

Abstract
Ablation of inhibitory neurons that produce AgRP, NPY and GABA in the arcuate nucleus (ARC) of adult mice results in starvation within ∼ 6 days. Viral-mediated inactivation of GABA biosynthesis in the ARC also promotes anorexia. Chronic subcutaneous delivery of bretazenil (a GABAA receptor partial agonist) for 11 days maintains feeding and survival following ablation of AgRP neurons, an effect that persists following cessation of drug delivery. Moreover, direct delivery of bretazenil into the parabrachial nucleus (PBN), a nucleus that relays gustatory and visceral sensory information, is sufficient to maintain feeding after AgRP neuron ablation, whereas delivery into other post-synaptic targets of AgRP neurons is ineffective. These results suggest that hyperactivity within the PBN following loss of GABAergic input from AgRP neurons promotes anorexia. We suggest that suppression of neuronal excitability within the PBN permits feeding and instatement of compensatory mechanisms that eventually allow mice to eat without GABA signaling from AgRP neurons.
Introduction
The agouti-related peptide (AgRP)-expressing neurons and the neighboring proopiomelanocortin (POMC)-expressing neurons that reside in the arcuate region of the hypothalamus integrate hormonal and neurotransmitter signals to modulate appetite and metabolism and thereby help maintain energy balance (Cone, 2005;Morton et al., 2006;Saper et al., 2002). Genetic, pharmacological and physiological data establish that enhanced melanocortin signaling by POMC neurons inhibits feeding while stimulating metabolism (Cone, 2005). AgRP neurons, which also produce neuropeptide Y (NPY) and γ-amino-butyric acid (GABA), send axons to many of the same brain regions as POMC neurons, where they antagonize the effects of melanocortin on post-synaptic cells (Broberger et al., 1998;Haskell-Luevano et al., 1999;Jacobowitz and O'Donohue, 1978;Watson et al., 1978).
The literature on regulation of body weight has emphasized the role of peptide hormones, neuropeptides and monoamines that act on membrane receptors to activate intracellular signaling cascades (Cone, 2005;Morton et al., 2006;Saper et al., 2002), with relatively little discussion of the role of neurotransmitters that regulate ion channels (Meister, 2007), despite the fact that both GABAA and GABAB receptor agonists enhance feeding by rodents and other animals (Cooper, 2005;Duke et al., 2006;Ebenezer and Prabhaker, 2007). Research on AgRP neurons was first directed towards the role of NPY and then, after the discovery of AgRP and its co-expression with NPY, focus turned to the complementary roles of these neuropeptides in regulation of feeding behavior (Broberger and Hokfelt, 2001;Flier, 2006;Kalra et al., 1999;Shutter et al., 1997). However, inactivation of the genes encoding NPY, AgRP or both had little effect on body weight regulation, suggesting that something else produced by these neurons was important (Erickson et al., 1996;Qian et al., 2002). Support for this idea came from experiments in which the AgRP neurons were genetically engineered to express the diphtheria toxin receptor (DTR), which allows their ablation by administration of diphtheria toxin (DT). Ablation of AgRP neurons in adult mice inhibits feeding and results in starvation within about 6 days of DT treatment (Gropp et al., 2005;Luquet et al., 2005), even in mice lacking functional Npy and Agrp genes (Phillips and Palmiter, 2008). Thus, the sudden loss of something other than NPY and AgRP promotes anorexia in this model. The starvation phenotype following AgRP neuron ablation is unaltered in the Ay genetic background, in which melanocortin signaling is blocked by ectopic production of agouti protein, indicating that starvation does not depend upon activation of the melanocortin signaling pathway (Wu et al., 2008a). Because AgRP neurons also express GABA (Cowley et al., 2001;Horvath et al., 1997), these observations led to the idea that GABA might be the critical transmitter produced by AgRP neurons. Support for this hypothesis comes from experiments in which GABA signaling by AgRP neurons was prevented by selective inactivation of the vesicular GABA transporter gene (Vgat); mice with constitutive inactivation of Vgat had a lean phenotype and were resistance to diet-induced obesity (Tong et al., 2008). This phenotype is reminiscent of the mild phenotype observed after ablation of AgRP neurons in neonatal mice (Luquet et al., 2005;Luquet et al., 2007), suggesting that compensation can occur when GABA production by AgRP neurons is compromised during early development.
As an alternative approach to examining the role of GABA signaling from AgRP neurons, we tested the ability of GABAA receptor agonists to maintain feeding after ablation of AgRP neurons. An extensive literature indicates that benzodiazepines can stimulate feeding and modulate taste reactivity – the stereotyped behaviors associated with different tastes (Berridge and Pecina, 1995;Cooper, 2005). Experiments in rats identified bretazenil as being particularly effective, in part, because it has less sedative activity than first-generation benzodiazepines like diazepam. Benzodiazepines were the most effective for modulating feeding when they were delivered into the 4th ventricle. Direct injection into various hindbrain regions indicated that the parabrachial nucleus (PBN) was an important site of action in mediating taste reactivity (Higgs and Cooper, 1996). The role of GABA in the hindbrain is particularly intriguing because ablation of AgRP neurons in adult mice not only reduces the motivation to initiate meals, but it also blocks consumption of liquid food delivered directly into the mouth (Wu et al., 2008a), and the latter is regulated by the hindbrain (Grill and Kaplan, 2002).
We previously demonstrated robust Fos gene activation in numerous post-synaptic targets of AgRP neurons following ablation of AgRP neurons (Wu et al., 2008b;Wu et al., 2008a). These findings suggested that lack of GABA signaling from AgRP neurons could lead to hyperactivity in post-synaptic targets. The resulting dysregulation caused by lack of GABA signaling from AgRP neurons could be responsible for the blockade of both appetitive and consummatory aspects of feeding (Wu et al., 2008a). Here we show that chronic stimulation of GABAergic signaling within a specific brain region allows feeding to continue after AgRP neuron ablation and adaptations that eventually allow mice to eat without AgRP neurons.
Results
Suppression of anorexia following ablation of AgRP neurons by chronic delivery of a GABAA receptor agonist
Mice in which the human DTR was targeted to the Agrp locus (AgrpDTR/+) were used for these studies. In agreement with previous results (Luquet et al., 2005;Wu et al., 2008a), administration of DT to these mice (2 intramuscular injections, 50 μg/kg, 2 days apart) results in progressive decline in food intake such that body weight falls 20% by 6-7 days, at which point the experiments are terminated for animal welfare considerations and to collect brain tissues for analysis (Figure 1A, B).
Figure 1. Chronic administration of bretazenil protects against starvation in adult mice after acute ablation of AgRP neurons.

(A) Percentage of initial body weight of DT-treated, AgrpDTR/+ mice after subcutaneous implantation of mini-osmotic pumps (mp) loaded with either bretazenil (n = 12) or vehicle (n = 10). DT was injected intramuscularly (im) twice as indicated by arrows. Minipumps were removed on Day 11 after drug solution was depleted.
(B) Liquid diet intake by the mice described in A.
(C) Licking activity of the mice described in A was plotted as average number of licks in 2-h bins. *, p < 0.01, ANOVA.
Error bars represent the standard error of mean (SEM).
We showed previously that ablation of AgRP neurons by this method results in robust induction of Fos mRNA in most post-synaptic targets of AgRP neurons and speculated that aberrant excitation of post-synaptic neurons could be responsible for the starvation phenotype (Wu et al., 2008a). To test this idea, Alzet minipumps were loaded with bretazenil, a GABAA receptor partial agonist, and implanted subcutaneously to deliver 750 ng/hr for 14 days. After 3 baseline days with the bretazenil-eluting minipump, the AgrpDTR/+ mice were treated with DT as before. In contrast to starvation normally observed, mice with the minipump transiently lost <10% of their body weight and their body weight returned to normal. Food consumption by the bretazenil-treated group fell for the first 6 days after DT treatment, but then also returned to normal over the next 12 days (Figure 1A, B). Remarkably, feeding continued even when the minipump was removed 11 days after initiation of DT treatment (Figure 1A, B). Licking activity at the liquid food dispenser persisted with a typical daily rhythm in the bretazenil- treated group; whereas in the absence of the agonist, licking ceased 7 days after DT treatment began (Figure 1C).
Bretazenil (0.2 mg/kg, ip) increased food consumption by wild-type mice during the first 4 hr, but 24-hr food intake was normal. Chronic delivery of bretazenil to wild-type animals for 10 days via a minipump had no effect on body weight (Figure S1 available online). Once-daily administration of bretazenil (0.2 mg/kg, ip) was ineffective at preventing starvation after AgRP neuron ablation, suggesting that chronic activation of GABAA receptors is an important aspect of the rescue strategy (Figure S2).
The number of meals was maintained by mice with the bretazenil-eluting minipump after ablation, whereas it gradually declined to nothing without drug treatment (Figure S3A). The meal size (number of licks per meal) persisted at a comparable level throughout the time course in bretazenil-treated group (Figure S3B) instead of increasing as expected for food-deprived animals (Wu et al., 2008a). Ablation of AgRP neurons also prevents normal food consumption even when food is delivered directly into the mouth via an intraoral fistula, indicating that consummatory aspects of feeding are also disrupted (Wu et al., 2008a). Chronic treatment with bretazenil restored intra-oral sucrose consumption to >80% of original after ablation of AgRP neurons (Figure S3C). Thus, both consummatory and appetitive responses are rescued by bretazenil treatment after AgRP neuron ablation.
Enhanced GABAA receptor signaling in hindbrain rescues feeding after ablation of AgRP neurons
To grossly determine where in the brain GABAA receptor activation is most important for maintenance of feeding, Alzet minipumps were implanted subcutaneously and connected to cannulas placed into either the 3rd or 4th ventricles. When bretazenil was delivered at 75 ng/hr into the 4th ventricle or subcutaneously it rescued feeding after AgRP neuron ablation; delivery of this dose into the 3rd ventricle retarded weight loss but did not prevent starvation (Figure 2A). Mice with delivery of bretazenil into the 3rd ventricle appeared sedated, which may have prevented adequate feeding. To minimize the sedative effects, the dose was reduced another 5 fold. Delivery of bretazenil (15 ng/hr) into either 3rd or 4th ventricle was sufficient to prevent starvation (Figure 2B) and sedation was no longer apparent. Delivery of bretazenil into the 4th ventricle at an even lower dose (7.5 ng/hr) still maintained body weight within acceptable limits; however, delivery of this dose into the 3rd ventricle delayed loss of body weight to 80% of original body weight (Figure 2C). A further 3-fold reduction of bretazenil delivery (2.5 ng/hr) was ineffective at preventing weight loss in the AgRP ablated mice (Figure 2C). These results indicate that delivery of an appropriate dose of bretazenil into either the 3rd or 4th ventricle can maintain adequate feeding, but the effect of the drug in the brainstem is marginally more effective than in the forebrain. Daily administration of bretazenil into the 4th ventricle (1 μg/day) significantly slowed the starvation phenotype but was unable to prevent it, again suggesting that chronic delivery of the drug is critical (Figure S4).
Figure 2. Minipump delivery of bretazenil into the 4th ventricle is more efficacious than delivery into the 3rd ventricle.

(A) Percentage of initial body weight of DT-treated, AgrpDTR/+ mice in which bretazenil (75 ng/hr; 1/10th of the dosage used in Figure 1) or vehicle was chronically administered either subcutaneously (sc) or directly into the 3rd (3v) or 4th (4v) ventricle.
(B) Percentage of initial body weight of DT-treated, AgrpDTR/+ mice in which bretazenil (15 ng/hr; 1/50th of the dosage used in Figure 1) or vehicle was chronically administered directly into the 3rd or 4th ventricle. Note that abrupt withdrawal of bretazenil delivered into the 4th ventricle at Day 4 resulted in more profound decline of body weight than that was shown by the AgRP-ablated mice infused with vehicle; *, p < 0.01, ANOVA.
(C) Percentage of initial body weight of DT-treated, AgrpDTR/+ mice in which bretazenil (7.5 or 2.5 ng/hr, 1/100th or 1/300th of the dosage used in Figure 1, respectively) or vehicle was chronically administered directly into either the 3rd or 4th ventricle.
N = 6 - 8 for each group. Error bars represent the SEM.
It is noteworthy that withdrawal of the minipump delivering bretazenil at 75 ng/hr into the 4th ventricle at day 4 after initiation of DT treatment resulted in precipitous loss of body weight (Figure 2B), whereas the mice continued to feed when the minipumps were removed at day 11 (Figure 1A, B).
To confirm that the action of bretazenil is mediated by GABAA receptors, we blocked the effect of bretazenil with flumazenil, a GABAA receptor antagonist. Mice with minipumps delivering bretazenil (750 ng/hr, sc) were treated with DT as above, but then flumazenil was introduced either peripherally (10 mg/kg, ip) or into the 3rd or 4th ventricle (3 μg) on a once-daily basis. Mice receiving either peripheral or the 4th ventricle delivery of the antagonist stopped feeding and rapidly lost body weight during the next 3 or 4 days, whereas mice with delivery of the flumazenil into the 3rd ventricle survived (Figure S5A, B). Dispensing the same dose of flumazenil into the 4th ventricle of DT-treated, wild-type mice had no effect on food intake or body weight, as expected since this drug only blocks the benzodiazepine site (Figure S5A, B). These results help establish the GABAA receptor as the critical target of bretazenil and suggest that GABA signaling in the brainstem is more important than in the forebrain for maintenance of feeding behavior in AgRP-ablated mice.
Activation of GABAA receptors suppresses Fos and astrocyte activity
Ablation of AgRP neurons leads to robust induction of Fos mRNA in many brain regions that are known post-synaptic targets of AgRP neurons compared to pair-fed, control mice (Wu et al., 2008b). We hypothesized that the inhibition of feeding after ablation of AgRP neurons is due to the dysregulation of critical feeding circuits and that bretazenil acts by reducing neuronal activation. Hence, we measured Fos gene activation by semi-automated in situ hybridization (Lein et al., 2007). Bretazenil (minipump at 750 ng/hr, sc) significantly reduced Fos expression in 5 of the 8 brain regions that are heavily innervated by AgRP fibers (Figures 3, S6). In the forebrain, Fos expression was reduced in the arcuate nucleus (ARC), paraventricular nucleus (PVN) and the lateral septum (LS), while in the hindbrain the reduction was striking in the parabrachial nucleus (PBN) and nucleus of the solitary tract (NTS). The LS and PBN were the only anatomic positions where Fos expression was reduced to the level observed in pair-fed mice (summarized in Figure 3Q). Delivery of bretazenil (75 ng/hr) into the 3rd ventricle suppressed Fos and astrocyte activation in the ARC and PVN but not in the PBN, while delivery of bretazenil into the 4th ventricle suppressed Fos activation in the PBN but not the ARC or PVN, indicating that appreciable amounts of the drug do not diffuse from one ventricle to another (Figure S7).
Figure 3. Fos activation after AgRP neuron ablation is suppressed in some brain regions by bretazenil treatment.
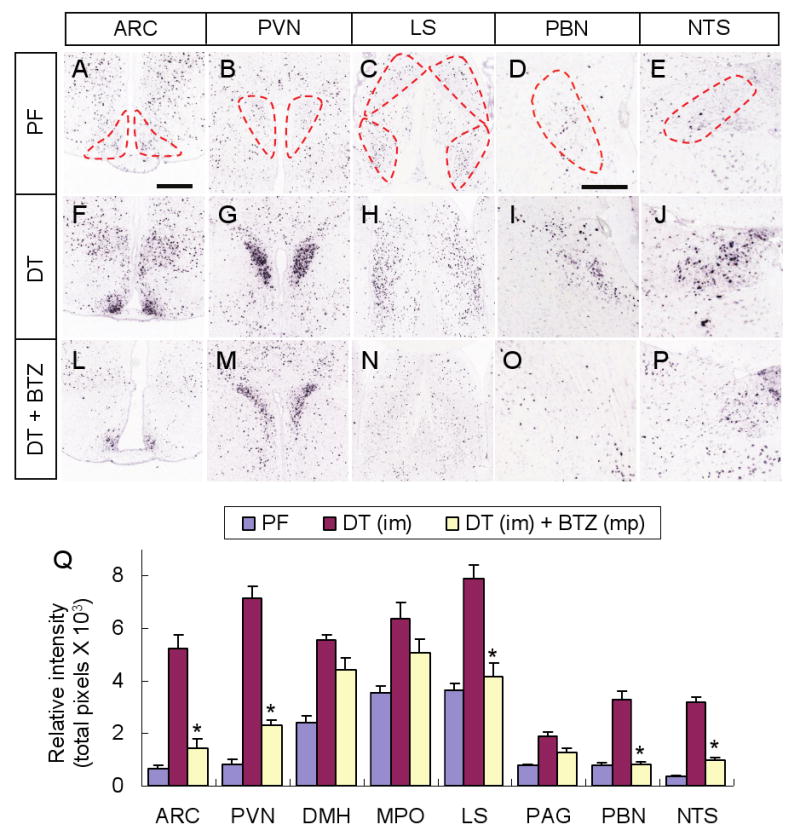
(A-E) Representative pictures of Fos in situ hybridization in post-synaptic regions of AgRP neurons including the ARC (A), PVN (B), LS (C), PBN (D), and NTS (E), in pair-fed AgrpDTR/+ mice after losing ∼20% of their initial body weight. Post-synaptic areas of AgRP neurons are denoted by dotted lines.
(F-J) Representative pictures of Fos in situ hybridization in the same post-synaptic regions of AgRP neurons in DT-treated, AgrpDTR/+ mice after losing ∼20% of their initial body weight.
(L-P) Representative pictures of Fos in situ hybridization in the same post-synaptic regions of AgRP neurons in DT-treated, AgrpDTR/+ mice with subcutaneous implantation of minipumps eluting bretazenil.
(Q) Quantified results for Fos in situ signals in selected post-synaptic regions of AgRP neurons of either pair-fed, DT-treated, or DT/bretazenil-treated, AgrpDTR/+ mice. See Supplementary Figure S6 for a complete set of images of Fos in situ hybridization in these regions. *, p < 0.01 between the DT-treated group and the DT/bretazenil-treated group for each respective area, ANOVA.
N = 4 - 6 per group. Scale bar (in A): A-C, F-H, and L-N, 400 μm; scale bar (in D): D, E, I, J, O, and P, 400 μm. Error bars in Q represent the SEM.
Ablation of AgRP neurons not only induces Fos expression in post-synaptic brain regions, but also activates astrocytes and microglia in most of the same regions (Wu et al., 2008b). Release of cytokines and other molecules by activated glia may contribute to the dysregulation of feeding. We found that chronic treatment of bretazenil suppressed activation of astrocytes (revealed by GFAP immunohistochemistry) without reducing microglial activation (revealed by IBA1 immunohistochemistry) in most post-synaptic targets of AgRP neurons (Figure S8). Moreover, like the effects on Fos activation, the suppression of astrocyte activation was restricted to the brain regions in proximity to the ventricle where bretazenil was delivered (Figure S8).
GABAA receptor signaling to the PBN is necessary to prevent anorexia
Based on the observation that Fos expression was reduced in 5 of the brain regions examined, we attempted to rescue feeding by minipump delivery of bretazenil (15 ng/hr) bilaterally into those selected brain regions. In the hindbrain, only delivery of bretazenil into the PBN achieved a full recovery of body weight following a transient loss of ∼10% during the time course of treatment; whereas delivery into the PAG or NTS was ineffective (Figure 4A). In the forebrain, delivery of bretazenil to the PVN was ineffective, but delivery into the LS prolonged survival about 1 week but the mice eventually lost 80% of their body weight (Figure 4B). These results indicate that stimulating GABAergic signaling in the PBN is sufficient to protect against starvation in AgRP neuron-ablated mice. The coordinates for bretazenil infusion into the PBN are shown in Figure 4C. In those cases where the cannulas were misplaced or one or both sides became clogged, anorexia was not prevented by bretazenil. The observation that delivery of bretazenil to the PAG was ineffective at preventing anorexia indicates that the effective diffusion range of bretazenil is less than 1.2 mm (the average distance from PAG to PBN). Likewise, delivery of bretazenil to the PBN suppressed Fos expression in the PBN but not in neighboring nuclei where Fos is activated after AgRP neuron ablation (data not shown).
Figure 4. Administration of bretazenil into the PBN prevents starvation in mice after ablation of AgRP neurons.
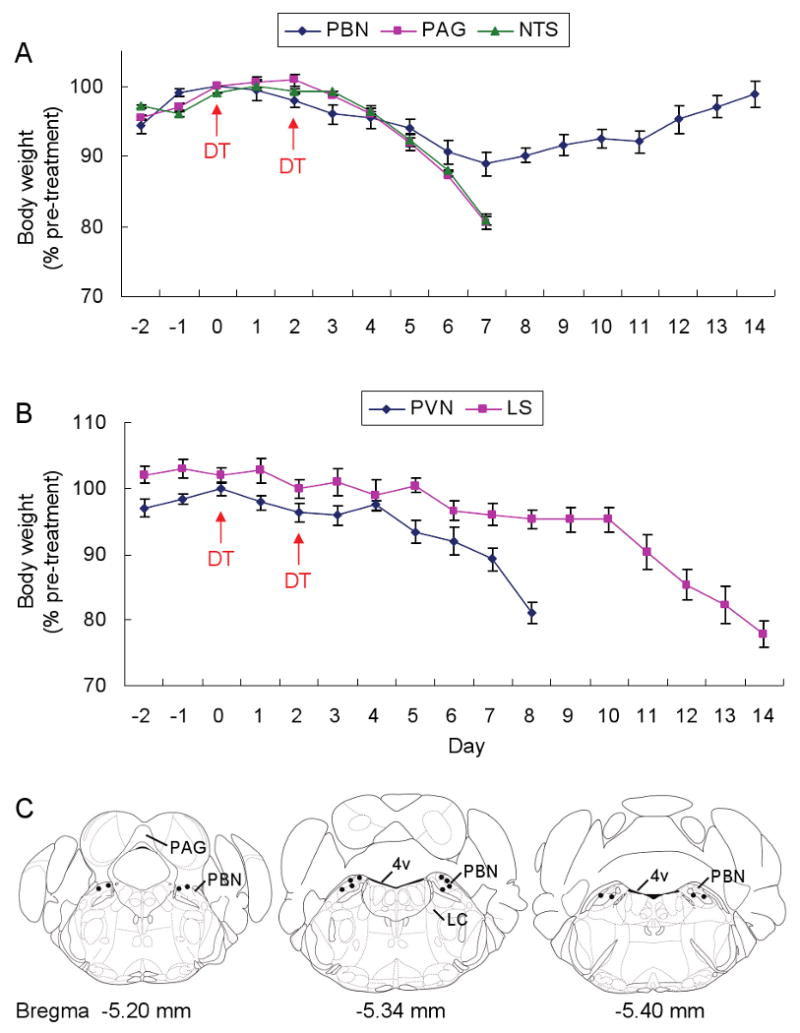
(A) Percentage of initial body weight of DT-treated, AgrpDTR/+ mice after chronic administration of bretazenil in 3 post-synaptic areas of AgRP neurons in the hindbrain (PBN, PAG, and NTS).
(B) Percentage of initial body weight of DT-treateted, AgrpDTR/+ mice after chronic administration of bretazenil in 2 forebrain regions (PVN and LS) with projections from AgRP neurons.
N = 6 – 8 per each group. Error bars represent the SEM.
(C) Coronal illustrations of mouse hindbrain regions; the dots represent effective infusion sites in the PBN area. LC, locus ceruleus; 4v, 4th ventricle.
To determine whether inhibition of GABAA-receptor signaling would lead to anorexia in normal mice, bicuculline, an antagonist that binds to the GABA site in the receptor, was infused into the PBN via bilateral cannulas for 4 days and then the pumps were disconnected. There was a dose-dependent inhibition of feeding and loss of body weight that rebounded when drug delivery ceased (Figure 5A, B). Thus, continual GABA signaling in the PBN is necessary to maintain feeding behavior and prevent anorexia.
Figure 5. Inhibition of GABAA receptors in the PBN or disruption of the neural inputs from AgRP neurons to the PBN produces anorexia.

(A) Percentage of initial body weight of C57BL/6 wild-type mice in which bicuculline (100, 300, and 1000 ng/day) was chronically infused to the PBN area through bilateral cannulae for 4 days and then discontinued.
(B) Intake of liquid diet by the mice described in A. *, p < 0.01, ANOVA between the groups of 100 ng/day and 1000 ng/day. Error bars represent the SEM. N = 5 – 6 mice per group.
(C) Percentage of initial body weight of AgrpDTR/+ and wild-type mice that received a single injection of DT into the PBN area (bilateral, 4 ng per side).
(D) Intake of liquid diet by the mice described in C.
To establish whether signaling from AgRP neurons to the PBN is required to maintain feeding, DT was injected into the PBN with the assumption the human DTR would be located throughout AgRP neurons, including their axons; thus, engaging DTR receptors on axonal processes would mediate cell death. Figure 5C, D shows that AgrpDTR mice injected bilaterally with a low dose of DT (4 ng per side) became anorexic, whereas injection of control mice with DT had no effect. Injection of the same dose of DT bilaterally into the LS did not lead to anorexia (data not shown).
AgRP neurons in the ARC are GABAergic
The in situ hybridization experiments included probes for Agrp and Gad1 mRNA. Gad1 encodes glutamate decarboxylase (GAD67), the major biosynthetic enzyme for GABA. Figure S9 reveals nearly complete loss of Agrp hybridization signal in the ARC of mice treated with DT, regardless of whether they had a bretazenil-eluting minipump. Thus, the rescue from starvation by bretazenil is not due to protection of AgRP neurons from DT-mediated ablation. Ablation of AgRP neurons with DT treatment resulted in 65% depletion of Gad1 mRNA in the ARC, but not in the dorsomedial hypothalamus (Figure 6A-D) indicating that AgRP neurons are the predominate population of GABAergic cells in the ARC.
Figure 6. Loss of GABAergic signaling by AgRP neurons promotes anorexia.
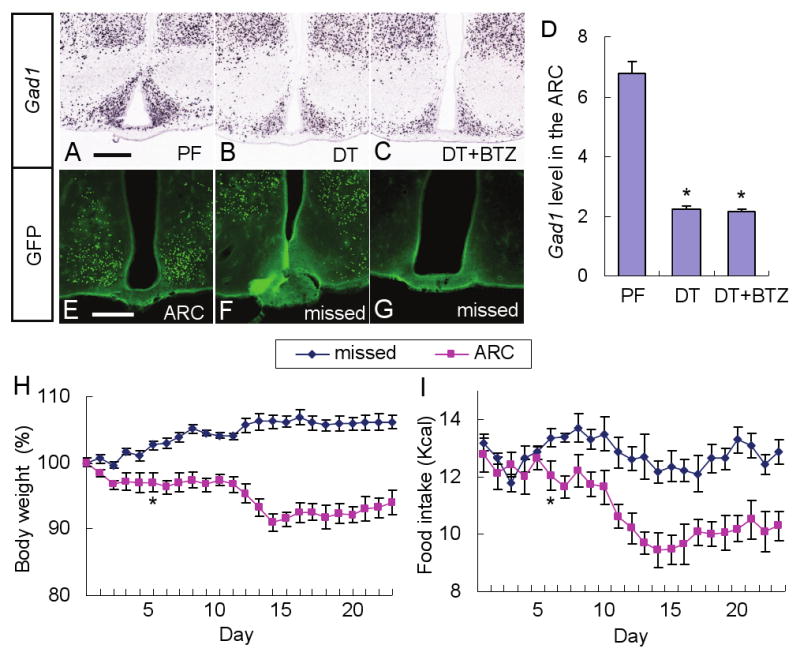
(A-C) Representative pictures of Gad1 in situ hybridization in the ARC of AgrpDTR/+ mice after either pair feeding (A), DT treatment (B), or DT treatment plus chronic administration of bretazenil (C).
(D) Quantified results for Gad1 in situ signals in the ARC of the mice described in A-C. *, p < 0.01, ANOVA. Error bars represent the SEM.
N = 6 mice per each group. Scale bar (in A): A-C, 400 μm.
(E-G) Representative pictures of GFP immunostaining showing the AAV1-Cre-GFP infected neurons located either precisely in the ARC (n = 4; E) or at nonspecific surrounding areas (F) or completely absent from region (G) (n = 12) in Gad1lox/lox, Gad2-/-, Rosa26fsLacZ/fsLacZ mice. Scale bar (in E): E-G-, 400 μm.
(H, I) Percentage change of body weight and intake of liquid diet by the mice with infection in the ARC (E) or missed (F,G). Statistical significance (*, p < 0.01, ANOVA) was obtained starting from Day 6 until end of the experiment. Error bars represent the SEM.
Inactivation of the Gad1 gene in the ARC of adult mice produces anorexia
The previous results suggest that loss of GABAA-receptor signaling within the PBN contributes to the severe anorexia observed after ablation of AgRP neurons. To examine whether the loss of GABA signaling from the ARC to the PBN is directly involved, we used an adeno-associated virus (AAV) strategy to inactivate GABA synthesis in the ARC. Mice with conditional Gad1 alleles (Gad1lox/lox), null alleles of Gad2 (Gad2-/-), and flox-stop Rosa26 reporter allele (G(t)RosafsLacZ/fsLacZ) were injected bilaterally in the ARC with AAV1-CreGFP. When the Cre-expressing virus was delivered precisely to the ARC as shown in Figure 6E (4 of 16 injected mice), food intake began to decline 5 days after viral injection and body weight fell significantly below the pre-injection weight, but the mice survived at least 23 days (Figure 6H, I). However, when viral transduction was either unilateral in the ARC, in more dorsal regions of the hypothalamus, or undetectable (12 of 16 injected mice) there was no effect on feeding behavior (Figure 6F-I). These results indicate that the inhibition of feeding is not an indirect consequence of viral delivery, but GABA synthesis by neurons that reside in the ARC is important for maintenance of feeding behavior.
AgRP neurons may directly innervate the Fos-positive cells in the PBN
Broberger et al (1998) established that the PBN is a prominent post-synaptic target of NPY fibers from hypothalamic NPY/AgRP neurons based on showing that all of the fiber staining was eliminated after destroying neurons in the ARC with mono-sodium glutamate treatment. To ascertain whether the Fos-positive cells in the PBN that arise after ablation of AgRP neurons are possibly direct targets of AgRP neurons, we stained sections including the PBN with antisera against Fos and NPY. This experiment was performed 4 days after the initial DT treatment, at a time when Fos was already activated but the AgRP/NPY neurons had not yet degenerated. Figure S10A, D-F shows that Fos-positive cells are in close proximity to NPY fibers, which supports the possibility that activation of Fos may be due to direct loss of GABA signaling from AgRP neurons onto select PBN neurons. The Fos-positive cells are in the lateral segment of the PBN, in the same region where Fos is activated by peritoneal injection of LiCl, a treatment that produces gastrointestinal malaise (Figure S10B, C). We have also observed a large, progressive increase of GAD67 staining in fibers throughout the PBN during the 6 days after treatment of AgrpDTR mice with DT that could reflect a compensatory increase by other GABAergic neurons (Figure S10G-I).
Discussion
Our results establish the importance of GABA signaling by AgRP neurons onto post-synaptic GABAA receptors for maintenance of feeding behavior. Two lines of evidence support this conclusion: starvation resulting from AgRP neuron ablation can be prevented by chronic delivery of a GABAA receptor agonist, and viral-mediated inactivation of GABA biosynthesis in the ARC inhibits feeding. Furthermore, ablation of AgRP neurons in mice lacking AgRP and NPY, the other known neuromodulators made by these neurons, results in starvation; hence, loss of these neuropeptides is not responsible for the starvation phenotype (Phillips and Palmiter, 2008).
We suggest that loss of GABA signaling from AgRP neurons onto post-synaptic GABAA receptors results in unopposed excitation of post-synaptic cells as revealed by Fos and astrocyte activation. Our observation that inactivation of GABA synthesis in the ARC leads to anorexia suggests that direct GABA signaling by AgRP neurons to critical brain regions is important to maintain feeding after AgRP neuron ablation; however, we cannot rule out the contribution by other neurons in the ARC that produce GABA such as the POMC neurons (Hentges et al., 2004). The anorexia observed with viral inactivation of GABA synthesis was more severe than that observed by Tong et al (2008) but still not as great as that observed with AgRP neuron ablation, presumably because only a fraction of the AgRP neurons was transduced by the virus.
We assume that chronic delivery of bretazenil during AgRP neuron ablation provides sufficient inhibitory tone to suppress the excitability of post-synaptic cells in critical feeding circuits, but suppression of cytokine production by astrocytes may also be involved. We predicted that those brain regions where Fos mRNA was lowered the most would reveal the most critical brain regions. We have identified two brain targets of AgRP neurons (PBN and LS) that appear to be particularly important. The most dramatic reduction of Fos expression was in PBN and that is the only brain region where direct delivery of bretazenil was able to prevent starvation, although delivery of bretazenil to the LS was partially effective. Furthermore, injection of DT into the PBN (but not the LS) produced anorexia indicating that signaling by AgRP neurons to this brain region is particularly important. Nevertheless, experiments involving chronic delivery of bretazenil to the 3rd or 4th ventricle suggest that excessive neuronal activity in both forebrain and hindbrain may contribute to the eating disorder. The pharmacology of the agonists and antagonists used in this study indicate that GABA signaling from AgRP neurons onto GABAA receptors is sufficient to prevent starvation, but activation of GABAB receptors may also be involved. The close apposition of axonal fibers from AgRP neurons with Fos-positive neurons in the lateral PBN suggests that there may be direct GABA signaling onto the critical PBN neurons.
The PBN is a major relay for gustatory and visceral information and mediates anorexia associated with malaise induced by intraperitoneal injection of LiCl or lipopolysaccharide – treatments that are used to mimic the effects of toxic or rancid foods and bacterial infections, respectively (Rinaman and Dzmura, 2007). Lesions of PBN prevent the acquisition of conditioned taste aversion (Trifunovic and Reilly, 2002) and conditioned taste preferences (Reilly and Trifunovic, 2000). The PBN is the most responsive brain region where benzodiazepine injection promotes taste reactivity (Berridge and Pecina, 1995). Direct injections of cannabinoid or mu-opioid receptor agonists into the PBN stimulate feeding (Chaijale et al., 2008;DiPatrizio and Simansky, 2008;Wilson et al., 2003). Thus, the PBN can process both aversive and appetitive ascending visceral information to modulate feeding behavior. LiCl-induced anorexia induces robust Fos expression in the PBN and other brain regions (Andre et al., 2007;Lamprecht and Dudai, 1995;Swank and Bernstein, 1994). The Fos-positive cells in the PBN after LiCl induction are in the same region of the PBN as those induced after AgRP neuron ablation. Thus, it is likely that loss of GABAergic input to the PBN after AgRP neuron ablation activates circuits that normally promote nausea-induced anorexia. Our findings that bretazenil infusion into the PBN prevents anorexia induced by AgRP neuron ablation, while infusion of bicuculline into the PBN of normal mice promotes anorexia are consistent with this hypothesis. The role played by the LS in feeding is more obscure. Because the LS relays signals related to sensory input and reward and projects to the lateral hypothalamus with a well-established role in feeding behavior (Bernardis and Bellinger, 1996), aberrant activity within the LS may produce anorexia via its projections to the lateral hypothalamus. Pharmacological, electrophysiological, and lesion experiments suggest a role of the LS in modulation of feeding (Scopinho et al., 2008) but the precise role of the GABAergic inputs from AgRP neurons to this brain region require further investigation.
In addition to the consensus view that AgRP neurons oppose melanocortin signaling by POMC neurons in pathways that regulate appetite and metabolism (Cone, 2005;Morton et al., 2006;Saper et al., 2002), we propose that there are some brain regions such as the PBN where AgRP neurons have largely melanocortin-independent effects. In our view (Figure 7), ablation (or inhibition) of AgRP neurons leads to activation of POMC neurons and the melanocortin-signaling pathway, as well as a pathway involving the PBN, both of which inhibit feeding. Activation of AgRP neurons has the opposite effect. The profound anorexia that ensues from activation of the PBN – perhaps mimicking severe gastrointestinal malaise or aversive gustatory input - could mask the role of AgRP neurons in regulating appetite and metabolism by counteracting melanocortin signaling. The transient ∼10% loss of body weight that still occurs when bretazenil is infused may reflect the melancortin-dependent role of AgRP neurons on feeding and metabolism. If so, this effect is gradually compensated because the mice eventually regain their body weight after ∼ 10 days, which may account for the relatively mild effect of chronic loss of GABA signaling by AgRP neurons (Tong et al., 2008).
Figure 7. Diagram illustrating how loss of GABAergic signaling from AgRP neurons leads to starvation.

AgRP and POMC neurons that reside in the ARC send axons to many of the same target areas, e.g. the PVN, MPO, PAG, NTS and PBN. Ablation (or inhibition) of inhibitory AgRP neurons leads to anorexia by two mechanisms. First, the loss of inhibition onto POMC neurons and their post-synaptic target cells stimulates the melanocortin system, which suppresses feeding. Second, the loss of GABAergic inhibition onto neurons in the PBN (and to a lesser extent the LS) mimics the activation of these neurons in response to gastrointestinal malaise, resulting in severe anorexia, an effect that is independent of melanocortin signaling. The severe anorexia can be prevented by infusing bretazenil, a GABAA receptor partial agonist, into the PBN which antagonizes Fos activation.
It is noteworthy that abrupt withdrawal of bretazenil during 4th ventricle delivery results in a more precipitous decline of body weight than that achieved by ablation of AgRP neurons alone. This result mimics the loss of body weight and appetite, a common symptom of the “benzodiazepine withdrawal syndrome” seen in patients who discontinue chronic use of benzodiazepines (Pecknold, 1993). In parallel with a clinical study showing that flumazenil treatment alleviated the withdrawal syndrome (Gerra et al., 2002), our results demonstrate that bretazenil-mediated restoration of food intake was completely abolished by the 4th ventricle administration of flumazenil. On the other hand, after cessation of chronic infusion of bicuculline into the PBN of wide-type mice, their anorexia switched to prolonged hyperphagia that was inversely proportional to the dosage of bicuculline. These complementary results illustrate robust dynamics and sensitivity of a hindbrain GABA signaling network that governs appetite and food palatability.
An unexpected finding is that feeding persists in AgRP neuron-ablated mice after the bretazenil-eluting minipumps are depleted or removed. This result is reminiscent of the observation that ablation of AgRP neurons in neonatal mice does not result in starvation (Luquet et al., 2005;Phillips and Palmiter, 2008). We suggested previously (Luquet et al., 2005) that ablation of AgRP neurons in neonates has a minimal effect on feeding because adaptations can take place during the 2 to 3 weeks between ablation and weaning - when AgRP neurons mature and independent feeding becomes important (Nilsson et al., 2005). We originally thought that the adaptations might involve establishing new neuronal circuits, but the present results suggest a different explanation. We now suggest that the hyperactive post-synaptic neurons adapt by reducing the effects of excitatory inputs and/or enhancing alternative inhibitory inputs (Horvath, 2006). The increase in GAD67 staining in the PBN after AgRP neuron ablation may reflect such compensatory changes. Synaptic plasticity could also involve changes in the number of excitatory and/or inhibitory synapses as well as changes in abundance and activity of various receptors, including ion channels and G-protein-coupled receptors. Examination of the mechanisms involved will depend on more precise identification of the post-synaptic neurons with elevated Fos signal. Our results suggest that these hypothetical adaptations are incomplete and mostly ineffective 6 days after DT treatment, but can be established within 11 days with the aid of chronic GABAA-receptor activation. In this view, bretazenil suppresses the excitability of post-synaptic neurons and reduces local gliosis, which in turn renders sufficient time and a favorable physiological setting for the adaptations to occur.
Methods
Animal Maintenance and Neuron Ablation
Mice were housed in a temperature- and humidity-controlled facility with a 12-hour light/dark cycle. All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Washington. In compliance with our approved protocol, all experiments were terminated if the body weight of mice fell to 80% of their original body weight. AgrpDTR/+ mice were generated by targeting the human diphtheria toxin receptor (heparin-binding epidermal growth factor) to the Agrp locus (Luquet et al., 2005). Gad1lox/lox mice (with loxP sites flanking exon 2) were generated as described (Chattopadhyaya et al., 2007). They were bred with Gad2-/- mice (Kash et al., 1997) and Gt(ROSA)26SorfsLacZ/fsLacZ mice (Soriano, 1999) to create the triple homozygotes that were used for viral injection experiments. Mice were group housed with standard chow diet (LabDiet 5053) and water provided ad libitum until the beginning of the experiments. To ablate AgRP neurons in adult mice, intramuscular injection of diphtheria toxin (two injections of 50 μg/kg, two days apart; List Biological Laboratories, Campbell, CA) in 6-week-old mice was performed (Luquet et al., 2005).
Viral injections
AAV1 virus expressing myc-nls-Cre-GFP fusion protein was kindly provided by James Allen (Univ. of Washington). The virus was produced by transfecting HEK cells and then purified from cell extracts by pelleting through sucrose followed by banding on a CsCl gradient. Mice were anesthetized and virus was injected bilaterally (1 μl of ∼109 particles/μl per each side) through a cannula-guided injector (31 gauge, Plastics One, Roanoke, VA) just above the ARC using coordinates ±0.5 mm (X axis), -1.4 mm (Y axis) and -6 mm (Z axis) (Paxinos and Frank, 2001). Brain samples from all mice were collected at the end of the experiment and proceeded with immunohistological analysis.
In Situ Hybridization
Brains were sectioned (25 μm coronal) and every 8th section was used for either Nissl staining or in situ hybridization with Agrp, Fos, or Gad1 using an automated procedure for hybridization and image capture. Materials and procedures concerning this high-throughput data generation process (riboprobe production, tissue processing, in situ hybridization, image capture and processing) have been described (Lein et al., 2007) and are available on the Allen Brain Atlas website (www.brain-map.org).
Drug treatments
Alzet 14-day mini-osmotic pumps (model 1002, Durect, Cupertino, CA) loaded with 100 μL of bretazenil (3 mg/ml in saline plus 10% DMSO, Sigma-Aldrich, St Louis, MO) were implanted subcutaneously on the back of anesthetized mice 3 days before DT treatment. These minipumps dispense 0.25 μL/hr. Alternatively, cannulas (28 gauge, Plastics One) were placed into either the 3rd or 4th ventricles under anesthesia and the subcutaneous Alzet minipumps were connected to the cannulas by tubing (PE60, Stoelting, Wood Dale, IL) that was threaded under the skin to help prevent the mice from dislodging it. For some experiments, the Alzet minipumps were connected to bilateral cannulas (28 gauge, Plastics One) directed to specific brain regions by using the following coordinates: the PVN, ±0.5 mm (X axis), -0.8 mm (Y axis) and -4.6 mm (Z axis); the LS, ±0.5 mm (X axis), 0.3 mm (Y axis) and -3.8 mm (Z axis); the PBN, ±1 mm (X axis), -5.3 mm (Y axis) and -3.3 mm (Z axis); the PAG, ±1 mm (X axis), -4.75 mm (Y axis) and -2.8 mm (Z axis); the NTS, ±1 mm (X axis), -7.4 mm (Y axis) and -4.5 mm (Z axis). Bretazenil used for central administration was prepared by diluting the stock solution (3 mg/ml in DMSO) in saline as indicated in the text and figure legends. For one experiment, the mice had a subcutaneous minipump loaded with bretazenil (3 mg/ml) and then flumazenil, a GABAA receptor antagonist (1.5 μl of 2 mg/ml in saline, Sigma-Aldrich), was injected either intraperitoneally, or into the 3rd or 4th ventricle via a cannula-guided injector (31 gauge, Plastics One) through a Hamilton syringe (size 25 μl, Hamilton, Reno, NV). Of note, flumazenil or vehicle was injected at ∼8:00 pm immediately before the mice entered active feeding phase. Bicuculline methiodide (a water soluble form of bicuculline, Sigma-Aldrich) was prepared in saline (170 μg/ml, 50 μg/ml, and 17 μg/ml) and loaded to Alzet mini pumps for central infusion. LiCl (0.15 M in saline, J.T. Baker) was injected intraperitoneally at a dose of 20 μl/g of body weight (0.12 g/kg), then after 24 h the brains were collected and processed for immunohistochemistry. The patency and placement of the bilateral minipump was verified at the end of each experiment by infusing a blue dye followed by histological analysis.
Food intake and body weight measurements
For feeding assays, mice were transferred to lickometer cages (Columbus Instruments, Columbus, OH) supplied with water and liquid diet (5LD-101, TestDiet, Richmond, IN; 1 Kcal/ml). The mice were allowed to acclimate for 3 days before initiating each experiment and data collection. Body weigh and total food intake were recorded every 24 hr. Licking activity at food and water dispensers was recorded continuously as described (Wu et al., 2008a). Licking data were grouped into 2-hr bins and further analyzed by using a program encoded by the Python software.
Intra-oral feeding studies
Intra-oral fistulas were implanted under anesthesia at the same time when inserting minipumps loaded with bretazenil. Consumption of 0.1 M sucrose solution was measured as described (Wu et al., 2008a).
Immunohistochemistry
Mice were sacrificed by CO2 asphyxiation and perfused transcardially with ice-cold PBS buffer containing 4% paraformaldehyde. Brains were dissected and post-fixed overnight at 4°C in the fixation buffer. Free-floating brain sections (30 μm) were washed in PBS and 0.1% Triton X-100 (PBST buffer) solution 3 × 15 min and then blocked with 3% normal donkey serum in PBST for 2 – 3 hr at room temperature. Rabbit anti-GFAP (1:3000 dilution; Novus Biologicals), rabbit anti-IBA1 (1:1000 dilution; Wako Chemicals USA), guinea pig anti-NPY (1:1000 dilution; Abcam), and rabbit anti-Fos (1: 1500 dilution; Millipore); monoclonal anti-GAD67 (1:1000 dilution, Sigma) were applied to the sections for overnight incubation at 4°C, followed by 3 × 15-min rinses in PBST. Finally, sections were incubated in Cy2- or Cy3-labeled secondary antibody (1:300 dilution, Jackson Immunolaboratory) before visualization. Images were captured using a digital camera mounted on a Leica TCS SP1 confocal microscope (Leica Microsystems USA); all paired photos were obtained through the same system setting. For each group of mice, at least 8 sections from 4 different mice were analyzed.
Data analyses
Quantification of Fos-positive cells was done using the NIH Image software (National Institutes of Health). Anatomical correlations of brain sections and delineation of individual nuclei were determined by comparing landmarks of Nissl staining images with those given in the stereotaxic atlas (Paxinos and Frank, 2001). From the anatomically matched sections, a region of interest of the same size was further defined. Meanwhile, an optimized threshold that can discern round Fos-positive nuclei from partially stained ones as well as background noise was preset for all measurement. The total number of pixels of Fos-positive cells inside the defined region was automatically recorded. Data sets collected from all experiments, unless otherwise stated, were analyzed by one-way ANOVA followed by Student-Newman-Keuls method for statistical significance and plotted as means ± standard error of mean (SEM). Post-hoc analysis was performed when group differences were significant by ANOVA at p < 0.05.
Supplementary Material
Acknowledgments
We thank Glenda Froelick and Betty Li for help with histology, Aundrea Rainwater for help with mouse breeding, Alex Scouras for help with computer code, James Allen for providing AAV1-Cre-GFP virus and the staff of the Allen Brain Research Institute for performing the in situ hybridization studies. We appreciate the helpful comments on the manuscript by Larry Zweifel and Ali Guler. This work was supported in part by NIH DA024908 to R.D.P.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Andre V, Dube C, Francois J, Leroy C, Rigoulot MA, Roch C, Namer IJ, Nehlig A. Pathogenesis and pharmacology of epilepsy in the lithium-pilocarpine model. Epilepsia. 2007;48 5:41–47. doi: 10.1111/j.1528-1167.2007.01288.x. [DOI] [PubMed] [Google Scholar]
- Bernardis LL, Bellinger LL. The lateral hypothalamic area revisited: ingestive behavior. Neurosci Biobehav Rev. 1996;20:189–287. doi: 10.1016/0149-7634(95)00015-1. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Pecina S. Benzodiazepines, appetite, and taste palatability. Neurosci Biobehav Rev. 1995;19:121–131. doi: 10.1016/0149-7634(94)00026-w. [DOI] [PubMed] [Google Scholar]
- Broberger C, Hokfelt T. Hypothalamic and vagal neuropeptide circuitries regulating food intake. Physiol Behav. 2001;74:669–682. doi: 10.1016/s0031-9384(01)00611-4. [DOI] [PubMed] [Google Scholar]
- Broberger C, Johansen J, Johansson C, Schalling M, Hokfelt T. The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci U S A. 1998;95:15043–15048. doi: 10.1073/pnas.95.25.15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaijale NN, Aloyo VJ, Simansky KJ. A naloxonazine sensitive (mu(1) receptor) mechanism in the parabrachial nucleus modulates eating. Brain Res. 2008 doi: 10.1016/j.brainres.2008.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di CG, Wu CZ, Knott G, Kuhlman S, Fu Y, Palmiter RD, Huang ZJ. GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron. 2007;54:889–903. doi: 10.1016/j.neuron.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- Cooper SJ. Palatability-dependent appetite and benzodiazepines: new directions from the pharmacology of GABA(A) receptor subtypes. Appetite. 2005;44:133–150. doi: 10.1016/j.appet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- DiPatrizio NV, Simansky KJ. Activating parabrachial cannabinoid CB1 receptors selectively stimulates feeding of palatable foods in rats. J Neurosci. 2008;28:9702–9709. doi: 10.1523/JNEUROSCI.1171-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke AN, Platt DM, Cook JM, Huang S, Yin W, Mattingly BA, Rowlett JK. Enhanced sucrose pellet consumption induced by benzodiazepine-type drugs in squirrel monkeys: role of GABAA receptor subtypes. Psychopharmacology (Berl) 2006;187:321–330. doi: 10.1007/s00213-006-0431-2. [DOI] [PubMed] [Google Scholar]
- Ebenezer IS, Prabhaker M. The effects of intraperitoneal administration of the GABA(B) receptor agonist baclofen on food intake in CFLP and C57BL/6 mice. Eur J Pharmacol. 2007;569:90–93. doi: 10.1016/j.ejphar.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–421. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- Flier JS. AgRP in energy balance: Will the real AgRP please stand up? Cell Metab. 2006;3:83–85. doi: 10.1016/j.cmet.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Gerra G, Zaimovic A, Giusti F, Moi G, Brewer C. Intravenous flumazenil versus oxazepam tapering in the treatment of benzodiazepine withdrawal: a randomized, placebo-controlled study. Addict Biol. 2002;7:385–395. doi: 10.1080/1355621021000005973. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Kaplan JM. The neuroanatomical axis for control of energy balance. Front Neuroendocrinol. 2002;23:2–40. doi: 10.1006/frne.2001.0224. [DOI] [PubMed] [Google Scholar]
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- Haskell-Luevano C, Chen P, Li C, Chang K, Smith MS, Cameron JL, Cone RD. Characterization of the neuroanatomical distribution of agouti-related protein immunoreactivity in the rhesus monkey and the rat. Endocrinology. 1999;140:1408–1415. doi: 10.1210/endo.140.3.6544. [DOI] [PubMed] [Google Scholar]
- Hentges ST, Nishiyama M, Overstreet LS, Stenzel-Poore M, Williams JT, Low MJ. GABA release from proopiomelanocortin neurons. J Neurosci. 2004;24:1578–1583. doi: 10.1523/JNEUROSCI.3952-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs S, Cooper SJ. Hyperphagia induced by direct administration of midazolam into the parabrachial nucleus of the rat. Eur J Pharmacol. 1996;313:1–9. doi: 10.1016/0014-2999(96)00446-3. [DOI] [PubMed] [Google Scholar]
- Horvath TL. Synaptic plasticity in energy balance regulation. Obesity (Silver Spring) 2006;14 5:228S–233S. doi: 10.1038/oby.2006.314. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Bechmann I, Naftolin F, Kalra SP, Leranth C. Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain Res. 1997;756:283–286. doi: 10.1016/s0006-8993(97)00184-4. [DOI] [PubMed] [Google Scholar]
- Jacobowitz DM, O'Donohue TL. alpha-Melanocyte stimulating hormone: immunohistochemical identification and mapping in neurons of rat brain. Proc Natl Acad Sci U S A. 1978;75:6300–6304. doi: 10.1073/pnas.75.12.6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr Rev. 1999;20:68–100. doi: 10.1210/edrv.20.1.0357. [DOI] [PubMed] [Google Scholar]
- Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A. 1997;94:14060–14065. doi: 10.1073/pnas.94.25.14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R, Dudai Y. Differential modulation of brain immediate early genes by intraperitoneal LiCl. Neuroreport. 1995;7:289–293. [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- Luquet S, Phillips CT, Palmiter RD. NPY/AgRP neurons are not essential for feeding responses to glucoprivation. Peptides. 2007;28:214–225. doi: 10.1016/j.peptides.2006.08.036. [DOI] [PubMed] [Google Scholar]
- Meister B. Neurotransmitters in key neurons of the hypothalamus that regulate feeding behavior and body weight. Physiol Behav. 2007;92:263–271. doi: 10.1016/j.physbeh.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- Nilsson I, Johansen JE, Schalling M, Hokfelt T, Fetissov SO. Maturation of the hypothalamic arcuate agouti-related protein system during postnatal development in the mouse. Brain Res Dev Brain Res. 2005;155:147–154. doi: 10.1016/j.devbrainres.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Frank KB. The Mouse Brain in Sterotaxic Coordinates. San Diego: Academic Press; 2001. [Google Scholar]
- Pecknold JC. Discontinuation reactions to alprazolam in panic disorder. J Psychiatr Res. 1993;27 1:155–170. doi: 10.1016/0022-3956(93)90025-w. [DOI] [PubMed] [Google Scholar]
- Phillips CT, Palmiter RD. Role of agouti-related protein-expressing neurons in lactation. Endocrinology. 2008;149:544–550. doi: 10.1210/en.2007-1153. [DOI] [PubMed] [Google Scholar]
- Qian S, Chen H, Weingarth D, Trumbauer ME, Novi DE, Guan X, Yu H, Shen Z, Feng Y, Frazier E, et al. Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol Cell Biol. 2002;22:5027–5035. doi: 10.1128/MCB.22.14.5027-5035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly S, Trifunovic R. Lateral parabrachial nucleus lesions in the rat: aversive and appetitive gustatory conditioning. Brain Res Bull. 2000;52:269–278. doi: 10.1016/s0361-9230(00)00263-x. [DOI] [PubMed] [Google Scholar]
- Rinaman L, Dzmura V. Experimental dissociation of neural circuits underlying conditioned avoidance and hypophagic responses to lithium chloride. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1495–R1503. doi: 10.1152/ajpregu.00393.2007. [DOI] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36:199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- Scopinho AA, Resstel LB, Correa FM. alpha(1)-Adrenoceptors in the lateral septal area modulate food intake behaviour in rats. Br J Pharmacol. 2008 doi: 10.1038/bjp.2008.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutter JR, Graham M, Kinsey AC, Scully S, Luthy R, Stark KL. Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and diabetic mutant mice. Genes Dev. 1997;11:593–602. doi: 10.1101/gad.11.5.593. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Swank MW, Bernstein IL. c-Fos induction in response to a conditioned stimulus after single trial taste aversion learning. Brain Res. 1994;636:202–208. doi: 10.1016/0006-8993(94)91018-9. [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008 doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic R, Reilly S. Medial versus lateral parabrachial nucleus lesions in the rat: effects on mercaptoacetate-induced feeding and conditioned taste aversion. Brain Res Bull. 2002;58:107–113. doi: 10.1016/s0361-9230(02)00766-9. [DOI] [PubMed] [Google Scholar]
- Watson SJ, Akil H, Richard CW, III, Barchas JD. Evidence for two separate opiate peptide neuronal systems. Nature. 1978;275:226–228. doi: 10.1038/275226a0. [DOI] [PubMed] [Google Scholar]
- Wilson JD, Nicklous DM, Aloyo VJ, Simansky KJ. An orexigenic role for mu-opioid receptors in the lateral parabrachial nucleus. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1055–R1065. doi: 10.1152/ajpregu.00108.2003. [DOI] [PubMed] [Google Scholar]
- Wu Q, Howell MP, Cowley MA, Palmiter RD. Starvation after AgRP neuron ablation is independent of melanocortin signaling. Proc Natl Acad Sci U S A. 2008a;105:2687–2692. doi: 10.1073/pnas.0712062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Howell MP, Palmiter RD. Ablation of neurons expressing agouti-related protein activates fos and gliosis in postsynaptic target regions. J Neurosci. 2008b;28:9218–9226. doi: 10.1523/JNEUROSCI.2449-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.