

Abstract
Isolates of baboon α-herpesvirus Papiine herpesvirus 2 (HVP2) exhibit one of two distinct phenotypes in mice: extremely neurovirulent or apathogenic. Previous studies implicated the type I interferon (IFN) response as being a major factor in controlling infection by apathogenic isolates. To further investigate the possibility that the host IFN-β response underlies the pathogenicity of the two HVP2 subtypes, the susceptibility of mice lacking the IFN-β receptor (IFNAR−/−) to infection was examined. Apathogenic isolates of HVP2 (HVP2ap) replicated in IFNAR−/− primary mouse dermal fibroblast (PMDF) cultures as well as neurovirulent (HVP2nv) isolates. IFNAR−/− mice were also susceptible to lethal infection by HVP2ap isolates. Unlike Balb/c or parental 129 mice, LD50 and ID50 values for HVP2ap were the same in IFNAR−/− mice indicating that in these mice infection always progressed to death. HVP2ap replicated in the skin at the site of inoculation and invaded dorsal root ganglia as efficiently as HVP2nv in IFNAR−/− mice. Since the virion host shutoff (vhs) protein encoded by the UL41 gene of herpes simplex virus has been implicated in circumventing the host IFN-β response and the phenotype of UL41 deletion mutants of HSV is very similar to that of HVP2ap isolates, the UL41 gene was deleted from HVP2nv (Δ41) and replaced with the UL41 ORF from HVP2ap (Δ41C). Like the parental HVP2nv virus, the Δ41C recombinant replicated efficiently in Balb/c PMDFs and did not induce a strong IFN-β response. The neuropathogenicity of the Δ41C recombinant was also the same as the parental HVP2nv virus in Balb/c mice, indicating that the vhs protein does not underlie the different neuropathogenic phenotype of HVP2ap and HVP2nv. In contrast, the Δ41 deletion virus induced a strong IFN-β response but was still able to undergo multiple rounds of replication in PMDF cultures, albeit at a slower pace than the parental HVP2nv. This was reflected in vivo as the Δ41 mutant had an LD50 equivalent to that of the parental HVP2nv virus although the time to death was longer. These results indicate that while the vhs protein is involved in preventing and/or suppressing an IFN-β response, it is not responsible for the ability of HVP2nv to overcome IFN-β induced resistance of uninfected cells and does not underlie the divergent pathogenicity of the two HVP2 subtypes in mice.
Keywords: Herpesvirus, interferon, pathogenesis, baboon, neurovirulence, innate immunity
Introduction
The baboon herpesvirus Papiine herpesvirus 2 (Herpesvirus papio 2; HVP2) is very closely related to both herpes simplex virus (HSV) of humans (Eberle and Hilliard, 1995; Huff and Barry, 2003; Keeble, 1960; Palmer, 1987; Weigler, 1992; Whitely and Hilliard, 2001) and Macacine herpesvirus 1 (monkey B virus; BV) of macaques. While BV causes a severe and usually fatal encephalomyelitis when transmitted to humans or other non-macaque primates (Huff and Barry, 2003; Loomis et al., 1981; Sabin and Wright, 1934; Thompson et al., 2000), there are no reported incidents of HVP2 infection or death in humans. However, in Balb/c mice one subtype of HVP2 (HVP2ap) does not produce clinical signs of disease and infection results in only minimal tissue destruction at both the site of inoculation and within both the peripheral and central nervous system (PNS and CNS). In contrast, the second subtype (HVP2nv) produces a fulminant, rapidly fatal CNS infection (Ritchey et al., 2002; Rogers et al., 2003; Rogers et al., 2006). HVP2 has however caused fatal infections in young baboons (Wolf et al., 2006) and an HVP2nv isolate was also recently reported to be the cause of a fatal neurological infection in a Colobus monkey (Troan et al., 2007). The clinicopathogenesis of HVP2nv in mice closely parallels what has been observed in human BV infections and provides an excellent model system for examining host-virus interactions within the context of cross-species infections (Rogers et al., 2006).
Within a particular species, it is the sum of multiple complex interactions between virus and host that determines the pathogenesis and clinical outcome of infection (Brandt, 2005; Enquist et al., 1998; Mossman and Ashkar, 2005; Rouse and Lopez, 1984). Thus, virtually any viral factor which affects the ability of a virus to replicate and/or spread within a host can be considered a virulence factor. In addition, both viral dose and route of inoculation can influence the ability of a virus to establish a productive infection within a given host (Breshears, Eberle, and Ritchey, 2005; Weeks et al., 2000). In HSV infection of mice, the efficiency of viral replication at the peripheral site of infection is directly related to the ability of the virus to enter sensory neurons and invade the CNS (Yamada et al., 1986). However, once a virus has gained entry to a host organism, the most important host determinant of pathogenicity is likely the host innate immune response (Mossman and Ashkar, 2005; Simons and Nash, 1984).
A critical component of the innate anti-viral response is production of the type I interferons α and β (IFN-α, IFN-β). IFN-α is rapidly produced by infected plasmacytoid dendritic cells (pDCs) or when they are stimulated by recognition of viral components via toll-like receptors (TLRs) either through endocytosis of viral particles or autophagy of infected cells (Lee et al., 2007). The ability of pDCs to mount an early IFN-α response is due to constitutive expression of the interferon regulatory factor (IRF) -7 while in other cell types, baseline IRF7 expression is weak and only moderately induced by viral infection (Dai et al., 2004; Lee et al., 2007).
IFN-β is rapidly induced in many cell types (including those infected by herpesviruses such as fibroblasts and epithelial cells) due to their constitutive expression of IRF-3. IFN-α and -β produced by infected cells binds to the IFN-α/β receptor (IFNAR) initiating a signal transduction cascade via the Janus associated kinase – signal transducer and activator of transcription (Jak-Stat) pathway (Darnell, Kerr, and Stark, 1994; Goodbourn, Didcock, and Randall, 2000). The end result of this pathway is that infected cells are recognized and destroyed while uninfected cells are protected from viral infection, thereby limiting the ability of the viral infection to spread. Although infected IFNAR−/− cells are capable of expressing IFN, the lack of the receptor for IFN-α/β prevents the signal transduction cascade via the Jak/Stat pathway, resulting in no amplification of the IFN-β response, and so neighboring uninfected cells remaining sensitive to viral infection.
The pathogenic phenotype of HVP2ap in mice is similar to that described for HSV: normal replication in vitro but reduced neuropathogenicity in vivo (Rogers et al., 2003). HVP2ap-infected primary mouse dermal fibroblast (PMDF) cultures have also been shown to produce more IFN-β than HVP2nv-infected PMDF cultures (Rogers, Black, and Eberle, 2007). Further, pretreatment of Balb/c PMDF cell cultures with exogenous murine IFN-β significantly decreased titers of HVP2ap but not HVP2nv, suggesting that HVP2nv is not effectively controlled by the mouse IFN-β response. Similar phenotypes have been observed with other alpha-herpesviruses and experimental animal models have proven invaluable in dissecting the mechanisms of numerous viral anti-IFN genes.
The virion host shutoff (vhs) protein of HSV1 and HSV2 encoded by the UL41 gene is probably the best known example of a herpesvirus protein responsible for abrogating the type I IFN response in infected cells (Duerst and Morrison, 2004; Korom, Wylie, and Morrison, 2008; Suzutani et al., 2000). The vhs protein is a component of the virion tegument, and so is released into the cytoplasm of cells immediately on infection. The HSV vhs has RNase activity and functions to degrade certain mRNAs in infected cells (Esclatine, Taddeo, and Roizman, 2004; Everly et al., 2002; Karr and Read, 1999). Late in infection the vhs protein interacts with the viral α-transducing factor VP16. This interaction reduces the RNase activity of vhs, allowing accumulation of viral transcripts (Lam et al., 1996; Schmelter et al., 1996; Strand and Leib, 2004). Deletion of the UL41 gene results in higher levels of IFN-β production in infected cell cultures and dramatically affects the pathogenicity of HSV in mice (Korom, Wylie, and Morrison, 2008; Smith, Ackland-Berglund, and Leib, 2000; Smith, Morrison, and Leib, 2001; Strand and Leib, 2004; Strelow and Leib, 1995; Strelow and Leib, 1996). Recently, it has been shown that selective mutation of the RNase activity of the HSV2 vhs affects pathogenicity of the virus (Korom, Wylie, and Morrison, 2008).
The availability of IFNAR−/− knockout mice which do not express the IFN-α/β receptor (Muller et al., 1994) provides an ideal model system for investigating the role for IFN-β in HVP2 cross-species infections. These studies were undertaken to assess the role of IFN-β and the HVP2 vhs in vivo.
Methods and Materials
Animals
129 mice (female and male) weighing 10–12 g were obtained from The Jackson Laboratory (Bar Harbor, ME). Two breeding pairs of IFNAR−/− mice were generously provided by Dr. H.W. Virgin (Washington University School of Medicine, St. Louis, MO) and were bred and raised at Oklahoma State University. The genotype and genetic background of these IFN-β receptor knock-out mice has been described (Muller et al., 1994). Mice were weaned and used for experiments at a weight of 10–12 grams (21–25 days of age). Balb/c mice were purchased from Charles River Labs.
Viruses and Cell Cultures
The origins and neuropathogenicity of the HVP2ap (A951 & OU2-5) and HVP2nv (OU1-76 & X313) strains used in this study have been described in detail elsewhere (Eberle et al., 1997a; Eberle et al., 1998; Eberle et al., 1995; Rogers et al., 2003). All virus stocks were grown and titrated in Vero cells. PMDF cultures from Balb/c, 129 and IFNAR−/− mice were prepared and cultured as described previously (Rogers, Black, and Eberle, 2007). Vero cells were originally obtained from the American Type Culture Collection (Rockville, MD, USA).
PMDF cultures in multi-well trays were mock infected with an uninfected Vero cell lysate or with virus as described (Rogers, Black, and Eberle, 2007). After 1 hour at 39°C, the inoculum was removed, cells were washed with warm PBS, and fresh media containing 2% FBS added. Cultures were photographed using a Nikon Eclipse TE-200 inverted fluorescent microscope (Melville, NY) with attached RS Photometrics digital camera prior to harvesting the cells for quantitation of infectious virus by standard plaque assay on Vero cells (Rogers et al., 2003). A commercial ELISA kit (LKT Laboratories; St. Paul, MN) was used to quantitate IFN-β levels in the extracellular media of infected cell cultures. All assays were performed on duplicate samples and were repeated at least twice. Levels of anti-HVP2 IgG in mouse serum were determined by ELISA as described (Ohsawa, Lehenbauer, and Eberle, 1999; Rogers et al., 2003).
Construction of Recombinant Viruses
Approximately 2 Kbp of sequences flanking each side of the UL41 ORF of HVP2nv strain OU1-76 were amplified by PCR and a GFP expression cassette (Black, Saliki, and Eberle, 2002) positioned between them in place of the UL41 ORF via restriction sites incorporated at the UL41 start and stop codons (Sph1 & Xba1, resp.). This construct was excised from the vector and transfected into Vero cells using the GenePorter reagent as recommended by the manufacturer (Genlantis; San Diego, CA). At 24 hrs post transfection, the cell culture was infected with HVP2nv (OU1-76) at an MOI = 1 PFU/cell, incubated at 37°C until CPE was complete to allow homologous recombination to occur between the construct and viral DNA, and the cultures harvested as a viral stock. Virus was then plated on Vero cells and observed at 48 hr PI under an inverted fluorescent microscope (Black, Saliki, and Eberle, 2002). GFP-positive plaques were picked and plaque purified four times. Deletion of the UL41 ORF and proper insertion of the GFP expression cassette in place of the UL41 ORF was confirmed by Southern blot analysis and PCR/sequencing (Eberle et al., 1997b). This UL41 gene deletion virus was designated HVP2 OU1-76Δ41.
Construction of a revertant (HVP2 OU1-76Δ41R) and a recombinant virus (HVP2 OU1-76Δ41C) followed the same basic procedure. Briefly, the UL41 ORF was amplified by PCR from the parental HVP2nv strain OU1-76 (to construct the revertant virus) or from HVP2ap strain OU2-5 (to construct the recombinant) and inserted into the flanking sequences used to construct the UL41 gene deletion virus using the same restriction sites. After transfection of the constructs into Vero cells, the cultures were infected with the GFP-positive HVP2 OU1-76Δ41 virus, GFP-negative plaques picked 48 hrs later, and the virus plaque purified. Confirmation of restoration of the UL41 ORF, its sequence integrity across the entire ORF, and deletion of the GFP expression cassette were confirmed by Southern blot analysis and PCR/sequencing.
Mouse Inoculations
Mice were infected by scarification on the hind flank as described previously (Rogers et al., 2006). Briefly, mice were shaved, an area of skin approximately 7 × 7 mm was lightly scarified by 5 × 5 scratches with a 22 gauge needle in a checkerboard pattern, and 10 μl of virus applied to the area and ‘rubbed in’ with a pipet tip. Sterile PBS was used as diluent for inoculum dilutions. Once infected, mice were observed twice daily for clinical signs of infection. All mice were humanely euthanized by isofluorane overdose when clinical signs of infection became severe or at termination of the experiment at 14 days post infection (DPI). Blood was collected by cardiac puncture at the time of death/euthanasia and the serum stored at −80°C.
Pathology and Virus Isolation
Following euthanasia, mice were opened with a midline abdominal incision and placed in buffered formalin for 24–48 hrs before tissues were collected for pathological examination. All histological procedures were conducted as previously described (Ritchey et al., 2002; Ritchey, Payton, and Eberle, 2005; Rogers et al., 2006). For virus isolation, tissues were sterilely dissected, placed in a sterile microfuge tube, weighed, and frozen at −80°C. For assay of infectious virus, sterile PBS was added to tissue samples, tissues were homogenized using a motorized pestle, insoluble debris removed by centrifugation at 14,000 × g for 2 min, and the clarified supernatant transferred to a clean tube. Infectious virus was quantitated by plaque assay on Vero cells and expressed as PFU/g of tissue.
Statistical Analyses
The 50% infectious dose (ID50) and the 50% lethal dose (LD50) were calculated by probit regression with PROC PROBIT in PC SAS Version 9.1 (SAS Institute, Cary, NC, USA). The values calculated were compared by methods developed for effective dosages (Ritchey, Payton, and Eberle, 2005; Rogers et al., 2006). The ID50 was defined by the presence of anti-HVP2 IgG in serum of mice that survived to at least 11 DPI. Mice that died prior to 11 DPI were assumed to be infected and were considered to be positive for calculation of ID50 values. LD50 values were based on animals that either died as a result of the infection or required euthanasia due to the severity of disease. Virus titers in tissues were analyzed to compare levels of HVP2nv vs. HVP2ap in different mouse strains and at different times PI by analysis of variance procedures with PROC MIXED in PC SAS Version 9.1 (SAS Institute, Cary, NC). Virus titer values were transformed using a log base 10 function prior to analysis. Significant differences in the simple effects of one factor given values of the other factors were determined by protected pair-wise t tests with a SLICE option in an LSMEANS statement.
Results
Previous experiments with Balb/c PMDF cultures suggested that IFN-β may differentially affect replication of the two HVP2 subtypes in vitro and that HVP2nv may be more efficient than HVP2ap at preventing and/or overcoming the expression of IFN-β (Rogers, Black, and Eberle, 2007). To further test the role of the IFN-β response in controlling HVP2 infection in the mouse model, PMDF cultures were prepared from parental 129 and IFNAR−/− knockout mice and the ability of HVP2ap and HVP2nv to replicate in them was assessed.
Replication of both subtypes of HVP2 was compared in PMDF cultures prepared from Balb/c, 129 and IFNAR−/− mice. Cultures were infected with two isolates each of HVP2ap or HVP2nv at an MOI of 0.25 PFU/cell and observed for plaque formation over 3 days. As shown in Figure 1, HVP2nv strains replicated equally well in PMDFs derived from all three mouse strains. Large plaques were evident by 24 hrs PI and the cultures were completely destroyed by 48 hrs PI. As described previously for Balb/c PMDFs (Rogers, Black, and Eberle, 2007), HVP2ap strains formed plaques in Balb/c and 129 cultures that were equivalent in size to those of HVP2nv strains at 24 hrs PI. However, HVP2ap plaques ceased to enlarge much after 24 hrs and by 72 hrs PI uninfected cells had filled in the site of the initial plaques. In contrast, HVP2ap strains replicated in IFNAR−/−PMDF cultures as efficiently as HVP2nv strains; plaques were evident at 24 hrs PI, and by 48 hrs PI the monolayers were completely involved.
Figure 1. Plaque formation by HVP2 subtypes in BALB/c, 129 and IFNAR−/− PMDF cultures.
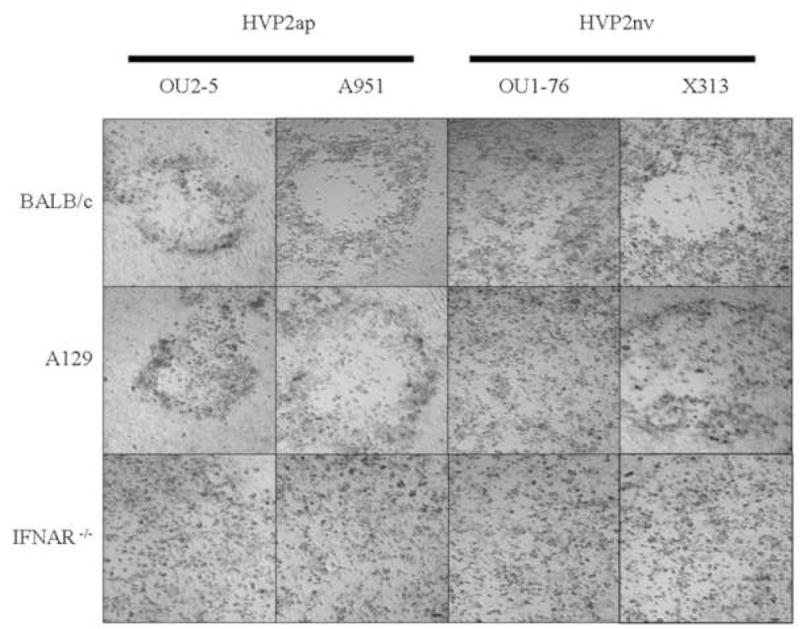
Confluent PMDF monolayers were inoculated at an MOI of 0.25 PFU/cell and photographed at 48 hrs PI. HVP2nv isolates produced complete CPE within 48 hrs in all three cell types. HVP2ap isolates produced discrete plaques by 24 hrs in Balb/c and 129 cell cultures, but these failed to enlarge with time. In contrast, HVP2ap isolates completely destroyed IFNAR−/− cultures by 48 hr PI.
To quantitate replication of the two HVP2 subtypes in PMDFs, cell cultures derived from the three mouse strains were infected as above and infectious virus quantitated by plaque assay at 48 hrs PI (Figure 2). Consistent with the observed formation of plaques, HVP2nv strains produced significantly more infectious virus than HVP2ap strains in both Balb/c and 129 PMDF cultures (p > 0.05). Although levels of infectious virus in HVP2ap infected IFNAR−/− cultures were approximately 10-fold less than in HVP2nv infected IFNAR−/− cultures, HVP2ap strains produced significantly more infectious virus in IFNAR−/− PMDF cultures than in Balb/c or 129 cultures (p > 0.05).
Figure 2. Replication of HVP2 subtypes in Balb/c, 129 and IFNAR−/− PMDF cultures.

Confluent Balb/c (light gray), 129 (dark gray) or IFNAR−/− (black) PMDF cultures were inoculated with HVP2ap isolates A951 and OU2-5 or HVP2nv isolates X313 and OU1-76 at an MOI of 0.25 PFU/cell. At 48 hrs PI, infected cells were scraped into the media and infectious virus quantitated. There was no significant difference in the amount of HVP2nv produced in the three cell lines while HVP2ap isolates produced significantly more infectious progeny virus in IFNAR−/− cells than in either Balb/c or 129 cells (as indicated by asterisks above the standard error bars).
We previously demonstrated that HVP2ap infected PMDF cultures derived from Balb/c mice produced higher levels of IFN-β than did HVP2nv infected cultures (Rogers, Black, and Eberle, 2007). Since IFNAR−/−mice lack the IFN-β receptor, uninfected cells in IFNAR−/− PMDF cultures should not be able to respond to the production of IFN-β by infected cells. PMDF cultures prepared from the three mouse strains were infected at an MOI = 1 PFU/cell and media assayed for IFN-β at 16 hrs PI. This MOI does not result in infection of 100% of cells, thus allowing amplification of the IFN-β response to occur. As shown in Figure 3, neither HVP2nv strain induced high levels of IFN-β in any of the three PMDF cultures. In contrast, HVP2ap strains induced high levels of IFN-β in both Balb/c and 129 PMDF cultures but not in IFNAR−/− PMDF cultures. These results suggest that most of the IFN-β response induced by HVP2ap infection is the result of the amplification of IFN-β expression by uninfected cells in the cultures rather than being due to expression of high levels of IFN-β by HVP2ap infected cells.
Figure 3. IFN-β production in HVP2ap and HVP2nv infected PMDF cultures.

PMDF cultures derived from three mouse strains were infected with HVP2nv strains OU1-76 (black) and X313 (hatched) or HVP2ap strains OU2-5 (light gray) and A951 (dark gray) at an MOI of 1.0 PFU/cell. Media was collected at 24 hrs PI and IFN-β quantitated by ELISA. In Balb/c and 129 cells, the IFN-β concentration was significantly higher in HVP2ap infected than in HVP2nv infected cultures (as indicated by asterisks above the standard error bars); there was no significant difference in IFN-β produced in IFNAR−/− cultures infected with the two HVP2 subtypes.
Based on the susceptibility of IFNAR−/− cells to infection by HVP2ap strains, IFNAR−/− mice should be susceptible to HVP2ap infection. To test this, mice were by infected skin scarification with HVP2ap strain OU2-5 or HVP2nv strain OU1-76. The results of these experiments are summarized in Table 1. The pathogenesis of HVP2ap and HVP2nv infections in 129 mice were the essentially same as described previously for Balb/c mice (Rogers et al., 2006). ID50 and LD50 values for HVP2nv were not significantly different, while the ID50 value for HVP2ap was significantly less that the LD50. The LD50 for HVP2ap strain OU2-5 in 129 mice (5.14 PFU) was also significantly lower than in Balb/c mice (>6.0 PFU). However, all 129 mice that died of HVP2ap infection did so at times far past the time at which mice die of acute CNS infection (5–6 DPI). At necropsy, all HVP2ap infected 129 mice that died <14 DPI showed signs of dehydration with distended bladders and impacted intestines. Histological examination of tissues revealed pathology of the mural ganglia characterized by mild to moderate infiltration of mononuclear cells. Immunohistochemical staining demonstrated the presence of viral antigen in these ganglia. These histological findings are similar to those previously reported in the large intestines and urinary bladders of HVP2 and Saimirine herpesvirus 1 infected Balb/c mice (Breshears, Eberle, and Ritchey, 2001; Breshears, Eberle, and Ritchey, 2005; Ritchey et al., 2002).
Table 1.
Pathogenesis of HVP2 in 129 and IFNAR−/− Mice
| Mouse Strain | Virus | Dose (PFU)1 | Group Size | Clinical Signs | Dead | Anti-Viral IgG2 | ID503 | LD503 | Time to Death4 |
|---|---|---|---|---|---|---|---|---|---|
| 129 | HVP2ap | 106 | 5 | 3 | 3 | 5/5 | 3.50a | 5.14b | 11.75a |
| 105 | 5 | 2 | 2 | 5/5 | |||||
| 104 | 5 | 3 | 2 | 3/5 | |||||
| 103 | 5 | 0 | 0 | 0/5 | |||||
| HVP2nv | 106 | 10 | 10 | 10 | 10 | 3.04a | 3.03a | 6.5b | |
| 105 | 10 | 10 | 10 | 10 | |||||
| 104 | 5 | 5 | 5 | 5 | |||||
| 103 | 5 | 2 | 2 | 2 | |||||
| 102 | 5 | 0 | 0 | 0 | |||||
| 101 | 5 | 0 | 0 | 0 | |||||
| IFNAR−/− | HVP2ap | 106 | 5 | 5 | 5 | 5/5 | 3.53a | 3.07a | 5.5b |
| 105 | 5 | 5 | 5 | 5/5 | |||||
| 104 | 5 | 5 | 5 | 5/5 | |||||
| 103 | 9 | 3 | 3 | 3/9 | |||||
| 102 | 10 | 1 | 1 | 0/95 | |||||
| 101 | 7 | 0 | 0 | 0/7 | |||||
| HVP2nv | 104 | 9 | 9 | 9 | 9/9 | 2.76 | 2.82 | 5.5 | |
| 103 | 8 | 5 | 5 | 5/75 | |||||
| 102.5 | 7 | 2 | 2 | 2/7 | |||||
| 102 | 4 | 0 | 0 | 0/4 |
Mice were inoculated by skin scarification as described (Rogers et al., 2006).
Sera from mice surviving >10 DPI were tested by ELISA. Mice that died ≤10 DPI were considered to be antibody positive for calculation of ID50 values.
Values with the same superscript lettering are not statistically different from each other at p = >0.05.
Average time to death (in days) for animals humanely euthanized before termination of the experiment at 14 DPI. Values with the same superscript lettering are not statistically different from each other at p = >0.05.
No serum was available for testing from one mouse in each of these groups.
The ID50 and LD50 of HVP2nv were not significantly different in IFNAR−/− and 129 mice (p > 0.05), indicating that the lack of the IFN-β receptor did not result in an increase in the neuropathogenicity of HVP2nv. In contrast, the LD50 for HVP2ap in IFNAR−/− mice was not significantly different from the LD50 for HVP2nv (p >0.05). Furthermore, HVP2ap infected IFNAR−/− mice died within the same timeframe as HVP2nv infected mice. As seen for HVP2nv infected mice, the LD50 and ID50 values for HVP2ap in IFNAR−/− mice were not significantly different from one another (p > 0.05). Histologically, IFNAR−/− mice infected with either HVP2nv or HVPap and 129 mice infected with HVP2nv exhibited lesions having similar quality and intensity. Cutaneous lesions were characterized by abrupt epidermal necrosis with multifocal ulceration and the exposed dermis overlain by a coagulum of fibrin and necrotic cell debris. Scattered remaining epidermal keratinocytes as well as follicular epithelial cells exhibited typical herpetic intranuclear inclusion bodies. The dermis contained a moderate to severe, locally extensive inflammatory infiltrate composed of neutrophils admixed with fewer macrophages and lymphocytes. Spinal cords exhibit multifocal to diffuse vacuolation of the neuropil, usually worse on the ipsilateral side, with numerous scattered (small) neurons predominantly in the ipsilateral dorsal grey horn exhibiting herpetic inclusion bodies. In lumbar segments, neurons within dorsal root ganglia and other paraspinal ganglia were often acutely necrotic or had intranuclear inclusion bodies. For the most part, although spinal cord lesions were replete with inclusion bodies, inflammation was minimal to non-existent. Spinal cord and skin sections collected at 14 DPI from 129 mice infected with HVP2ap exhibited no significant microscopic lesions.
In Balb/c mice HVP2ap invades the CNS, but does so much less effectively than HVP2nv; HVP2ap takes longer to appear in the CNS, is present in the CNS in lesser amounts, and does not induce as strong an inflammatory response in the CNS (Rogers et al., 2006). However, the in vivo results above suggest that the kinetics of HVP2ap neuropathogenesis in IFNAR−/− mice should be similar to that of HVP2nv. To test this, groups of six mice were infected and sacrificed at 2, 3 and 4 dpi, three being processed for histopathology and three for virus quantitation. The results are summarized in Table 2. Differences in titers of the two viruses in the skin of 129 infected mice were statistically significant (p = >0.05) only at 3 DPI when HVP2ap titers decrease more than HVP2nv titers. Although HVP2nv titers were generally higher than HVP2ap titers, the differences at days 2 and 4 were not statistically significant due to the small group size and variation among individual mice. However, comparison of virus titers in the skin at 2, 3 and 4 DPI revealed that while there was a significant increase in HVP2nv titers each day, there was no statistically significant increase in HVP2ap titers from 2–4 DPI. Based on histopathological examination, skin lesions were subjectively more severe in 129 mice inoculated with HVP2nv than HVP2ap, but this could reflect variability in sampling of the inoculation site rather than definitive differences in skin lesion severity. Thus, replication in the skin of 129 mice at the site of inoculation was not dramatically different for the two HVP2 subtypes up to 4 dpi.
Table 2.
Comparative Pathogenesis of HVP2ap and HVP2nv in 129 and IFNAR−/− Mice1
| 2 DPI |
3 DPI |
4 DPI |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Histopathology | Virus Isolation | Histopathology | Virus Isolation | Histopathology | Virus Isolation | |||||||||
| Mouse Strain |
Virus | Mouse No. |
Skin | Lumbar DRG |
Skin | Lumbar DRG |
Skin | Lumbar DRG |
Skin | Lumbar DRG |
Skin | Lumbar DRG |
Skin | Lumbar DRG |
| 129 | HVP2nv | 1 | 1+ | 0 | 4.3e4 | 0 | 3+ | 2+ | 1.9e4 | 1.0e4 | 4+ | 4+ | 2.0e4 | 4.4e3 |
| 2 | 1+ | 0 | 6.2e3 | 0 | 4+ | 0 | 2.7e4 | 1.3e3 | 1+ | 2+ | 6.5e5 | 2.0e5 | ||
| 3 | 2+ | 0 | 5.7e4 | 0 | 4+ | 2+ | 2.1e4 | 5.0e3 | 0 | 3+ | 8.0e4 | 2.0e5 | ||
| Mean±SEM | 3.5±1.5e4 | 0 | 2.2±0.3e4 | 5.4±2.5e3 | 2.5±2.0e5 | 1.3±0.6e5 | ||||||||
| HVP2ap | 1 | 1+ | 0 | 2.2e3 | 0 | 1+ | 0 | 3.5e2 | 0 | 0 | 2+ | 3.8e4 | 0 | |
| 2 | 2+ | 0 | 6.2e3 | 0 | 3+ | 0 | 2.9e3 | 0 | 1+ | 1+ | 5.9e4 | 5.1e3 | ||
| 3 | 0 | 0 | 1.5e4 | 0 | 2+ | 1+ | 9.0e3 | 1.4e3 | NS* | 2+ | 4.3e4 | 6.2e2 | ||
| Mean±SEM | 7.8±3.8e3 | 0 | 4.1±2.6e3 | 4.7±4.5e2 | 4.7±0.6e4 | 1.9±1.6e3 | ||||||||
| IFNAR−/− | HVP2nv | 1 | 3+ | 0 | 6.7e6 | 0 | 4+ | 1+ | 1.4e6 | 0 | 4+ | 3+ | 1.4e6 | 9.6e5 |
| 2 | 0 | 0 | 2.7e6 | 0 | 3+ | 1+ | 1.1e6 | 4.9e5 | 4+ | 4+ | 5.4e5 | 6.5e5 | ||
| 3 | 1+ | 0 | 1.0e6 | 0 | 2+ | 0 | 1.6e5 | 6.7e3 | 3+ | 1+/- | 1.2e6 | 1.3e6 | ||
| Mean±SEM | 3.5±1.7e6 | 0 | 8.9±3.7e5 | 1.7±1.6e5 | 1.0±0.3e6 | 9.7±1.9e5 | ||||||||
| HVP2ap | 1 | 3+ | 0 | 2.1e6 | 0 | 4+ | 2+ | 5.8e5 | 4.3e3 | 4+ | 3+ | 2.3e6 | 1.3e6 | |
| 2 | 2+ | 0 | 3.1e6 | 0 | 4+ | 0 | 7.4e5 | 5.7e3 | 4+ | 2+ | 2.4e5 | 3.4e5 | ||
| 3 | 2+ | 0 | 1.3e6 | 0 | 2+ | 0 | 1.4e6 | 1.0e3 | 4+ | 2+ | 1.4e6 | 2.6e5 | ||
| Mean±SEM | 2.2±0.5e6 | 0 | 9.1±2.5e5 | 3.7±1.4e3 | 1.3±0.6e6 | 6.3±3.3e5 | ||||||||
NS, no sample.
Groups of 6 mice were infected by skin scarification. At each time point, 3 mice were used to quantitate infectious virus in the skin or lumbar spinal cord and associated DRGs, and 3 mice were used for histopathological examination.
When virus levels in the lumbar spinal cord and associated DRGs of 129 mice were compared, titers of HVP2nv were significantly higher than HVP2ap levels at 3 and 4 DPI (p = >0.05). Microscopic lesions in the CNS and DRGs were also more severe in HVP2nv infected 129 mice, with larger numbers of necrotic neurons and neurons with intranuclear inclusion bodies being present than were observed in HVP2ap infected mice. In the CNS, the varying severity of microscopic lesions between HVP2 subtypes was consistent with the observed difference in virus titers. These results indicate that in 129 mice HVP2nv is better able to invade and/or replicate in the CNS than is HVP2ap.
Results were very different in IFNAR−/− mice. In the skin at the site of inoculation there was not a statistically significant increase in viral titers of either virus between 2 and 4 DPI, nor were titers of HVP2nv significantly different from those of HVP2ap at any time point. While levels of HVP2ap and HVP2nv in the skin were equivalent in IFNAR−/− mice, there was a significant difference in the amount of each virus present in the skin of 129 vs. IFNAR−/− mice at all time points. The same was true for virus levels in the CNS of IFNAR−/− mice: while titers of both HVP2ap and HVP2nv increased dramatically between 2 and 4 DPI, titers of the two virus subtypes were not significantly different from each other. At each time point, microscopic skin lesions in the skin and CNS were of similar character and severity between the two virus subtypes. The lack of variation in lesion severity in IFNAR−/− mice is consistent with the lack of difference between tissue virus titers in IFNAR−/−mice. Thus, consistent with the equivalent LD50 values for the two HVP2 subtypes in IFNAR−/− mice (Table 1), there was no apparent difference in the ability of HVP2ap and HVP2nv to replicate in the skin at the site of inoculation or to invade the CNS in IFNAR−/− mice.
Comparison of virus levels in tissues of 129 vs. IFNAR−/− mice revealed that the in the skin, titers of both viruses were significantly higher at all time points in IFNAR−/− mice (p = >0.05). When titers of virus in the CNS were compared, titers of HVP2nv at 3 and 4 DPI were not significantly different in the two mouse strains. However, CNS titers of HVP2ap were significantly higher (p = >0.05) in IFNAR−/− as compared to 129 mice at 3 and 4 DPI. Thus, the ability of HVP2ap to replicate locally in the skin to levels equivalent to that of HVP2nv appears to correlate with the ability of the virus to effectively invade the CNS.
In HSV, the vhs protein encoded by the UL41 gene has been shown to be important in abrogating the IFN-β response of cells following infection, presumably through the ability of the vhs protein to degrade some cellular mRNAs (Kwong and Frenkel, 1987; Kwong and Frenkel, 1989; Murphy et al., 2003). The reduced pathogenicity of HSV UL41 deletion mutants in mice (Smith, Ackland-Berglund, and Leib, 2000; Smith, Morrison, and Leib, 2001; Strelow, Smith, and Leib, 1997; Strelow and Leib, 1995; Suzutani et al., 2000) is similar to that of HVP2ap strains, raising the possibility that differences in the UL41 gene might underlie the dichotomous neuropathogenicity of the two HVP2 subtypes. To address this possibility the UL41 ORF of four HVP2 strains (2 apathogenic & 2 neurovirulent) was sequenced and the predicted amino acid sequences aligned to identify substitutions that correlated with the neurovirulence phenotype of the isolates (Figure 4). There were six amino acid residues that showed variation. Although two of the variations were unique to a single HVP2 isolate, four correlated with the neurovirulence phenotype. None of these four substitution sites were located in a highly conserved region of the vhs polypeptide. Two were conservative substitutions (Ile ↔ Leu and Ala ↔ Thr) and two were non-conservative substitutions (both Leu ↔ Pro). The two Leu ↔ Pro substitutions were located very near each other. Since proline residues induce turns in the polypeptides, it is possible that the Leu/Pro substitutions ‘offset’ any effect via their close proximity in the peptide. Thus, there did not appear to be any major difference in the vhs sequences of HVP2ap and HVP2nv isolates.
Figure 4. Location of variable amino acid residues in the HVP2 UL41 (vhs) polypeptide.

AA sequences of four HVP2 strains (HVP2ap A951 & OU2-5 and HVP2nv OU1-76 & X313) were aligned to identify residues that varied among isolates. The sequences for HSV1 and HSV2 are included for reference. AA residues conserved in all sequences are indicated by lack of shading. Residues that vary between HSV and HVP2 are indicated by gray/black shading. The four potential HVP2 subtype-specific substitutions are indicated by a solid triangle below the sequences and the two isolate-specific substitutions are indicated by an open triangle below them.
To experimentally assess the role of the UL41 gene in pathogenicity of HVP2 isolates, the UL41 ORF was deleted from a neurovirulent strain of HVP2 (designated Δ41) and then replaced with the UL41 ORF from either an apathogenic virus (Δ41C, a recombinant) or the parent neurovirulent virus (Δ41R, a revertant). Based on one-step growth curves, there were no differences in the ability of any of these viruses to replicate in Vero cells (Figure 5A). While degradation of host cell β-actin mRNA occurred in cells infected with any of the viruses containing an intact UL41 ORF (wt, Δ41C & Δ41R), β-actin mRNA degradation was more efficient in cells infected with the HVP2ap parental virus than in cells infected with HVP2nv parent virus, and β-actin mRNA degradation by the Δ41C recombinant and the Δ41R revertant reflected that of the parent virus their UL41 ORFs were derived from (Figure 5B). The recombinant Δ41C replicated in PMDF cultures as efficiently as the HVP2nv parent and Δ41R revertant viruses, destroying the entire monolayer within 48 hrs. In contrast, virus lacking the UL41 ORF (Δ41) did not exhibit significant degradation of β-actin mRNA and grew much slower in PMDFs than the other viruses. However, unlike HVP2ap plaques which ceased to expand after 24 hrs and were eventually overgrown by uninfected cells (Rogers, Black, and Eberle, 2007), HVP2nvΔ41 plaques continued to slowly enlarge in size up to 6 days PI when the experiment was terminated.
Figure 5. Characterization of UL41 mutants.
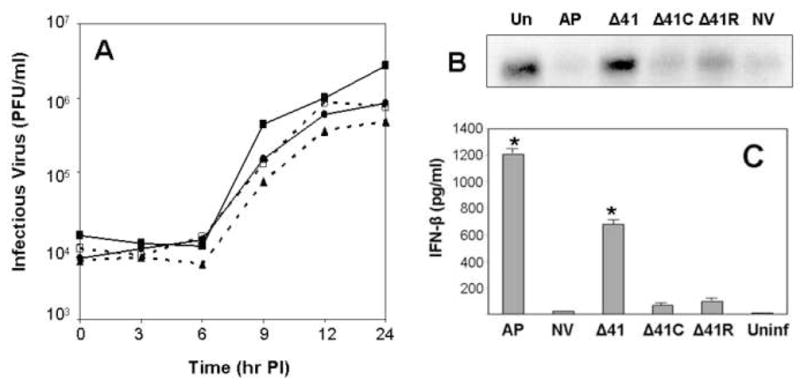
One-step growth curves performed in Vero cells are shown in A. Viruses included the parental HVP2nv (■——-■), HVP2nv Δ41 (▲………▲), HVP2nv Δ41C (□----□) and HVP2nv Δ41R (●——-●). Northern blot of β-actin mRNA in infected cells is shown in B. Total RNA was isolated from cells at 4 hrs PI, 15 μg loaded in each lane, and northern blotting performed using the Vero cell β-actin gene as probe. IFN-β production in PMDF cultures is shown in C. Cultures were infected with the indicated viruses at an MOI = 1 PFU/cell and the medium assayed for IFN-β at 24 hrs PI. IFN-β levels that were significantly different (p = >0.05) from the level induced by the parental HVP2nv virus are indicated by an asterisk above the standard error bar.
The ability of the UL41 mutants to prevent an IFN-β response was compared by assaying the medium from infected PMDF cultures at 24 hrs PI (Figure 5C). Cultures infected with the Δ41 mutant produced high levels of IFN-β typical of HVP2ap strains. Cultures infected with the recombinant Δ41C produced low levels of IFN-β comparable to those produced by wt HVP2nv and Δ41R revertant infected cultures. These results indicate that while the vhs protein is not responsible for the difference in the ability of HVP2ap and HVP2nv strains to prevent an IFN-β response by the host cell, its absence does allow the host cell to mount an effective IFN-β response following infection.
Since the Δ41C recombinant virus grows as well as the parental HVP2nv virus in PMDF cultures and as virus replication in PMDF cultures correlates with the neurovirulence of HVP2 isolates (Rogers, Black, and Eberle, 2007), the Δ41C recombinant should have a neurovirulence phenotype similar to the parental HVP2nv and Δ41R revertant viruses. To test this, Balb/c mice were infected with each virus by skin scarification. The results are summarized in Table 3. As expected, the LD50 of the revertant virus (Δ41R) was not significantly different from that of the parental HVP2nv virus. Most mice rapidly developed paresis of the ipsilateral hind foot by 5–6 days PI, followed by paralysis and euthanasia within 24 hrs. Mice infected with the same dose of the Δ41C recombinant virus followed a similar pathogenic progression of infection and time course of disease. Differences in the LD50 and mean time to death of the wt HVP2nv, Δ41C recombinant and Δ41R revertant viruses were not statistically significant but were different from that of HVP2ap. Mice inoculated with the Δ41 deletion mutant exhibited a somewhat delayed development of paresis of the ipsilateral hind foot, and paresis did not always progress to full paralysis of the affected limb. As time progressed, skin lesions at the site of inoculation in Δ41 infected mice continued to develop and spread. Despite the prolonged disease progression, the LD50 of the Δ41 deletion mutant was not significantly different from that of the other HVP2nv viruses but was significantly lower than that of HVP2ap (p = >0.05). Thus, while deletion of the UL41 ORF did qualitatively affect the neuropathogenicity of HVP2nv, it did not severely reduce neuropathogenicity of the virus as has been reported for HSV1 and HSV2 (Korom, Wylie, and Morrison, 2008; Strand and Leib, 2004; Strelow and Leib, 1995).
Table 3.
Pathogenesis of HVP2 UL41 Mutants in Balb/c Mice
| Dose (PFU) | OU1-76 | OU2-5 | Δ41 | Δ41C | Δ41R |
|---|---|---|---|---|---|
| 106 | ND2 | 1/101 | ND | ND | ND |
| 105 | 8/8 | 0/8 | 10/10 | 10/10 | 5/5 |
| 104 | 6/8 | 0/8 | 8/12 | 9/12 | 10/12 |
| 103 | 2/12 | ND | 1/11 | 1/12 | 3/12 |
| 102 | 0/8 | ND | 0/5 | 0/5 | 0/5 |
| LD503 | 103.6 | >106 | 103.9 | 103.7 | 103.4 |
Number of mice that developed clinical signs of fatal CNS infection in each group.
ND, not done
The LD50 for HVP2ap OU2-5 was significantly different (p = >0.05) from the LD50 of all other viruses (which were not significantly different from each other).
Histologically, skin and spinal cord lesions in mice infected with the UL41 mutants were qualitatively and quantitatively similar to those previously described in Balb/c mice infected with wt HVP2nv isolates. Briefly, there was epithelial necrosis and ulceration with herpetic inclusions present in sloughed epithelial cells in the crusts as well as in scattered remnant epidermal cells and follicular epithelial cells. The dermis exhibited moderate to severe disseminated mononuclear and neutrophilic dermatitis. Immunostaining revealed conspicuous HVP2 antigen confined to the epithelium. In the lumbar spinal cord, there was multifocal to disseminated vacuolation of the ipsilateral white matter in all funiculi, most usually severe in the dorsal and lateral funiculus (Figure 6). There was also mild, mononuclear inflammation in the ipsilateral dorsal grey horn and funiculus with numerous conspicuous herpetic inclusions within small neurons and glial cells. The only quantitative difference among the mutants was that extension of the inflammation to the brain only occurred in wt HVP2nv, Δ41C and Δ41R; there were no brain lesions observed in 33 brains examined from Δ41 infected mice, even in those inoculated with high doses of virus.
Figure 6. Comparison of spinal cord lesions in mice infected with UL41 mutants.
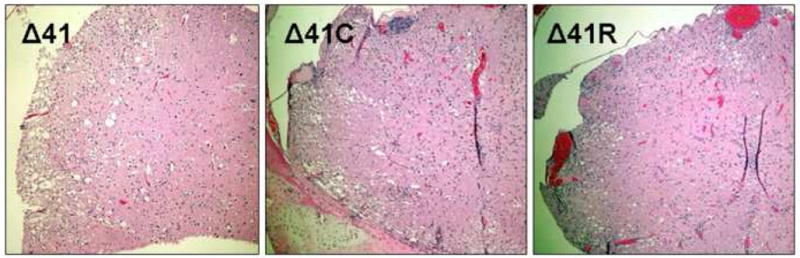
Lumbar spinal cord lesions in Balb/c mice infected with the deletion mutant Δ41, the recombinant Δ41C or the Δ41R revertant exhibit roughly the same quantity and quality of histological lesions characterized by multifocal to disseminated white matter vacuolation, mild inflammation primarily in the dorsal grey horn and funiculus, and numerous herpetic inclusion bodies (not visible at this magnification). H & E stain; bar = 260 μm.
Discussion
In their natural host, acute infection by HSV and related primate herpesviruses is normally followed by the establishment of a latent infection within the PNS that persists for the life of the host. In contrast to the usually mild, self-limiting infections that occur in the natural host, when these viruses infect a non-natural host they can produce severe, frequently fatal infections that involve the CNS such as occurs when BV infects humans. The host and viral mechanisms that allow these viruses to enter and spread via lytic infection within the CNS in a non-natural host rather than establishing a latent infection in the PNS as occurs in the natural host are largely unknown. A more thorough understanding of virus/host interactions which determine the serious outcome of cross-species or zoonotic infections is critical.
With regards to pathogenicity, the HVP2nv-mouse model closely resembles human BV infections. Although HVP2nv is not neurovirulent in its natural baboon host and only rarely causes severe infections in baboons (Rogers et al., 2005), HVP2nv isolates are extremely neuropathogenic in mice. In contrast, mice are able to very efficiently control infections by naturally occurring HVP2ap isolates. Previous studies indicate that control of HVP2ap infections in mice begins locally at the site of inoculation and continues even within the CNS (Rogers et al., 2006). Since the pathogenesis of HVP2nv and HVP2ap infection in mice diverges well before an adaptive immune response develops, innate immunity likely plays a major role in effecting the divergent pathogenicity of the two HVP2 subtypes. IFN-β appears to be involved in controlling HVP2ap infections since HVP2nv isolates do not induce and/or overcome an IFN-β response, and are thus not as susceptible to IFN-β in vitro as HVP2ap isolates are (Rogers, Black, and Eberle, 2007). The experiments presented here extend these observations and confirm a critical role for the type I IFN response in controlling HVP2ap infections in vivo.
Other investigators have observed that HSV1 and HSV2 UL41 mutants exhibit reduced neurovirulence in the mouse model, but in IFNAR−/− mice their neurovirulence is comparable to that of the wild-type virus (Leib et al., 1999; Murphy et al., 2003). Consistent with these HSV studies, HVP2ap isolates are just as neuropathogenic as HVP2nv isolates in IFNAR−/− mice as evidenced by similar LD50 values and similar levels of virus in the skin at early times PI. The relationship between the ID50 and LD50 for HVP2ap isolates in IFNAR−/− mice also reflects that of HVP2nv isolates in 129 and Balb/c mice: the ID50 and LD50 values are not significantly different, indicating that when HVP2ap establishes an infection in IFNAR−/− mice it inexorably progresses to death of the host. Similarly, temporal progression of virus from the peripheral site of inoculation into the CNS is similar for both HVP2 subtypes in IFNAR−/− mice. Since the level of virus in the lumbar spinal cord and associated DRGs represents the earliest point in the pathogenic process that is demonstrably different for HVP2ap and HVP2nv in wild-type 129 mice, it is likely that the ability of the host to delay and/or lessen entry of virus into the CNS plays a critical role in the dichotomous outcome of infection with the two HVP2 subtypes. In contrast, studies with HSV have shown that replication of the virus at the inoculation site appears to be important in that this replication amplifies the amount of virus thereby increasing the likelihood of viral entry into unmyelinated nerve fibers present in the skin to provide access to the CNS (Cunningham et al., 2006; Mossman and Ashkar, 2005; Yamada et al., 1986). While differences in the amount of HVP2ap vs. HVP2nv in skin were statistically significant only at 3 DPI, levels of HVP2ap were lower than those of HVP2nv at 2 and 4 DPI. Given the small group size used (3 mice/group), it is possible that that were larger group sizes used these differences may have been significant. If so, our results would be consistent with the results of other investigators studying HSV pathogenesis which suggest that reduction of virus replication at the site of inoculation is critical in determining the outcome of the infection.
Another observation from these experiments was that the inability of the host to mount a type I IFN response did not have a major effect on the temporal or spatial progression of HVP2nv infection at early times PI. The only noticeable difference was that in IFNAR−/− mice higher titers of HVP2nv were present in skin at the inoculation site at 2 dpi than in 129 mice. However, HVP2ap titers at 2 dpi at the site of inoculation were also higher in the knock-out mice vs. wild-type indicating that higher viral titers at this time is a function of the mouse genotype rather than being peculiar to HVP2nv. This suggests that an intact type I IFN response does not interfere with the ability of HVP2nv to invade and replicate within the CNS. The higher titers of HVP2nv in skin of IFNAR−/− mice at 2 DPI suggests that the early IFN response at the site of inoculation may nonetheless have a dampening effect on HVP2nv replication in the skin at very early times after infection. This would again be consistent with the hypothesis that controlling the initial replication and amplification of virus at the site of inoculation is a critical factor in the ability of the host to prevent virus from gaining access to the CNS.
In previous experiments pre-treatment of PMDF cultures with recombinant murine IFN-β did not effectively protect cells from HVP2nv infection, suggesting that it may be the induction of the IFN system rather than the production of type I IFNs which is important for controlling HVP2 infections (Rogers, Black and Eberle, 2007). In addition, while we have not detected IFN-α induction by HVP2 in vitro, IFNAR loss would affect both IFN-β and IFN-α induced pathways of antiviral immunity. As such, the importance of IFN-α in controlling HVP2 infection in vivo can not be ruled out. While type I IFNs are important for the early, innate response to viral infection they also contribute to the antiviral immune response by stimulating the cytotoxic activity of natural killer cells, signaling for dendritic cell maturation, and promoting various T-cell functions, including expansion of the memory population (Garcia-Sastre and Biron, 2006). Further studies are needed to determine the exact IFN mechanism that so efficiently controls HVP2ap while allowing CNS invasion by HVP2nv.
In HSV1 and HSV2 the UL41 gene encoding the vhs protein has been shown to play a major role in determining the neurovirulence of the virus. UL41 mutants of HSV replicate normally in tissue culture but are impaired in their ability to invade the CNS from peripheral sites and are defective in their ability to spread within the CNS of mice (Korom, Wylie, and Morrison, 2008; Smith, Ackland-Berglund, and Leib, 2000; Smith, Morrison, and Leib, 2001; Strelow, Smith, and Leib, 1997; Strelow and Leib, 1995). This is very similar to what is observed for apathogenic isolates of HVP2. However, replacement of the HVP2nv UL41 ORF with an HVP2ap UL41 ORF did not affect the ability of the virus to infect mice and invade the CNS, nor did it result in high levels of IFN-β production in infected PMDF cultures. This demonstrates that the UL41 protein alone is not responsible for the disparate neurovirulence of the two HVP2 subtypes.
Although the UL41 gene does not appear to underlie the neuropathogenic differences of HVP2ap and HVP2nv isolates, it does appear to play a role in allowing HVP2 to prevent the host IFN-β response. Deletion of the HVP2nv UL41 gene resulted in a strong IFN-β response in infected PMDF cultures. Consistent with this, HVP2nvΔ41 replicated poorly in PMDFs while HVP2ap abortively infects PMDFs. HVP2nvΔ41 also displayed reduced pathogenesis in mice much as described for HSV UL41 deletion mutants. While HVP2nvΔ41 can invade the CNS, it does not produce the rapid and severe CNS disease observed in mice infected with wt HVP2nv. Since HVP2nvΔ41 infected PMDF cultures produce high levels of IFN-β comparable to those induced by HVP2ap isolates, the UL41 gene does appear to be involved in preventing or suppressing the IFN-β response. The recombinant HVP2nvΔ41C carrying an HVP2ap UL41 gene behaved similar to the wt HVP2nv in all respects, including suppression of the host IFN-β response. This demonstrates that the vhs protein itself is not functionally different in apathogenic and neurovirulent HVP2 isolates. This apparent inconsistency could be explained by the interaction of the vhs protein with some other viral factor(s) to suppress the IFN-β response, this second factor being the one actually responsible for the disparate pathogenic phenotype of HVP2 isolates.
The interplay between large, complex herpesviruses and the intricate workings of the host immune system is understandably convoluted. In addition to vhs, α-herpesviruses encode numerous other IFN antagonists which most likely interact with multiple IFN induction pathways in the host. To further complicate matters, during HSV infection pathogen recognition and subsequent type I IFN production has been shown to be both cell-type and time-dependent (Rasmussen et al., 2007). While pDCs are responsible via Toll-like receptor 9 for early production of IFN in response to HSV infection in vivo, macrophages and fibroblasts also produce type I IFNs later in infection. The production of type I IFNs in these cell types is dependent on both viral entry and replication, and is ablated in cells unable to signal through the mitochondrial antiviral signaling (MAVS) protein pathway. Therefore, interference with both the MAVS- and TLR9-dependent pathways at multiple times PI may be necessary for HVP2nv to ensure sufficient replication in the dermatome for entry into the CNS.
The fact that the LD50 for HVP2ap isolates in IFNAR−/− mice was the same as the LD50 for HVP2nv strains suggests that the type I IFN response may be the primary if not sole facet of the host innate response involved in the initial control of HVP2ap infection. The basis for the divergent neuropathogenicity of HVP2nv vs. HVP2ap isolates in mice thus appears to lie in the ability of the HVP2nv isolates to prevent and/or overcome the host type I IFN response and/or downstream host effector mechanisms which are controlled and initiated via the type I IFN pathway. The UL41-encoded vhs protein gene does appear to be directly involved in preventing the IFN-β response in that deletion of the UL41 gene from HVP2nv results in IFN-β production following infection. However, since the HVP2nvΔ41 deletion mutant can replicate in PMDF cells and mice despite the induction of an IFN-β response, the vhs protein does not appear to be the primary factor responsible for the ability of HVP2nv to overcome the IFN-β response. It remains to be seen how HVP2nv isolates accomplish this and why HVP2ap isolates are unable to do so.
Acknowledgments
This work was supported in part by grants P40 RR12317, R24 RR16556 and T35 RR07061-13 from the Public Health Service.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Black DH, Saliki JT, Eberle R. Development of a green fluorescent protein reporter cell line to reduce biohazards associated with detection of infectious Cercopithecine herpesvirus 1 (monkey B virus) in clinical specimens. Compar Med. 2002;52(6):534–42. [PubMed] [Google Scholar]
- Brandt CR. The role of viral and host genes in corneal infection with herpes simplex virus type 1. Exptl Eye Res. 2005;80(5):607–21. doi: 10.1016/j.exer.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Breshears MA, Eberle R, Ritchey JW. Characterization of gross and histological lesions in Balb/c mice experimentally infected with Herpesvirus saimiri 1 (HVS1) J Compar Pathol. 2001;125(1):25–33. doi: 10.1053/jcpa.2001.0473. [DOI] [PubMed] [Google Scholar]
- Breshears MA, Eberle R, Ritchey JW. Temporal progression of viral replication and gross and histological lesions in Balb/c mice inoculated epidermally with Saimiriine herpesvirus 1 (SaHV-1) J Compar Path. 2005;133(2–3):103–13. doi: 10.1016/j.jcpa.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Cunningham AL, Diefenbach RJ, Miranda-Saksena M, Bosnjak L, Kim M, Jones C, Douglas MW. The cycle of human herpes simplex virus infection: virus transport and immune control. J Infect Dis 194 Suppl. 2006;1:S11–8. doi: 10.1086/505359. [DOI] [PubMed] [Google Scholar]
- Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J Immunol. 2004;173(3):1535–48. doi: 10.4049/jimmunol.173.3.1535. [DOI] [PubMed] [Google Scholar]
- Darnell JE, Kerr IM, Stark GR. Transcriptional activation in response to IFNs and other extracellular signalling proteins. Science. 1994;264:248–254. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Duerst RJ, Morrison LA. Herpes simplex virus 2 virion host shutoff protein interferes with type I interferon production and responsiveness. Virol. 2004;322(1):158–67. doi: 10.1016/j.virol.2004.01.019. [DOI] [PubMed] [Google Scholar]
- Eberle R, Black DH, Blewett EL, White GL. Prevalence of Herpesvirus papio 2 in baboons and identification of immunogenic viral polypeptides. Lab Anim Sci. 1997a;47(3):256–62. [PubMed] [Google Scholar]
- Eberle R, Black DH, Lehenbauer TW, White GL. Shedding and transmission of baboon Herpesvirus papio 2 (HVP2) in a breeding colony. Lab Anim Sci. 1998;48(1):23–28. [PubMed] [Google Scholar]
- Eberle R, Black DH, Lipper S, Hilliard JK. Herpesvirus papio 2, an SA8-like alpha-herpesvirus of baboons. Arch Virol. 1995;140(3):529–45. doi: 10.1007/BF01718429. [DOI] [PubMed] [Google Scholar]
- Eberle R, Hilliard JK. The simian herpesviruses: a review. Infect Agents Dis. 1995;4:55–70. [PubMed] [Google Scholar]
- Eberle R, Tanamachi B, Black D, Blewett EL, Ali M, Openshaw H, Cantin EM. Genetic and functional complementation of the HSV1 UL27 gene and gB glycoprotein by simian alpha-herpesvirus homologs. Arch Virol. 1997b;142(4):721–36. doi: 10.1007/s007050050114. [DOI] [PubMed] [Google Scholar]
- Enquist LW, Husak PJ, Banfield BW, Smith GA. Infection and spread of alphaherpesviruses in the nervous system. Adv Virus Res. 1998;51:237–347. doi: 10.1016/s0065-3527(08)60787-3. [DOI] [PubMed] [Google Scholar]
- Esclatine A, Taddeo B, Roizman B. The UL41 protein of herpes simplex virus mediates selective stabilization or degradation of cellular mRNAs. PNAS. 2004;101(52):18165–70. doi: 10.1073/pnas.0408272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everly DN, Feng P, Mian IS, Read GS. mRNA degradation by the virion host shutoff (vhs) protein of herpes simplex virus: genetic and biochemical evidence that vhs is a nuclease. J Virol. 2002;76:8560–8571. doi: 10.1128/JVI.76.17.8560-8571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312(5775):879–82. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Goodbourn S, Didcock L, Randall RE. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J Gen Virol. 2000;81(Pt 10):2341–64. doi: 10.1099/0022-1317-81-10-2341. [DOI] [PubMed] [Google Scholar]
- Huff JL, Barry PA. B-virus (Cercopithecine herpesvirus 1) infection in humans and macaques: potential for zoonotic disease. Emerg Infect Dis. 2003;9:246–250. doi: 10.3201/eid0902.020272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karr BM, Read GS. The virion host shutoff function of herpes simplex virus degrades the 5′ end of a target mRNA before the 3′ end. Virol. 1999;264(1):195–204. doi: 10.1006/viro.1999.9986. [DOI] [PubMed] [Google Scholar]
- Keeble SA. B virus infection in monkeys. Ann NY Acad Science. 1960;85:960–969. doi: 10.1111/j.1749-6632.1960.tb50016.x. [DOI] [PubMed] [Google Scholar]
- Korom M, Wylie KM, Morrison LA. Selective ablation of virion host shutoff protein RNase activity attenuates herpes simplex virus 2 in mice. J Virol. 2008;82(7):3642–53. doi: 10.1128/JVI.02409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong AD, Frenkel N. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. PNAS. 1987;84(7):1926–30. doi: 10.1073/pnas.84.7.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong AD, Frenkel N. The herpes simplex virus virion host shutoff function. J Virol. 1989;63:4834–4839. doi: 10.1128/jvi.63.11.4834-4839.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam Q, Smibert CA, Koop KE, Lavery C, Capone JP, Weinheimer SP, Smiley Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 1996;15:2575–2581. [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science (New York, N Y) 2007;315(5817):1398–401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses In vivo. J Exp Med. 1999;189:663–672. doi: 10.1084/jem.189.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis MR, O’Neill T, Bush M, Montali RJ. Fatal herpesvirus infection in patas monkeys and a black and white colobus monkey. J Am Vet Med Assoc. 1981;179:1236–1239. [PubMed] [Google Scholar]
- Mossman KL, Ashkar AA. Herpesviruses and the innate immune response. Viral Immunology. 2005;18(2):267–81. doi: 10.1089/vim.2005.18.267. [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–21. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Murphy JA, Duerst RJ, Smith TJ, Morrison LA. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J Virol. 2003;77(17):9337–45. doi: 10.1128/JVI.77.17.9337-9345.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa K, Lehenbauer TW, Eberle R. Herpesvirus papio 2: a safer and sensitive alternative for serodiagnosis of B virus infection in macaque monkeys. Lab Anim Sci. 1999;49:605–616. [PubMed] [Google Scholar]
- Palmer AE. Herpesvirus simiae: historical perspective. J Med Primatol. 1987;16:99–130. [PubMed] [Google Scholar]
- Rasmussen SB, Sorensen LN, Malmgaard L, Ank N, Baines JD, Chen ZJ, Paludan SR. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J Virol. 2007;81(24):13315–24. doi: 10.1128/JVI.01167-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchey JW, Ealey KA, Payton M, Eberle R. Comparative pathology of infections with baboon and African green monkey alpha-herpesviruses in mice. J Compar Pathol. 2002;127:150–161. doi: 10.1053/jcpa.2002.0575. [DOI] [PubMed] [Google Scholar]
- Ritchey JW, Payton ME, Eberle R. Clinicopathological characterization of monkey B virus (Cercopithecine herpesvirus 1) infection in mice. J Compar Pathol. 2005;132:202–217. doi: 10.1016/j.jcpa.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Rogers KM, Black DH, Eberle R. Primary mouse dermal fibroblast cell cultures as an in vitro model system for the differential pathogenicity of cross-species herpesvirus papio 2 infections. Arch Virol. 2007;152(3):543–52. doi: 10.1007/s00705-006-0865-1. [DOI] [PubMed] [Google Scholar]
- Rogers KM, Ealey KA, Ritchey JW, Black DH, Eberle R. Pathogenicity of different baboon Herpesvirus papio 2 isolates is characterized by either extreme neurovirulence or complete apathogenicity. J Virol. 2003;77:10731–10739. doi: 10.1128/JVI.77.20.10731-10739.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers KM, Ritchey JW, Payton M, Black DH, Eberle R. Neuropathogenesis of Herpesvirus papio 2 in Mice Parallels Cercopithecine herpesvirus 1 (B Virus) Infections in Humans. J Gen Virol. 2006;87:267–276. doi: 10.1099/vir.0.81476-0. [DOI] [PubMed] [Google Scholar]
- Rogers KM, Wolf RF, White GL, Eberle R. Experimental infection of baboons (Papio cynocephalus anubis) with apathogenic and neurovirulent subtypes of Herpesvirus papio 2. Compar Med. 2005;55(5):425–430. [PubMed] [Google Scholar]
- Rouse BT, Lopez C. Immunobiology of herpes simplex virus infection. CRC Press; 1984. [Google Scholar]
- Sabin AB, Wright AM. Acute ascending myelitis following a monkey bite, with isolation of a virus capable of reproducing the disease. J Expl Med. 1934;59:115–136. doi: 10.1084/jem.59.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelter J, Knez J, Smiley JR, Capone JP. Identification and characterization of a small modular domain in the herpes simplex virus host shutoff protein sufficient for Interaction with VP16. J Virol. 1996;70:2124–2131. doi: 10.1128/jvi.70.4.2124-2131.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons A, Nash AA. Zosteriform spread of herpes simplex virus as a model of recrudescence and its use to investigate the role of immune cells in prevention of recurrent disease. J Virol. 1984;52:816–821. doi: 10.1128/jvi.52.3.816-821.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TJ, Ackland-Berglund CE, Leib DA. Herpes simplex virus virion host shutoff (vhs) activity alters periocular disease in mice. J Virol. 2000;74:3598–3604. doi: 10.1128/jvi.74.8.3598-3604.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TJ, Morrison LA, Leib DA. Pathogenesis of herpes simplex virus type 2 virion host shutoff (vhs) mutants. J Virol. 2001;76:2054–2061. doi: 10.1128/jvi.76.5.2054-2061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand SS, Leib DA. Role of the VP16-binding domain of vhs in viral growth, host shutoff activity, and pathogenesis. J Virol. 2004;78(24):13562–72. doi: 10.1128/JVI.78.24.13562-13572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelow L, Smith T, Leib D. The virion host shutoff function of herpes simplex virus type 1 plays a role in corneal invasion and functions independently of the cell cycle. Virol. 1997;231(1):28–34. doi: 10.1006/viro.1997.8497. [DOI] [PubMed] [Google Scholar]
- Strelow LI, Leib DA. Role of the viron host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J Virol. 1995;69:6779–6786. doi: 10.1128/jvi.69.11.6779-6786.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelow LI, Leib DA. Analysis of conserved domains of UL41 of herpes simplex virus type 1 in virion host shutoff and pathogenesis. J Virol. 1996;70(8):5665–7. doi: 10.1128/jvi.70.8.5665-5667.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzutani T, Nagamine M, Shibaki T, Ogasawara M, Yoshida I, Daikoku T, Nishiyama Y, Azuma M. The role of the UL41 gene of herpes simplex virus type 1 in evasion of non-specific host defence mechanisms during primary infection. J Gen Virol. 2000;81(Pt 7):1763–71. doi: 10.1099/0022-1317-81-7-1763. [DOI] [PubMed] [Google Scholar]
- Thompson SA, Hilliard JK, Kittel D, Lipper S, Giddens WE, Black DH, Eberle R. Retrospective analysis of an outbreak of B virus in a colony of DeBrazza’s monkeys (Cercopithecus neglectus) Compar Med. 2000;50:649–657. [PubMed] [Google Scholar]
- Troan BV, Perelygina L, Patrusheva I, van Wettere A, Hilliard J, Loomis M, De Voe R. Naturally Transmitted Herpesvirus papio 2 Infection in a Black and White Colobus Monkey. JAVMA. 2007;231:1878–1883. doi: 10.2460/javma.231.12.1878. [DOI] [PubMed] [Google Scholar]
- Weeks BS, Ramchandran RS, Hopkins JJ, Friedman HM. Herpes simplex virus type-1 and -2 pathogenesis is restricted by the epidermal basement membrane. Arch Virol. 2000;145:385–396. doi: 10.1007/s007050050030. [DOI] [PubMed] [Google Scholar]
- Weigler BJ. Biology of B virus in macaque and human hosts: a review. Clin Infect Dis. 1992;14:555–567. doi: 10.1093/clinids/14.2.555. [DOI] [PubMed] [Google Scholar]
- Whitely RJ, Hilliard JK. Cercopithecine herpesvirus (B virus) In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott Williams and Wilkins; Philadelphi, PA: 2001. pp. 2835–2848. [Google Scholar]
- Wolf RF, Rogers KM, Blewett EL, Fakhari F, Hill CA, Kosanke SD, White GL, Eberle R. A naturally occurring fatal case of Herpesvirus papio 2 pneumonia in an infant baboon (Papio cynocephalus anubis) Compar Med. 2006;45(1):42–46. [PubMed] [Google Scholar]
- Yamada M, Arao Y, Uro F, Nii S. Mechanisms of difference in pathogenicity between two variants of a laboratory strain of herpes simplex virus type 1. Microb Immunol. 1986;30:1259–1270. doi: 10.1111/j.1348-0421.1986.tb03058.x. [DOI] [PubMed] [Google Scholar]