

Summary
Although action potentials are typically generated in the axon initial segment (AIS), the timing and pattern of action potentials is thought to depend on inward current originating in somatodendritic compartments. Using 2-photon imaging, we show that T- and R-type voltage-gated Ca2+ channels are co-localized with Na+ channels in the AIS of dorsal cochlear nucleus interneurons, and that activation of these Ca2+ channels is essential to the generation and timing of action potential bursts known as complex spikes. During complex spikes, where Na+-mediated spikelets fire atop slower depolarizing conductances, selective block of AIS Ca2+ channels delays spike timing and raises spike threshold. Furthermore, AIS Ca2+ channel block can decrease the number of spikelets within a complex spike, and even block single, simple spikes. Similar results were found in cortex and cerebellum. Thus, voltage-gated Ca2+ channels at the site of spike initiation play a key role in generating and shaping spike bursts.
Keywords: action potential, axon initial segment, complex spike, electrophysiology, Ca2+ imaging, Ca2+ channels
Introduction
Periods of high-frequency action potential discharges, termed bursts, are a mechanism for encoding neural information within spike trains. Burst firing is an integral component of a variety of neural computations, including sensory processing, attention, circuit-wide oscillations in activity rates, and synaptic plasticity (reviewed in: Krahe and Gabbiani, 2004; Lisman, 1997). Action potentials within bursts often fire atop long-lasting depolarizations. Present evidence suggests that these long depolarizations are generated exclusively by intrinsic and synaptic conductances in somatodendritic compartments. These slow waveforms then trigger action potential generation in the proximal part of the axon, the axon initial segment (AIS) (Bean, 2007).
The AIS is a specialized cellular compartment that has been identified as the site of action potential initiation in a wide variety of cells (Coombs et al., 1957; Goldberg et al., 2008; Khaliq and Raman, 2006; Mainen et al., 1995; Martina et al., 2000; Palmer and Stuart, 2006; Shu et al., 2007; Stuart et al., 1997). It is characterized by a high density of Na+ channels anchored to a thick cytoskeleton network (Catterall, 1981; Kole et al., 2008; Palay et al., 1968; Wollner and Catterall, 1986). Recent studies indicate that the AIS is also enriched with K+ channels, and that the availability of Na+ and K+ conductances in the AIS depends on recent activity (Goldberg et al., 2008; Kole et al., 2007). While the presence of these channels clearly facilitates generation of individual fast spikes in axons, it remains unclear what ionic mechanisms exist that allow somatically triggered spike bursts to invade axons and lead to a reliable and potent cluster of synaptic signals.
We examined initial segment Ca2+ signals in cartwheel cells, Purkinje-like interneurons found in the dorsal cochlear nucleus, a cerebellum-like auditory brainstem nucleus (Oertel and Young, 2004). A particular advantage of cartwheel cells as a model preparation is that their local, compact axonal arbor allows one to image Ca2+ in all compartments, from dendritic spine to axonal bouton. Cartwheel cells fire both Na+-mediated simple spikes and complex spikes, a form of burst firing where Na+-dependent action potentials termed spikelets ride on slower, Ca2+ depolarizations (Kim and Trussell, 2007; Molitor and Manis, 2003; Schmolesky et al., 2002). Spike timing in cartwheel cells plays a key role in triggering synaptic plasticity (Tzounopoulos et al., 2004). Their complex spikes can be evoked by somatic depolarization, allowing a high level of experimental control over complex spike patterns. Using Ca2+ and Na+ imaging, we found that T- and R-type voltage-gated Ca2+ channels were present in the AIS. Ca2+ channels were co-distributed with Na+ channels, and served to boost membrane depolarization over the entire time course of a complex spike. Moreover, activation of AIS Ca2+ channels determined both the probability of spiking and spike timing. Similar results were found in cortical layer 5b pyramidal cells and cerebellar Purkinje cells, suggesting that this is a common feature of bursting cells. Thus, these data support a model in which AIS Ca2+ currents modify somatodendritic excitatory signals and thereby provide the final determination of the properties of a spike burst.
Results
Na+ and Ca2+ signals in cartwheel cells
We performed whole-cell recordings from cartwheel cells in mouse dorsal cochlear nucleus and assayed spike-evoked Na+ and Ca2+ transients in various cellular compartments using 2-photon laser scanning microscopy. Previous studies show that cartwheel cells have a typical AIS, with an electron dense plasma membrane undercoating, fascicles of microtubules, and a myelin sheath that does not form until 30–35 μm from the hillock (Wouterlood and Mugnaini, 1984). A hallmark of the AIS in other cells is a high density of Na+ channels (Catterall, 1981; Wollner and Catterall, 1986). Studies in pyramidal and Purkinje neurons utilizing low-affinity Na+ dyes have shown that spike-triggered Na+ transients are largest in the AIS, with little to no signal observed in other cellular compartments (Kole et al., 2008; Lasser-Ross and Ross, 1992). Here, we recorded from cartwheel cells with patch pipettes loaded with the volume marker Alexa 594 and the Na+ indicator CoroNa. Cell morphology was assessed by visualizing Alexa 594 after the two dyes filled the neuron (see Methods; Fig. 1A). Linescans were made in various cellular structures, and Na+ transients were measured in response to a train of 12 simple spikes at 100 Hz (Fig. 1B). Large Na+ signals were observed in the first 25–30 μm of axon, consistent with the presence of a high density of Na+ channels in the AIS. Little to no Na+ signal was detected in the soma, dendrites, or axonal boutons (Fig. 1C–D). Because the complex spike of cartwheel cells is driven by a Ca2+ dependent voltage waveform (Kim and Trussell, 2007; Molitor and Manis, 2003; Schmolesky et al., 2002), we next asked if voltage-gated Ca2+ channels might also concentrate in the AIS. To test this, we replaced CoroNa with the Ca2+ indicator Fluo-5F (Fig. 2). Trains of simple spikes evoked Ca2+ transients in cartwheel cell dendrites and axonal boutons, consistent with previous results (Roberts et al., 2008). Surprisingly, Ca2+ transients in the proximal axon were as large as those observed in boutons and dendrites, and appeared to be strictly confined to the AIS (peak train ΔG/R, AIS: 6.16 ± 0.52%, n = 26 sites, 7 cells; bouton: 6.07 ± 0.85%, n = 8 boutons, 4 cells; dendrite: 6.50 ± 0.33%, n = 16 sites, 6 cells). Similar relationships were observed when only single spikes were used to evoke Ca2+ transients (Fig. 2D; peak single spike ΔG/R, AIS: 1.86 ± 0.16%, bouton: 1.60 ± 0.39%, dendrite: 1.00 ± 0.38%). ΔG/R values in the AIS correspond to [Ca2+] elevations of 1.1 to 3.6 μM for single spikes and trains, respectively.
Fig. 1. Na+ transients in cartwheel cells.

(A) Maximum intensity montage of cartwheel cell. Arrowheads correspond to quantification in (C–D). Black: soma, red: axonal shaft, cyan: boutons, orange: dendrite. Red box: site of linescan highlighted in (B).
(B) Top: somatic current drives 12 simple spikes at 100 Hz. Middle: Na+ in initial segment visualized with CoroNa, morphology visualized with Alexa 594. Bottom: Na+ transients (grey) are computed as change in green fluorescence (G; CoroNa) over red (R; Alexa). Decay was fit with single exponential (black).
(C) Maximal ΔG/R from 100 Hz train at points shown for cell in (A).
(D) Summary ΔG/R across 4 cells (3–7 linescans/bin). Bars are SEM.
Fig. 2. Ca2+ transients in cartwheel cells.

(A) Maximum intensity projection of cartwheel local axonal field. Arrowheads correspond to quantification in (C–D). Black: soma, red: axonal shaft, cyan: boutons. Red box: site of linescan highlighted in (B).
(B) Top: somatic current drives 6 simple spikes at 50 Hz. Middle: Ca2+ in initial segment visualized with Fluo-5F, morphology visualized with Alexa 594. Bottom: Ca2+ transients (grey) are computed as change in green fluorescence (G; Fluo-5F) over red (R; Alexa). Decay was fit with single exponential (black).
(C) Maximal ΔG/R from 50 Hz train for cell in (A) at points along axon.
(D) Summary ΔG/R across 6 cells (4–11 linescans/bin). Closed circles: 50 Hz train, open: single spikes. Linescans were performed through distal dendrite for comparison. Bars are SEM.
To determine if Na+ and Ca2+ signals were co-distributed in the AIS, we performed simultaneous Na+ and Ca2+ imaging in mice expressing GFP in cartwheel cells (Fig. 3A, Oliva et al., 2000; Roberts et al., 2008). Cells with intact axons were identified using GFP and patched with an internal solution containing CoroNa and a red Ca2+ indicator, X-rhod-5F. A linescan site was selected while imaging GFP using 970 nm light, and then the laser was re-tuned to 770 nm. Both CoroNa and X-rhod-5F were easily excited at this wavelength, but GFP was not (see Methods). Thus, we were able to separately assay morphology or Na+ on the same green channel by changing the excitation wavelength. Trains of 12 simple spikes at 100 Hz elicited both Ca2+ and Na+ transients (Fig. 3B). Linescans were made in the soma, dendrite, and along the first 40 μm of the axon (n = 7 cells, 4–11 linescans/5 μm bin). When CoroNa and X-rhod-5F signals were scaled to their respective maxima, we found that Ca2+ and Na+ signals covaried >10 μm from the axon hillock (Fig. 3C). Further, point by point comparisons of Na+ and Ca2+ signals across all axonal imaging sites were correlated positively (Fig. 3D; n = 39 sites, 4 cells, R2 = 0.4, p < 0.0001, linear regression). As in experiments examining Ca2+ or Na+ alone, Ca2+ transients in the dendrites were as large as those found in the AIS, but Na+ signals were near zero (Fig. 3C, n = 7 sites), suggesting that the indicators used were selective for their respective ion.
Fig. 3. Simultaneous Na+ and Ca2+ imaging in GFP+ cartwheel cells.

(A) Maximum intensity projection of two GFP+ cartwheel cells. Red arrowheads trace axons.
(B) Top: somatic current drives 12 simple spikes at 100 Hz. Bottom: example of simultaneous Ca2+ (X-rhod-5F) and Na+ (CoroNa) transients 17 μm from axon hillock. Decay was fit with single exponential (black).
(C) Na+ (CoroNa, open circles) and Ca2+ (X-rhod-5F, closed circles) transients in soma (black), axon (red) and dendrites (orange). Bars are SEM.
(D) Na+ transient amplitude vs. Ca2+ transient amplitude at each imaging location in axon. n = 39 sites from 4 cells.
While Ca2+ and Na+ signals co-varied in the distal AIS, Ca2+ transients were larger within the first 10 μm of the axon and in the soma (Fig. 3C). This could reflect a higher relative density of Ca2+ channels in the early portion of the AIS, or be due to diffusion of either Ca2+ or Ca2+-bound dye from the somatodendritic domain. To test the latter, we isolated the Ca2+-mediated component of complex spikes by blocking Na+ channels with tetrodotoxin (TTX, Fig. 4). The resulting depolarizations yield large somatodendritic Ca2+ transients (Molitor and Manis, 2003). Under these conditions, Ca2+ signals were observed only within the first 10 μm of axon, and their amplitude fell exponentially with distance from the hillock (Fig. 4B–D, length constant: 3.9 ± 0.2 μm, n = 5 cells), consistent with diffusion of Ca2+ or Ca2+-bound dye, or passive depolarization of the most proximal part of the AIS that in turn opened voltage-gated Ca2+ channels. These data also suggest that the distal AIS was electrically isolated from the somatodendritic domain, and that Ca2+ channels in this region required local depolarization through Na+ channels for activation.
Fig. 4. . AIS Ca2+ requires active depolarization of the AIS.

(A) Maximum intensity projection of cartwheel cell detailing axon initial segment. Arrowheads correspond to Ca2+ transients and peak ΔG/R amplitudes in (B) and (C), respectively.
(B) Ca2+ transients (middle) are aligned to the max dV/dt of initial underlying complex spike waveform isolated in 500 nM TTX (top). ΔG/R at points along axon (bottom). Colors correspond to traces above and sites in (A). Grey line: single exponential fit. Experiments performed in 2.4 mM external Ca2+.
(C) Summary of decay of peak Ca2+ transient with distance from axon hillock (n = 5 cells).
(D) Space constant of Ca2+ decay. Bars are SEM.
The AIS contains T- and R-type Ca2+ channels
To identify the Ca2+ channel classes mediating Ca2+ influx in the AIS, we examined Ca2+ transients resulting from trains of simple spikes in the presence of various Ca2+ channel antagonists (Fig. 4). Linescans were made 15.7 ± 0.2 μm from the axon hillock to minimize non-axonal Ca2+ sources (see Fig. 4). Ca2+ transients were reduced to 41.9 ± 2.6% of baseline levels by 3 μM mibefradil, a T-type Ca2+ channel antagonist (Fig. 5A, E; n = 5 cells, p < 0.0001, one sample t-test). Transients were further reduced by the R-type blocker SNX-482 (Fig. 5B, E; 500 nM; SNX-482 alone: 79.7 ± 3.3%, n = 3, p < 0.05; SNX-482 + mibefradil: 11.0 ± 3.9%, n = 4, p < 0.001, one sample t-test). Low concentrations of Ni2+ (50 μM), specific for R- and T-type channels (Kampa et al., 2006; Magee and Johnston, 1995), reduced transients to 44.3 ± 5.8% of baseline (n = 4, p < 0.01, one sample t-test). L- and N-type blockers had no effect on AIS Ca2+ (10 μM nifedipine: 100.4 ± 3.9%, n = 5, p = 0.9; 3 μM ω-conotoxin-GVIA: 100.3 ± 2.2%, n = 4, p = 0.9, one sample t-test). ω-agatoxin-TK (150 nM), a potent P/Q-channel blocker, largely blocked Ca2+ transients in boutons (Fig. 5C, E; 10.6 ± 3.2%, n = 6 boutons, 4 cells, p < 0.0001, one sample t-test). AIS Ca2+ transients were larger in agatoxin (115.2 ± 3.2%, n = 5 cells, p < 0.01, one sample t-test) and spike decay was slightly slower, probably reflecting indirect block of BK channels that repolarize spikes (Berkefeld et al., 2006). No alterations in spike shape were observed with other antagonists. Similar reductions in Ca2+ signals were observed when only one simple spike was used (% of control signals: mibefradil: 39.2 ± 5.7%, n = 5, p < 0.001; SNX-482: 76.4 ± 6.2%, n = 3, p = 0.06; SNX-482 + mibefradil: 11.4 ± 2.1%, n = 4, p < 0.001; Ni2+: 42.8 ± 7.3%, n = 4, p <0.01; nifedipine: 99.1 ± 2.5%, n = 5, p = 0.7; ω-conotoxin-GVIA: 103 ± 7.5%, n = 4, p = 0.7; ω-agatoxin-TK: 126.6 ± 13.9%, n = 5, p = 0.12, one sample t-test). Thus, AIS Ca2+ influx was mediated by a mix of T- and R-type Ca2+ channels, and one spike was sufficient to drive Ca2+ influx.
Fig. 5. AIS contains T- and R-type Ca2+ channels.

(A) Trains of simple spikes (top) drive Ca2+ transients in AIS (bottom), measured 15 μm from hillock. Dark grey: control. Light grey: mibefradil in bath. Black: fits to peak.
(B) Mibefradil + SNX-482 largely abolish Ca2+ transient.
(C) ω-agatoxin reduces bouton Ca2+ transient (bottom), but not AIS Ca2+ (middle). Shades in (B–C) as in (A).
(D) T-channel currents (IT, middle) isolated by voltage steps from −100 to −60 mV (top) produce a Ca2+ transient in AIS (bottom).
(E) Summary of Ca2+ transient amplitudes in the presence of various voltage-gated Ca2+ channel antagonists, normalized to control levels. Asterisk: p < 0.05, one sample t-test. Bars are SEM.
To confirm the presence of T-type Ca2+ channels in the AIS, we made voltage-clamp recordings and isolated T-channel currents with voltage steps from −100 to −60 mV while imaging AIS Ca2+ (Fig. 5D). Ca2+ signals rose rapidly in response to a transient T-channel current, and then continued to rise more slowly for the duration of the voltage step (IT transient: −113 ± 30 pA, τactivation: 1.85 ± 0.20 ms, peak ΔG/R: 3.6 ± 0.8%, n = 5 cells), suggesting that T-channels were tonically active at −60 mV, though the tonic current was masked by outward leak currents.
Impact of AIS Ca2+ on complex spikes
The fact that subtle changes in spike repolarization in the presence of ω-agatoxin altered Ca2+ levels in the AIS suggests that the slow depolarization underlying complex spikes could significantly influence AIS Ca2+ levels during bursts. The depolarizing boost provided by this Ca2+ could in turn influence the timing or generation of spikelets riding atop the slow depolarization. To determine whether Ca2+ channels were activated during the entire complex spike, or only during strong, spikelet-mediated depolarizations, we compared the amount of Ca2+ observed in the AIS during a complex spike to what would be expected from a burst of simple spikes at the same timing. In presynaptic boutons, invading action potentials typically drive uniform increases in local Ca2+, and Ca2+ transients resulting from bursts of action potentials can be predicted reliably by the linear sum of individual events offset by their relative timing (Brenowitz and Regehr, 2007; Koester and Johnston, 2005; Koester and Sakmann, 2000; Roberts et al., 2008; Bischofberger et al., 2002; Geiger and Jonas, 2000). We found that for trains of simple spikes (6 at 50 Hz), this relationship held true both in cartwheel cell boutons (Fig. 6A, E; observed/expected Ca2+ transient: 1.02 ± 0.02, n = 8 boutons, 4 cells, p = 0.35, one sample t-test), and in the AIS [Fig. 6B, E; 1.01 ± 0.01, n = 26 sites (0–22 μm from hillock), 7 cells, p = 0.33, one sample t-test]; however, complex spikes resulted in larger AIS Ca2+ transients than would be expected by the simple linear sum of Ca2+ from its constituent spikelets (Fig. 6C, E; 1.35 ± 0.05, n = 26 sites, p < 0.0001, one sample t-test). The presence of a larger-than-predicted Ca2+ signal indicates that Ca2+ influx must have occurred both during and between spikes. The latter probably reflected channel activity associated with the slow depolarization that drives spikelet generation. This supralinear Ca2+ level was relatively constant throughout the AIS (Fig. 6E), but returned to unity in boutons (Fig. 6D–E; 0.99 ± 0.01, n = 8, p = 0.35, one sample t-test), suggesting that all spikelets were transformed into separate propagating action potentials(Roberts et al., 2008). Observed/predicted Ca2+ values were not calculated for axonal sites between the AIS and boutons due to the low signal/noise of single spike responses in that area. Complex spikes also evoked supralinear Ca2+ transients in cartwheel cell dendrites (Fig. 6E train: 0.98 ± 0.01, p = 0.06; complex spike: 1.32 ± 0.04, p < 0.0001, n = 16 sites, 6 cells, one sample t-test), consistent with findings from a study of burst firing in cortical pyramidal cells (Kampa and Stuart, 2006).
Fig. 6. Complex spike-triggered axonal Ca2+ transients.
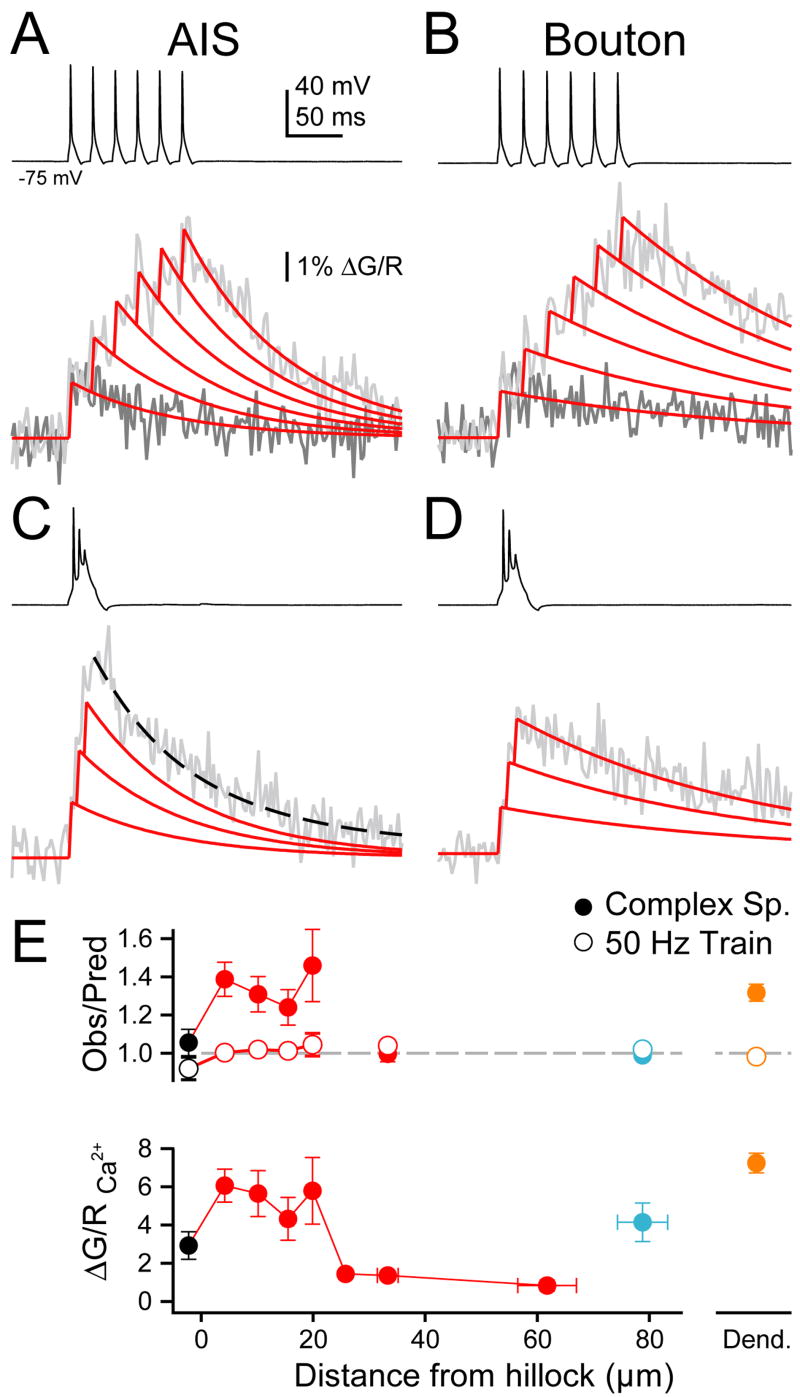
(A) Train (black) and corresponding Ca2+ imaging in AIS. Dark grey, Ca2+ from single spike. Light grey, Ca2+ from train. Red, linear sum of fit of single spike transient, temporally offset to match train timing. Scale applies to (A–D).
(B) Train in Bouton. Color code as in (A).
(C) Complex spike in AIS. Red solid: expected fit. Black dash: actual fit.
(D) Complex spike in bouton.
(E) Top, observed Ca2+ amplitude divided by predicted amplitude from linear sum for train and complex spike. Bottom, maximum ΔG/R values for complex spikes along axon. Color code as in Fig. 1. Bars are SEM.
These results indicate that the depolarization underlying a complex spike is sufficient to activate AIS Ca2+ channels. To determine the role of these channels during complex spikes, we blocked AIS T-type channels with a local puff of mibefradil immediately before eliciting a complex spike with 2–3 spikelets. Since complex spike dynamics change over the course of a whole-cell recording (Kim and Trussell, 2007), control complex spikes were interleaved with complex spikes paired with mibefradil puffs (mib-CS; 2s between complex spikes, then a 45s delay to allow for mibefradil wash-out). SNX-482 was not included since it does not wash out over these time scales. Mibefradil was dissolved in a HEPES-based vehicle and loaded into one side of a theta (double-barrel) pipette. The other side was loaded only with the vehicle, and the assembly was connected to a Picospritzer allowing independent control of either barrel of the pipette. Neurons with axons well separated from their dendritic field were selected and the puff pipette was placed within 2 μm of the axon, 10–12 μm from the hillock (Fig. 7A).
Fig. 7. AIS T-type channels shape complex spikes.

(A) Maximum intensity projection of cartwheel cell showing placement of puffer pipette near AIS.
(B) Complex spikes evoked via somatic depolarization paired with mibefradil puffs onto AIS.
(C) Vehicle puffs onto AIS (left) and mibefradil puffs onto proximal dendrite (right) did not alter complex spikes. Insets in (B–C): schematic cartwheel cell detailing pipette placement.
(D) Phase plane plot of complex spikes shown in (B). Arrow shows spike threshold for first spikelet. Numbers correspond to spikelet sequence in complex spike. Color code as in (B).
(E) Relative timing of spike peak for all conditions (n = 5 cells). AIS mibefradil puffs delay spike timing.
(F) Relative threshold Vm for spikelets 1 and 2 of complex spike (n = 5). Threshold is more depolarized with AIS mibefradil puffs. Asterisk: p < 0.05, ANOVA. Bars are SEM.
We found that T-type channel block principally affected two properties of complex spikes activated by a current step: spikelet timing and spikelet threshold. We calculated changes by comparing relative spikelet timing or threshold membrane voltage across pairs of control and mib-CSs (>10 trials/cell, n = 5 cells). Spikelet peaks were delayed by mibefradil, with later spikelets exhibiting greater delays (Fig. 7B, E; 1st spikelet: 1.45 ± 0.30 ms, 2nd: 2.36 ± 0.50, 3rd: 3.30 ± 0.66; p < 0.001, ANOVA vs. vehicle puff). These changes in spikelet timing and inter-spikelet interval resulted from changes in spikelet threshold, as mibefradil increased the amount of depolarization required to reach threshold (Fig. 7D, F; 1st spikelet: 4.46 ± 1.12 mV, 2nd: 2.48 ± 1.62; p < 0.001) and delayed threshold onset (Supplemental Fig. 1; 1st spikelet: 1.44 ± 0.29 ms, 2nd: 1.92 ± 0.41; p < 0.001). To test whether these changes were due to specific effects of mibefradil on the AIS, we moved the puff pipette next to a proximal dendrite, ~10 μm from its somatic origin. Mibefradil puffs onto the proximal dendrite had no effect on complex spikes (Fig. 7C, E–F; timing, 1st spikelet: −0.01 ± 0.04 ms, 2nd: −0.05 ± 0.02, 3rd: −0.15 ± 0.003, p > 0.05; threshold Vm, 1st spikelet: 0.03 ± 0.03 mV, 2nd: 0.18 ± 0.13, p > 0.05, ANOVA). Therefore, the changes in spikelet dynamics resulted from a block of AIS T-type channels.
Mibefradil puffs moved the tissue close to the pipette, making it difficult to simultaneously image Ca2+ transients in the AIS. We therefore employed a complementary technique to determine the extent to which AIS Ca2+ signaling was reduced with local Ca2+ channel blockade. A microelectrode containing Ni2+ was placed next to the axon. Control complex spikes were then interleaved with complex spikes paired with Ni2+ iontophoretic pulses. The iontophoretic current was adjusted to approximate the changes in spikelet timing observed with mibefradil. As in mibefradil puff experiments, both spikelet timing and spikelet threshold were altered (Fig. 8A, C–D; timing, 1st spikelet: 1.63 ± 0.25 ms, 2nd: 2.84 ± 0.49, 3rd: 3.81 ± 0.95; threshold Vm, 1st spikelet: 4.02 ± 0.88 mV, 2nd: 3.40 ± 0.51; n = 9–11 spikelets/group, 11 cells, p < 0.001 for all groups, one sample t-test). We imaged the axon directly adjacent to the iontophoretic pipette and found that complex spike-evoked Ca2+ transients were reduced to 51.2 ± 2.4% of baseline levels in Ni2+ (n = 9, p < 0.0001, one sample t-test); however, it was unclear what proportion of this decrease was due to the increase in inter-spike intervals and concomitant reduction in Ca2+ transient summation (Fig. 8B, E). To evaluate changes in Ca2+ signaling independent of changes in spike timing, we replaced complex spikes with trains of 6 simple spikes at 50 Hz while maintaining the timing and intensity of the iontophoretic pulse. Train-evoked Ca2+ transients were reduced to 53.0 ± 4.1% of baseline in the AIS (Fig. 8B, E; n = 10, p < 0.0001, one sample t-test), suggesting that reductions observed with complex spikes were primarily due to Ca2+ channel block. The effective concentration of Ni2+ during these experiments was under 50 μM, as local block reduced Ca2+ transients less than bath application of 50 μM Ni2+ (Fig. 5E). We next performed linescans through the soma and a proximal dendrite and found that Ca2+ signals were not altered by the application of Ni2+ to the AIS (Fig. 8B, E; soma: 100.4 ± 1.9% of control, n = 10, p = 0.8; dendrite: 100.7 ± 1.4%, n = 10, p = 0.6, one sample t-test). Combined with mibefradil puff experiments, these results suggest that Ca2+ channel block was localized to the AIS, and that the changes in spike timing and threshold were due to a ~50% reduction in local Ca2+.
Fig. 8. AIS Ni2+ iontophoresis alters complex spikes.

(A) Complex spikes evoked via somatic depolarization paired with Ni2+ iontophoresis onto AIS. Black: control. Grey: Ni2+. Inset: schematic cartwheel cell detailing pipette placement.
(B) 50 Hz simple spike trains and corresponding Ca2+ transients in AIS, soma, and proximal dendrite in control conditions (black) and when paired with AIS Ni2+ iontophoretic injection (grey).
(C) Relative timing of spike peak for all conditions (n = 11 cells). Ni2+ delays spike timing.
(D) Relative threshold Vm for spikelets 1 and 2 of complex spike (n = 11). Threshold is more depolarized with Ni2+.
(E) Normalized ΔG/R for Ni2+ iontophoresis compared to control conditions (n = 10). Relative amplitudes are shown for complex spikes (CS) in the AIS, and trains in the AIS, soma, and proximal dendrite. Asterisk: p < 0.05, one sample t-test. Bars are SEM.
AIS Ca2+ current regulates spike generation
Since spike threshold is more depolarized when AIS T-type channels are blocked, it is likely that T-type channel activation can gate spike generation from what would otherwise be a subthreshold stimulus. In a subset of Ni2+ iontophoretic experiments, we substituted EPSP-like somatic current injections for square waves to better mimic parallel fiber activity. EPSP-like stimuli were based on recorded parallel fiber EPSPs, and allowed us to depolarize the AIS in a controlled, highly repeatable fashion (Roberts et al., 2008). Stimulus amplitude was first adjusted to just suprathreshold levels for a single, simple spike. When paired with Ni2+, spikes were blocked (Fig. 9A–B; control spikes/stim: 1.00 ± 0.00, Ni2+: 0.06 ± 0.06; p < 0.03, Wilcoxon signed rank test, n = 6 cells). We then increased the stimulus amplitude to elicit complex spikes with 2–3 spikelets. Again, Ni2+ reduced the number of spikelets evoked with each stimulus (Fig. 9A, C; control: 2.11 ± 0.12, Ni2+: 1.24 ± 0.13; p < 0.01, Wilcoxon signed rank test, n = 9 cells).
Fig. 9. AIS T-channel block reduces the probability of spiking.

(A) EPSP-like injected waveform (top) of variable amplitude drives 1, 2, or 3 spikes (bottom, black). Interleaved trials paired with Ni2+ iontophoresis show reduced numbers of spikes (grey).
(B–C) Summary for experiments where single simple spikes (B) or complex spikes (C) were evoked. Dots are averages from 9–16 trails/cell (n = 6 simple, 9 complex). Lines connect conditions within a cell.
(D) View of area highlighted by dashed box in (A). “Ctrl-Ni2+” is a subtraction of the two traces.
(E) Lines are average Ctrl-Ni2+ traces for each cell where single spikes were examined (n = 6, see Methods). Grey bar: Average 2×standard deviation (2×SD) of baseline. Arrow in (D–E): stimulus onset.
(F) Time Ctrl-Ni2+ trace emerged from noise (2×SD), relative to stimulus onset (n = 6). Bars are SEM.
To determine how quickly Ca2+ channels were activated with these stimuli, we examined the timecourse of subthreshold depolarization in control and Ni2+-paired conditions where single spikes were blocked by Ni2+. The average difference between conditions was calculated for each cell (Ctrl-Ni2+: Fig. 9D). Differences between control and Ni2+-paired depolarizations were evident within 0.77 ± 0.07 ms of stimulus onset (Fig. 9D–F; n = 6 cells). By the time spike threshold was reached in control conditions, Ni2+-paired depolarizations were reduced 4.8 ± 0.4 mV (measured for each sweep; n = 46 sweeps, 6 cells). Therefore, AIS Ca2+ channels not only influence spikelet timing within complex spikes, they can also provide depolarization necessary for spike generation, and do so within 1 ms of stimulus onset.
Does AIS Ca2+ drive Ca2+-activated K+ channels?
In addition to contributing to local depolarization, Ca2+ can activate big and small conductance K+ channels, BK and SK. While both SK and BK are expressed in cartwheel cells (Kim and Trussell, 2007), it is unclear whether they co-localize with Ca2+ channels in the AIS. To test this, we first examined aspects of complex spike waveforms that could change if Ca2+ channel block by mibefradil or Ni2+ were also indirectly blocking BK/SK signaling. Block of BK channels by bath application of iberiotoxin (IbTX) slows spike repolarization rates in cartwheel cells (Kim and Trussell, 2007); however, repolarization rates were not altered by AIS Ca2+ block with mibefradil or Ni2+ (Supplemental Fig. 1). During complex spikes, bath application of IbTX or the SK antagonist apamin depolarizes inter-spikelet repolarization potentials (Kim and Trussell, 2007). As with spike repolarization rates, inter-spikelet repolarization was not affected by AIS Ca2+ channel block (Supplemental Fig. 1); however, this could reflect a balance between reductions in both Ca2+ influx and K+ efflux with the block of Ca2+ and BK/SK channels, resulting in no net change in repolarization potential. Therefore, we repeated local Ni2+ iontophoresis experiments, but blocked SK and BK by bath application of 100 nM apamin and 100 nM IbTX, respectively. If AIS Ca2+ activated BK and SK channels, these conditions should reveal the isolated contribution of reduced Ca2+ influx to inter-spikelet repolarization potential. Bath application of apamin and IbTX produced characteristic changes to action potentials evoked with long step somatic current injections (Supplemental Fig. 2A, C). Complex spikes with 2–3 spikelets could still be evoked with short step currents, and when paired with AIS Ni2+ iontophoresis, spikelet timing was delayed (Supplemental Fig. 2B, D–E; e.g., 1st spikelet delay: 1.80 ± 0.30 ms in apamin, n = 8; 2.14 ± 0.41 ms in IbTX, n = 7; p > 0.2 for all conditions vs. Ni2+ iontophoresis alone, unpaired t-test), but inter-spikelet repolarization potentials were not altered (Supplemental Fig. 2B, D, F; e.g., relative repolarization following 1st spikelet: 0.11 ± 0.19 mV in apamin, n = 8; 0.57 ± 0.26 mV in IbTX, n = 7; mV, n = 11 for Ni2+ iontophoresis alone; p > 0.3 vs. apamin and IbTX, unpaired t-test). Thus, AIS Ca2+ did not appear to drive local BK or SK activity in a way that significantly altered simple or complex spike initiation.
Bursts in other bursting cell types are also shaped by AIS Ca2+ channels
Spike bursts are common to many cell types. To determine if AIS Ca2+ channels have a general role in regulating bursts, we replicated key aspects of this study in two other cell types from other, non-auditory brain regions: primary somatosensory cortex layer 5b pyramidal cells, and cerebellar Purkinje cells (Fig. 10A). Similar to cartwheel cells, both pyramidal and Purkinje cell action potentials initiate in the axon (Davie et al., 2008; Khaliq and Raman, 2006; Palmer and Stuart, 2006), and spike bursts drive elevations in dendritic Ca2+ (Kampa and Stuart, 2006; Sugimori and Llinas, 1990). Ca2+ transients in response to trains of action potentials (6 × 50 Hz) were observed in the AIS of both cells (Fig. 10B, Pyramidal: ΔG/R: 5.39 ± 0.99%, recorded 15.2 ± 1.4 μm from axon hillock, n = 6 cells; Purkinje: ΔG/R: 4.89 ± 0.73%, 16.3 ± 1.8 μm from hillock, n = 5 cells). These Ca2+ transients could be reduced to 53 ± 6% and 53 ± 5% of baseline when trains were paired with local Ni2+ iontophoresis in pyramidal and Purkinje cells, respectively (n = 5 for both cell types). As in cartwheel cells, pairing Ni2+ iontophoresis with spike bursts (2–3 spikes/burst) raised spike threshold and delayed spike timing (Fig. 10C–E; Pyramidal cell: relative spike timing delay, 1st spike: 2.07 ± 0.39 ms, p < 0.01; 2nd spike: 3.48 ± 0.60 ms, p < 0.001; relative threshold depolarization, 1st spike: 2.86 ± 0.37 mV, p < 0.001; 2nd spike: 2.63 ± 0.41 mV, p < 0.001, n = 8 cells; Purkinje cell: relative spike timing delay, 1st spike: 0.86 ± 0.12 ms, p < 0.001; 2nd spike: 1.49 ± 0.28 ms, p < 0.01; relative threshold depolarization, 1st spike: 2.77 ± 0.80 mV, p < 0.05; 2nd spike: 3.26 ± 1.08 mV, p < 0.05, n = 6 cells, one sample t-test). In pyramidal cells, we found that this effect was specific to AIS Ca2+ channel block; Ni2+ iontophoresis onto neighboring basal dendrites had no effect on the timing of spikes within bursts (relative spike timing, 1st spike: 0.05 ± 0.10 ms; 2nd spike: −0.15 ± 0.17 ms; relative threshold, 1st spike: −0.15 ± 0.16 mV, 2nd spike −0.18 ± 0.19 mV; p > 0.4 for all conditions, one sample t-test; dendrites were 20 ± 4 μm from axon, n = 4 dendrites, 2 cells). Thus, initial segment Ca2+ channels have similar roles in both excitatory and inhibitory cell types found in diverse brain regions.
Fig. 10. AIS Ca2+ channels alter spike bursts in pyramidal and Purkinje cells.
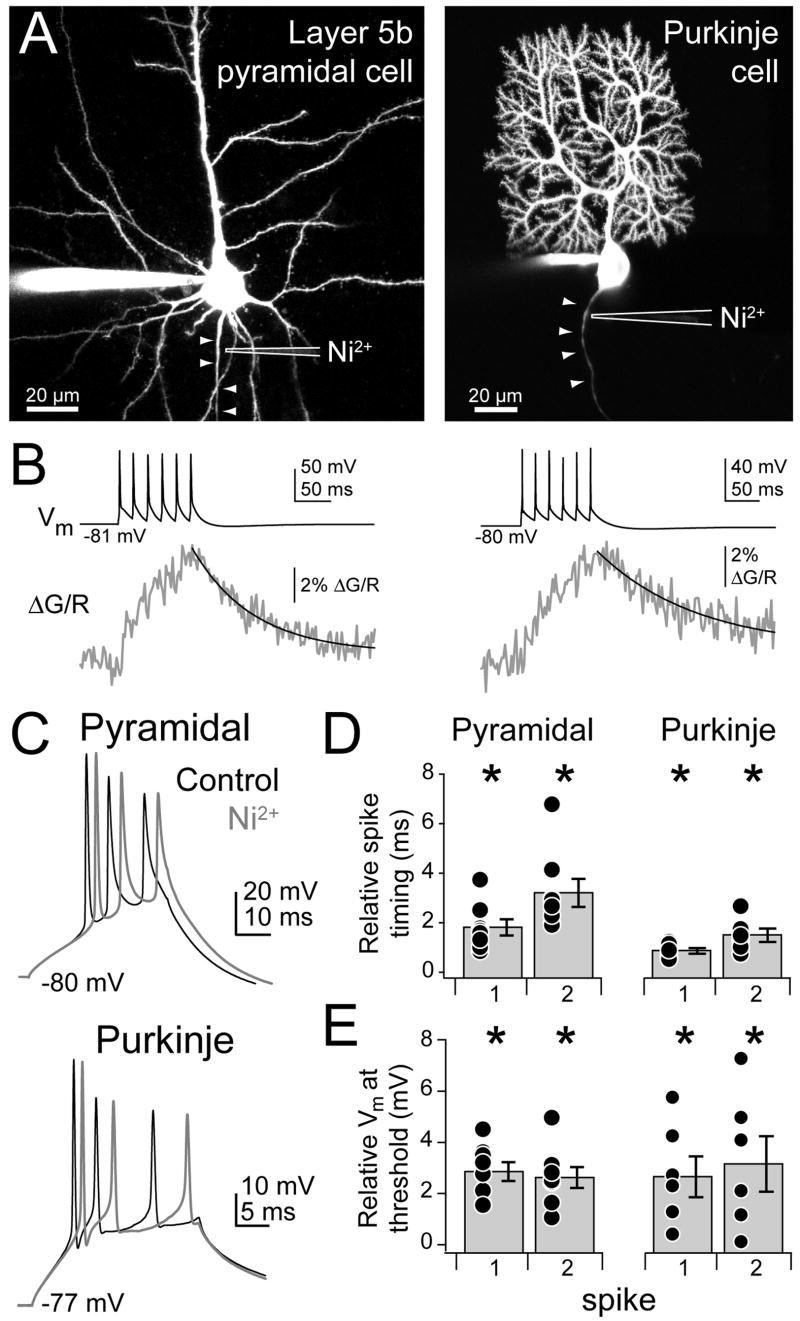
(A) Maximum intensity projections of cortical layer 5b pyramidal cell (left) and cerebellar Purkinje cell (right). Arrowheads follow axon. Iontophoretic pipette containing Ni2+ is outlined in white.
(B) Train of spikes (top) drives Ca2+ transients in AIS (bottom), measured 15 μm from hillock for pyramidal (left) and Purkinje cells (right).
(C) Spike bursts evoked via somatic depolarization paired with Ni2+ iontophoresis onto AIS in both cell types. Black: control. Grey: Ni2+.
(D) Relative timing of spike peak. Ni2+ delays spike timing.
(E) Relative threshold Vm. Threshold is more depolarized with Ni2+. Asterisk: p < 0.05, one sample t-test. n = 8 pyramidal cells, 6 Purkinje cells. Bars are SEM.
Discussion
These results show that voltage-gated Ca2+ channels localized to the initial segment have a substantial impact on the timing and generation of action potentials. We estimate that Ca2+ transients during activity reflected a 1.1 to 3.2 μM increase in local Ca2+ with single and complex spikes, respectively, and that Ca2+ current contributed substantially to local membrane depolarization. When Ca2+ channels were blocked with T- and R-type channel antagonists, spike timing within bursts was delayed, threshold was elevated, and spike probability dropped. Since Ca2+ levels were reduced by only ~50% while localizing pharmacological effects to the AIS, the overall impact of AIS Ca2+ channels could be larger than that reported here.
This work suggests that the initial segment is the site of action potential initiation in cartwheel cells, as Na+ channels were concentrated there and because manipulations of AIS Ca2+ affect both single and complex spikes. This conclusion is consistent with recent work in cerebellar Purkinje cells indicating that all complex spike spikelets originate in the axon (Davie et al., 2008). Experimental and theoretical data suggest that the AIS—and in some cases, the distal end of the AIS—is the site of action potential generation (Khaliq and Raman, 2006; Mainen et al., 1995; Palmer and Stuart, 2006); therefore, currents present there have the greatest influence on spike initiation. We found Ca2+ transients throughout the AIS, including the distal end (Figs. 2–3). Ca2+ transients during complex spikes exceeded those expected from the simple sum of Ca2+ entry from the same number of similarly timed simple spikes by at least 35%, suggesting that AIS Ca2+ channels were sensitive to subthreshold depolarizations such as the underlying component of complex spikes. This likely underestimates the relative contribution of the underlying complex spike depolarization to total AIS Ca2+, since later spikelets within complex spikes were not as large as the simple spikes used to generate estimates. Thus, subthreshold depolarizations have a significant influence on action potential initiation through activation of AIS Ca2+ channels.
Recent work in cortical pyramidal cells has shown that AIS Na+ channels mediate local subthreshold depolarizations that precede action potential initiation (Kole and Stuart, 2008). Here, we found that blocking Na+ channels with TTX had a profound effect on AIS Ca2+ channel signaling. In TTX, the Ca2+-mediated component of complex spikes could still be observed in the somatodendritic domain, but Ca2+ transient amplitude in the AIS fell exponentially with distance from the hillock (Fig. 4). This suggests that without subthreshold Na+ channel activity, the AIS is electrotonically isolated from the somatodendritic domain. AIS Ca2+ channels therefore may require subthreshold Na+ current for activation, and thus the two may act in synergy to drive membrane potential to action potential threshold. What remains unclear is whether this synergistic action not only promotes generates of spikelets in the AIS but also the conduction of full amplitude, propagating spikes in the axon (Roberts et al., 2008).
Properties of initial segment Ca2+ channels
In cartwheel cells, AIS Ca2+ transients were reduced by mibefradil, Ni2+, and SNX-482, but not by nifedipine, conotoxin, or ω-agatoxin (Figs. 5, 7–8), suggesting that the AIS contains only R- and T-type Ca2+ channels. SNX-482, a selective antagonist of α1E-containing R-type (Cav2.3) channels, blocked ~20% of the local Ca2+ signal. Ca2+ transients were further reduced with bath application of mibefradil, which is most selective for T-type channels (Cav3.X) (McDonough and Bean, 1998), but can also block R-type channels at low concentrations and other voltage gated channel types at higher concentrations (Perez-Reyes, 2003). To confirm the presence of low-voltage activated T-channels, we made voltage steps to only −60 mV, a stimulus which evoked both a transient inward current and a Ca2+ signal in the AIS. Co-application of mibefradil and SNX-482 did not block AIS Ca2+ transients completely. This small, residual Ca2+ transient could arise from several sources, including toxin-insensitive R-type channels (Metz et al., 2005; Tottene et al., 2000) or diffusion of Ca2+ or Ca2+-bound dye from the soma (Fig. 4).
Both bath application of 50 μM Ni2+ and local Ni2+ iontophoresis reduced AIS Ca2+ transients by ~50% in cartwheel cells. Given that similar reductions were observed with Ni2+ iontophoresis in pyramidal and Purkinje cells, and that Ni2+ is relatively specific for T- and R-type Ca2+ channels at this concentration (Magee and Johnston, 1995), these channel subtypes are likely present in the AIS of these other cell types. Direct pharmacological analysis will be required to determine whether other Ca2+ channel subtypes are present in the AIS of pyramidal and Purkinje cells.
A major feature of T-type channels is that while they activate quickly, they inactivate rather slowly, providing an ideal mechanism to exert control over relatively long duration events like spike bursts. By analyzing the kinetics of subthreshold depolarizations in the presence of local Ni2+, we found that Ca2+ channels activated within 1 ms of stimulus onset, and boosted somatically recorded membrane potential ~5 mV by spike onset (Fig. 9). This initial current was likely mediated by either Cav3.1 or 3.2 channels. Activation kinetics of T-channel currents isolated by voltage-clamp steps from −100 to −60 mV were similar to those isolated in HEK cells (Lee et al., 1999). Later current could incorporate Cav3.3 channels, which are slower to activate, and Cav2.3 R-type channels, which require more depolarization to open (Catterall et al., 2005).
The largest effects of AIS Ca2+ channel block were on the timing and threshold of spikes triggered by current steps, which could be due to changes in the rate and amplitude of membrane depolarization, or Ca2+-dependent modifications of local Na+ channels. Calmodulin-regulated Nav1.6 channels are expressed in the AIS (Boiko et al., 2003); however, Ca2+ alters their inactivation rates, not threshold or activation rates (Herzog et al., 2003). AIS Ca2+ did not appear to activate BK or SK channel. BK channels typically co-localize with P/Q channels (Berkefeld et al., 2006), which were not present in the AIS (Fig. 4), and have dramatic effects on spike repolarization rates, which were not altered with local mibefradil or Ni2+ application (Supplemental Fig. 1). SK channels, which help shape inter-spike repolarizations, are often linked to T-type Ca2+ channels (Cueni et al., 2008; Wolfart and Roeper, 2002), but inter-spike repolarization levels were not altered by SK or BK antagonists (Supplemental Fig. 2). Therefore, we suggest that initial segment Ca2+ channels largely provide a depolarizing drive, but it remains unknown whether AIS Ca2+ drives any second messenger pathways, at these or other timescales.
Ca2+ channels are not typically thought to be concentrated in the initial segment, and while immunohistochemistry suggests that T-type channels are expressed in axon, signal levels are too weak to draw a definitive conclusion (McKay et al., 2006). T- and R- channel histology rarely shows labeling beyond the soma and proximal dendrites, suggesting that current antibodies are not sensitive enough to identify channels throughout all neural arbors (Day et al., 1996). In contrast, Ca2+ imaging techniques readily detect Ca2+ influx through pharmacologically identified low-threshold channels throughout dendritic trees (Goldberg et al., 2004), and axonal Ca2+ transients have been shown in a variety of neurons, including cerebellar Purkinje, cortical pyramidal—both in this report and in others—and dorsal root ganglion cells (Callewaert et al., 1996; Luscher et al., 1996; Schiller et al., 1995). Indeed, Ca2+ imaging combined with channel pharmacology might provide a more accurate assessment of Ca2+ channel localization than currently available Ca2+ channel antibodies.
Bath application of R- and T-channel antagonists alters spike threshold in cartwheel cells, reduces the number of spikelets in cartwheel and cerebellar Purkinje cells, and can even prevent complex spike initiation at the onset of step depolarizations (Cavelier et al., 2008; Kim and Trussell, 2007). We obtained similar results while only blocking AIS Ca2+ channels. This suggests that some results previously attributed to somatodendritic Ca2+ channels might be mediated at least in part by Ca2+ channels in the initial segment. For example, cells of the cerebellar nuclei fire characteristic rebound bursts following strong hyperpolarizations, a phenomenon known to require T-type channels (Aizenman and Linden, 2000). AIS T-type channels in close proximity to the spike initiation zone would be ideally suited to drive this rebound firing. Furthermore, while we have shown that AIS Ca2+ is important for other cells that fire bursts (Fig. 10), they could also play important roles in cells that have Ca2+-mediated after depolarizations, or Ca2+ channel-mediated oscillations (Bean, 2007; Cueni et al., 2008; Metz et al., 2005). Careful dissection of AIS and somatodendritic Ca2+ channel contributions would be required to determine if this is the case in these cell types.
Functional implications for bursting cells
An intriguing aspect of these results is that the AIS incorporates low-threshold Ca2+ channels that are selectively recruited from more hyperpolarized membrane potentials. Cartwheel cells operate over a broad range of resting membrane potentials, and can be quiescent or spontaneously active in vitro and in vivo (Davis and Young, 1997; Kim and Trussell, 2007). Complex spikes are evoked preferentially at the onset of long current steps only when the basal membrane potential is well hyperpolarized, presumably reflecting the availability of low threshold T-channels (Kim and Trussell, 2007). Further, T-channel-dependent complex spikes evoked from hyperpolarized membrane potentials tend to have more spikelets with shorter inter-spikelet intervals compared to complex spikes evoked from depolarized levels (Kim and Trussell, 2007). Similarly, bursts in many cell types, including thalamic relay and pyramidal cells, vary based on recent membrane potential and spiking activity (Destexhe et al., 1998; Williams and Stuart, 1999). Thus, Ca2+ channels localized to the initial segment could help encode the recent voltage history of a neuron in the patterns within spike bursts and have a significant impact on information processing and synaptic plasticity (Harris et al., 2001; Krahe and Gabbiani, 2004; Lisman, 1997; Magee and Johnston, 1997).
Experimental procedures
Electrophysiology
All procedures were conducted in accordance with OHSU IACUC guidelines. Following anesthesia, coronal brainstem slices (210 μm) were made from P17-21 ICR (Harlan, Indianapolis, IN) mice or GIN mice expressing GFP in cartwheel cells. GIN mice were phenotyped as described previously (Roberts et al., 2008). Cutting solution contained (in mM): 130 NaCl, 3 KCl, 2.4 CaCl2, 1.3 MgSO4, 1.2 KH2PO4, 20 NaHCO3, 3 Na-HEPES, 10 glucose; bubbled with 5%CO2/95%O2; ~305 mOsm, 33–34 °C. Following cutting, slices were incubated for 1 hr at 33 °C, then at room temperature until recording. Cartwheel cells were identified based on their dendritic morphology and their ability to fire complex spikes in response to somatic depolarization (Wouterlood and Mugnaini, 1984; Zhang and Oertel, 1993). Recordings were made at 33 °C. For recording, CaCl2 was reduced to 1.0 mM and MgSO4 was raised to 2.7 mM to maintain the divalent concentration, unless otherwise noted. This Ca2+ concentration is lower than used previously (Roberts et al., 2008), and may account for more reliable spikelet propagation. DNQX (10 μM), strychnine (500 nM), and SR95531 (20 μM) were added to block synaptic activity. Neurons were visualized with infrared gradient contrast microscopy (Dodt et al., 2002). Patch electrodes (Schott 8250 glass, 2–3 MΩ tip resistance, <10 MΩ series resistance) were filled with an intracellular solution containing (in mM): 113 K-Gluconate, 9 HEPES, 4.5 MgCl2, 0.1 EGTA, 14 Tris2-phosphocreatine, 4 Na2-ATP, 0.3 tris-GTP; ~290 mOsm, pH: 7.22. Internal solution was supplemented with Ca2+- and Na+-sensitive dyes as described below. Dyes were allowed to equilibrate >15 min before linescans were performed. Electrophysiological data were recorded at 50 kHz and filtered at 10 kHz using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA), and acquired with an ITC-18 interface (Instrutech, Port Washington, NY) using custom software written in Igor Pro (Wavemetrics, Lake Oswego, OR; software: DE Feldman). Vm was held <−75 mV with constant current injection. Action potentials were evoked via somatic depolarization (e.g., simple spikes: 1–2 nA for 2 ms), often followed by hyperpolarizing steps to prevent complex spike generation (2–400 pA for 10 ms). Data were corrected for a measured junction potential of 12 mV.
Two-photon imaging
A Ti:sapphire pulsed laser (Chameleon Ultra II; Coherent, Auburn, CA) powered a Prairie Technologies Ultima two-photon imaging system (Middleton, WI). Laser intensity was modulated with a Pockels cell (350–80 LA; Conoptics, Danbury, CT). Epi- and transfluorescence signals were captured through a 60×, 0.9 NA objective and a 1.4 NA oil immersion condenser (Olympus, Melville, NY). Fluorescence was split into red and green channels using dichroic mirrors and band-pass filters (epi: 575 DCXR, HQ525/70, HQ607/45; trans: T560LPXR, ET510/80, ET620/60; Chroma, Rockingham, VT), then focused on multialkali photomultiplier tubes (R9110; Hamamatsu, Bridgewater, NJ). Data were collected in linescan mode (2.2 ms/line, including mirror flyback). Ca2+ and Na+ transient peaks were calculated from exponential fits of the fluorescence decay following stimulus offset. Unless otherwise noted, dendritic linescans were made across processes >30 μm from the center of the soma, where the process width was relatively constant.
Ca2+ imaging
EGTA was omitted from the internal solution and replaced with the Ca2+ sensor Fluo-5F (250 μM, green) and a volume marker Alexa 594 (20 μM, red). The laser was tuned to 810 nm. Data are presented as averages of 20–40 events per site, and expressed as Δ(G/R)/(G/R)max*100, where (G/R)max was the maximal fluorescence in saturating Ca2+ (2 mM; Yasuda et al., 2004). [Ca2+] was estimated assuming a KD of 585 nM for Fluo-5F (Brenowitz and Regehr, 2007).
Na+ imaging
500 μM CoroNa green (stock: 50 mM in DMSO, final [DMSO]: 1%) and 20 μM Alexa 594 were added to the internal, and the laser was tuned to 810 nm. Data are presented as averages of 40–80 events per site, and expressed as Δ(G/R)*100. (G/R)max was not determined since CoroNa cannot be saturated in physiological [Na+] (Meier et al., 2006).
Simultaneous Na+ and Ca2+ imaging
Experiments were performed in GIN mice (all others in ICR mice). EGTA was excluded from the internal and replaced with 500 μM CoroNa green and the red Ca2+ sensor X-rhod-5F (250 μM). Gross cell morphology was mapped before patching with the laser tuned to 970 nm. The laser was re-tuned to 770 nm for Na+/Ca2+ imaging. GFP fluorescence at 770 nm was 4% of its maximum at 970 nm, and undetectable at the laser intensities used for Na+/Ca2+ imaging. Data are presented as ΔF/Fo for each channel, with Fo defined as fluorescence levels 0–400 ms before stimulus onset.
Pharmacology
Ca2+ channel blockers were bath applied (recirculation volume: 20–30 mL). Bovine serum albumin (0.1 mg/mL) was added to the Ringer before introducing drugs to minimize nonspecific peptide binding. Drugs were allowed to equilibrate for >15 min before data collection. Bath concentrations (in μM): 3 mibefradil, 0.15 Ω-agatoxin-TK, 10 nifedipine, 3 ω-conotoxin-GVIA, 0.5 SNX-482, 50 Ni2+, 0.1 apamin, 0.1 iberiotoxin.
Voltage-clamp isolation of T-channel currents
Internal solution contained (in mM): 110 CsMeSO3, 40 HEPES, 1 KCl, 4 NaCl, 4 Mg-ATP, 10 Na-phosphocreatine, 0.4 Na2-GTP, 0.5 Fluo-5F, 0.02 Alexa-594; ~290 mOsm, pH: 7.22, voltages adjusted for 10 mV junction potential. Cartwheel cells were identified based on morphology, and AIS linescans were made 9.4 ± 1.2 μm from the axon hillock. T-channels were activated with voltage steps from −100 to −60 mV. Leak currents were subtracted using the P/4 protocol (Bezanilla and Armstrong, 1977) with +10 mV steps from −80 mV. Experiments were performed in the presence of 500 nM TTX, 20 μM SR95531, 500 nM strychnine, 10 mM DNQX, and 2.4 mM external Ca2+.
Local mibefradil puffing
Internal solution was the same as in Ca2+ imaging experiments. A theta pipette (tip diameter <1 μm) was filled on one side with a vehicle solution plus 10 μM Alexa 594, and on the other side with vehicle plus 100 μM mibefradil. Vehicle solution contained (in mM): 142 NaCl, 3 KCl, 1.0 CaCl2, 2.7 MgSO4, 1.2 KH2PO4, 10 Na-HEPES, 10 glucose; pH: 7.2, ~305 mOsm. Chambers were sealed with epoxy and coupled to a single channel Picospritzer (Pressure System IIe, Toohey Co, Fairfield NJ) via a switch. The puff pipette was placed within 2 μm of a neurite, 10–12 μm from the hillock or dendritic origin. A control complex spike was followed 2s later by a complex spike paired with a puff (2 psi, 50–100 ms duration, offset 10 ms before somatic current injection). These pairs of complex spikes were evoked every 45s. Comparisons were made between pairs, and then averaged across trials (>10 trails/condition). Spike timing was defined as the time of the peak of the action potential, and threshold Vm was defined as the maximum of the 3rd derivative before action potential peak (Henze and Buzsaki, 2001). Spike threshold was not calculated for the 3rd spikelet because it often lacked a clear threshold inflection.
Local Ni2+ iontophoresis
Internal solution was the same as in Ca2+ imaging experiments. KH2PO4 was removed from the Ringer to prevent Ni3(PO4)2 precipitation (KCl was increased by 1.2 mM). A borosilicate pipette (15 MΩ tip resistance, −4 to −6 nA backing current) containing 1 M NiCl2 and 50 μM Alexa 594 in water was placed close to the axon, 10 μm from the hillock. Ni2+ iontophoretic pulses were paired with somatic depolarization (100–200 nA, 25–40 ms, offset 5–10 ms before somatic depolarization) and interleaved with control pulses as described above. Ni2+ pulses had no effect on baseline G/R levels. In some experiments, an EPSP-like current injection based on the kinetics of parallel fiber-evoked EPSPs was used as described previously (Roberts et al., 2008) according to the function:
where Amp was the peak EPSP amplitude (0.5–5 nA), τrise = 0.3 ms, and τdecay = 1.59 ms. Ni2+-induced changes in subthreshold depolarizations were considered significant when the difference between control and Ni2+-paired stimuli (Ctrl-Ni2+) was greater than 2 × standard deviation of the baseline voltage, 0–5 ms before stimulus onset.
Pyramidal and Purkinje cell experiments
For pyramidal cell experiments, oblique slices (300 μm thick, P17-18 ICR mice) containing one barrel in layer 4 from each whisker row were made through primary somatosensory cortex, 50° from the midsagittal plane (“across row” slices, Bender et al., 2003). For Purkinje cell experiments, parasagittal slices containing cerebellar vermi were made (300 μm thick, P14 ICR mouse). Chilled cutting/storage solution contained (in mM): 87 NaCl, 25 NaHCO3, 25 glucose, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2. Following cutting, slices were incubated for 30 min at 32 °C, then at room temperature until use. Layer 5b pyramidal cells within the whisker barrel subfield were identified based on their morphology, ability to fire spike bursts, and laminar position. For cortical experiments, DNQX, but not strychnine or SR-95531, was added to the bath. All other procedures were as in cartwheel cell experiments.
Chemicals
CoroNa green, Fluo-5F pentapotassium salt, X-rhod-5F tripotassium salt, and Alexa Fluor 594 hydrazide Na+ salt were from Invitrogen (Carlsbad, CA). TTX, strychnine, SR95531, and DNQX were from Ascent Scientific (Weston-Super-Mare, UK). ω-agatoxin-TK, ω-conotoxin-GVIA, apamin, iberiotoxin, and SNX-482 were from Peptides International (Louisville, KY). All others were from Sigma-Aldrich (St. Louis, MO).
Statistics
All data are shown as mean ± standard error (SEM). Statistical tests are noted in text (significance: α < 0.05). ANOVAs were followed with a Fisher’s PLSD post-hoc test.
Supplementary Material
Acknowledgments
We are grateful to D. Chiu, J. Christie, M. Herman, Y. Kim, S. Kuo, C. Jahr, and A. Reigel for technical assistance, and to V. Bender, I. Raman, and M. Roberts for extensive discussions and comments on this manuscript. GIN mice were provided by Sascha du Lac. Supported by NIH grants DC004450 and NS028901 (LOT), NIH training grants NS007381 and DK007680, and a Tartar Trust fellowship (KJB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizenman CD, Linden DJ. Rapid, synaptically driven increases in the intrinsic excitability of cerebellar deep nuclear neurons. Nat Neurosci. 2000;3:109–111. doi: 10.1038/72049. [DOI] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Bender KJ, Rangel J, Feldman DE. Development of columnar topography in the excitatory layer 4 to layer 2/3 projection in rat barrel cortex. J Neurosci. 2003;23:8759–8770. doi: 10.1523/JNEUROSCI.23-25-08759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, et al. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006;314:615–620. doi: 10.1126/science.1132915. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Geiger JR, Jonas P. Timing and efficacy of Ca2+ channel activation in hippocampal mossy fiber boutons. J Neurosci. 2002;22:10593–10602. doi: 10.1523/JNEUROSCI.22-24-10593.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci. 2003;23:2306–2313. doi: 10.1523/JNEUROSCI.23-06-02306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG. Reliability and heterogeneity of calcium signaling at single presynaptic boutons of cerebellar granule cells. J Neurosci. 2007;27:7888–7898. doi: 10.1523/JNEUROSCI.1064-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaert G, Eilers J, Konnerth A. Axonal calcium entry during fast ‘sodium’ action potentials in rat cerebellar Purkinje neurones. J Physiol. 1996;495(Pt 3):641–647. doi: 10.1113/jphysiol.1996.sp021622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Localization of sodium channels in cultured neural cells. J Neurosci. 1981;1:777–783. doi: 10.1523/JNEUROSCI.01-07-00777.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- Cavelier P, Lohof AM, Lonchamp E, Beekenkamp H, Mariani J, Bossu JL. Participation of low-threshold Ca2+ spike in the Purkinje cells complex spike. Neuroreport. 2008;19:299–303. doi: 10.1097/WNR.0b013e3282f4cb96. [DOI] [PubMed] [Google Scholar]
- Coombs JS, Curtis DR, Eccles JC. The interpretation of spike potentials of motoneurones. J Physiol. 1957;139:198–231. doi: 10.1113/jphysiol.1957.sp005887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueni L, Canepari M, Lujan R, Emmenegger Y, Watanabe M, Bond CT, Franken P, Adelman JP, Luthi A. T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat Neurosci. 2008;11:683–692. doi: 10.1038/nn.2124. [DOI] [PubMed] [Google Scholar]
- Davie JT, Clark BA, Hausser M. The origin of the complex spike in cerebellar Purkinje cells. J Neurosci. 2008;28:7599–7609. doi: 10.1523/JNEUROSCI.0559-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KA, Young ED. Granule cell activation of complex-spiking neurons in dorsal cochlear nucleus. J Neurosci. 1997;17:6798–6806. doi: 10.1523/JNEUROSCI.17-17-06798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day NC, Shaw PJ, McCormack AL, Craig PJ, Smith W, Beattie R, Williams TL, Ellis SB, Ince PG, Harpold MM, et al. Distribution of alpha 1A, alpha 1B and alpha 1E voltage-dependent calcium channel subunits in the human hippocampus and parahippocampal gyrus. Neuroscience. 1996;71:1013–1024. doi: 10.1016/0306-4522(95)00514-5. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Neubig M, Ulrich D, Huguenard J. Dendritic low-threshold calcium currents in thalamic relay cells. J Neurosci. 1998;18:3574–3588. doi: 10.1523/JNEUROSCI.18-10-03574.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt HU, Eder M, Schierloh A, Zieglgansberger W. Infrared-guided laser stimulation of neurons in brain slices. Sci STKE. 2002;2002:PL2. doi: 10.1126/stke.2002.120.pl2. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Jonas P. Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58:387–400. doi: 10.1016/j.neuron.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JH, Lacefield CO, Yuste R. Global dendritic calcium spikes in mouse layer 5 low threshold spiking interneurones: implications for control of pyramidal cell bursting. J Physiol. 2004;558:465–478. doi: 10.1113/jphysiol.2004.064519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Hirase H, Leinekugel X, Henze DA, Buzsaki G. Temporal interaction between single spikes and complex spike bursts in hippocampal pyramidal cells. Neuron. 2001;32:141–149. doi: 10.1016/s0896-6273(01)00447-0. [DOI] [PubMed] [Google Scholar]
- Henze DA, Buzsaki G. Action potential threshold of hippocampal pyramidal cells in vivo is increased by recent spiking activity. Neuroscience. 2001;105:121–130. doi: 10.1016/s0306-4522(01)00167-1. [DOI] [PubMed] [Google Scholar]
- Herzog RI, Liu C, Waxman SG, Cummins TR. Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J Neurosci. 2003;23:8261–8270. doi: 10.1523/JNEUROSCI.23-23-08261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa BM, Letzkus JJ, Stuart GJ. Requirement of dendritic calcium spikes for induction of spike-timing-dependent synaptic plasticity. J Physiol. 2006;574:283–290. doi: 10.1113/jphysiol.2006.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa BM, Stuart GJ. Calcium spikes in basal dendrites of layer 5 pyramidal neurons during action potential bursts. J Neurosci. 2006;26:7424–7432. doi: 10.1523/JNEUROSCI.3062-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci. 2006;26:1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Trussell LO. Ion channels generating complex spikes in cartwheel cells of the dorsal cochlear nucleus. J Neurophysiol. 2007;97:1705–1725. doi: 10.1152/jn.00536.2006. [DOI] [PubMed] [Google Scholar]
- Koester HJ, Johnston D. Target cell-dependent normalization of transmitter release at neocortical synapses. Science. 2005;308:863–866. doi: 10.1126/science.1100815. [DOI] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics associated with action potentials in single nerve terminals of pyramidal cells in layer 2/3 of the young rat neocortex. J Physiol 529 Pt. 2000;3:625–646. doi: 10.1111/j.1469-7793.2000.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ. Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci. 2008;11:178–186. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- Kole MH, Letzkus JJ, Stuart GJ. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron. 2007;55:633–647. doi: 10.1016/j.neuron.2007.07.031. [DOI] [PubMed] [Google Scholar]
- Kole MH, Stuart GJ. Is action potential threshold lowest in the axon? Nat Neurosci. 2008 doi: 10.1038/nn.2203. [DOI] [PubMed] [Google Scholar]
- Krahe R, Gabbiani F. Burst firing in sensory systems. Nat Rev Neurosci. 2004;5:13–23. doi: 10.1038/nrn1296. [DOI] [PubMed] [Google Scholar]
- Lasser-Ross N, Ross WN. Imaging voltage and synaptically activated sodium transients in cerebellar Purkinje cells. Proc Biol Sci. 1992;247:35–39. doi: 10.1098/rspb.1992.0006. [DOI] [PubMed] [Google Scholar]
- Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci. 1999;19:1912–1921. doi: 10.1523/JNEUROSCI.19-06-01912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- Luscher C, Lipp P, Luscher HR, Niggli E. Control of action potential propagation by intracellular Ca2+ in cultured rat dorsal root ganglion cells. J Physiol. 1996;490(Pt 2):319–324. doi: 10.1113/jphysiol.1996.sp021146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol. 1995;487(Pt 1):67–90. doi: 10.1113/jphysiol.1995.sp020862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Joerges J, Huguenard JR, Sejnowski TJ. A model of spike initiation in neocortical pyramidal neurons. Neuron. 1995;15:1427–1439. doi: 10.1016/0896-6273(95)90020-9. [DOI] [PubMed] [Google Scholar]
- Martina M, Vida I, Jonas P. Distal initiation and active propagation of action potentials in interneuron dendrites. Science. 2000;287:295–300. doi: 10.1126/science.287.5451.295. [DOI] [PubMed] [Google Scholar]
- McDonough SI, Bean BP. Mibefradil inhibition of T-type calcium channels in cerebellar purkinje neurons. Mol Pharmacol. 1998;54:1080–1087. doi: 10.1124/mol.54.6.1080. [DOI] [PubMed] [Google Scholar]
- McKay BE, McRory JE, Molineux ML, Hamid J, Snutch TP, Zamponi GW, Turner RW. Ca(V)3 T-type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur J Neurosci. 2006;24:2581–2594. doi: 10.1111/j.1460-9568.2006.05136.x. [DOI] [PubMed] [Google Scholar]
- Meier SD, Kovalchuk Y, Rose CR. Properties of the new fluorescent Na+ indicator CoroNa Green: comparison with SBFI and confocal Na+ imaging. J Neurosci Methods. 2006;155:251–259. doi: 10.1016/j.jneumeth.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Metz AE, Jarsky T, Martina M, Spruston N. R-type calcium channels contribute to after depolarization and bursting in hippocampal CA1 pyramidal neurons. J Neurosci. 2005;25:5763–5773. doi: 10.1523/JNEUROSCI.0624-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitor SC, Manis PB. Dendritic Ca2+ transients evoked by action potentials in rat dorsal cochlear nucleus pyramidal and cartwheel neurons. J Neurophysiol. 2003;89:2225–2237. doi: 10.1152/jn.00709.2002. [DOI] [PubMed] [Google Scholar]
- Oertel D, Young ED. What’s a cerebellar circuit doing in the auditory system? Trends Neurosci. 2004;27:104–110. doi: 10.1016/j.tins.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Jiang M, Lam T, Smith KL, Swann JW. Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J Neurosci. 2000;20:3354–3368. doi: 10.1523/JNEUROSCI.20-09-03354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palay SL, Sotelo C, Peters A, Orkand PM. The axon hillock and the initial segment. J Cell Biol. 1968;38:193–201. doi: 10.1083/jcb.38.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Stuart GJ. Site of action potential initiation in layer 5 pyramidal neurons. J Neurosci. 2006;26:1854–1863. doi: 10.1523/JNEUROSCI.4812-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Roberts MT, Bender KJ, Trussell LO. Fidelity of complex spike-mediated synaptic transmission between inhibitory interneurons. J Neurosci. 2008;28:9440–9450. doi: 10.1523/JNEUROSCI.2226-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Helmchen F, Sakmann B. Spatial profile of dendritic calcium transients evoked by action potentials in rat neocortical pyramidal neurones. J Physiol. 1995;487(Pt 3):583–600. doi: 10.1113/jphysiol.1995.sp020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmolesky MT, Weber JT, De Zeeuw CI, Hansel C. The making of a complex spike: ionic composition and plasticity. Ann N Y Acad Sci. 2002;978:359–390. doi: 10.1111/j.1749-6632.2002.tb07581.x. [DOI] [PubMed] [Google Scholar]
- Shu Y, Duque A, Yu Y, Haider B, McCormick DA. Properties of action-potential initiation in neocortical pyramidal cells: evidence from whole cell axon recordings. J Neurophysiol. 2007;97:746–760. doi: 10.1152/jn.00922.2006. [DOI] [PubMed] [Google Scholar]
- Stuart G, Schiller J, Sakmann B. Action potential initiation and propagation in rat neocortical pyramidal neurons. J Physiol. 1997;505(Pt 3):617–632. doi: 10.1111/j.1469-7793.1997.617ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimori M, Llinas RR. Real-time imaging of calcium influx in mammalian cerebellar Purkinje cells in vitro. Proc Natl Acad Sci U S A. 1990;87:5084–5088. doi: 10.1073/pnas.87.13.5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottene A, Volsen S, Pietrobon D. alpha(1E) subunits form the pore of three cerebellar R-type calcium channels with different pharmacological and permeation properties. J Neurosci. 2000;20:171–178. doi: 10.1523/JNEUROSCI.20-01-00171.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO. Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci. 2004;7:719–725. doi: 10.1038/nn1272. [DOI] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Mechanisms and consequences of action potential burst firing in rat neocortical pyramidal neurons. J Physiol . 1999;521(Pt 2):467–482. doi: 10.1111/j.1469-7793.1999.00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfart J, Roeper J. Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci. 2002;22:3404–3413. doi: 10.1523/JNEUROSCI.22-09-03404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollner DA, Catterall WA. Localization of sodium channels in axon hillocks and initial segments of retinal ganglion cells. Proc Natl Acad Sci U S A. 1986;83:8424–8428. doi: 10.1073/pnas.83.21.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouterlood FG, Mugnaini E. Cartwheel neurons of the dorsal cochlear nucleus: a Golgi-electron microscopic study in rat. J Comp Neurol. 1984;227:136–157. doi: 10.1002/cne.902270114. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE. 2004;2004:pl5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]
- Zhang S, Oertel D. Cartwheel and superficial stellate cells of the dorsal cochlear nucleus of mice: intracellular recordings in slices. J Neurophysiol. 1993;69:1384–1397. doi: 10.1152/jn.1993.69.5.1384. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.