 cells: potential role in cell survival following oxidative insults. J. Biol. Chem. 277, 9614-9621. [DOI] [PubMed] [Google Scholar]
- Turjanski, A. G., Vaque, J. P. and Gutkind, J. S. (2007). MAP kinases and the control of nuclear events. Oncogene 26, 3240-3253. [DOI] [PubMed] [Google Scholar]
- Wang, X. and Tournier, C. (2006). Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 18, 753-760. [DOI] [PubMed] [Google Scholar]
- Wang, X., Merritt, A. J., Seyfried, J., Guo, C., Papadakis, E. S., Finegan, K. G., Kayahara, M., Dixon, J., Boot-Handford, R. P., Cartwright, E. J. et al. (2005). Targeted deletion of mek5 causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Mol. Cell. Biol. 25, 336-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., Finegan, K. G., Robinson, A. C., Knowles, L., Khosravi-Far, R., Hinchliffe, K. A., Boot-Handford, R. P. and Tournier, C. (2006a). Activation of extracellular signal-regulated protein kinase 5 downregulates FasL upon osmotic stress. Cell Death Differ. 13, 2099-2108. [DOI] [PubMed] [Google Scholar]
- Wang, Y., Su, B. and Xia, Z. (2006b). Brain-derived neurotrophic factor activates ERK5 in cortical neurons via a Rap1-MEKK2 signaling cascade. J. Biol. Chem. 281, 35965-35974. [DOI] [PubMed] [Google Scholar]
- Wilson, E. M. and Rotwein, P. (2006). Control of MyoD function during initiation of muscle differentiation by an autocrine signaling pathway activated by insulin-like growth factor-II. J. Biol. Chem. 281, 29962-29971. [DOI] [PubMed] [Google Scholar]
- Wilson, E. M., Hsieh, M. M. and Rotwein, P. (2003). Autocrine growth factor signaling by insulin-like growth factor-II mediates MyoD-stimulated myocyte maturation. J. Biol. Chem. 278, 41109-41113. [DOI] [PubMed] [Google Scholar]
- Wu, Z., Woodring, P. J., Bhakta, K. S., Tamura, K., Wen, F., Feramisco, J. R., Karin, M., Wang, J. Y. and Puri, P. L. (2000). p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol. Cell. Biol. 20, 3951-3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, C., Luo, H., Lee, J. D., Abe, J. and Berk, B. C. (2001). Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J. Biol. Chem. 276, 10870-10878. [DOI] [PubMed] [Google Scholar]
- Yan, L., Carr, J., Ashby, P. R., Murry-Tait, V., Thompson, C. and Arthur, J. S. (2003). Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev. Biol. 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, G., Bao, Z. Q. and Dixon, J. E. (1995). Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 270, 12665-12669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.